

Graphical abstract

Keywords: Ribonuclease, Deadenylase, Caf1/CNOT7, PARN, Mg2+ dependent nuclease
Abstract
Eukaryotic mRNA contains a 3′ poly(A) tail, which plays important roles in the regulation of mRNA stability and translation. Well-characterized enzymes involved in the shortening of the poly(A) tail include the multi-subunit Ccr4-Not deadenylase, which contains the Caf1 (Pop2) and Ccr4 catalytic components, and poly(A)-specific ribonuclease (PARN). Two Mg2+ ions present in the active sites of these ribonucleases are required for RNA cleavage. Here, we report the discovery, synthesis and biochemical profiling of purine-2,6-dione derivatives as (sub)micromolar inhibitors of Caf1.
In eukaryotic cells, cytoplasmic mRNA is characterized by the presence of a 3′ poly(A) tail. The median length of the tail varies from 27 to 28 nucleotides in yeast to 60–100 nucleotides in mammalian cells.1,2 The tail is important for the control of gene expression: enzymatic shortening of the poly(A) tail (deadenylation) can initiate mRNA degradation and repress translation.3 An important enzyme involved in cytoplasmic deadenylation is the multi-component Ccr4-Not complex.4,5 In addition to six non-catalytic subunits, the complex contains two subunits with ribonuclease activity: both Caf1 and Ccr4 display Mg2+-dependent 3′–5′ exo-ribonuclease activity with a preference for poly(A). However, whereas the enzymatic activity of Caf1 is associated with an RNAse D/DEDD (Asp-Glu-Asp-Asp) domain,6–8 the enzymatic activity of Ccr4 is provided by an EEP (endonuclease-exonuclease-phosphatase) domain.9,10
The analysis of genetically modified mice has identified the importance of Ccr4-Not subunits in the regulation of physiological functions such as bone formation and male fertility11,12 as well as obesity and heart disease.13,14 Moreover, mutations in CNOT3 are frequently identified in acute lymphoblastic leukemia.15 Currently, it is unclear whether the ribonuclease activities of the Ccr4-Not complex are involved in the regulation of these processes and to what extent the Caf1 and Ccr4 subunits have unique roles, or cooperate in deadenylation.16–18 Because the structural complexity of the Ccr4-Not deadenylase is a significant barrier to distinguishing between catalytic and structural roles of the complex, additional tools to study Ccr4-Not function are required. In particular, cell-permeable small-molecule inhibitors that selectively inhibit the enzyme activities of Caf1 or Ccr4 are desirable as a complementary approach to RNAi and genetic techniques. Such molecules will be highly useful as pharmacological tools to study the involvement of the catalytic activity of Ccr4-Not in physiological processes, and contribute to the evaluation of this complex as a potential therapeutic target.
While nucleoside analogues have been reported as inhibitors of the poly(A)-specific ribonuclease PARN, whose DEDD-type nuclease domain is closely related to that of Caf1,19,20 non-nucleoside inhibitors have only been reported for a limited number of Mg2+-dependent ribonucleases. These include influenza RNA endonuclease (required for viral transcription) and human immunodeficiency virus (HIV) RNAse H (an RNA endonuclease involved in the degradation of RNA strands of RNA:DNA hybrids).21,22 In addition, we recently reported the discovery of non-nucleoside inhibitors of Caf1, which were identified by screening a compound library.22 Here, we describe an alternative approach, based on the discovery of N-hydroxyimide compounds as inhibitors of HIV RNAse H and human flap endonuclease FEN1, a structure-specific DNA endonuclease.23,24 It was suggested that these compounds inhibited enzyme activity by coordination of the two divalent metal ions required for catalysis (Fig. 1A). Interestingly, N-hydroxyimides were also identified as inhibitors of the influenza RNA endonuclease PA.21 Based on the distance between the Mg2+ ions in the active site of the Caf1 ribonuclease (3.9–4.0 Å; Fig. 2a and b),25 we hypothesized that compounds containing this moiety may adopt a similar binding mode in the active site of Caf1, thereby blocking substrate binding.
Figure 1.

Discovery of 3-hydroxy-pyrimidine-2,4-dione compounds as inhibitors of the Caf1 ribonuclease. (A) Possible interaction modes of 3-hydroxy-pyrimidine-2,4-dione compounds with two divalent metal ions in the active site of Caf1. (B) Structure and activity of compounds selected from the Open Chemical Repository Collection (NCI, Bethesda). IC50 values refer to inhibition of Caf1. Also indicated is the standard error of the mean (n = 3).
Figure 2.

Molecular modelling of inhibitors in the active site of Caf1. Complex crystal structures of human Caf1 (a; PDB 4GMJ.B)25 and PARN (b; PDB 2A1R)30 show that the cognate poly(A) ligand (stick models) likely binds in a similar manner to the active site (protein surface with Mg2+ ions as green spheres), despite the fact that the active sites in Caf1 and PARN have somewhat different shapes. In (c) it can be seen that the top-ranking poses of the most potent compounds 5j (magenta), 8e (cyan), and 8j (green) adopt very similar conformations upon docking to the 4GMJ.B model, whereas several poses with similar scores were recorded for lead compound 5a (yellow), whereof only some (one shown) were similar to the bound conformations of the more potent compounds. Probable polar binding contacts of the most potent compound 8j with the Caf1 active site include a cation–π interaction between one of the Mg2+ ions and the phenyl ring of the inhibitor, as well as H-bonds and polar interactions involving the 3° amine and one of the carbonyl O and the OH of the N-hydroxyimide group as indicated (d; dashed lines). Furthermore, 8j makes numerous van-der-Waals contacts with residues (labelled in d) of the active site cavity.
We therefore searched the Open Chemical Repository Collection, a diverse set of more than 200,000 compounds (Developmental Therapeutics Program, National Cancer Institute, Bethesda), for the presence of N-hydroxyimides. To limit the number of compounds, we focused on 3-hydroxypyrimidine-2,4-diones and obtained seven compounds for further analysis. Following LC–MS and solubility analysis, five compounds were evaluated for their ability to inhibit Caf1 activity using a fluorescence-based biochemical assay (Fig. 1B).22 Interestingly, whereas compound NSC-86353 was found to inhibit Caf1 (IC50 = 22.8 ± 0.1 μM), substitution of the benzyl moiety with a methyl group at the N7 position (NSC-85703) reduced the inhibitory activity >10-fold.
Based on this observation, we first explored the activity of 7-substituted 1-hydroxy-purine-2,6-diones 5 further. The synthesis of these compounds was accomplished by amination (with amines 1) of ethyl 2-bromoacetate to provide the N-substituted ethyl 2-aminoacetate intermediates 2 (Scheme 1). These were N-alkylated with (ethoxymethylene)cyanamide26 and the resulting cyanoformamidine adducts were cyclised in situ27 with KOBut to the 3-subsituted ethyl 5-aminoimidazole-4-carboxylates 3. The 1-allyloxy-purinedione system of the bicyclic compounds 4 was then formed by cyclisation of intermediates 3 with the aid of carbonyldiimidazole (CDI) and O-allylhydroxylamine.28 The target compounds 5 were obtained following reductive deprotection of precursors 4.29
Scheme 1.

Synthesis of 7-substituted 1-hydroxy-3,7-dihydro-1H-purine-2,6-diones. Reagents and conditions: (a) ethyl 2-bromoacetate, CHCl3, rt, 2 h (37–79%); (b) (ethoxymethylene)cyanamide, THF, Δ, 16 h, then KOBut, EtOH, Δ, 2 h (46–80% over 2 steps); (c) CDI, PhMe, Δ, 2 h, then O-allylhydroxylamine, aq NaOH, EtOH, Δ, 2 h (21–61% over 2 steps); (d) Pd(OAc)2, PPh3, HCOOH, EtOH–H2O (8:2), 80 °C, 2 h (23–55%); (e) CDI, PhMe, Δ, 2 h, then O-benzylhydroxylamine, aq NaOH, EtOH, Δ, 2 h (74% over 2 steps); (f) R2X (X = Br or I), K2CO3, DMF, 80 °C, 2–12 h, (67–98%); (g) H2, 10% (w/w) Pd(C), CH2Cl2–MeOH (1:9) (33–53%); (h) urea, 2-methoxyethanol, 190 °C, 24 h, then aq NaOH, Δ, 3 h (33%). For definitions of R1 and R2 refer to Tables 1–3.
Starting with 7-benzylpurinedione 5a (Table 1), we hypothesised that the N7-benzyl substituent binds into the hydrophobic Caf1 active site pocket delineated by residues Phe156, Leu209, and His225 (Fig. 2c and d).31–33 Based on this hypothesis, we introduced comparatively bulky lipophilic groups at N7. Replacement of the benzyl with a cyclohexyl group (5b) reduced activity nearly 3-fold, in line with what we observed upon substituting the benzyl with a simple methyl group (see above). More conservative replacements of the benzyl with pyridylmethyl groups afforded active compounds, especially for the 2- and 3-pyridylmethyl analogues 5c and 5d, whereas the 4-pyridylmethyl isomer 5e was less active. Lengthening of the alkyl linker between the phenyl group and N7 from CH2 (5a) to (CH2)2 (5j) and (CH2)3 (5f) showed maximal activity for the phenethyl derivative 5j. This observation is in line with the modelled Caf1 binding poses of compounds containing the phenethyl group (Fig. 2c). These poses suggest an optimal cation–π interaction between one of the Mg2+ ions and the phenyl ring in the case of a 2-carbon linker, while maintaining hydrophobic interactions of the phenyl ring with Leu209. Similar dual interactions with the Caf1 active site are not possible with 1- and 3-carbon linkers between the phenyl ring and N7. Replacement of the benzyl group with a long-chain lipophilic substituent (5g) was not productive. On the other hand, insertion of an O atom into the linker (5i), or replacement of the phenyl group in 5j with a 2-thienyl (5h) or 2-pyridyl (5k) group, retained most of the activity.
Table 1.
Activity of 7-substituted 1-hydroxy-3,7-dihydro-1H-purine-2,6-diones 5
| Cmpd | 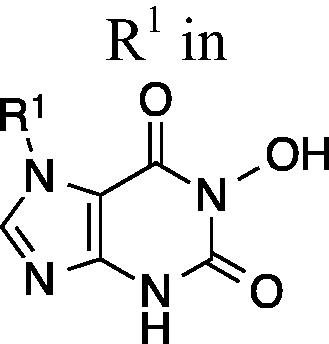 |
IC50 (μM)a (Caf1) | IC50 (μM)a (PARN) |
|---|---|---|---|
| 5a | 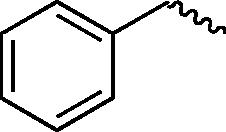 |
10.4 ± 0.4 | 84.1 ± 6.7 |
| 5b | 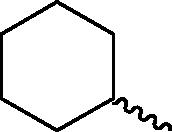 |
28.2 ± 7.6 | n.d. |
| 5c | 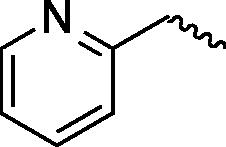 |
10.6 ± 2.7 | 119 ± 25 |
| 5d | 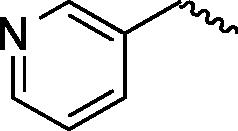 |
6.6 ± 0.7 | 125 ± 32 |
| 5e | 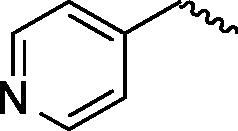 |
23.3 ± 2.2 | 245 ± 20 |
| 5f | 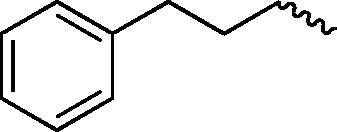 |
13.3 ± 2.3 | n.d. |
| 5g | 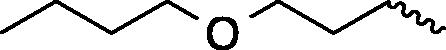 |
20.6 ± 5.8 | n.d. |
| 5h | 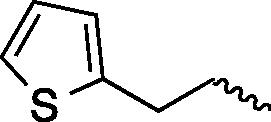 |
3.6 ± 1.1 | n.d. |
| 5i | 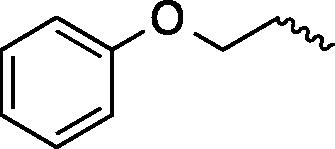 |
10.6 ± 4.3 | n.d. |
| 5j | 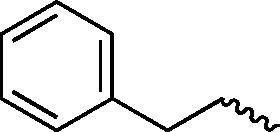 |
1.5 ± 0.3 | n.d. |
| 5k | 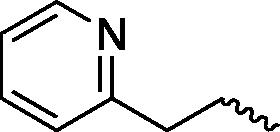 |
4.0 ± 1.1 | n.d. |
IC50 values were determined using a fluorescence-based biochemical assay as described.22 Also indicated are the standard errors of the means (n = 3). N.d., not determined.
We next turned our attention to the purine N3-substituent in the context of the optimal 7-phenethyl group (Table 2). Here we envisaged that a comparatively bulky substituent may make favourable contacts with the Caf1 active site pocket lined by residues Phe43, Pro44, Ser112, and Leu116 (Fig. 2c and d). The 3,7-disubstituted 1-hydroxypurine-2,6-diones 8 were prepared from the O-benzyl precursor 6, obtained by cyclisation of imidazole 3 (R1 = (CH2)2Ph) with O-benzylhydroxylamine,34 by successive alkylation with appropriate alkyl halides and hydrogenolysis of products 7 (Scheme 1). Introduction of a methyl (8a), ethyl (8c), n-pentyl (8d), isohexyl (8e), n-nonyl (8f), n-decyl (8g), or n-dodecyl group (8h) at N3 showed optimal activity for the isohexyl congener 8e, although its activity was not improved with respect to the parent compound 5j. However, replacement of the isohexyl group with the 1-(N,N-dimethylamino)prop-3-yl group (8j) resulted in a further affinity enhancement and afforded the most potent compound in our series with activity at the submicromolar level.35 Again this potency gain can be rationalised based on docking studies, which suggest that the protonated (at physiological pH) 3° amine group in 8j is H-bonded to the amide carbonyl O of Pro44, while maintaining most of the hydrophobic interactions of the N3-substituent compared with the isohexyl group in 8e (Fig. 2d).
Table 2.
Activity of 3-substituted-1-hydroxy-7-phenethyl-3,7-dihydro-1H-purine-2,6-diones 8
| Cmpd | 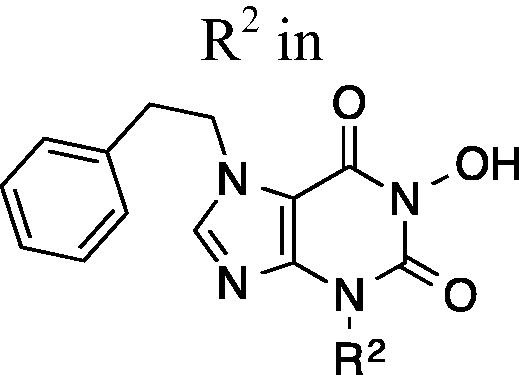 |
IC50a (μM) (Caf1) | IC50a (μM) (PARN) |
|---|---|---|---|
| 8a | CH3 | 9.3 ± 1.7 | n.d. |
| 8b | CH2(CH2)2Ph | 2.1 ± 0.3 | n.d. |
| 8c | CH2CH3 | 12.2 ± 3.5 | n.d. |
| 8d | CH2(CH2)3CH3 | 4.8 ± 1.2 | n.d. |
| 8e | 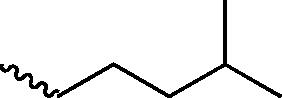 |
1.7 ± 0.4 | n.d. |
| 8f | CH2(CH2)7CH3 | 9.9 ± 3.6 | n.d. |
| 8g | CH2(CH2)8CH3 | 20.7 ± 6.7 | n.d. |
| 8h | CH2(CH2)10CH3 | 29.5 ± 22.4 | n.d. |
| 8i | CH2Ph | 14.5 ± 0.9 | n.d. |
| 8j | 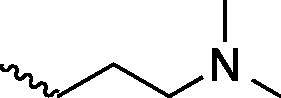 |
0.59 ± 0.11 | 23.9 ± 3.7 |
| 8k | 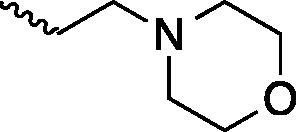 |
8.7 ± 0.3 | n.d. |
IC50 values were determined using a fluorescence-based biochemical assay as described.22 Also indicated are the standard errors of the means (n = 3). N.d., not determined.
According to our binding hypothesis (Fig. 2c) the N-hydroxyimide function, especially the C6- and N-linked O atoms, present in all compound of the series discussed thus far, is important for polar interactions with the Mg2+ ions coordinated in the active site of Caf1. Additionally, we propose that the N-hydroxyl function H-bonds to the side-chain carboxyl of Asp61. To probe this hypothesis we tested compounds containing modified N-hydroxyimide substructures (Table 3). To this end, a purine-2,6-dione derivative (9) lacking the N-hydroxy group of compounds 5 and 8 was obtained from intermediate 3 (R1 = Bn) by cyclisation with urea under basic conditions (Scheme 1).34 6-Hydroxy-1-phenethyl-1,4-dihydro-5H-imidazo[4,5-b]pyridin-5-one 13 was obtained from the nitroimidazole precursor 10 (Scheme 2).36 A dichloromethyl group was introduced into this compound at the 5-position using a vicarious nucleophilic procedure with chloroform and KOBut, followed by hydrolysis of the dichloromethyl group to the aldehyde and reduction of the nitro function to the aminoaldehyde 11.37 This intermediate was then cyclised with ethyl allyloxyacetate to the imadazopyridinone 12,38 which was deprotected to afford target compound 13. Blocking of the N-hydroxyl function with an allyl group in the synthesis intermediate 4a, as well as removing the N-hydroxyl group in 9, abolished activity completely, as expected. Furthermore, omission of the C6-carbonyl oxygen in the hydroxy-imidazopyridinone 13 reduced activity significantly, pointing to an important contribution of this carbonyl group to binding, as suggested by our docking studies.
Table 3.
Activity of 7-substituted 1-hydroxy-3,7-dihydro-1H-purine-2,6-diones with modified N-hydroxyimide function
| Cmpd | Structure | IC50a (μM) (Caf1) |
|---|---|---|
| 4a | 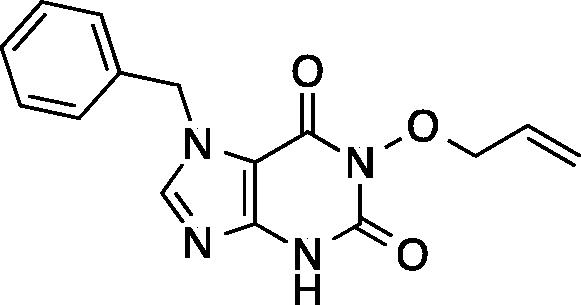 |
>1000 |
| 9 | 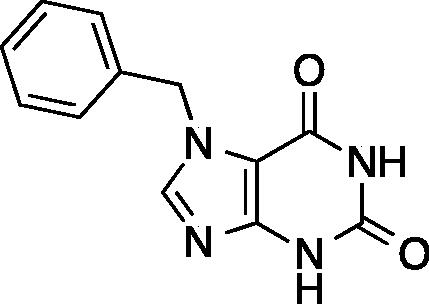 |
>1000 |
| 13 | 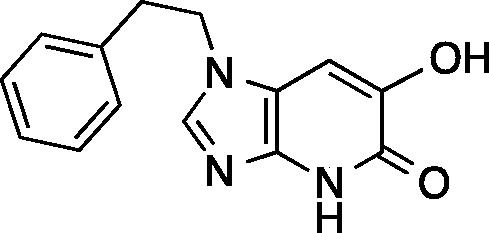 |
15.1 ± 0.3 |
IC50 values were determined using a fluorescence-based biochemical assay as described.22 Also indicated are the standard errors of the means (n = 3).
Scheme 2.

Synthesis of 6-hydroxy-1-phenethyl-1,4-dihydro-5H-imidazo[4,5-b]pyridin-5-one 13. Reagents and conditions: (a) CHCl3, KOBut, THF–DMF (2:1), −100 °C, 15 min, then CaCO3, H2O, 75 °C, 48 h, then H2, 10% (w/w) Pd(C), EtOAc–MeOH–AcOH (18:1:1), 55 psi, 18 h (60% over 3 steps); (b) ethyl allyloxyacetate, (Me3Si)2NLi, THF, −78 °C to rt over 16 h (36%); (c) Pd(OAc)2, PPh3, HCOOH, EtOH–H2O (8:2), 80 °C, 2 h (33%).
The selectivity of the compounds was determined by comparing their activities against Caf1 and two related deadenylase enzymes: the PARN ribonuclease, which has a conserved nuclease domain similar to that of Caf1, and Ccr4, whose biological function is related to Caf1, but which contains a dissimilar EEP-type nuclease domain.22 Compounds with N7 substituents containing an aromatic group linked via a single carbon spacer (5a and 5c–e) selectively inhibited Caf1 (Fig. 3; Table 1). By contrast, the more potent compounds 5h, 5j and 5k with N7 substituents containing an aromatic group linked via a two-carbon spacer facilitating optimal cation–π interactions displayed reduced selectivity and also inhibited the related deadenylase PARN. Selectivity was not improved by the introduction of N3 allyl substituents (8a–8f). However, the most potent compound 8j containing an N3-1-(N,N-dimethylamino)prop-3-yl group displayed reduced activity versus PARN, suggesting that substituents in this position can contribute to selectivity (Table 2). None of the compounds tested displayed activity towards the EEP-type deadenylase Ccr4.
Figure 3.

Inhibition of the ribonuclease activities of (A) Caf1, (B) PARN and (C) Ccr4 by 3,7-N-substituted-1-hydroxy-3,7-dihydro-1H-purine-2,6-diones. Reactions contained no enzyme (control), complete reactions (enzyme) or complete reactions in the presence of the indicated compound. Compounds were used at 30 μM (5a–e, 8a–d, 8f), 10 μM (5h–k), or 3 μM (8e, 8j). Error bars represent the standard deviation.
The molecules described here are structurally unrelated to the inhibitors of Caf1 that we described before and which were identified using a combined approach involving virtual and compound library screening.22 Compound 8j is significantly more potent (>20-fold) than all previously identified compounds, although the potency of the lead compound NSC-86353 (5a) is comparable to that of NCC-00037292 (IC50 = 14.6 ± 3.1 μM), which was reported previously.22 To the best of our knowledge, no other inhibitors of the Caf1 deadenylase have been described to date.
In summary, we discovered purine-2,6-dione derivatives as inhibitors of the ribonucleases Caf1 and PARN. Compound 8j was the most potent compound with a >10-fold increased activity as compared to compound 5a, which was identified as the initial hit compound. In addition to polar interactions involving the N-hydroxyimide moiety, we propose that a cation–π interaction contributes to coordination of the Mg2+ ions. We conclude that these compounds will be useful for the biochemical analysis of deadenylase enzymes and contribute to the design of improved inhibitors of the Caf1 and PARN ribonucleases with increased potency and selectivity.
Acknowledgements
This work was supported by the Medical Research Council (grant G1100205), the Islamic Development Bank (scholarship to MM), and the Schlumberger Foundation (Faculty for the Future award to BA).
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmcl.2015.07.095. These data include MOL files and InChiKeys of the most important compounds described in this article.
Contributor Information
Peter M. Fischer, Email: peter.fischer@nottingham.ac.uk.
G. Sebastiaan Winkler, Email: sebastiaan.winkler@nottingham.ac.uk.
Supplementary data
The following ZIP file contains the MOL files of the most important compounds referred to in this article.
ZIP file containing the MOL files of the most important compounds in this article.
References and notes
- 1.Chang H., Lim J., Ha M., Kim V.N. Mol Cell. 2014;53:1044. doi: 10.1016/j.molcel.2014.02.007. [DOI] [PubMed] [Google Scholar]
- 2.Subtelny A.O., Eichhorn S.W., Chen G.R., Sive H., Bartel D.P. Nature. 2014;508:66. doi: 10.1038/nature13007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goldstrohm A.C., Wickens M. Nat Rev Mol Cell Biol. 2008;9:337. doi: 10.1038/nrm2370. [DOI] [PubMed] [Google Scholar]
- 4.Collart M.A., Panasenko O.O. Gene. 2012;492:42. doi: 10.1016/j.gene.2011.09.033. [DOI] [PubMed] [Google Scholar]
- 5.Wahle E., Winkler G.S. Biochim Biophys Acta. 2013;1829:561. doi: 10.1016/j.bbagrm.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 6.Moser M.J., Holley W.R., Chatterjee A., Mian I.S. Nucleic Acids Res. 1997;25:5110. doi: 10.1093/nar/25.24.5110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Daugeron M.-C., Mauxion F., Seraphin B. Nucleic Acids Res. 2001;29:2448. doi: 10.1093/nar/29.12.2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tucker M., Valencia-Sanchez M.A., Staples R.R., Chen J., Denis C.L., Parker R. Cell. 2001;104:377. doi: 10.1016/s0092-8674(01)00225-2. [DOI] [PubMed] [Google Scholar]
- 9.Chen J., Chiang Y.C., Denis C.L. EMBO J. 2002;21:1414. doi: 10.1093/emboj/21.6.1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tucker M., Staples R.R., Valencia-Sanchez M.A., Muhlrad D., Parker R. EMBO J. 2002;21:1427. doi: 10.1093/emboj/21.6.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berthet C., Morera A.M., Asensio M.J., Chauvin M.A., Morel A.P., Dijoud F., Magaud J.P., Durand P., Rouault J.P. Mol Cell Biol. 2004;24:5808. doi: 10.1128/MCB.24.13.5808-5820.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakamura T., Yao R., Ogawa T., Suzuki T., Ito C., Tsunekawa N., Inoue K., Ajima R., Miyasaka T., Yoshida Y., Ogura A., Toshimori K., Noce T., Yamamoto T., Noda T. Nat Genet. 2004;36:528. doi: 10.1038/ng1344. [DOI] [PubMed] [Google Scholar]
- 13.Morita M., Oike Y., Nagashima T., Kadomatsu T., Tabata M., Suzuki T., Nakamura T., Yoshida N., Okada M., Yamamoto T. EMBO J. 2011;30:4678. doi: 10.1038/emboj.2011.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neely G.G., Kuba K., Cammarato A., Isobe K., Amann S., Zhang L., Murata M., Elmen L., Gupta V., Arora S., Sarangi R., Dan D., Fujisawa S., Usami T., Xia C.P., Keene A.C., Alayari N.N., Yamakawa H., Elling U., Berger C., Novatchkova M., Koglgruber R., Fukuda K., Nishina H., Isobe M., Pospisilik J.A., Imai Y., Pfeufer A., Hicks A.A., Pramstaller P.P., Subramaniam S., Kimura A., Ocorr K., Bodmer R., Penninger J.M. Cell. 2010;141:142. doi: 10.1016/j.cell.2010.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Keersmaecker K., Atak Z.K., Li N., Vicente C., Patchett S., Girardi T., Gianfelici V., Geerdens E., Clappier E., Porcu M., Lahortiga I., Luca R., Yan J., Hulselmans G., Vranckx H., Vandepoel R., Sweron B., Jacobs K., Mentens N., Wlodarska I., Cauwelier B., Cloos J., Soulier J., Uyttebroeck A., Bagni C., Hassan B.A., Vandenberghe P., Johnson A.W., Aerts S., Cools J. Nat Genet. 2013;45:186. doi: 10.1038/ng.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aslam A., Mittal S., Koch F., Andrau J.C., Winkler G.S. Mol Biol Cell. 2009;20:3840. doi: 10.1091/mbc.E09-02-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mittal S., Aslam A., Doidge R., Medica R., Winkler G.S. Mol Biol Cell. 2011;22:748. doi: 10.1091/mbc.E10-11-0898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Winkler G.S., Balacco D.L. Front. Genet. 2013;4:296. doi: 10.3389/fgene.2013.00296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Balatsos N.A., Vlachakis D., Maragozidis P., Manta S., Anastasakis D., Kyritsis A., Vlassi M., Komiotis D., Stathopoulos C. Biochemistry. 2009;48:6044. doi: 10.1021/bi900236k. [DOI] [PubMed] [Google Scholar]
- 20.Vlachakis D., Pavlopoulou A., Tsiliki G., Komiotis D., Stathopoulos C., Balatsos N.A., Kossida S. PLoS ONE. 2012;7:e51113. doi: 10.1371/journal.pone.0051113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parkes K.E., Ermert P., Fassler J., Ives J., Martin J.A., Merrett J.H., Obrecht D., Williams G., Klumpp K. J Med Chem. 2003;46:1153. doi: 10.1021/jm020334u. [DOI] [PubMed] [Google Scholar]
- 22.Maryati M., Kaur I., Gopal J., Olotu-Umoren L., Oveh B., Hashmi L., Fischer P.M., Winkler G.S. Nucleic Acids Res. 2014;42:e30. doi: 10.1093/nar/gkt972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klumpp K., Hang J.Q., Rajendran S., Yang Y., Derosier A., Wong Kai In P., Overton H., Parkes K.E., Cammack N., Martin J.A. Nucleic Acids Res. 2003;31:6852. doi: 10.1093/nar/gkg881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tumey L.N., Bom D., Huck B., Gleason E., Wang J., Silver D., Brunden K., Boozer S., Rundlett S., Sherf B., Murphy S., Dent T., Leventhal C., Bailey A., Harrington J., Bennani Y.L. Bioorg Med Chem Lett. 2005;15:277. doi: 10.1016/j.bmcl.2004.10.086. [DOI] [PubMed] [Google Scholar]
- 25.Petit A.P., Wohlbold L., Bawankar P., Huntzinger E., Schmidt S., Izaurralde E., Weichenrieder O. Nucleic Acids Res. 2012;40:11058. doi: 10.1093/nar/gks883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Benko, Z.; Boebel, T.; Breaux, N.; Bryan, K.; Davis, G.; Epp, J.; Lorsbach, B.; Martin, T.; Meyer, K.; Nader, B.; Owen, W.; Pobanz, M.; Ruiz, J.; Smith, F.; Sullenberger, M.; Webster, J.; Yao, C.; Young, D. WO2009094442A2, 2009.
- 27.He R., Ching S.M., Lam Y. J Comb Chem. 2006;8:923. doi: 10.1021/cc060092+. [DOI] [PubMed] [Google Scholar]
- 28.Tumey, L. N.; Bennani, Y.; Huck, B.; Bom, D. C. WO2006014647A2, 2006.
- 29.Vutukuri D.R., Bharathi P., Yu Z., Rajasekaran K., Tran M.-H., Thayumanavan S. J Org Chem. 2003;68:1146. doi: 10.1021/jo026469p. [DOI] [PubMed] [Google Scholar]
- 30.Wu M., Reuter M., Lilie H., Liu Y., Wahle E., Song H. EMBO J. 2005;24:4082. doi: 10.1038/sj.emboj.7600869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Docking was performed using AutoDock 4.2 and AutoDockTools 4.2 (ADT). The model from 4GMJ.B was protonated and partial charges were assigned (including for the two Mg2+ ions in the active site). Grid preparation, docking, and analysis was carried out from within ADT, using default parameters and settings.
- 32.Morris G.M., Goodsell D.S., Halliday R.S., Huey R., Hart W.E., Belew R.K., Olson A.J. J Comput Chem. 1998;19:1639. [Google Scholar]
- 33.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. J Comput Chem. 2009;30:2785. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tang J., Maddali K., Metifiot M., Sham Y.Y., Vince R., Pommier Y., Wang Z. J Med Chem. 2011;54:2282. doi: 10.1021/jm1014378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.8j: 1H NMR (400 MHz, DMSO-d6): δ 1.76 (m, 2H, (CH3)2NCH2CH2), 2.10, (s, 6H, (CH3)2N), 2.23 (t, J = 7.1 Hz, 2H, (CH3)2NCH2), 3.10 (t, J = 7.4 Hz, 2H, PhCH2), 4.00 (t, J = 7.3 Hz, 2H, (CH3)2N(CH2)2CH2), 4.46 (t, J = 7.4 Hz, 2H, BnCH2), 7.15–7.30 (m, 5H, ArH), 7.85 (s, 1H, purine 8-H); 13C NMR (75 MHz, DMSO-d6): δ 26.0, 36.8, 41.9, 45.5, 56.7, 106.0, 127.1, 128.9, 129.1, 138.0, 142.9, 147.2, 149.8, 152.9, 162.1. LC–MS: >95%, calcd for C18H24N5O3 [M+H]+ 358.19, found 358.3.
- 36.Almeida, L.; Chuaqui, C. E.; Ioannidis, S.; Peng, B.; Su, M. WO2009150462A1, 2009.
- 37.Reayi A., Hosmane R.S. J Med Chem. 2004;47:1044. doi: 10.1021/jm0304257. [DOI] [PubMed] [Google Scholar]
- 38.Duplantier A.J., Becker S.L., Bohanon M.J., Borzilleri K.A., Chrunyk B.A., Downs J.T., Hu L.-Y., El-Kattan A., James L.C., Liu S., Lu J., Maklad N., Mansour M.N., Mente S., Piotrowski M.A., Sakya S.M., Sheehan S., Steyn S.J., Strick C.A., Williams V.A., Zhang L. J Med Chem. 2009;52:3576. doi: 10.1021/jm900128w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
ZIP file containing the MOL files of the most important compounds in this article.