

Abstract
Many longstanding questions about dynamics of virus-cell interactions can be answered by combining fluorescence imaging techniques with fluorescent protein (FP) tagging strategies. Successfully creating a FP fusion with a cellular or viral protein of interest first requires selecting the appropriate FP. However, while viral architecture and cellular localization often dictate the suitability of a FP, a FP's chemical and physical properties must also be considered. Here, we discuss the challenges of and offer suggestions for identifying the optimal FPs for studying the cell biology of viruses.
INTRODUCTION
Statements such as “my fluorescent virus can barely replicate” and “I cannot see my fluorescent protein fusion” are, unfortunately, not uncommon complaints about fusion proteins made with fluorescent proteins (FPs). In some cases, FPs are inappropriate for the system or question. Other times, FPs are incorporated in a haphazard manner. Finally, FPs may not perform as advertised or the investigator may not read the “fine print.” In this review, we describe how to avoid common FP problems and how to select appropriate FPs for FP fusions.
With multiple fluorescent labeling strategies available, why use FP tags? Generally, FPs have many desirable characteristics. They are genetically encoded, have sufficiently short sequences for incorporation into many viral genomes, and enable site-specific labeling of a viral or cellular protein of interest. Dye-labeling techniques often randomly label proteins and require extracellular delivery, and dye-labeled proteins cannot be delivered into subcellular organelles, such as the endoplasmic reticulum. Difficulties frequently arise when investigators expect FPs to be inert, well behaved in all environments, and provide a bright signal. While few, if any, FPs satisfy every item on a wish list, FPs are undeniably powerful cell biology tools. For a detailed list of virus-relevant methods that exploit FPs, see reference 1 (especially Tables 1 and 2). For basic considerations for developing FP fusion proteins, see reference 2.
PHYSICAL PROPERTIES OF FLUORESCENT PROTEINS
First, let us consider the general properties of FPs. They have short primary sequences but fold into proteins that are not small (∼5 nm in diameter) (3). FPs are evolved as soluble cytoplasmic proteins, with only the environmental considerations related to the pH-neutral cytoplasm as a selective pressure. Taken together, these properties suggest significant concerns when using FPs, such as the potential for steric hindrance in fusion proteins and for the performance of FPs in subcellular compartments. The photophysical characteristics of FPs range tremendously in brightness and photostability. Unless the investigator is studying isolated FPs or using advanced microscopy techniques, including total internal-reflection fluorescence (TIRF) (4) and photoactivated localization microscopy (PALM) (5), it is difficult to detect the signal of a single FP in the presence of the cellular autofluorescence background (2). The photophysical properties of FPs suggest that many low-abundance proteins, such as many kinases or a range of viral proteins (6), may not be appropriate targets for FP tagging and standard imaging approaches in cells. Either such targets may require nonphysiologic overexpression, or this may not be a problem if the proteins are concentrated and confined in a cellular compartment or as part of a virus.
Virus structure and assembly present conformational challenges. The diversity of viral architecture precludes a one-size-fits-all viral protein FP fusion strategy. During assembly, viral components are often packed into confined spaces. A 5-nm-diameter FP may be too large to incorporate as a viral capsid or matrix fusion protein into an intact virus. Figure 1A illustrates a relative-size comparison of green FP (GFP) with the coat protein of tobacco mosaic virus and influenza virus hemagglutinin membrane glycoprotein. A FP may disrupt viral protein folding and/or confound proper viral assembly. Such interference can decrease FP-labeled virus infectivity (7). Figure 1B illustrates a hypothetical example of a GFP fusion with the C terminus of a major capsid protein, Vp54 of paramecium bursaria chlorella virus type 1. The size of the capsid protein monomer is similar to that of a FP molecule, and the presence of a FP could interfere with the highly symmetrical packing of trimeric capsomers. One solution has been to coexpress both tagged and untagged versions of the viral protein (8). Smaller amounts of FP-tagged proteins can still be sufficient for a bright fluorescent signal, while untagged proteins provide more space for the incorporated FP fusions. Another strategy is to identify sites other than the N or C terminus that would better tolerate insertion of a FP insertion or smaller FP alternatives. Zheng and Kielian successfully used structural and mutagenesis information to identify a region in Sindbis virus capsid amenable to insertion of a 12-amino-acid (aa) tetracysteine consensus peptide sequence and ReAsH fluorescent dye labeling (7). The resulting virus exhibited normal ultrastructure and infectivity, and both the virus and capsid protein could be imaged in live cells. Unfortunately, the FlAsH/ReAsH system is incompatible with use in the secretory pathway, as the cysteines become oxidized and cannot be labeled (9). Alternatively, split FPs enable insertions as small as 10 aa and have been used in influenza A virus studies (10). To observe fluorescence, the remainder of the FP must be supplied in trans and then bind irreversibly to form a complete FP. As with the dyes, FP fragment binding depends on the accessibility of the binding motif.
FIG 1.
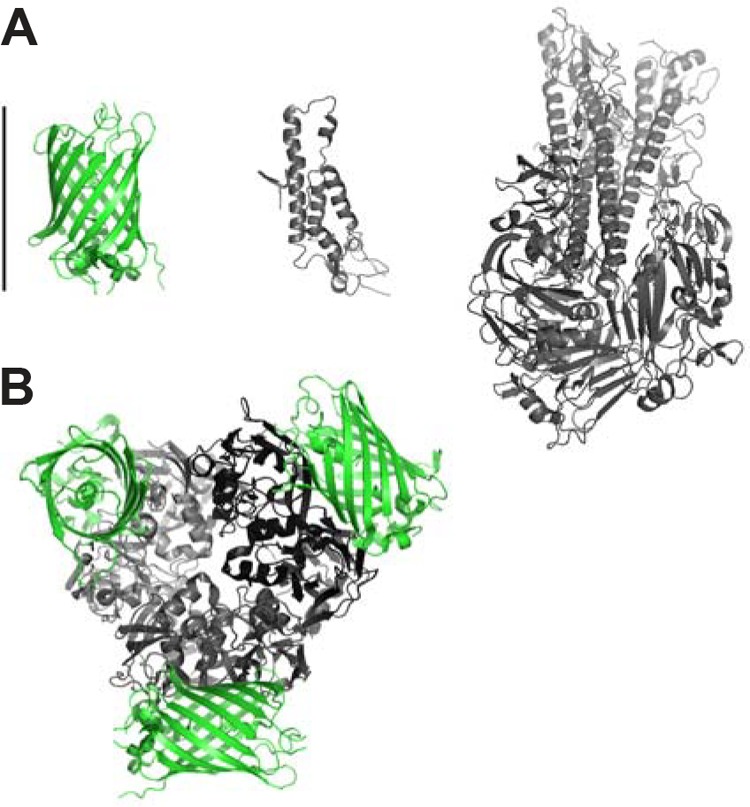
Size comparison of GFP with viral proteins illustrates the potential effect of steric hindrance. (A) Size comparison of GFP (PDB code 2B3P), the coat protein of tobacco mosaic virus (2OM3), and 1918 influenza virus hemagglutinin membrane glycoprotein (1RUZ) relative to 5-nm scale bar. (B) Hypothetical fusion protein of a major capsid protein, Vp54 of paramecium bursaria chlorella virus type 1 (1M4X), with GFP. The trimeric capsomer and associated GFP molecules represent the potential restrictions of a viral FP fusion protein.
THE OLIGOMERIC TENDENCY OF FLUORESCENT PROTEINS
A major concern with FP fusion proteins is the tendency to form dimers. This is not a problem for a typical soluble fusion protein at low expression levels or a transcriptional reporter, but for proteins that incorporate into highly packed oligomers, such as a trimeric envelope protein or a capsid composed of hundreds of subunits, FP oligomerization becomes a significant concern. When a fusion protein no longer tumbles in 720° of free rotation in solution and is restricted to 360°, effective FP concentrations increase, raising the probability of collision and oligomerization. Such interactions can disrupt protein function, drive nonnative interactions, and grossly distort cellular architecture.
Members of the GFP family have a hydrophobic interface that can be modified (A206K) to prevent FPs from dimerizing (11). FPs not derived from the GFP family, including many commonly utilized red FPs (RFPs), were derived from obligate dimers and tetramers (2). We examined RFPs, including: mCherry, TagRFP, mKate2, mRuby2, and Fusion Red (12, 13), in a live cell assay (14) that assesses the propensity of FPs to oligomerize. Importantly, reportedly monomeric TagRFP, mRuby2, and mKate2 all formed inappropriate oligomers, as did mCherry, but to a much more limited degree (15, 16).
When FP tagging is appropriate for a study, which of the dozens of FPs should be used? The cellular environment(s) dictates the suitability of most FPs (17). In the cytoplasm, there are a number of excellent FP options for fusion proteins, including monomerized (V206K) superfolder GFP, mTurquoise, Cerulean3, enhanced blue FP 2 (EBFP2), TagBFP, mNeonGreen, and monomerized Venus (A206K) (13, 18). Several orange and red FPs are extremely bright and work well as transcriptional reporters but present serious problems for fusion proteins, including long maturation times and a tendency toward oligomerization and stickiness (see above). Red and orange FPs can work well for soluble monomeric cytoplasmic fusion proteins and reporters of changes in cell localization, i.e., apoptosis reporters of caspase cleavage of Asp-Glu-Val-Asp (DEVD)-containing peptides (19).
THE IMPACT OF THE CELLULAR ENVIRONMENT ON FLUORESCENT PROTEINS
Different subcellular environments can seriously hobble FPs. For example, endocytosed viruses encounter a progressively decreasing luminal pH. Most compartments of the secretory pathway are acidic. pH significantly impacts FP brightness. The pKa value of a FP refers to the pH at which the brightness of the FP is half of its possible maximum. Brightness further decreases as pH decreases. Thus, a bright FP in the cytoplasm (i.e., a yellow FP [YFP]) with a pKa value of 6.9 would be dark inside a late endosome (pH 4.5 to 5).
Viral secretory proteins traverse the secretory pathway, an oxidizing environment and site of posttranslational modifications, including disulfide bond formation and N- and O-linked glycosylation. Either modification can induce gross misfolding and produce a population of nonfluorescent misfolded FPs (15, 20). To protect against these cellular environments, we recommend selecting FPs with low pKa values and no or few residues subject to posttranslational modifications, i.e., cysteines and N-linked glycosylation consensus sequences (N-X-S/T).
For expressing proteins in oxidizing cellular compartments (secretory pathway, inner membrane space of mitochondria, and periplasm of Gram-negative bacteria), we recommend the “mox” FPs we recently developed, including moxsynGFP, moxCerulean3, moxVenus, and moxBFP (15). These genuinely monomeric FPs lack cysteines and, thus, cannot form disulfide bonds. moxFPs can be paired (moxBFP with moxsynGFP or moxVenus and moxCerulean3 with moxVenus) for robust two-color imaging with standard fluorescence microscope filter sets. The low pKa value of moxCerulean3 (3.8) ensures fluorescence in the acidic late endocytic pathway. mCherry, mCerulean3, and mTurquoise2 (21) have similar or even lower pKa values (3.1 to 4.5). We note that mCherry is resistant to destruction in the lysosome and that mCherry fusions can accumulate in lysosomes, while other FP fusions also traffic to lysosomes via the secretory pathway or autophagy and are destroyed in the proteolytic lysosomal environment (15). Finally, the monomeric moxFPs are especially well suited for fusions with integral membrane proteins.
There is a growing awareness of environmental impacts on FP functionality. If you have been using FPs from over a decade ago, we strongly encourage you to upgrade your FPs. There are several FP options that are significantly improved for the needs of most virologists. Importantly, when considering the latest FPs, closely look at the potential for posttranslational modifications, whether the FPs have been convincingly demonstrated to be monomeric, and whether the FPs would be functional in your cellular compartment of interest. Investigators who need even more colors, specific properties, or FP alternatives should frequently check for reports of new FPs in the literature. With an improved palette of environmentally inert FPs, future studies of fluorescent viruses look very bright.
ACKNOWLEDGMENTS
We sincerely apologize to all whose work could not be cited due to space restrictions.
This work was supported by grants from the National Institute of General Medical Sciences (NIGMS) (grant 1R01GM10599-01) and from the Marion Bessin Liver Center Imaging and Cell Structure Core, supported by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (grant P30DK041296) (E.L.S).
A provisional patent application has been filed covering part of the moxFPs described in this manuscript (L.M.C. and E.L.S). Albert Einstein College of Medicine and E.L.S. have licensed technology described in this manuscript to Lucigen Corp.
The content is solely our responsibility and does not necessarily represent the official views of the NIGMS, the NIDDK, or the NIH.
We declare that we have no competing financial interests.
REFERENCES
- 1.Sun E, He J, Zhuang X. 2013. Live cell imaging of viral entry. Curr Opin Virol 3:34–43. doi: 10.1016/j.coviro.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Snapp E. 2005. Design and use of fluorescent fusion proteins in cell biology. Curr Protoc Cell Biol Chapter 21:Unit 21.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang F, Moss LG, Phillips GN Jr. 1996. The molecular structure of green fluorescent protein. Nat Biotechnol 14:1246–1251. doi: 10.1038/nbt1096-1246. [DOI] [PubMed] [Google Scholar]
- 4.Axelrod D. 2001. Total internal reflection fluorescence microscopy in cell biology. Traffic 2:764–774. doi: 10.1034/j.1600-0854.2001.21104.x. [DOI] [PubMed] [Google Scholar]
- 5.Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. 2006. Imaging intracellular fluorescent proteins at nanometer resolution. Science 313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 6.Loret S, Guay G, Lippé R. 2008. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J Virol 82:8605–8618. doi: 10.1128/JVI.00904-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zheng Y, Kielian M. 2013. Imaging of the alphavirus capsid protein during virus replication. J Virol 87:9579–9589. doi: 10.1128/JVI.01299-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vogel M, Diez M, Eisfeld J, Nassal M. 2005. In vitro assembly of mosaic hepatitis B virus capsid-like particles (CLPs): rescue into CLPs of assembly-deficient core protein fusions and FRET-suited CLPs. FEBS Lett 579:5211–5216. doi: 10.1016/j.febslet.2005.08.044. [DOI] [PubMed] [Google Scholar]
- 9.Adams SR, Campbell RE, Gross LA, Martin BR, Walkup GK, Yao Y, Llopis J, Tsien RY. 2002. New biarsenical ligands and tetracysteine motifs for protein labeling in vitro and in vivo: synthesis and biological applications. J Am Chem Soc 124:6063–6076. doi: 10.1021/ja017687n. [DOI] [PubMed] [Google Scholar]
- 10.Avilov SV, Moisy D, Munier S, Schraidt O, Naffakh N, Cusack S. 2012. Replication-competent influenza A virus that encodes a split-green fluorescent protein-tagged PB2 polymerase subunit allows live-cell imaging of the virus life cycle. J Virol 86:1433–1448. doi: 10.1128/JVI.05820-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zacharias DA, Violin JD, Newton AC, Tsien RY. 2002. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science 296:913–916. doi: 10.1126/science.1068539. [DOI] [PubMed] [Google Scholar]
- 12.Siegel AP, Baird MA, Davidson MW, Day RN. 2013. Strengths and weaknesses of recently engineered red fluorescent proteins evaluated in live cells using fluorescence correlation spectroscopy. Int J Mol Sci 14:20340–20358. doi: 10.3390/ijms141020340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Day RN, Davidson MW. 2009. The fluorescent protein palette: tools for cellular imaging. Chem Soc Rev 38:2887–2921. doi: 10.1039/b901966a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Costantini LM, Fossati M, Francolini M, Snapp EL. 2012. Assessing the tendency of fluorescent proteins to oligomerize under physiologic conditions. Traffic 13:643–649. doi: 10.1111/j.1600-0854.2012.01336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Costantini LM, Baloban M, Markwardt ML, Rizzo M, Guo F, Verkhusa VV, Snapp EL. 2015. A palette of fluorescent proteins optimized for diverse cellular environments. 6:7670 Nat Commun doi: 10.1038/ncomms8670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang L, Pike D, Sleat DE, Nanda V, Lobel P. 2014. Potential pitfalls and solutions for use of fluorescent fusion proteins to study the lysosome. PLoS One 9:e88893. doi: 10.1371/journal.pone.0088893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Costantini LM, Snapp EL. 2013. Fluorescent proteins in cellular organelles: serious pitfalls and some solutions. DNA Cell Biol 32:622–627. doi: 10.1089/dna.2013.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shaner NC, Lambert GG, Chammas A, Ni Y, Cranfill PJ, Baird MA, Sell BR, Allen JR, Day RN, Israelsson M, Davidson MW, Wang J. 2013. A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat Methods 10:407–409. doi: 10.1038/nmeth.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bhola PD, Simon SM. 2009. Determinism and divergence of apoptosis susceptibility in mammalian cells. J Cell Sci 122:4296–4302. doi: 10.1242/jcs.055590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grotzke JE, Lu Q, Cresswell P. 2013. Deglycosylation-dependent fluorescent proteins provide unique tools for the study of ER-associated degradation. Proc Natl Acad Sci U S A 110:3393–3398. doi: 10.1073/pnas.1300328110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goedhart J, van Weeren L, Hink MA, Vischer NOE, Jalink K, Gadella TWJ. 2010. Bright cyan fluorescent protein variants identified by fluorescence lifetime screening. Nat Methods 7:137–139. doi: 10.1038/nmeth.1415. [DOI] [PubMed] [Google Scholar]