

ABSTRACT
Enterovirus 71 (EV71) recruits various cellular factors to assist in the replication and translation of its genome. Identification of the host factors involved in the EV71 life cycle not only will enable a better understanding of the infection mechanism but also has the potential to be of use in the development of antiviral therapeutics. In this study, we demonstrated that the cellular factor 68-kDa Src-associated protein in mitosis (Sam68) acts as an internal ribosome entry site (IRES) trans-acting factor (ITAF) that binds specifically to the EV71 5′ untranslated region (5′UTR). Interaction sites in both the viral IRES (stem-loops IV and V) and the heterogeneous nuclear ribonucleoprotein K homology (KH) domain of Sam68 protein were further mapped using an electrophoretic mobility shift assay (EMSA) and biotin RNA pulldown assay. More importantly, dual-luciferase (firefly) reporter analysis suggested that overexpression of Sam68 positively regulated IRES-dependent translation of virus proteins. In contrast, both IRES activity and viral protein translation significantly decreased in Sam68 knockdown cells compared with the negative-control cells treated with short hairpin RNA (shRNA). However, downregulation of Sam68 did not have a significant inhibitory effect on the accumulation of the EV71 genome. Moreover, Sam68 was redistributed from the nucleus to the cytoplasm and interacts with cellular factors, such as poly(rC)-binding protein 2 (PCBP2) and poly(A)-binding protein (PABP), during EV71 infection. The cytoplasmic relocalization of Sam68 in EV71-infected cells may be involved in the enhancement of EV71 IRES-mediated translation. Since Sam68 is known to be a RNA-binding protein, these results provide direct evidence that Sam68 is a novel ITAF that interacts with EV71 IRES and positively regulates viral protein translation.
IMPORTANCE The nuclear protein Sam68 is found as an additional new host factor that interacts with the EV71 IRES during infection and could potentially enhance the translation of virus protein. To our knowledge, this is the first report that describes Sam68 actively participating in the life cycle of EV71 at a molecular level. These studies will not only improve our understanding of the replication of EV71 but also have the potential for aiding in developing a therapeutic strategy against EV71 infection.
INTRODUCTION
Hand-foot-and-mouth disease (HFMD) is a common viral illness in infants and children. The disease causes fever and skin rash or blister-like eruptions (1). HFMD is caused by viruses that belong to the Enterovirus genus of the Picornavirus family, which mainly includes coxsackieviruses and echoviruses (1, 2). Enteroviruses, especially enterovirus 71 (EV71), have been associated with HFMD and may lead to disease with severe effects on morbidity and mortality (3) and a high rate of neurological complications, such as aseptic meningitis, acute flaccid paralysis, and fatal neurogenic pulmonary edema (4, 5). The first EV71 strain (BrCr-CA-70) was isolated in 1970 in California and reported in 1974. Since then, HFMD outbreaks caused by EV7 have been reported from countries as diverse as the United Kingdom, Australia, Sweden, Bulgaria, Japan, Hong Kong, Taiwan, and Malaysia (6). Although molecular diagnostic methods of detecting EV71 have recently been developed (7–9), no specific therapy has been developed to treat this virus disease, partly due to the fact that the molecular mechanism by which EV71 induces infection remains elusive.
EV71 is a small, nonenveloped virus with a genome size of about 7,400 nucleotides (nt). The virus has a single-stranded positive-sense RNA containing a single open reading frame (ORF), flanked by a highly structured 5′ untranslated region (5′UTR) and a 3′UTR with a poly(A) tail. The ORF encodes a polyprotein that, following viral protease-mediated co- and posttranslational processing, gives rise to four structural proteins (VP1, VP2, VP3, and VP4) and seven nonstructural proteins (NSPs) (2A, 2B, 2C, 3A, 3B, 3C, and 3D) (6). EV71 enters into cells via specific receptors: human P-selectin glycoprotein ligand 1 and scavenger receptor B2 (10, 11). After infection of the host cells, the EV71 genome, which lacks a 5′ cap but has an internal ribosome entry site (IRES) in its 5′UTR, is translated in a cap-independent manner into a single polyprotein, which is subsequently processed by the virus-encoded proteases 2Apro and 3Cpro into the structural capsid proteins and the nonstructural proteins. The latter are involved mainly in the replication and translation of the viral RNA (12–15). During virus replication, the genomic RNA not only directs the synthesis of the viral polyprotein but also serves as the template for RNA synthesis as well as packing into virions. These processes must be regulated by viral and host cell factors for efficient replication of the virus. Studies of other picornaviruses, including poliovirus, have revealed that the processes of translation and RNA replication cannot occur simultaneously on the same RNA molecule, indicating that there may be a molecular switch to shut down RNA replication and allow initiation of translation (16–18). The IRES-mediated initiation of translation allows viral RNA translation while host cell translation is shut down during infection. Virus translation appears to be mediated by interactions resulting from cellular and viral factors binding to the virus 5′UTR (19, 20). Lin et al. (21) identified 12 cellular proteins that interact with the 5′UTR of EV71. Among these proteins, heterogeneous nuclear ribonucleoprotein K (hnRNP K), polypyrimidine tract-binding protein (PTB), poly(rC)-binding protein 1 (PCBP1), PCBP2, the autoantigen La, and upstream N-ras protein (Unr) had previously been shown to interact with the 5′UTRs of various picornaviruses and to regulate virus translation or replication.
Lin et al. (21) have proved that hnRNP K is enriched in the cytoplasm where EV71 virus replication occurs so as to interact with the EV71 5′UTR and participate in virus replication. Yang et al. (22) found that hnRNP K interacts specifically with the 68-kDa Src-associated protein in mitosis (Sam68) under basal conditions. Sam68 belongs to the signal transduction and activation of RNA (STAR) protein family and the hnRNP K homology (KH) domain family of RNA-binding proteins (23). The KH domain is the second most prevalent RNA binding motif in proteins (24). Sam68 has been suggested to participate in cell cycle and signaling, cell growth, alternative splicing (25), and virus replication (26). Sam68 is also involved in several RNA metabolic processes, such as pre-mRNA splicing and trafficking (27). The Sam68 protein is composed of 443 amino acids and contains one KH domain and several proline-rich sequences that are the sites of protein-protein interactions with SH3- and WW domain-containing proteins (28). Sam68 is not a shuttling protein and is confined to the nucleus as a result of a specialized domain identified as the C-terminal 24 amino acids termed the nuclear localization signal (NLS) (29). Interestingly, Sam68 relocalized from the nucleus to the cytoplasm and interacted with the poliovirus RNA polymerase during poliovirus infection (30). The binding of Sam68 to the foot-and-mouth disease virus (FMDV) IRES during infection could potentially enhance translation of the viral RNA (31). Furthermore, Sam68 can significantly enhance the translation of retrovirus genes by marking the viral RNA transcripts (32). In our study, we have demonstrated that Sam68 was translocated to the cytoplasm and colocalized with EV71 during virus infection. These observations, in combination with the known RNA- and protein-binding properties of Sam68, led us to test the hypothesis that Sam68 has a functional role in the replication of EV71. The results of the electrophoretic mobility shift assay (EMSA) presented in this paper showed that Sam68 could bind to the EV71 5′UTR of the genome. We also found that Sam68 interacted with the IRES of the EV71 genome and facilitated the translation of viral RNA. These results indicated that Sam68 plays multiple important roles in the life cycle of EV71.
MATERIALS AND METHODS
Cells and virus.
Human rhabdomyosarcoma (RD) cells were cultured in minimum essential medium (MEM). HeLa and human glioma U251 cells were maintained in Dulbecco's modified Eagle's medium (DMEM). Both MEM and DMEM contained 10% fetal bovine serum (FBS), 100 μg/ml penicillin, and 100 μg/ml streptomycin. All cells were cultured at 37°C in a humidified atmosphere containing 5% CO2. EV71 virus (BrCr-Tr strain; GenBank accession number AB204852.1) was kindly provided by Qi Jin (Institute of Pathogen Biology, Chinese Academy of Medical Science & Peking Union Medical College, Beijing, China). For EV71 purification, the virus was inoculated onto confluent monolayer RD cells for 2 h at 37°C with occasional shaking. The cells were then incubated in MEM supplemented with 2.5% FBS until an 80% cytopathic effect (CPE) was reached. After an initial centrifugation at 2,000 × g for 5 min, the cell pellet was subjected to three cycles of freezing and thawing, followed by centrifugation to collect the supernatant. Polyethylene glycol (Mr, 6,000) was added to the supernatant to a final concentration of 8% for 2 h, followed by centrifugation at 12,000 × g for 2 h. The precipitated virus was resuspended in RNase-free double-distilled H2O (ddH2O), and aliquots were stored at −70°C (33).
Sam68 protein and antibodies.
Glutathione S-transferase (GST)-tagged full-length Sam68 (P01) was purchased from Abnova. Mouse anti-EV71 VP1 monoclonal antibody (MAB979) was purchased from Millipore. Rabbit monoclonal anti-Sam68 (2981-1) antibody was purchased from Epitomics. Mouse monoclonal anti-PCBP2 (ab110200) and mouse monoclonal anti-poly(A)-binding protein (anti-PABP) (ab6126) was purchased from Abcam. Mouse anti-Flag monoclonal antibody (M2008) was purchased from Abmart. Mouse β-actin monoclonal antibody (ZM-0001), goat anti-mouse secondary antibodies conjugated to horseradish peroxidase (HRP) (ZB-2305), goat anti-rabbit secondary antibodies (ZB-2301) used for Western blotting, goat anti-rabbit secondary antibodies conjugated to fluorescein isothiocyanate (FITC) (ZF-0311), and goat anti-mouse secondary antibodies conjugated to tetramethyl rhodamine isothiocyanate (TRITC) (ZF-0313) used for immunofluorescence were all purchased from Beijing Zhongshan Biotech, China.
Plasmid construction and in vitro transcription of RNA probes.
To construct full-length and different truncated forms of Sam68, the corresponding DNA templates were amplified and cloned into the KpnI and EcoRI sites of the pcDNA3.0-Flag vector, resulting in the corresponding Flag fusion proteins. To prepare the T7-EV71-5′UTR and its various deletion constructs, cDNA fragments of the corresponding 5′UTR sequences were amplified by reverse transcription-PCR (RT-PCR) using an EV71 sense primer containing the T7 promoter sequence (TAATACGACTCACTATAG) and cloned into the pMD19-T vector (TaKaRa) by TA cloning. As these cDNA sequences are located downstream of the T7 polymerase promoter, the corresponding RNAs can be produced and labeled with [α-32P]UTP or bio-16-UTP using the MEGAscript T7 kit (Ambion), according to the protocol provided by the manufacturer.
Electrophoretic mobility shift assays.
RNA binding reactions and electrophoretic mobility shift assays (EMSAs) were performed as described previously but with some modifications (34). The purified RNA probes were refolded by melting at 80°C, followed by gradual cooling down to room temperature. 32P-labeled RNA (40 to 100 nM) was incubated with the indicated amount of purified Sam68 in an RNA binding buffer (5 mM HEPES [pH 7.5], 25 mM KCl, 2 mM MgCl2, 0.1 mM EDTA, 3.8% glycerol, 2 mM dithiothreitol [DTT], 0.5 mg/ml bovine serum albumin [BSA], 0.5 mg/ml heparin, 0.25 mg/ml Escherichia coli tRNA, and 5 U of RNasin RNase inhibitor [Promega]) for 30 min at 30°C. The resulting ribonucleoprotein (RNP) complexes were analyzed by electrophoresis in 5% native polyacrylamide (29:1 acrylamide-bisacrylamide) gels containing 5% glycerol and 0.5× Tris-borate EDTA for 1 h. RNAs were detected by exposing the dried gels to a phosphorimager screen.
Biotin RNA pulldown assay, RNA-protein coimmunoprecipitation, and RT-PCR.
Cells were washed two times with phosphate-buffered saline (PBS) and disrupted with radioimmunoprecipitation assay (RIPA) lysis buffer (150 mM NaCl, 50 mM Tris-HCl [pH 8.0], 1% Nonidet P-40, and 0.5% deoxycholate supplemented with 1 minitablet of Complete protease inhibitor cocktail per 50 ml buffer). Cell lysates were centrifuged at 12,000 × g for 10 min to harvest supernatants for further analysis. Protein assays were performed on all supernatants by using the Bradford method. For biotin RNA pulldown assay, the harvested cell extracts were incubated at 37°C with 12.5 pmol biotinylated EV71-5′UTR RNA probe in an RNA binding mixture buffer (5 mM HEPES [pH 7.5], 40 mM KCl, 0.1 mM EDTA, 2 mM MgCl2, 2 mM DTT, 1 U RNasin, and 0.25 mg/ml heparin) for 15 min in a total volume of 30 μl, then added to 400 μl streptavidin MagneSphere paramagnetic particles (Promega), and incubated for 10 min at room temperature to allow binding. After the protein-RNA probe complexes were washed three times with RNA binding buffer without heparin, 30 μl of 2× SDS-PAGE loading buffer (100 mM Tris [pH6.8], 4% SDS, 20% glycerol, 200 mM DTT, 0.2% bromophenol blue) was added to the beads and incubated for 10 min at room temperature to dissociate the proteins from the RNA. The samples of eluted proteins were then boiled, subjected to 15% SDS-PAGE, and analyzed by Western blotting.
For RNA-protein coimmunoprecipitation analysis, cell extracts prepared as described above were preincubated with protein A-agarose on ice for 1 h to bind nonspecific protein. The nonspecific protein complexes were pelleted by centrifugation at 1,000 × g at 4°C for 10 min. The supernatant was recovered, and 100-μl samples were each diluted with 450 μl of RIPA lysis buffer and then either added to 5 μl of the indicated antibody or 5 μl of buffer containing no antibody before being incubated on ice for 2 h. Prewashed protein A-agarose (100 μl) in PBS (agarose-PBS [50:50]) was then added to each sample and incubated on ice for an additional hour. RNA-protein coimmunoprecipitation complexes were pelleted by centrifugation at 1,000 × g at 4°C for 5 min and washed three times with lysis buffer. Each pellet was resuspended in 400 μl of proteinase K buffer (100 mM Tris-HCl [pH 7.5], 12.5 mM EDTA, 150 mM NaCl, and 1% SDS) and incubated with 100 mg of predigested proteinase K for 30 min at 37°C. RNA was extracted from the samples with TRIzol reagent (Invitrogen) and dissolved in 20 μl diethyl pyrocarbonate (DEPC)-treated H2O. RT-PCR of the RNA was performed using a PrimeScript one-step RT-PCR kit (TaKaRa) and primers specific to EV71 5′UTR (ATTGAGCTAGTTAGTAGTCCTCCG and AATAGCTCTGTTTGACACTGGATGG). RT-PCR using primers specific to ribosomal protein S16 (RPS16) (GCGCGGTGAGGTTGTCTAGTC and GAGTTTTGAGTCACGATGGGC) served as a control.
Confocal microscopy analysis.
Cells were cultured to 80% confluence on 9-mm glass coverslips (Thomas Scientific) in 48-well plates and incubated with purified EV71 at the indicated multiplicity of infection (MOI). At the indicated time points, the cells were prepared for microscopy as follows. The cells were fixed with 4% paraformaldehyde for 15 min, quenched with 100 mM glycine for 15 min, permeabilized with 0.1% Triton X-100 for 15 min, and blocked with PBS containing 10% goat serum plus 1% BSA for 2 h. The cells were washed three times with PBS and incubated with either diluted rabbit anti-Sam68, mouse anti-PCBP2, mouse anti-PABP, or mouse anti-EV71 VP1 monoclonal antibodies, followed by either goat anti-rabbit secondary antibody conjugated to FITC (green) or goat anti-mouse secondary antibody conjugated to TRITC (red). The cells were then washed extensively with PBS before being incubated for 10 min in PBS containing 1% BSA and 0.1% 4′,6′-diamidino-2-phenylindole (DAPI) to stain the nuclei. The cells were examined and images were captured using 100× objectives with a confocal microscope (Leica SP8). The images were refined and figures were generated using Adobe Photoshop software (Adobe Systems, San Jose, CA).
Effects of RNA interference and overexpression of Sam68 on virus replication and virus yield.
Two human RNA interference (RNAi) targeting sequences were each cloned into the RNAi-Ready pSIREN-RetroQ to generate the corresponding RNAi-ready pSIREN-RetroQ-Sam68 (Sam68 shRNA). The RNAi targeting sequences were Sam68 ShRNA#1 (GGACCACAAGGGAATACAATC) and Sam68 ShRNA#2 (GCATCCAGAGGATACCTTTGC) (synthesized by Invitrogen) as described previously (35). shGFP (shRNA against green fluorescent protein [GFP]) (Invitrogen) was used as the negative-control shRNA. Plasmid transfections were carried out using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Briefly, cells at 80% confluence were transfected with plasmids using Lipofectamine 2000 that was diluted in serum-free Opti-MEM (Invitrogen). Five hours later, the medium was replaced with fresh serum-containing medium. The cells were then either transfected with Sam68 shRNA (or control shRNA) and screened in 2 μg/ml puromycin (Sigma) or transfected with Flag-Sam68 (or Flag-EV) and screened in 400 μg/ml antibiotic G418 (Sigma). Expression of Sam68 was detected using rabbit monoclonal anti-Sam68 antibody. To assess the effects of the shRNA treatments and overexpression of Sam68 on virus replication, cell monolayers transfected with the respective plasmids were infected with EV71 at an MOI of 1. Following virus absorption for 1 h, the inoculum was removed, and new medium was added to the cells. The virus-infected cells were harvested after incubation for 24 h at 37°C. Virus proteins in infected cells were detected by Western blotting using mouse EV71 VP1 monoclonal antibodies, and virus titers were determined by plaque assay as previously described (36). The 50% tissue culture infective dose (TCID50) per milliliter was obtained by plotting the plaque assay results in a logarithmic scale using Microsoft Excel (Microsoft). All assays were performed three times.
Luciferase reporter assay.
A dual-luciferase reporter assay kit (Promega) was used. A bicistronic reporter plasmid, pRFH-EV71-5′UTR, containing the EV71 IRES between Renilla luciferase (RLuc) and firefly luciferase (FLuc) was constructed by ligating a NotI-EV71 5′UTR-NotI fragment into pRHF. Cells in which Sam68 was knocked down or overexpressed as described above were transfected with either pRFH-EV71-5′UTR or pRFH using Lipofectamine 2000. At 48 h after cotransfection, the cells were harvested and examined for either reduction or overexpression of Sam68 by Western blotting. The activities of the RLuc and FLuc reporter genes were measured using a Veritas luminometer (Promega). As the expression of RLuc is driven by the cytomegalovirus (CMV) promoter while the expression of FLuc is dependent on EV71 IRES activity, the ratio of the FLuc expression level to the RLuc expression level in a sample represents the relative translation efficiency of the EV71 IRES of that sample. For capped bicistronic mRNA containing a poly(A) tail reporter or monocistronic RNA reporter assay, cells transfected with the corresponding plasmid were cultured for 3 days and then transfected with RNA. After 6 h posttransfection, cells were harvested, lysed, and Western blotted for Sam68, Flag, and β-actin.
Semiquantitative and quantitative reverse transcription-PCR.
Total RNA was reverse transcribed into cDNA by using a reverse transcription system (Promega). Semiquantitative reverse transcription-PCR (semi-qPCR) was performed using a PrimeScript one-step RT-PCR kit (TaKaRa). Quantitative reverse transcription-PCR (qRT-PCR) was carried out by using an ABI 7500 real-time PCR system with Power SYBR green master mix (Applied Biosystems). The PCR was set up under the following thermal cycling conditions: 95°C for 10 min, followed by 45 cycles, with 1 cycle consisting of 95°C for 15 s and 60°C for 1 min. Fluorescence signals were recorded by the instrument during the extension phase of each PCR cycle. The semi-qPCR and qRT-PCR were performed by using the following primer pairs: EV71 VP1 forward primer (AGTATGATTGAGACTCGGTG) and reverse primer (GCGACAAAAGTGAACTCTGC) and β-actin forward primer (GAACCCTAAGGCCAACCGTGAA) and reverse primer (CTCAGTAACAGTCCGCCTAGAA). All samples were run in triplicate, and the experiment was repeated three times. The relative mRNA level of EV71 VP1 was expressed as a percentage change relative to the value of the corresponding control.
Western blot and protein-protein coimmunoprecipitation.
Western blot analysis was performed as described previously (37). Briefly, proteins were separated in 15% gradient SDS-polyacrylamide gels and transferred to Hybond-P polyvinylidene difluoride (PVDF) membranes (GE). The membranes were blocked for 2 h with 5% nonfat dry milk solution in Tris-buffered saline containing 0.5% Tween 20 (TBST). They were then blotted with the required specific primary antibodies, followed by incubation with the corresponding secondary antibodies conjugated to horseradish peroxidase. Cellular β-actin was employed as an internal loading control protein. The blots were developed with the Western Lightning chemiluminescence kit (NC4109) by following the manufacturer's protocol (Pierce, USA). For protein-protein immunoprecipitation, which was done at 4°C, cell lysates (0.5 mg) were precleared with 20 μl of protein A/G-Sepharose beads (Abmart) for 60 min. Nonspecific complexes were pelleted by centrifugation at 10,000 × g at 4°C for 10 min. The supernatants were removed and incubated with either 2.5 μg of anti-Sam68 antibody or the isotype control IgG for 60 min before the addition of 20 μl of protein A/G-Sepharose beads and incubated for another 60 min in an end-over-end rotor. The immunoprecipitates were pelleted and washed three times with RIPA buffer. After the final wash, the pellet was resuspended in 40 μl of 2× SDS-PAGE loading buffer and boiled for 10 min before being analyzed by Western blotting.
Statistical analysis.
All data were analyzed by SAS software pack (SAS Institute, Cary, NC, USA). Comparisons between individual data points were made using Student's t test, and P < 0.05, P < 0.01, and P < 0.001 were regarded as statistically significant. Data are shown as means ± standard deviations.
RESULTS
Studies on the interaction of Sam68 with EV71 5′UTR in vitro.
EMSA experiments showed that Sam68 specifically interacted with the EV71 5′UTR. As shown in Fig. 1A, Sam68 bound to the 32P-labeled EV71 5′UTR, slowing its migration in a nondenaturing gel but not to the control RNA (an unrelated RNA sequence). The 5′UTR of EV71 contains both a cloverleaf structure (nt 1 to 90) and an IRES (nt 91 to 745), as predicted by the MFold software (38, 39). As shown in Fig. 1B, a 32P-labeled RNA probe containing the nucleotide sequence from nt 91 to 745 of the EV71 5′UTR was able to bind to Sam68, while another probe containing the sequence from nt 1 to 90 of the EV71 5′UTR did not bind to Sam68. These results indicated that it was the IRES of EV71 5′UTR that interacted with Sam68.
FIG 1.
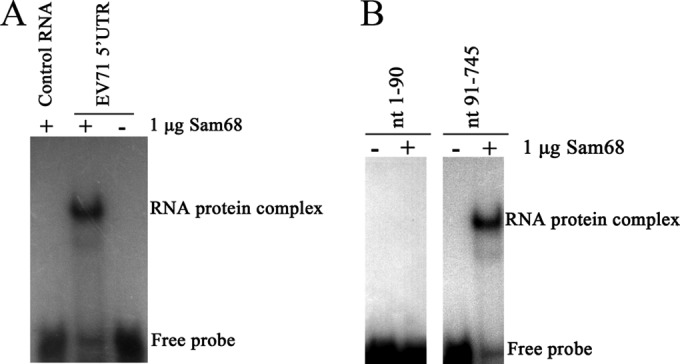
Binding of Sam68 to the IRES of EV71 5′UTR. (A) The binding of Sam68 to the EV71 5′UTR is specific. The EV71 5′UTR and control RNA were labeled with [α-32P]UTP (0.2 × 104 to 1 × 104 cpm per reaction mixture) and incubated at 30°C for 30 min either alone (−) or with 1 μg of Sam68 protein (+). The ribonucleoprotein complexes were resolved by an EMSA. (B) Sam68 specifically binds to the IRES of the EV71 5′UTR. Nucleotides 1 to 90 (nt 1-90) and nt 91-745 (IRES) were labeled with [α-32P]UTP (0.2 × 104 to 1 × 104 cpm per reaction mixture), and an EMSA was carried out as described above for panel A. The positions of the RNA-protein complex and free probe are shown to the right of the gels.
Identification of the Sam68 binding sites in the EV71 IRES.
To further identify the RNA sequences in the EV71 IRES that bound to Sam68, serial deletions to individually knock out each of the six stem-loops of the IRES were constructed. Each deletion was made so as not to disrupt the secondary structure of the remaining RNA sequence (Fig. 2A). PCR was carried out to amplify the cDNA of the EV71 IRES with various deletions. RNA probes corresponding to these deletion constructs for use in the EMSA were then synthesized and labeled at their 5′ ends with [α-32P]UTP. As shown in Fig. 2B, Sam68 did not interact with the cloverleaf structure (nt 1 to 90) of the 5′UTR, which is consistent with the result presented in Fig. 1B. For sequences within the IRES region, Sam68 did not interact with stem-loop II (nt 129 to 167), stem-loop III (nt 193 to 228), and stem-loop VI (nt 566 to 636). However, it slowed the migration of RNA probes corresponding to nt 91 to 745, nt 167 to 445, nt 167 to 561, nt 167 to 636, and nt 167 to 745, indicating that the Sam68 protein specifically interacted with stem-loops IV and V. These results, plus the fact that the binding of Sam68 to the FMDV IRES during infection could potentially enhance translation of the viral RNA (31), suggested that Sam68 might play important roles in the translation of EV71 proteins.
FIG 2.
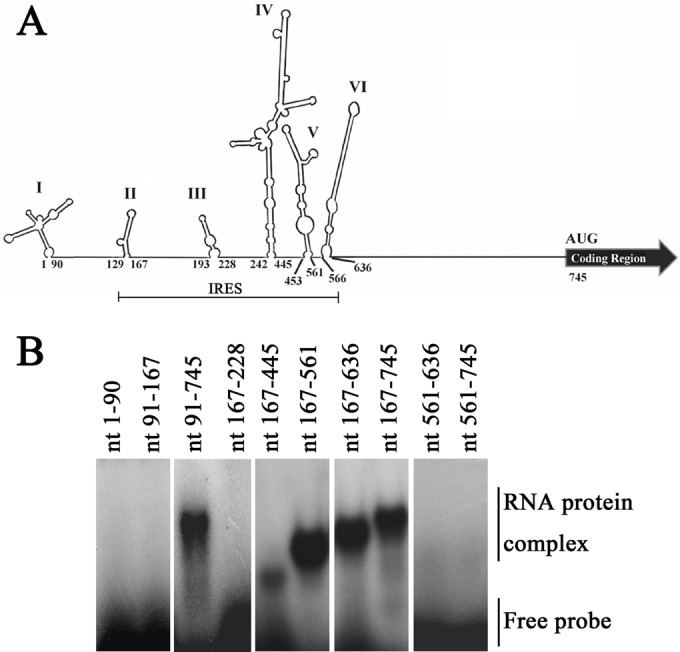
Identification of Sam68 binding sites in the EV71 IRES. (A) The secondary structure of the 5′ UTR was predicted by MFold software (47). The first and the last nucleotides in each stem-loop are numbered as indicated. Stem-loops I to VI are shown. (B) A serial deletion to eliminate each of the six stem-loops of the IRES was constructed. RNA probes were labeled with [α-32P]UTP (0.2 × 104 to 1 × 104 cpm per reaction mixture), and an EMSA was performed as described in the legend to Fig. 1A. Sam68 interacts with stem-loops IV and V, but it did not bind to other regions of the IRES.
Identification of the regions in the Sam68 protein that interact with EV71 IRES.
Sam68 is a nuclear RNA-binding protein containing two putative RNA binding domains, namely, a RGG box and a KH domain flanked by NK (N-terminal region of KH) and CK (C-terminal region of KH) regions (24, 40). A NLS has been mapped to the C-terminal tyrosine-rich region of Sam68 (29). To identify which of these domains within Sam68 is involved in the interaction with EV71 5′UTR, full-length and different truncated forms of DNA constructs of Sam68 were generated to express the corresponding Sam68 proteins fused with the Flag tag (Fig. 3A). These Sam68 plasmids were transfected individually into HeLa cells, and the cell lysates were utilized in the RNA-protein pulldown experiments to test their ability to interact with the EV71 5′UTR. The levels of expression of Sam68 (Fig. 3B, lane 13) and its truncated forms (Fig. 3B, lanes 1, 4, 7, and 10) in the lysates were detected by Western blotting using anti-Flag antibody. The results of the pulldown assays (Fig. 3B) showed that the streptavidin beads captured the biotinylated EV71 5′UTR and its associated full-length Sam68 (lane 15), as well as one truncated form of Sam68 (nt 96 to 257) (lane 9), but not the other truncated forms. These results suggested that Sam68 interacted with EV71 5′UTR through a region of Sam68 (nt 96 to 257) which is the KH RNA-binding domain flanked by NK and CK regions.
FIG 3.

Regions in the Sam68 protein interacting with EV71 5′UTR. (A) Schematic diagrams of Sam68 and its various truncated mutants. The KH domain is indicated by a dark gray box, and the NK domain and CK domain are indicated by light gray boxes. The other domains are indicated by white boxes. Four truncated forms, Sam68(1-157) (nt 1 to 157 of Sam68), Sam68(157-256), Sam68(96-279), and Sam68(256-443), and full-length Sam68(1-443) were generated and fused with Flag at their N-terminal ends. (B) Expression of full-length Sam68 and its truncated forms in HeLa cells and mapping the interacting regions in Sam68 with EV71 5′UTR. Plasmids that carried full-length Sam68 (lane 13) and various truncated forms of Sam68 (lanes 1, 4, 7, and 10) were transfected into HeLa cells. A Western blot using anti-Flag antibody was employed to examine protein expression. Cell extracts from transfected cells were collected 48 h posttransfection and then incubated either with or without biotinylated EV71 5′UTR. Streptavidin beads were used in the pulldown assay, and the complex was dissolved for Western blot analysis. The amount of “input” was exactly the same (100%) as the total amount of material used in the binding reaction mixture. The positions of molecular mass markers (M) (in kilodaltons) are indicated to the left of the blot.
Relocalization of Sam68 to the cytoplasm during EV71 infection.
Sam68 is normally localized in the nucleus, while EV71 replication occurs in the cytoplasm. The above specific interaction of Sam68 with the EV71 5′UTR plus the fact that some viruses in the Picornaviridae family, such as FMDV, poliovirus, and rhinovirus, have been shown to trigger the accumulation of Sam68 in the cytoplasm during infection (30, 31) led us to investigate whether the intracellular location of Sam68 changes as a result of EV71 infection. We compared the distributions of Sam68 in mock-infected and EV71-infected cells at defined times 8 h postinfection (hpi). RD cells and U251 cells were infected with EV71 at an MOI of 3, and their subcellular distribution of the Sam68 protein was analyzed by fluorescence confocal microscopy (at 8 hpi). EV71-infected cells were identified by fluorescence staining with antibody against EV71, and DAPI was used to stain the nuclei. The results showed that in the absence of EV71 infection, Sam68 protein was mainly localized in the nuclei of the cells, while EV71 infection redistributed the Sam68 protein to the cytoplasm, both in RD and U251 cells (Fig. 4A). The percentage of cells with Sam68 relocalization was increased to 47% in RD cells and 64% in U251 cells (Fig. 4B). These data suggested that EV71 infection induced the redistribution of Sam68 from the nucleus to the cytoplasm and that Sam68 was retained in the cytoplasm during EV71 infection.
FIG 4.
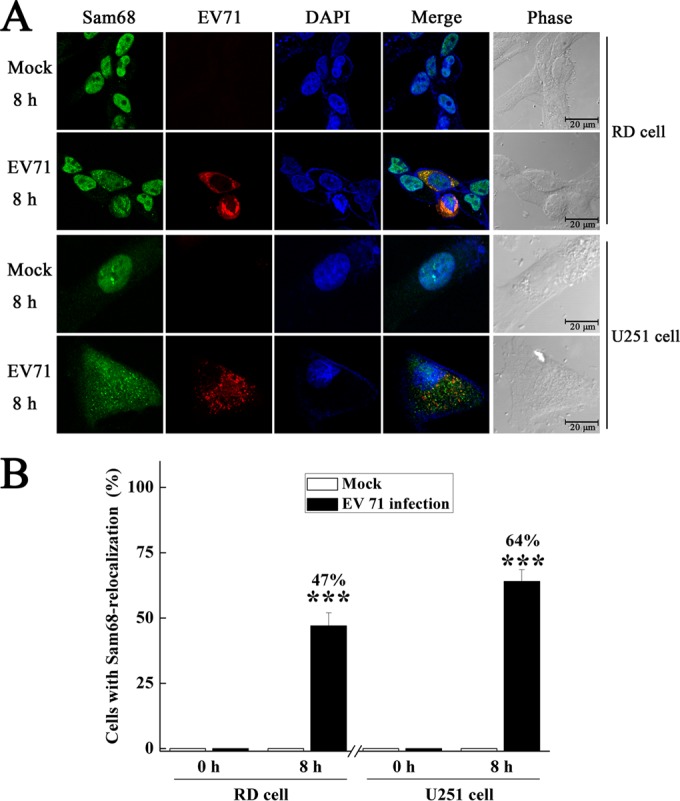
Redistribution of Sam68 to the cytoplasm of EV71-infected cells. RD cells or U251 cells were mock infected or infected with EV71 at an MOI of 3 for 8 h and then fixed and stained with antibodies directed against Sam68 and viral protein VP1. The leftmost or first column (Sam68) shows cells examined with a FITC filter. The second column (EV71) shows cells examined with a TRITC filter. The middle column presents staining of the same field with a 4′,6′-diamidino-2-phenylindole (DAPI) filter. The fourth column (Merge) shows the merged FITC, TRITC, and DAPI fields. The last or rightmost column presents the phase of the cells. (B) Quantification of cells with Sam68 relocalization either mock infected or infected with EV71 in panel A, which was expressed as a percentage of the total number of cells. Values are means plus standard deviations (error bars). Values that are significantly different (P < 0.001) from the value for mock-infected cells by Student's t test are indicated (***).
Sam68 interacts with cellular PCBP2 and PABP.
PCBP2 and PABP have previously been demonstrated to interact with portions of the EV71 genome (21) and the genomes of other picornaviruses during RNA replication (20, 41). In our study, we tested whether Sam68 could interact with PCBP2 and PABP to facilitate virus replication during virus infection. As shown in Fig. 5A, confocal microscopy analysis revealed that PCBP2 was predominantly perinuclear cytoplasmic with some nuclear fluorescence in mock-infected cells. However, PCBP2 was mainly redistributed to the cytoplasm with little to no nuclear fluorescence at 5 hpi. The change in subcellular distribution of PCBP2 was mirrored by Sam68. At the same time, some overlap (fluorescent puncta) was observed in EV71-infected cells, which implies that there may be some interaction between Sam68 and PCBP2 during virus infection. Unlike PCBP2, PABP was exclusively cytoplasmic in the cells tested whether the cells were infected with EV71 or mock infected. Although the distribution of PABP in the cytoplasm was not altered during EV71 infection, the diffuse PABP-specific fluorescence coalesced into discrete puncta at 5 hpi. Overlap of the PABP-specific puncta with Sam68-specific cytoplasmic puncta in EV71-infected cells was also observed (Fig. 5B). Given the data from the confocal microscopy experiments, we also investigated whether Sam68 coprecipitated with cellular proteins PCBP2 and PABP in the context of EV71 infection. Immunoprecipitation reactions were conducted with protein A/G beads coupled with anti-Sam68 in EV71-infected HeLa cell lysates probed for the presence of cellular proteins using anti-PABP or anti-PCBP2 antibody. As shown in Fig. 5C, immunoprecipitation reactions using anti-Sam68 coprecipitated PABP as early as 6 hpi at an MOI of 10. In contrast, coimmunoprecipitation experiments using anti-Sam68 antibodies could precipitate PCBP2 at any time point tested in EV71-infected cells. Despite almost no overlap of fluorescence observed between Sam68 and PCBP2 in mock-infected cells, Sam68 could coprecipitate with PCBP2 in mock-infected cells, which indicates that Sam68 does indeed interact with endogenous PCBP2 in basal conditions.
FIG 5.
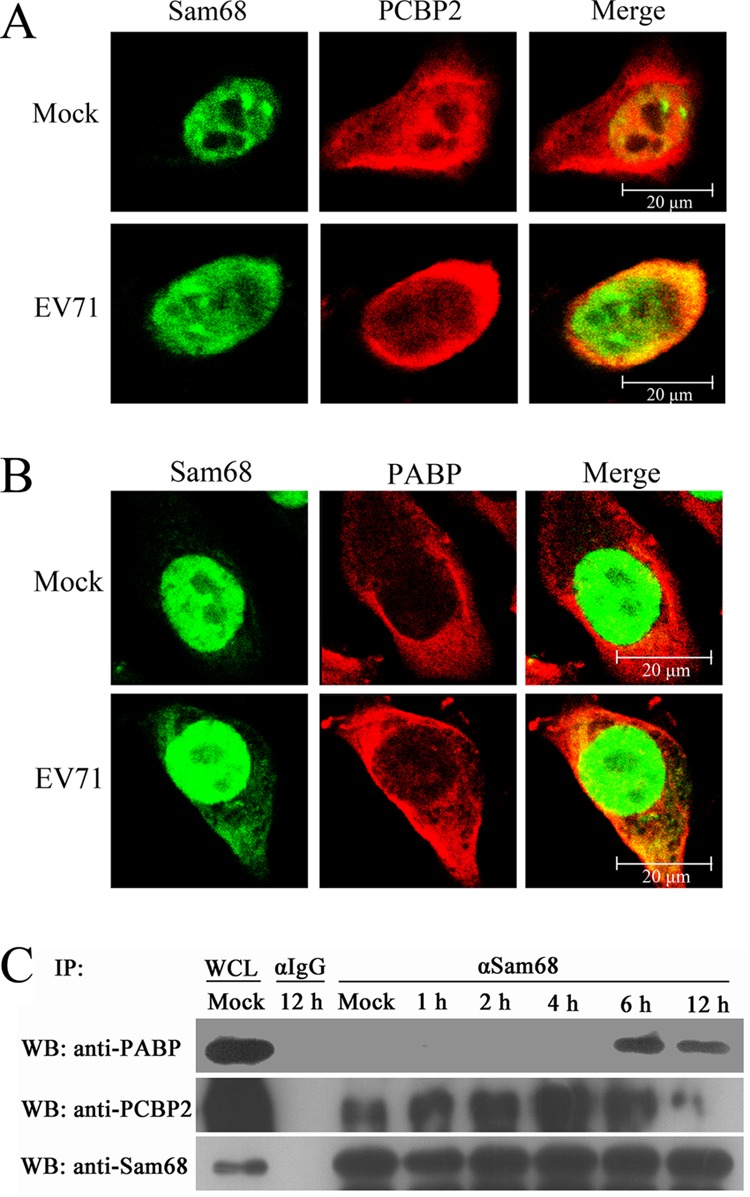
Sam68 overlaps with PCBP2 and PABP. Mock-infected and EV71-infected HeLa cells at 5 hpi were simultaneously probed with mouse anti-PCBP2 (A), mouse anti-PABP (B), and rabbit anti-Sam68 antibodies, followed by goat anti-rabbit secondary antibodies conjugated to FITC (green) and goat anti-mouse secondary antibodies conjugated to TRITC (red). Nuclear material was stained with DAPI (blue). (C) Sam68 coprecipitates with PCBP2 and PABP. Mock-infected HeLa cells or cells infected with EV71 at an MOI of 10 were harvested at 1, 2, 4, 6, and 12 hpi. Whole-cell lysates (WCL) were immunoprecipitated (IP) with agarose beads coupled to anti-Sam68 antibody (α-Sam68). Bound protein was eluted using a low-pH solution (pH 2.5), and the collected samples were analyzed by Western blotting (WB) probing with anti-PCBP2 and anti-PABP.
Studies on the association of Sam68 with EV71 5′UTR in EV71-infected cells.
To determine whether Sam68 associates with the EV71 5′UTR in EV71-infected cells, RD cells or HeLa cells incubated with EV71 at an MOI of 3 for 8 h were examined. The cells were lysed, and the RNA-Sam68 protein complexes were immunoprecipitated by either antibody specific to Sam68 or isotype anti-IgG. The RNA isolated from these immunoprecipitates was subjected to RT-PCRs using primers specific to either EV71 5′UTR or RPS16. The results showed that immunoprecipitation with Sam68 antibodies coprecipitated EV71 5′UTR, but not RPS16 (Fig. 6), confirming that Sam68 specifically associated with EV71 5′UTR in EV71-infected cells. No RT-PCR band representing either EV71 5′UTR or RPS16 was detected in immunoprecipitates obtained with the isotype IgG antibody or without antibody. Similarly, using H2O as a template in the RT-PCR negative control also did not produce any specific band. The results in Fig. 6 and 1 demonstrated that Sam68 specifically associates with EV71 5′UTR both in vivo and in vitro, respectively.
FIG 6.
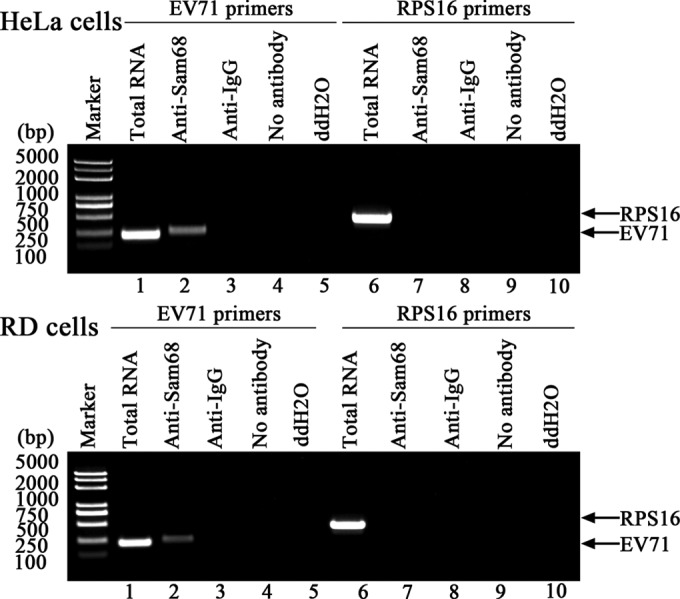
The 5′UTR of EV71 was pulled down with Sam68 from EV71-infected cell extracts. After HeLa cells or RD cells were infected with EV71 at an MOI of 3 for 12 h, the cell extract was collected and immunoprecipitated with either anti-Sam68 antibody or isotype anti-IgG. Following washing and dissociation, the RNA was extracted using TRIzol reagent and subjected to RT-PCR using primers that were specific to either EV71 5′UTR or ribosomal protein S16 (RPS16). RNA was also extracted from cell lysates without immunoprecipitation (lanes 1 and 6) and used as RT-PCR controls. Anti-Sam68 antibody was incubated with 100 mg of infected-cell lysate and then subjected to RNA extraction and RT-PCR analysis (lanes 2 and 7). Corresponding precipitations with isotype anti-IgG or without antibody were performed and used as negative controls (lanes 3, 4, 8, and 9). Double-distilled H2O was substituted as a template and used as a RT-PCR negative control (lanes 5 and 10). Sam68 protein binds to the EV71 5′UTR, but not to the control RNA (RPS16), suggesting that the binding is specific.
Effects of Sam68 expression levels on EV71 protein synthesis.
Sam68 is a multifunctional RNA-binding protein that has been implicated in both transcriptional and posttranscriptional regulation of gene expression (42, 43). Sam68 also plays important roles in the life cycle of FMDV and participates in the translation of virus proteins (31). These facts led us to investigate the importance of Sam68 in the life cycle of EV71, especially in synthesis of virus protein. Knockdown of Sam68 expression using Sam68 shRNA and overexpression of Sam68 in RD cells were employed for these studies. The cells were mock infected or infected with EV71 at an MOI of 1 for 24 h at 37°C. Subsequently, the samples were harvested, and the resulting cell lysates were analyzed by Western blotting and probing with anti-Sam68, anti-EV71 VP1, and anti-β-actin antibodies. The results obtained with anti-Sam68 antibody confirmed that Sam68 shRNA reduced the endogenous concentration of Sam68, while it was overexpressed in RD cells transfected with Flag-Sam68 (Fig. 7). The blot, which was probed with anti-EV71 VP1 antibody to determine whether the reduction or increase of Sam68 affected the production of the virus protein, showed that Sam68 shRNA constructs negatively impacted the production of a virus protein such that it was not detected on Western blots compared to cells transfected with control shRNA or untransfected cells (Fig. 7A, top right Western blots). In contrast, overexpression of Sam68 increased the synthesis of virus protein (Fig. 7B, top right Western blots). In a separate experiment to determine the effects of knockdown or overexpression of Sam68 on virus production in RD cells, the results showed that transfection of Sam68 shRNA molecules reduced the TCID50 by approximately 2 log units relative to cells transfected with control shRNA constructs (Fig. 7A, right graph). Conversely, overexpression of Sam68 significantly increased the TCID50 by 2 log units in RD cells (Fig. 7B, right graph). Similar results were obtained when these experiments were repeated in HeLa cells (Fig. 7A and B, left graphs). These results led us to conclude that Sam68 plays a vital role in the synthesis of EV71 protein, as evidenced from the strong effects that perturbations in Sam68 expression had on the levels of virus protein and on virus titer.
FIG 7.

Effects of downregulation and upregulation of Sam68 on EV71 replication. (A) HeLa cells and RD cells were transfected with either Sam68 shRNA, control shRNA, or no RNA for 72 h and then infected with EV71 at an MOI of 1 for 24 h. The concentrations of endogenous Sam68 and virus VP1 in each sample were evaluated by Western blotting probing with anti-Sam68 and anti-EV71 VP1 antibody. The Western blot was also probed with anti-β-actin antibody to normalize loading between lanes. The supernatants were collected, and the virus titers were determined using the plaque assay previously described. (B) HeLa cells and RD cells were transfected with either Flag-EV or Flag-Sam68 or without Flag for 48 h and then infected with EV71 (MOI of 1) for 24 h. The expression levels of Sam68 and EV71 VP1, as well as the virus titers, were determined as described above for panel A.
Studies on the role of Sam68 on IRES-dependent translation in EV71-infected cells.
IRES-mediated initiation of translation allows the translation of the virus RNA upon EV71 infection (39). Other studies have also shown that Sam68 can significantly enhance the translation of retrovirus genes by “marking” the viral RNA transcripts (32, 44–46). In order to investigate the role of Sam68 on IRES-dependent translation in EV71-infected cells, the effects of knockdown and overexpression of Sam68 on EV71 IRES activity in HeLa and RD cells transfected with either pRHF or pRHF-5′UTR were studied using the dual-luciferase reporter assay kit described in Materials and Methods. The results showed that knockdown of endogenous Sam68 protein with Sam68 shRNA decreased the EV71 IRES activity to 58% (P < 0.01) of that of the shRNA control in HeLa cells and to 53% (P < 0.01) of the control in RD cells (Fig. 8A). In contrast, overexpression of Sam68 protein significantly increased EV71 IRES activity both in HeLa cells to 147% (P < 0.01) of that of the control cells and in RD cells to 152% (P < 0.01) of that of the control cells. The CMV-driven transcript is fundamentally distinct from the viral transcription product, which led us to perform direct transfection of bicistronic reporter mRNA. The results of direct transfection of bicistronic reporter mRNA indicated that Sam68 knockdown decreased the EV71 IRES activity to 79% (P < 0.01) of that of the shRNA control in HeLa cells and to 76% (P < 0.01) of the control in RD cells (Fig. 8B). In contrast, overexpression of Sam68 protein significantly increased EV71 IRES activity in HeLa cells to 121% (P < 0.01) of that of the control cells and in RD cells to 125% (P < 0.01) of that of the control cells. In addition, monocistronic mRNAs that contained the EV71 5′UTR were alternatively transfected into cells, and the FLuc activity was measured after cells were either knocked down or overexpressed. The knockdown of endogenous Sam68 protein decreased EV71 IRES activity to 53% (P < 0.01) of that of the control in HeLa cells and to 51% (P < 0.01) of that of the control in RD cells. The overexpression of Sam68 increased EV71 IRES activity to 133% (P < 0.01) of that of the control in HeLa cells and to 141% (P < 0.01) of that of the control in RD cells (Fig. 8C). These data suggested that the nuclear protein Sam68 positively regulated the translation of EV71 RNA in an IRES-dependent manner during EV71 infection.
FIG 8.

Effects of downregulation and upregulation of Sam68 on EV71 IRES activity. (A) Schematic diagram of bicistronic reporter plasmids pRHF and pRHF-5′UTR. Plasmid expresses bicistronic mRNA, consisting of the RLuc gene at the first cistron and the EV71-5′UTR and the FLuc gene at the second cistron (CMV). A hairpin is inserted downstream of the first cistron to prevent ribosome read-through. HeLa cells and RD cells were either transfected with control shRNA or Sam68 shRNA and screened in 2 μg/ml puromycin or transfected with Flag-EV or Flag-Sam68 and screened in 400 μg/ml G418. The pRHF or pRHF-5′UTR plasmid was then transfected into the cells. At 48 h after cotransfection, the RLuc and FLuc activities in cell lysates were analyzed. The bars in the histogram represent FLuc/RLuc activity (as percentages). Experiments were performed in triplicate to obtain the bar graph. Western blotting was utilized to analyze the expression levels of Sam68 and cellular β-actin. (B) Schematic diagram of capped bicistronic mRNAs containing a poly(A) tail. The FLuc/RLuc activities and expression levels of Sam68 were determined as described above for panel A. (C) Schematic diagram of monocistronic reporter plasmids EV71 5′UTR-FLuc. Cells were transfected with EV71 5′UTR-FLuc mRNA, and FLuc activity was assayed as described above for panel A. Values that are significantly different (P < 0.01) by Student's two-tailed unpaired t test are indicated (**).
Effects of downregulation of Sam68 on the replication of EV71 genome.
To examine whether Sam68 plays any roles in viral RNA synthesis, RD cells and HeLa cells were transfected for 48 h with either Sam68 shRNA or control shRNA or not transfected and then infected with EV71 at an MOI of 3. Viral RNA was extracted at various times postinfection, and RT-PCR using a specific primer against EV71 VP1 gene was employed to measure viral RNA synthesis. Cellular β-actin RNA was also amplified to serve as an internal control. The results showed that although Sam68 was significantly knocked down by its specific Sam68 shRNA compared to cells treated with control shRNA, the amount of EV71 genome in the Sam68 shRNA-treated cells was similar to those in the shRNA-treated and untransfected control cells (Fig. 9A). To further identify a role of Sam68 in EV71 genome replication, the EV71 mRNA level in each sample (at 24 hpi) was measured by qRT-PCR and normalized to that of cellular β-actin. The results showed that neither the EV71 mRNA levels in the control shRNA nor that of the untransfected cells differed from that of Sam68 shRNA-treated cells (Fig. 9B). Both these results suggested that silencing of Sam68 did not have any significant inhibitory effect on the accumulation of EV71 genome.
FIG 9.
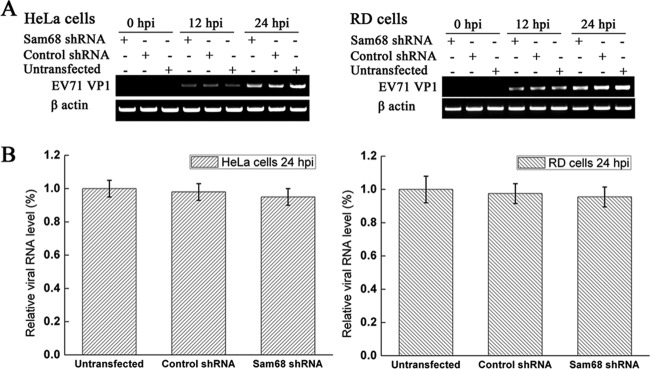
Effects of downregulation of Sam68 on the replication of the EV71 genome. (A) HeLa cells and RD cells were untransfected or transfected with the indicated Sam68 shRNAs or control shRNA for 48 h. Cells were then either mock infected or infected with EV71 (MOI of 3) for 24 h, and then the cellular RNA and viral genome were extracted. EV71 VP1 mRNA was measured by RT-PCR (semi-qPCR) for 20 cycles, and cellular β-actin mRNA levels were used as loading controls. (B) EV71 VP1 mRNA was quantified by qRT-PCR at 24 hpi, and cellular β-actin mRNA was used as an internal control. The mRNA level of EV71 VP1 was calculated relative to mRNA of β-actin gene, and the value of the untransfected cells was set at 1. Data were from three independent experiments, and the values are means ± standard deviations (SD) (error bars).
DISCUSSION
As a consequence of the limited coding capacity of its genome, a virus has to use a variety of cellular proteins and alter its cellular location frequently to complete its life cycle (21, 39, 47). Knowledge of the interactions between a virus and its host factors could offer insights into viral and cellular functions and provide new antiviral targets for disease control. The identification of such interactions and the associated host factors is thus a major frontier in virology (48, 49). In this study, we provided new evidence indicating that the host factor, nuclear protein Sam68, was relocalized to the cytoplasm during EV71 infection and interacted with the IRES of EV71 5′UTR. Sam68 has almost no effect on the replication of the virus genome. However, knockdown of Sam68 repressed the synthesis of virus protein and reduced EV71 titer. Conversely, overexpression of Sam68 increased the expression of virus protein and EV71 yield, suggesting that Sam68 might be a host factor involved in the positive regulation of IRES-dependent translation of EV71 RNA during infection. These observations, in conjunction with previous reports demonstrating that FMDV (31) and poliovirus (30) induced the redistribution of Sam68 and its involvement in the life cycles of these viruses suggested that Sam68 might have two roles in EV71 infection: Sam 68 may play one role by positively regulating viral RNA translation to support virus replication when redistributed to cytoplasm (Fig. 10) and may have a second role involved in signal transduction due to its protein- and RNA-binding properties (50).
FIG 10.
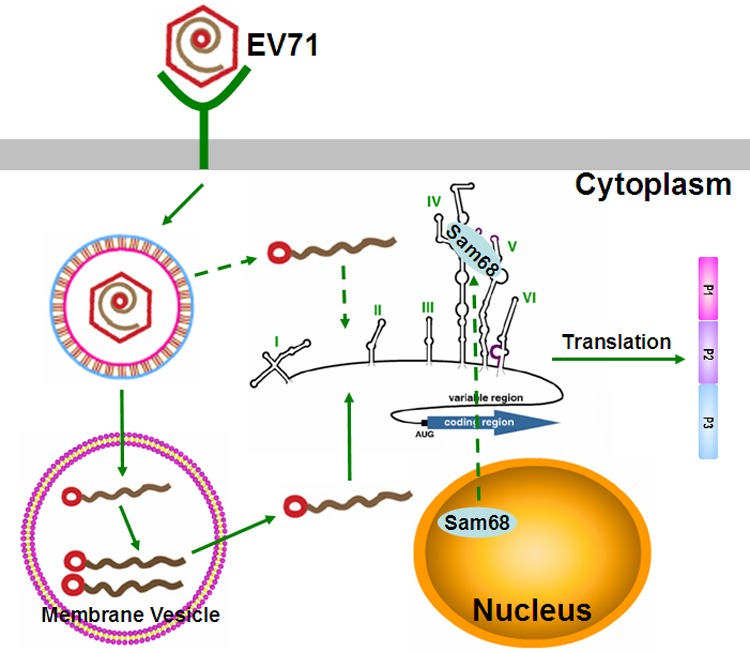
Schematic representation of the nuclear protein Sam68 involved in viral protein translation. EV71 binds to a cellular receptor, and the genome is released into the cytoplasm. EV71 infection triggered the redistribution of Sam68 to the cytoplasm, and then Sam68 binds to 5′UTR of genomic RNA and positively regulates EV71 IRES activity to promote the synthesis of EV71 proteins. The polyprotein is co- and posttranslationally processed to produce the various precursors and processed proteins that are needed for EV71's life cycle.
During EV71 infection, the viral proteases 2A and 3C cleave cellular proteins, including the translation initiation factor eIF4G, causing rapid termination of the host's cap-dependent translation and initiation of IRES-mediated viral RNA translation (51). IRES-dependent translation depends on both canonical translation initiation factors and IRES-specific trans-acting factors (ITAFs) which interact with various IRES elements to regulate their activities by affecting ribosome recruitment or modifying the structure of the IRES itself (39, 47, 52). Sam68 can now be added to the list of noncanonical host factors already reported that interact with the IRES elements of various picornaviruses (13, 53), such as FMDV. In order to investigate regulation of the EV71 IRES-dependent translation by Sam68, the FLuc reporter plasmid was used to evaluate the EV71 IRES activity. The FLuc/RLuc ratio obtained following DNA transfection may also be related to the use of a cryptic promoter in the IRES sequence. The results of EV71 IRES activity were consistent with those of a bicistronic construct in FMDV (31). With regard to this study, we found that Sam68 specifically interacted with two stem-loop regions of the EV71 IRES (Fig. 2B). More importantly, the interaction between Sam68 and EV71 IRES made some positive contribution to IRES activity. This can be interpreted from the decreased IRES-driven reporter gene expression in both HeLa cells and RD cells, along with the reduced cellular levels of Sam68. Our findings from the in vivo assay suggested that overexpression of Sam68 could significantly enhance the synthesis of EV71 virus protein and virus titer. The significant reduction in virus protein and titer resulting from the knockdown of available Sam68 implied that the roles of Sam68 in the EV71 life cycle may not be limited to augmentation of viral RNA translation. In addition to stem-loop V (nt 453 to 561), Sam68 also interacted with nt 242 to 445 of the EV71 5′UTR. Based on the RNA secondary structure predicted by Mfold software, nt 242 to 445 may form stem-loop IV within the IRES, which is crucial for viral protein synthesis. All the results revealed that knocking down Sam68 expression reduced EV71 IRES activity, while the upregulation of Sam68 expression increased EV71 IRES activity (Fig. 8). These results further suggested that Sam68 is involved in IRES-dependent translation of virus protein.
It has been reported that Sam68 takes part in the formation of both nuclear and cytosolic multimolecular complexes with viral RNA (26, 32, 46, 54) and that Sam68 could directly interact with the 40S ribosomal protein S3a (RPS3A) and the 60S acidic ribosomal protein P0 (RPLP0) (55). The translation initiation factor eIF3 and the ribosomal proteins L7, S3, S4, and S7 could also be involved in the mechanism of Sam68-dependent regulation of mRNA translation (56–58). In addition, we found that Sam68 could interact with cellular factors, such as PCBP2 and PABP, to facilitate virus replication. We therefore wonder whether Sam68 facilitates intramolecular annealing reactions in EV71 IRES for the latter to form a proper and stable structure required for the binding of the 40S ribosome and thus promotes the IRES-dependent translation of the EV71 genome. The formation of such a structure may also have a role in recruiting cellular proteins needed for the replication of EV71.
After confirming the interaction between EV71 5′UTR and Sam68 in vitro by an EMSA, their association with each other in vivo was further demonstrated. Our work utilized a biotinylated RNA-protein pulldown assay to provide a comprehensive view of different truncated isoforms of Sam68 that were associated with the EV71 5′UTR. The streptavidin beads captured the biotinylated EV71 5′UTR and its associated full-length Sam68 (nt 1 to 443), as well as one truncated form of Sam68 (nt 96 to 257), but not the other truncated forms tested. It demonstrated that the binding of Sam68 to EV71 5′UTR was specific. The binding of Sam68 to biotinylated EV71 5′UTR was not competitively eliminated by nonspecific RNA. Our results suggested that Sam68 interacted with EV71 5′UTR via regions that at least included the KH, NK, and CK RNA-binding domains. Although the KH domain is conserved in the STAR proteins and is the major determinant for binding to RNA (59), the NK region and CK region are requisite for the binding of Sam68 to EV71 5′UTR. In order to further investigate the binding of Sam68 to EV71 5′UTR during virus infection, immunoprecipitates were pulled down from EV71-infected cells with anti-Sam68 antibody and then subjected to RT-PCR to detect the existence of the 5′UTR. As expected, immunoprecipitation with anti-Sam68 antibodies coprecipitated EV71 5′UTR, but not the control RNA (RPS16), suggesting that the association of Sam68 with EV71 5′UTR in EV71-infected cells was specific. These results confirmed that Sam68 was associated with EV71 5′UTR through a distinct interaction both in vitro and in vivo.
Sam68 is a nuclear RNA-binding protein implicated in various aspects of RNA metabolism, including splicing, nuclear export, somatodendritic transport, and translation (32, 43). Sam68 is localized primarily in the nucleus and dictated by a nonconventional NLS embedded in the last 24 amino acids of the C-terminal region of the polypeptide (29). However, EV71 replication occurs in the cytoplasm and is dependent on multiple host factors. Since the Sam68 protein accumulates mostly in the nuclear compartment, an important question is how could a nucleus-localized host protein interact with EV71 5′UTR in the cytoplasm and modulate virus replication. This led us to investigate the possibility for the redistribution of Sam68 to the cytoplasm and its involvement in the virus life cycle. Similar to other picornavirus infections, such as those of FMDV (31) and poliovirus (30), our work indeed revealed that Sam68 was localized mainly in the nucleus in mock-infected cells, whereas the protein was found mainly in the cytoplasm of EV71-infected cells at 8 hpi (Fig. 4). This is consistent with previous reports showing that other host factors, such as hnRNP K (21), FBP1, FBP2 (47), and hnRNP A1 (39), were found to redistribute from the nucleus to the cytoplasm and associate with the EV71 IRES following infection with EV71. The ability of a host factor to control IRES-dependent translation initiation is dependent on its subcellular localization (60). Thus, we hypothesized that the redistribution of Sam68 to the cytoplasm is an absolute requirement for IRES-dependent translation. Therefore, we conclude that Sam68 promotes EV71 replication via binding to the 5′UTR and positively regulates translation of virus protein.
Although the current data provide a hypothetical model for the “hijacking” of Sam68 by EV71, many questions remain unanswered, and the model will certainly undergo further refinement. The exact mechanism of redistribution of Sam68 has yet to be elucidated. It would be of interest to further determine whether EV71 infection affects the nuclear import and/or export signaling pathways that control the subcellular redistribution of Sam68. Although we found that Sam68 specifically interacted with two regions of the EV71 IRES, nt 242 to 445 and nt 453 to 561, with the KH domain, the precise mechanism by which Sam68 enhances the translation of virus protein needs to be delineated. In addition, considering the diverse cellular functions attributed to Sam68 as well as the significant reduction in virus amplification in cells treated with Sam68 shRNA constructs, it is likely that the “hijacked” Sam68 may fulfill multiple distinct roles in the life cycle of EV71, all of which need further investigation.
In conclusion, we have identified the nuclear protein Sam68 as an additional new host factor that is redistributed to the cytoplasm during EV71 infection. These data indicated that the binding of Sam68 to the EV71 IRES during infection could potentially enhance the translation of virus protein. To our knowledge, this is the first report that describes Sam68 actively participating in the life cycle of EV71 in molecular details. These studies will not only improve our understanding of the replication of EV71 but they also have the potential to help develop a therapeutic strategy against EV71 infection.
ACKNOWLEDGMENTS
This work was supported by grants from the National Basic Research Program of China (973 Program) (grants 2011CB504703 and 2010CB530102) and the National Natural Science Foundation of China (NSFC) (grants 81321063, 31370201, 31300145, and 31270211).
We are grateful to Paul Chu (Institute of Microbiology, Chinese Academy of Sciences) for critical reading of the manuscript. We also thank Fulian Liao for assistance with cell culture, Xiaolan Zhang for assistance with confocal microscope analysis, and Weihua Zhuang for kindly helping with manuscript preparation.
We declare that we have no conflicts of interest.
REFERENCES
- 1.Kushner D, Caldwell BD. 1996. Hand foot-and-mouth disease. J Am Podiat Med Assn 86:257–259. doi: 10.7547/87507315-86-6-257. [DOI] [PubMed] [Google Scholar]
- 2.Miller GD, Tindall JP. 1968. Hand-foot-and-mouth disease. JAMA 203:827–830. [PubMed] [Google Scholar]
- 3.Sato C, Syoji M, Ueki Y, Sato Y, Okimura Y, Saito N, Kikuchi N, Yagi T, Numakura H. 2006. Isolation of enterovirus 71 from patients with hand, foot and mouth disease in a local epidemic on March 2006, in Miyagi prefecture, Japan. Jpn J Infect Dis 59:348. [PubMed] [Google Scholar]
- 4.Cardosa MJ, Perera D, Brown BA, Cheon D, Chan HM, Chan KP, Cho H, McMinn P. 2003. Molecular epidemiology of human enterovirus 71 strains and recent outbreaks in the Asia-Pacific region: comparative analysis of the VP1 and VP4 genes. Emerg Infect Dis 9:461–468. doi: 10.3201/eid0904.020395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McMinn P, Lindsay K, Perera D, Chan HM, Chan KP, Cardosa MJ. 2001. Phylogenetic analysis of enterovirus 71 strains isolated during linked epidemics in Malaysia, Singapore, and Western Australia. J Virol 75:7732–7738. doi: 10.1128/JVI.75.16.7732-7738.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown BA, Oberste MS, Alexander JP, Kennett ML, Pallansch MA. 1999. Molecular epidemiology and evolution of enterovirus 71 strains isolated from 1970 to 1998. J Virol 73:9969–9975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van der Sanden S, van Eek J, Martin DP, van der Avoort H, Vennema H, Koopmans M. 2011. Detection of recombination breakpoints in the genomes of human enterovirus 71 strains isolated in the Netherlands in epidemic and non-epidemic years, 1963-2010. Infect Genet Evol 11:886–894. doi: 10.1016/j.meegid.2011.02.011. [DOI] [PubMed] [Google Scholar]
- 8.Fujimoto T, Yoshida S, Munemura T, Taniguchi K, Shinohara M, Nishio O, Chikahira M, Okabe N. 2008. Detection and quantification of enterovirus 71 genome from cerebrospinal fluid of an encephalitis patient by PCR applications. Jpn J Infect Dis 61:497–499. [PubMed] [Google Scholar]
- 9.Wang X, Zhu JP, Zhang Q, Xu ZG, Zhang F, Zhao ZH, Zheng WZ, Zheng LS. 2012. Detection of enterovirus 71 using reverse transcription loop-mediated isothermal amplification (RT-LAMP). J Virol Methods 179:330–334. doi: 10.1016/j.jviromet.2011.11.019. [DOI] [PubMed] [Google Scholar]
- 10.Yamayoshi S, Yamashita Y, Li JF, Hanagata N, Minowa T, Takemura T, Koike S. 2009. Scavenger receptor B2 is a cellular receptor for enterovirus 71. Nat Med 15:798–801. doi: 10.1038/nm.1992. [DOI] [PubMed] [Google Scholar]
- 11.Nishimura Y, Shimojima M, Tano Y, Miyamura T, Wakita T, Shimizu H. 2009. Human P-selectin glycoprotein ligand-1 is a functional receptor for enterovirus 71. Nat Med 15:794–797. doi: 10.1038/nm.1961. [DOI] [PubMed] [Google Scholar]
- 12.Bedard KM, Semler BL. 2004. Regulation of picornavirus gene expression. Microbes Infect 6:702–713. doi: 10.1016/j.micinf.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 13.Lin JY, Chen TC, Weng KF, Chang SC, Chen LL, Shih SR. 2009. Viral and host proteins involved in picornavirus life cycle. J Biomed Sci 16:103. doi: 10.1186/1423-0127-16-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pilipenko EV, Viktorova EG, Guest ST, Agol VI, Roos RP. 2001. Cell-specific proteins regulate viral RNA translation and virus-induced disease. EMBO J 20:6899–6908. doi: 10.1093/emboj/20.23.6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zoll J, Heus HA, van Kuppeveld FJM, Melchers WJG. 2009. The structure-function relationship of the enterovirus 3′-UTR. Virus Res 139:209–216. doi: 10.1016/j.virusres.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 16.Paul AV, Yang CF, Jang SK, Kuhn RJ, Tada H, Nicklin M, Krausslich HG, Lee CK, Wimmer E. 1987. Molecular events leading to poliovirus genome replication. Cold Spring Harb Symp 52:343–352. doi: 10.1101/SQB.1987.052.01.039. [DOI] [PubMed] [Google Scholar]
- 17.Wimmer E, Kuhn RJ, Pincus S, Yang CF, Toyoda H, Nicklin MJH, Takeda N. 1987. Molecular events leading to picornavirus genome replication. J Cell Sci Suppl 7:251–276. [DOI] [PubMed] [Google Scholar]
- 18.Wimmer E, Nomoto A. 1993. Molecular biology and cell-free synthesis of poliovirus. Biologicals 21:349–356. doi: 10.1006/biol.1993.1095. [DOI] [PubMed] [Google Scholar]
- 19.Gamarnik AV, Andino R. 2000. Interactions of viral protein 3CD and poly(rC) binding protein with the 5′ untranslated region of the poliovirus genome. J Virol 74:2219–2226. doi: 10.1128/JVI.74.5.2219-2226.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herold J, Andino R. 2001. Poliovirus RNA replication requires genome circularization through a protein-protein bridge. Mol Cell 7:581–591. doi: 10.1016/S1097-2765(01)00205-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin JY, Li ML, Huang PN, Chien KY, Horng JT, Shih SR. 2008. Heterogeneous nuclear ribonuclear protein K interacts with the enterovirus 71 5′ untranslated region and participates in virus replication. J Gen Virol 89:2540–2549. doi: 10.1099/vir.0.2008/003673-0. [DOI] [PubMed] [Google Scholar]
- 22.Yang HP, Reddy TR, Truong KT, Suhasini M, Wong-Staal F. 2002. Functional interaction of Sam68 and heterogeneous nuclear ribonucleoprotein K. Oncogene 21:7187–7194. doi: 10.1038/sj.onc.1205759. [DOI] [PubMed] [Google Scholar]
- 23.Fumagalli S, Totty NF, Hsuan JJ, Courtneidge SA. 1994. A target for Src in mitosis. Nature 368:871–874. [DOI] [PubMed] [Google Scholar]
- 24.Burd CG, Dreyfuss G. 1994. Conserved structures and diversity of functions of RNA-binding proteins. Science 265:615–621. doi: 10.1126/science.8036511. [DOI] [PubMed] [Google Scholar]
- 25.Paronetto MP, Cappellari M, Busa R, Pedrotti S, Vitali R, Comstock C, Hyslop T, Knudsen KE, Sette C. 2010. Alternative splicing of the cyclin D1 proto-oncogene is regulated by the RNA-binding protein Sam68. Cancer Res 70:229–239. doi: 10.1158/0008-5472.CAN-09-2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soros VB, Carvajal HV, Richard S, Cochrane AW. 2001. Inhibition of human immunodeficiency virus type 1 Rev function by a dominant-negative mutant of Sam68 through sequestration of unspliced RNA at perinuclear bundles. J Virol 75:8203–8215. doi: 10.1128/JVI.75.17.8203-8215.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matter N, Herrlich P, Konig H. 2002. Signal-dependent regulation of splicing via phosphorylation of Sam68. Nature 420:691–695. doi: 10.1038/nature01153. [DOI] [PubMed] [Google Scholar]
- 28.Macias MJ, Wiesner S, Sudol M. 2002. WW and SH3 domains, two different scaffolds to recognize proline-rich ligands. FEBS Lett 513:30–37. doi: 10.1016/S0014-5793(01)03290-2. [DOI] [PubMed] [Google Scholar]
- 29.Ishidate T, Yoshihara S, Kawasaki Y, Roy BC, Toyoshima K, Akiyama T. 1997. Identification of a novel nuclear localization signal in Sam68. FEBS Lett 409:237–241. doi: 10.1016/S0014-5793(97)00455-9. [DOI] [PubMed] [Google Scholar]
- 30.McBride AE, Schlegel A, Kirkegaard K. 1996. Human protein Sam68 relocalization and interaction with poliovirus RNA polymerase in infected cells. Proc Natl Acad Sci U S A 93:2296–2301. doi: 10.1073/pnas.93.6.2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lawrence P, Schafer EA, Rieder E. 2012. The nuclear protein Sam68 is cleaved by the FMDV 3C protease redistributing Sam68 to the cytoplasm during FMDV infection of host cells. Virology 425:40–52. doi: 10.1016/j.virol.2011.12.019. [DOI] [PubMed] [Google Scholar]
- 32.He JJ, Henao-Mejia J, Liu Y. 2009. Sam68 functions in nuclear export and translation of HIV-1 RNA. RNA Biol 6:384–386. doi: 10.4161/rna.6.4.8920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wong WR, Chen YY, Yang SM, Chen YL, Horng JT. 2005. Phosphorylation of PI3K/Akt and MAPK/ERK in an early entry step of enterovirus 71. Life Sci 78:82–90. doi: 10.1016/j.lfs.2005.04.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Andino R, Rieckhof GE, Baltimore D. 1990. A functional ribonucleoprotein complex forms around the 5′ end of poliovirus RNA. Cell 63:369–380. doi: 10.1016/0092-8674(90)90170-J. [DOI] [PubMed] [Google Scholar]
- 35.Song LB, Wang L, Li Y, Xiong HP, Wu JH, Li J, Li MF. 2010. Sam68 up-regulation correlates with, and its down-regulation inhibits, proliferation and tumourigenicity of breast cancer cells. J Pathol 222:227–237. doi: 10.1002/path.2751. [DOI] [PubMed] [Google Scholar]
- 36.Rieder E, Henry T, Duque H, Baxt B. 2005. Analysis of a foot-and-mouth disease virus type A(24) isolate containing an SGD receptor recognition site in vitro and its pathogenesis in cattle. J Virol 79:12989–12998. doi: 10.1128/JVI.79.20.12989-12998.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lau C, Wang X, Song L, North M, Wiehier S, Proud D, Chow CW. 2008. Syk associates with clathrin and mediates phosphatidylinositol 3-kinase activation during human rhinovirus internalization. J Immunol 180:870–880. doi: 10.4049/jimmunol.180.2.870. [DOI] [PubMed] [Google Scholar]
- 38.Zuker M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin JY, Shih SR, Pan MJ, Li C, Lue CF, Stollar V, Li ML. 2009. hnRNP A1 interacts with the 5′ untranslated regions of enterovirus 71 and Sindbis virus RNA and is required for viral replication. J Virol 83:6106–6114. doi: 10.1128/JVI.02476-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sanchez-Margalet V, Najib S. 1999. p68 Sam is a substrate of the insulin receptor and associates with the SH2 domains of p85 PI3K. FEBS Lett 455:307–310. doi: 10.1016/S0014-5793(99)00887-X. [DOI] [PubMed] [Google Scholar]
- 41.Lawrence P, Rieder E. 2009. Identification of RNA helicase A as a new host factor in the replication cycle of foot-and-mouth disease virus. J Virol 83:11356–11366. doi: 10.1128/JVI.02677-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Busa R, Paronetto MP, Farini D, Pierantozzi E, Botti F, Angelini DF, Attisani F, Vespasiani G, Sette C. 2007. The RNA-binding protein Sam68 contributes to proliferation and survival of human prostate cancer cells. Oncogene 26:4372–4382. doi: 10.1038/sj.onc.1210224. [DOI] [PubMed] [Google Scholar]
- 43.Fu K, Sun X, Zheng WX, Wier EM, Hodgson A, Tran DQ, Richard S, Wan FY. 2013. Sam68 modulates the promoter specificity of NF-kappaB and mediates expression of CD25 in activated T cells. Nat Commun 4:1909. doi: 10.1038/ncomms2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Coyle JH, Guzik BW, Bor YC, Jin L, Eisner-Smerage L, Taylor SJ, Rekosh D, Hammarskjold ML. 2003. Sam68 enhances the cytoplasmic utilization of intron-containing RNA and is functionally regulated by the nuclear kinase Sik/BRK. Mol Cell Biol 23:92–103. doi: 10.1128/MCB.23.1.92-103.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bolinger C, Boris-Lawrie K. 2009. Mechanisms employed by retroviruses to exploit host factors for translational control of a complicated proteome. Retrovirology 6:8–27. doi: 10.1186/1742-4690-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reddy TR, Tang HL, Xu WD, Wong-Staal F. 2000. Sam68, RNA helicase A and Tap cooperate in the post-transcriptional regulation of human immunodeficiency virus and type D retroviral mRNA. Oncogene 19:3570–3575. doi: 10.1038/sj.onc.1203676. [DOI] [PubMed] [Google Scholar]
- 47.Lin JY, Li ML, Shih SR. 2009. Far upstream element binding protein 2 interacts with enterovirus 71 internal ribosomal entry site and negatively regulates viral translation. Nucleic Acids Res 37:47–59. doi: 10.1093/nar/gkn901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grdzelishvili VZ, Garcia-Ruiz H, Watanabe T, Ahlquist P. 2005. Mutual interference between genomic RNA replication and subgenomic mRNA transcription in brome mosaic virus. J Virol 79:1438–1451. doi: 10.1128/JVI.79.3.1438-1451.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hao LH, Sakurai A, Watanabe T, Sorensen E, Nidom CA, Newton MA, Ahlquist P, Kawaoka Y. 2008. Drosophila RNAi screen identifies host genes important for influenza virus replication. Nature 454:890–895. doi: 10.1038/nature07151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang H, Cong H, Song L, Tien P. 2014. The nuclear protein Sam68 is redistributed to the cytoplasm and is involved in PI3K/Akt activation during EV71 infection. Virus Res 180:1–11. doi: 10.1016/j.virusres.2013.11.020. [DOI] [PubMed] [Google Scholar]
- 51.Thompson SR, Sarnow P. 2003. Enterovirus 71 contains a type I IRES element that functions when eukaryotic initiation factor eIF4G is cleaved. Virology 315:259–266. doi: 10.1016/S0042-6822(03)00544-0. [DOI] [PubMed] [Google Scholar]
- 52.Shih SR, Stollar V, Li ML. 2011. Host factors in enterovirus 71 replication. J Virol 85:9658–9666. doi: 10.1128/JVI.05063-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Belsham GJ, Sonenberg N. 2000. Picornavirus RNA translation: roles for cellular proteins. Trends Microbiol 8:330–335. doi: 10.1016/S0966-842X(00)01788-1. [DOI] [PubMed] [Google Scholar]
- 54.Reddy TR, Xu W, Mau JKL, Goodwin CD, Suhasini M, Tang H, Frimpong K, Rose DW, Wong-Staal F. 1999. Inhibition of HIV replication by dominant negative mutants of Sam68, a functional homolog of HIV-1 Rev. Nat Med 5:635–642. doi: 10.1038/9479. [DOI] [PubMed] [Google Scholar]
- 55.Cappellari M, Bielli P, Paronetto MP, Ciccosanti F, Fimia GM, Saarikettu J, Silvennoinen O, Sette C. 2014. The transcriptional co-activator SND1 is a novel regulator of alternative splicing in prostate cancer cells. Oncogene 33:3794–3802. doi: 10.1038/onc.2013.360. [DOI] [PubMed] [Google Scholar]
- 56.Paronetto MP, Zalfa F, Botti F, Geremia R, Bagni C, Sette C. 2006. The nuclear RNA-binding protein Sam68 translocates to the cytoplasm and associates with the polysomes in mouse spermatocytes. Mol Biol Cell 17:14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Paronetto MP, Messina V, Bianchi E, Barchi M, Vogel G, Moretti C, Palombi F, Stefanini M, Geremia R, Richard S, Sette C. 2009. Sam68 regulates translation of target mRNAs in male germ cells, necessary for mouse spermatogenesis. J Cell Biol 185:235–249. doi: 10.1083/jcb.200811138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grange J, Belly A, Dupas S, Trembleau A, Sadoul R, Goldberg Y. 2009. Specific interaction between Sam68 and neuronal mRNAs: implication for the activity-dependent biosynthesis of elongation factor eEF1A. J Neurosci Res 87:12–25. doi: 10.1002/jnr.21824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lukong KE, Richard S. 2003. Sam68, the KH domain-containing superSTAR. Biochim Biophys Acta 1653:73–86. [DOI] [PubMed] [Google Scholar]
- 60.Lewis SM, Holcik M. 2008. For IRES trans-acting factors, it is all about location. Oncogene 27:1033–1035. doi: 10.1038/sj.onc.1210777. [DOI] [PubMed] [Google Scholar]