

ABSTRACT
Development of a vaccine to prevent congenital cytomegalovirus infection is a major public health priority. Live vaccines attenuated through mutations targeting viral mechanisms responsible for evasion of host defense may be both safe and efficacious. Safety and vaccine efficacy were evaluated using a guinea pig cytomegalovirus (GPCMV) model. Recombinant GPCMV with a targeted deletion of gp145 (designated Δ145), a viral protein kinase R (PKR) inhibitor, was generated. Attenuation was evaluated following inoculation of 107 PFU of Δ145 or parental virus into guinea pigs immunosuppressed with cyclophosphamide. Efficacy was evaluated by immunizing GPCMV-naive guinea pigs twice with either 105 or 106 PFU of Δ145, establishing pregnancy, and challenging the guinea pigs with salivary gland-adapted GPCMV. The immune response, maternal viral load, pup mortality, and congenital infection rates in the vaccine and control groups were compared. Δ145 was substantially attenuated for replication in immunocompromised guinea pigs. Vaccination with Δ145 induced enzyme-linked immunosorbent assay (ELISA) and neutralizing antibody levels comparable to those achieved in natural infection. In the higher- and lower-dose vaccine groups, pup mortality was reduced to 1/24 (4%) and 4/29 (14%) pups, respectively, whereas it was 26/31 (81%) in unvaccinated control pups (P < 0.0001 for both groups versus the control group). Congenital infection occurred in 20/31 (65%) control pups but only 8/24 (33%) pups in the group vaccinated with 106 PFU (P < 0.05). Significant reductions in the magnitude of maternal DNAemia and pup viral load were noted in the vaccine groups compared to those in the controls. Deletion of a GPCMV genome-encoded PKR inhibitor results in a highly attenuated virus that is immunogenic and protective as a vaccine against transplacental infection.
IMPORTANCE Previous attempts to develop successful immunization against cytomegalovirus have largely centered on subunit vaccination against virion proteins but have yielded disappointing results. The advent of bacterial artificial chromosome technologies has enabled engineering of recombinant cytomegaloviruses (CMVs) from which virus genome-encoded immune modulation genes have been deleted, toward the goal of developing a safe and potentially more efficacious live attenuated vaccine. Here we report the findings of studies of such a vaccine against congenital CMV infection based on a virus with a targeted deletion in gp145, a virus genome-encoded inhibitor of protein kinase R, using the guinea pig model of vertical CMV transmission. The deletion virus was attenuated for dissemination in immunocompromised guinea pigs but elicited ELISA and neutralizing responses. The vaccine conferred protection against maternal DNAemia and congenital transmission and resulted in reduced viral loads in newborn guinea pigs. These results provide support for future studies of attenuated CMV vaccines.
INTRODUCTION
Infection with human cytomegalovirus (HCMV) causes considerable morbidity and occasional mortality in immunocompromised solid organ transplant and hematopoietic stem cell transplant patients, HIV-infected individuals, and newborns that acquire infection in utero (1, 2). Due to the lifelong morbidity associated with congenital CMV infection, a preconception vaccine capable of preventing virus transmission to the fetus would provide a highly cost-effective public health advance (3). Unfortunately, the lack of clear immunological correlates of protective immunity has hampered development of an HCMV vaccine. In spite of this uncertainty, there is evidence that virus-neutralizing antibody responses targeting viral envelope glycoproteins, as well as cellular immune responses (CD4+ and CD8+) targeting multiple structural and regulatory proteins, play important roles in protection against acquisition and reactivation of infection (4–7). Recombinant vaccines employing a variety of expression strategies have focused on the immunodominant glycoprotein B (gB), as well as the major CD8+ T-cell target pp65, in clinical trials. These vaccines have demonstrated various levels of protection against HCMV infection and/or disease in both immunocompetent women (8) and immunocompromised solid organ and hematopoietic stem cell transplant recipients (9, 10). To date, no clinical trials have been powered to assess the efficacy of any vaccine for protection against congenital infection.
In addition to subunit vaccine studies, live attenuated vaccine approaches have also been pursued. The Towne vaccine, attenuated by extensive passage (>125 times) in tissue culture, is the best-studied live HCMV vaccine. In studies with a total of nearly 1,000 human subjects, the Towne vaccine has been evaluated for safety and efficacy in renal transplant recipients (11, 12) and HCMV-seronegative mothers of young children who were actively shedding virus (13). Because of inadequate efficacy in clinical trials, efforts to improve the immunogenicity of the Towne vaccine have included the generation of chimeric viruses containing both Towne sequences and sequences from the less attenuated Toledo strain (14), the evaluation of prime-boost strategies (15), and the coadministration of Towne vaccine with recombinant interleukin-12 (16). The molecular basis of attenuation of the Towne vaccine is uncertain, although a mutation in the Towne UL130 gene coding sequence that abrogates the synthesis of a functional UL130 protein (17), a component of the pentameric complex (PC) of HCMV proteins that have recently received considerable attention as potential HCMV subunit vaccine candidates, has been described (18). Other mutations in the Towne strain likely contribute to its attenuation when used as a vaccine, although the molecular basis of attenuation remains poorly understood. Given this uncertainty about the basis of attenuation, a live HCMV vaccine based on the targeted deletion of specific viral genes important for pathogenesis and/or immune modulation would be desirable. Such an approach to vaccine design could help assuage some of the regulatory and safety issues inherent in live virus vaccination (19, 20).
Because of the species specificity of cytomegaloviruses, experimental HCMV vaccines cannot be evaluated for efficacy against congenital infection in animal models. However, guinea pig cytomegalovirus (GPCMV) is able to cross the placenta to establish congenital infection and in many respects recapitulates the pathogenesis observed for HCMV infection in infants. Several GPCMV proteins have been identified to be immunologically important targets for modeling vaccine development, including gB and pp65 homologs (21, 22). The live attenuated vaccine approach has also been modeled in the guinea pig, in which GPCMV vaccines generated using bacterial artificial chromosome (BAC)-based mutagenesis strategies have been evaluated (23, 24). One limitation of these studies is that the BAC containing the GPCMV genome was found on sequence analysis to contain a 4-bp deletion that frameshifts the gp129 open reading frame (ORF) (25), hence rendering viruses reconstituted from the BAC unable to assemble the GPCMV homolog of the HCMV pentameric complex (PC), composed of GPCMV proteins gH, gL, gp129, gp131, and gp133. In this respect, live GPCMV vaccines derived from the N13R10 BAC have an attenuated parental genetic background similar to that of the Towne vaccine. In spite of this limitation, BAC-based mutagenesis of the GPCMV genome has allowed elucidation of the functional significance of deletion of several GPCMV immune modulation genes, and these engineered viruses have in turn been evaluated as potential vaccine candidates offering protection against congenital infection in this model.
Among the virus genome-encoded immune evasion genes with potential application for vaccine development are those that encode inhibitors of protein kinase R (PKR). PKR is an intracellular host factor that senses viral infection through binding to double-stranded RNA (dsRNA) and subsequently inhibits viral replication via global repression of protein synthesis. HCMV encodes two well-characterized PKR antagonists, IRS1 and TRS1, which inhibit PKR activation by binding to both dsRNA and PKR (26–28). Murine cytomegalovirus also encodes two proteins, M142 and M143, which form a complex that binds dsRNA and prevents activation of the PKR pathway (29–32). Recently, a similar function was attributed to gp145, a protein encoded by the gp145 ORF of GPCMV. In a proteomic screen, gp145 was found to bind dsRNA. That it functions as a PKR antagonist was subsequently suggested by its ability both to rescue the replication of a mutant vaccinia virus that lacks the vaccinia PKR inhibitor and to counteract the effects of guinea pig PKR in cotransfection assays (33).
For many pathogenic viruses, deletion of PKR antagonists attenuates viral replication by permitting PKR to shut down protein synthesis at late stages of the infectious cycle when dsRNA has accumulated. In herpesviruses, immediate early or early viral proteins are still synthesized, although their levels are somewhat reduced (27, 34). Because live vaccines comprised of viruses lacking PKR antagonists alone (35) or in combination with other viral gene deletions (36) have been shown to be safe and efficacious in other systems, we hypothesized that targeted deletion of PKR antagonists from a live cytomegalovirus vaccine could be an effective strategy for attenuation that would provide the safety of an inactivated virus vaccine yet retain the immunogenicity of a live vaccine. We therefore generated a recombinant GPCMV deleted of gp145 and evaluated its safety, immunogenicity, and efficacy as a live vaccine in the context of the congenital infection challenge model.
MATERIALS AND METHODS
Guinea pigs.
Outbred Hartley guinea pigs were purchased from Elm Hill Laboratories (Chelmsford, MA). All animals used for challenge studies were confirmed to be GPCMV seronegative by enzyme-linked immunosorbent assay (ELISA) (37), and approval for the study was obtained from the Institutional Animal Care and Use Committee at the University of Minnesota, Minneapolis, MN. GPCMV-seropositive animals obtained from a commercial supplier were bled, and sera were used as a source for ELISA and neutralization antibody studies of naturally seropositive animals, as described below.
Virus and cells.
A salivary gland (SG)-passaged GPCMV stock used in animal studies was prepared by sequential passage of GPCMV (strain 22122; American Type Culture Collection [ATCC]) in strain 2 guinea pigs as previously described (38). Cell culture propagation of virus was carried out in guinea pig lung (GPL) fibroblast cells (ATCC CCL158) maintained in F-12 medium supplemented with 10% fetal calf serum (FCS; Fisher Scientific), 10,000 IU/liter penicillin, 10 mg/liter streptomycin (Gibco-BRL), and 0.075% NaHCO3 (Gibco-BRL) or as described previously (33). Growth curves and viral titers were determined as described previously (39).
For neutralization assays, a green fluorescent protein (GFP)-expressing virus, vJZ848, was generated. Briefly, plasmid pKTS839 containing nucleotides 3059 to 4836 of GPCMV strain 22122 (GenBank accession number KC503762) was digested with BglII and ligated to a 2.3-kb BamHI fragment from plasmid pQ106 (40) containing gpt/GFP expression cassettes to generate plasmid pKTS848. Plasmid DNA was linearized with XbaI and then transfected into GPL cells, which were subsequently infected with SG-passaged GPCMV and subjected to selection with xanthine and mycophenolic acid as previously described (41). Clonal recombinant virus was generated by multiple rounds of selection and limiting dilution as described elsewhere (41). The genome structure of vJZ848 was confirmed by restriction endonuclease digestion, PCR, and targeted sequencing. Previous GFP-tagged GPCMVs contain 4-bp or 1.6-kb deletions that impact the gp129/gp133 locus and that confer an in vivo attenuation phenotype (25, 42). Virus vJZ848 retained the 1.6-kb locus, demonstrated no loss of the 1.6-kb locus after over 10 passages in GPL cells (as assessed by PCR), and maintained the wild-type sequence, as demonstrated by sequence analysis of PCR-amplified DNA from multiple serial passages for the gp129, gp131, and gp133 ORFs encoding components of the GPCMV pentameric complex (43, 44) (data not shown).
Deletion of gp145, assessment of genome structure, and generation of the Δ145 vaccine.
BAC construct N13R10 contains the complete GPCMV strain 22122 genome, while the BAC N2 contains a 17.9-kb deletion (39). BAC N2 with a targeted deletion of gp145 (N2-Δ145) and N13R10 with a targeted deletion of gp145 (N13R10-Δ145 or Δ145) were constructed using a linear recombination approach to result in the substitution of a kanamycin resistance cassette (Kn) for nucleotides 224206 to 226104 (GenBank accession number KM384022 [25]) comprising the gp145 open reading frame (Fig. 1A). A primer pair was next utilized in order to amplify by PCR Kn from plasmid pEPKanS, producing a PCR product comprised of Kn flanked by GPCMV homology sequences (which are underlined below). The primers (and their sequences) employed were as follows: GP145 forward (5′-CACCCGGATACAGTCCGCGTCACCGCGCGCGGCCCCCCGCGCCGACTCCCGCCTCCCGGATCGCGGCCCCGACGTTAGGGATAACAGGGTAATCGATTT-3′) and GP145 rev (5′-CGCCGCGCCAGAATCGAACGATATAGTCGGTGATCTCACGGATCGCATCGACCGATTCCAGACAGATACGGAAAGGCCAGTGTTACAACCAATTAACC-3′). Linear recombination between the PCR product and BAC N2 or N13R10 was conducted as previously described (45). Candidate clones were screened by PCR to identify clones with the proper insertion of Kn using primers gp145F (ACAGCAAGGTCCCTCTGAGC), gp145R (TATAGTCGGTGATCTCACGG), KanaF (CGGATTCAGTCGTCACTCAT), and KanaR (GCGAGCCCATTTATACCCAT). PCRs were performed, using 1 ng of purified BAC DNA with GoTaq Long PCR master mix (Promega), for 35 cycles of denaturation for 30 s at 94°C, primer annealing for 15 s at 57°C, and extension for 2.5 min at 72°C, followed by a final extension at 72°C for 7 min.
FIG 1.

Construction and in vitro characterization of recombinant viruses lacking gp145. (A) HindIII restriction map of BAC N13R10 (top) with the HindIII-I fragment expanded below. In BACs N13R10-Δ145 and N2-Δ145, replacement of gp145 with a Kn marker cassette results in the loss of the 8.8-kb HindIII-I fragment and creation of novel 3.4- and 4.5-kb HindIII fragments. Arrows indicate the positions of PCR primers gp145F/gp145R (black) and KanaF/KanaR (blue), used for confirmation of the genome structure in panel C. (B) Confirmation of recombinant BAC-derived constructs by restriction pattern analysis. DNA from parental and Δ145 mutant BACs was subjected to restriction digestion with HindIII, EcoRI, XbaI, or XhoI and analyzed by agarose gel electrophoresis. White asterisks indicate restriction fragments that are diagnostic for the Δ145 mutation: for HindIII, 8.8 kb lost and 3.4 and 4.5 kb gained; for EcoRI, 38.9 kb lost and 38.0 kb gained (differences are not resolvable under the gel conditions used in the present study); for XbaI, 6.9 kb lost and 6.0 kb gained; and for XhoI, 10.5 kb lost and 7.2 kb gained. (C) PCR was used to confirm the genome structure of Δ145 in the stock used for in vivo studies. An aliquot of the Δ145 stock was used to infect GPL cells, and infected cell DNA was isolated and PCR amplified. Primer pairs gp145F/KanaR (left) and gp145R/KanaF (right) were predicted to amplify 647-bp and 320-bp products, respectively, from Δ145 DNA and should not amplify wild-type DNA from ATCC or SG-passaged stocks. Primer pair gp145F/gp145R (bottom) was predicted to amplify a 1,446-bp product from Δ145 DNA and a 2,370-bp product from both wild-type DNAs.
BAC clones were analyzed by restriction endonuclease digestion with HindIII, EcoRI, XbaI, and XhoI to confirm that only the predicted restriction polymorphisms were present (Fig. 1B). A clone with the predicted gp145 deletion identified by these analyses was designated N13R10-Δ145. A similar recombinant, constructed in the N2 BAC (46), was also generated and designated N2-Δ145. GFP-positive viruses N2 and N2-Δ145 were reconstituted by transfection of their respective BAC DNAs into GPL cells without cre-mediated excision of the BAC origin, while viruses N13R10 and Δ145 were reconstituted by cotransfection of BAC N13R10 or N13R10-Δ145 DNA, respectively, with plasmid pCre, followed by limiting dilution isolation of GFP-negative viruses (from which the BAC origin was excised) as described previously (46).
The Δ145 virus was expanded in GPL cells to produce a large stock for vaccine and pathogenesis studies. Given the reduced replication capacity of the Δ145 virus (see Fig. 3), viral stocks were concentrated by centrifugation using 10-fold more material from tissue culture flasks in order to achieve an adequately concentrated stock of virus for vaccine studies. An aliquot from the final stock was used to infect GPL cells, and the Δ145 genome structure was again confirmed by PCR as described above using 1 ng purified infected cell DNA.
FIG 3.

Growth characteristics of Δ145 virus and effects of deleting gp145 on eIF2α phosphorylation. (A) GPL cells were infected with gp145-null virus Δ145 or parental virus N13R10 at MOIs of 0.1 or 0.01, and virus titers in the culture supernatants were determined up to 23 days postinfection. (B) GPL cell monolayers were infected with GFP-tagged gp145-null virus N2-Δ145 or parental virus N2 at an MOI of 0.001. One day later, six GFP-positive cells infected with each virus were marked and photographed. Markings were used to locate and photograph the same foci on subsequent days (the numbers of days are indicated in white). One representative series of photographs is shown for N2 (left). One N2-Δ145-infected cell spread to form a focus of GFP-positive cells (center). The other five N2-Δ145-infected cells failed to spread and GFP regressed to undetectable levels by 6 to 21 days postinfection; the regression of one representative N2-Δ145-infected cell is shown on the right. (C) GPLs were treated with 10 units/ml of murine beta interferon overnight and then mock treated or infected (MOI = 3) with Δ145 or N13R10. After 24 and 48 h, eIF2α phosphate (eIF2α-P), total eIF2α, and actin levels were detected by immunoblot assays.
Northern and reverse transcriptase (RT)-PCR analyses.
Transcription of the gp145 gene in N13R10- and Δ145-infected cells was examined by Northern blotting. GPL cells were infected at a multiplicity of infection (MOI) of 5, and total RNA was harvested from these and mock-infected cells at 72 h postinfection (hpi) using a Direct-zol RNA MiniPrep kit (Zymo Research). For Northern analysis, equal quantities of total RNA were resolved on a denaturing formaldehyde agarose gel and transferred to a positively charged nylon membrane (Roche). To generate a double-stranded DNA (dsDNA) probe specific to gp145, pKTS786 (33) was digested with HindIII and XbaI, and the resulting 1,995-bp fragment, which included the entire gp145 ORF, was gel purified and labeled using an Amersham ECL direct nucleic acid labeling and detection system (GE Healthcare). For analysis of the gp147 transcript, primers 5′-GGTCTCGTCGGTTTGAAGTT-3′ and 5′-GGCAACGGAACGTACTTAGAA-3′ were used to amplify wild-type GPCMV DNA using Taq polymerase, and the resulting 299-nucleotide gp147-specific fragment was gel purified for use as a probe in Northern blot assays. The hybridization of each specific probe to the nylon membranes was detected using an Amersham ECL Prime Western blotting detection reagent (GE Healthcare).
To examine the impact of the gp145 deletion on the transcription of adjacent ORFs, RT-PCR of RNA purified from Δ145- and N13R10-infected cells was performed (see Fig. 2B). RNA purified from mock-infected cells was used as a control. Nucleic acid was extracted from cells infected with N13R10 or N13R10-Δ145 or mock-infected GPL cells at 72 hpi using the Direct-zol RNA MiniPrep kit (Zymo Research) according to the manufacturer's instructions. cDNA was synthesized from 1,000 ng of total RNA using a QuantiTect reverse transcription kit (Qiagen) with oligo(dT). Conventional PCR was carried out using cDNA as the template and GoTaq (Promega). The primer pairs used for these PCRs were as follows: for gp144, CB94 (5′-GACTGCTGAAGTGGTACG-3′) and CB95 (5′-GGGTACAGCGAGAAGACC-3′); for gp145, CB96 (5′-ATTCCGGCCTGACGTTTC-3′) and CB97 (5′-CGGTCCATGATCTTCAGC-3′); and for gp146, CB98 (5′-GTTCGTTTCATACGAGACC-3′) and CB99 (5′-GCTCCGGCAGATCCAG-3′). The expected sizes of the PCR amplification products for gp144, gp145, and gp146 were 537 bp, 511 bp, and 486 bp, respectively. The conditions for the gp145 PCR were initial denaturation at 95°C for 10 min, followed by 95°C for 30 s, 55°C for 30 s, and 72°C for 30 s for a total of 35 cycles. The same conditions were used for gp144 and gp146, but 40 cycles were used. All reactions employed a final elongation step at 72°C for 7 min. The PCR product was run in a 1.0% agarose gel and visualized by ethidium bromide staining.
FIG 2.
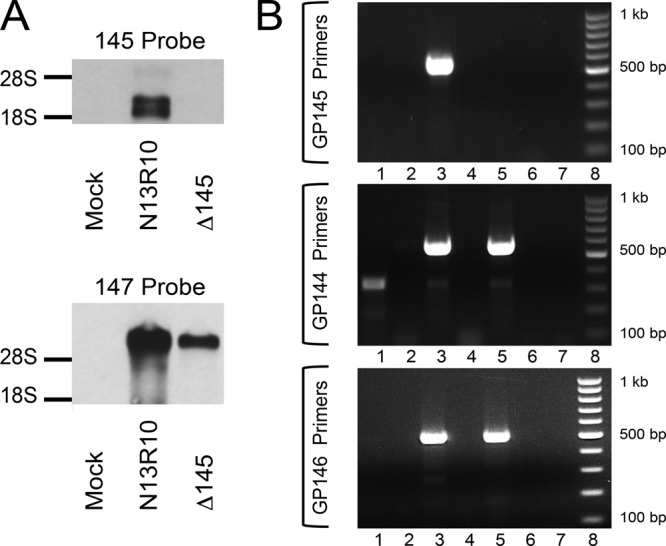
Transcriptional analyses of N13R10 and Δ145 viruses. (A) Northern blot showing a dsDNA probe specific to gp145 (top) or gp147 (bottom) hybridized to RNA purified from mock-, N13R10-, or Δ145-infected GPL cells at 72 hpi. The locations of rRNAs (visualized by ethidium bromide staining) are indicated. (B) RT-PCR analysis of RNA from N13R10- or Δ145-infected cells. Oligo(dT)-primed RNA from mock-infected and virus-infected cells was harvested at 72 hpi and subjected to PCR using primers specific to gp145 (top), gp144 (middle), or gp146 (bottom). Lanes 1, RNA from mock-infected cells plus RT; lanes 2, RNA from mock-infected cells with no RT; lanes 3, RNA from N13R10-infected cells plus RT; lanes 4, RNA from N13R10-infected cells with no RT; lanes 5, RNA from Δ145-infected cells plus RT; lanes 6, RNA from Δ145-infected cells with no RT; lanes 7, no-template PCR control; lanes 8, molecular size markers (100-bp ladder).
Animal pathogenicity studies and vaccination.
Viral challenge studies were performed in cyclophosphamide-treated immunocompromised guinea pigs as previously described (47). Nonpregnant Hartley guinea pigs were treated with 100 mg of cyclophosphamide per kg of body weight by intraperitoneal (i.p.) injection 1 day prior to viral challenge and again 14 days after challenge. Three groups of four animals each were challenged with the Δ145 or N13R10 virus or sham infected with phosphate-buffered saline (PBS). Viruses were administered subcutaneously (s.c.) at a dose of 1 × 107 PFU. Animal weights were monitored every 3 to 5 days, and blood samples were obtained at days 7 and 14 postinoculation for viral load determination by quantitative real-time PCR (qPCR). Liver and spleen samples for qPCR were collected upon necropsy on day 35 (Table 1).
TABLE 1.
Viral loads in cyclophosphamide-treated guinea pigs challenged with N13R10 or Δ145 virus
| Challenge virus | Mean ± SEM level of DNAemia (no. of genome copies/ml) |
Mean ± SEM tissue viral load (no. of genome copies/mg) |
||
|---|---|---|---|---|
| Day 7a | Day 14 | Liverb | Spleenc | |
| N13R10 | 6,328 ± 985 | 573 ± 473 | 92 ± 26 | 430 ± 371 |
| Δ145 | <100d | <100d | <1.0d | <1.0d |
P < 0.0001 versus Δ145.
P = 0.002 versus Δ145.
P = 0.03 versus Δ145.
Limit of detection of PCR assay.
For vaccination/challenge studies, young weanling female Hartley guinea pigs were divided into 3 groups of 8 animals each. Animals in group 1 were immunized with two s.c. injections of 106 PFU Δ145 virus separated by 1 month. Group 2 animals were immunized with two s.c. injections of 105 PFU of Δ145 virus separated by 1 month. The control group (n = 8) was unimmunized. Blood was obtained 21 days after each vaccine dose. Study animals were placed with seronegative breeder males 1 month after completion of the immunization series and examined weekly for evidence of pregnancy. At mid- to late gestation (40 to 45 days), dams were challenged with 105 PFU of SG-passaged GPCMV and observed daily until delivery. Animals that failed to become pregnant (one animal each in groups 1 and 2 and two animals in group 3) were included in the immunogenicity comparisons but not in the pregnancy outcome analyses. Following delivery, tissue was immediately harvested from stillborn pups or within 72 h postdelivery from live-born pups. There were 7, 7, and 6 evaluable pregnancies in groups 1, 2, and 3, respectively.
ELISA and neutralization and Western blot assays.
GPCMV-specific serum IgG titers were determined by ELISA using protocols that have been previously described (21). ELISA titers were defined as the reciprocal of the highest dilution that produced an absorbance of at least 0.10 and twice the absorbance of a negative-control antigen prepared from uninfected GPL cells. Titers of <20 were assigned a value of 10 for statistical comparison (21). The GFP-tagged recombinant vJZ848 virus was used for neutralization assays, using previously published protocols (37). Neutralizing titers were defined as the dilution resulting in a ≥50% reduction in GFP-positive foci. ELISA and neutralization assays using six serum samples from GPCMV-seropositive animals obtained from the commercial supplier were also performed in a separate experiment to compare the vaccine-induced responses with those observed in naturally infected animals. All neutralizing titers were determined in assays with guinea pig lung cells (ATCC CCL158). Analyses of phosphorylation of the α subunit of eukaryotic initiation factor 2 (eIF2α) were conducted using immunoblot methods and antibodies as previously described (24, 30) following spin inoculation (700 × g, 1 h, room temperature) of GPL cells pretreated with mouse beta interferon (10 units/ml).
qPCR analysis.
Maternal blood was obtained on days 7, 14, and 21 postchallenge with SG-passaged GPCMV and analyzed for viral load by qPCR as described previously (41). Briefly, DNA was extracted either from 100 μl citrated maternal blood (MagNA Pure LC DNA isolation kit I; Roche) or from tissues, using 0.1 g of homogenized samples of liver, lung, or spleen tissue (MagNA Pure LC DNA isolation kit II; Roche). Amplification primers GP83TM_F1 (5′-CGTCCTCCTGTCGGTCAAAC-3′) and GP83TM_R1 (5′-CTCCGCCTTGAACACCTGAA-3′) were used at a final concentration of 0.4 μM, while the GP83 hydrolysis probe (FAM-CGCCTGCATGACTCACGTCGA-BHQ1, where FAM is 6-carboxyfluorescein and BHQ1 is black hole quencher 1) was used at 0.1 μM. PCR was performed using a LightCycler 480 real-time PCR system (Roche) under the following conditions: 40°C for 10 s, 95°C for 15 min, and 45 amplification cycles of 95°C for 10 s, 56°C for 15 s, and 72°C for 10 s. Data were analyzed with LightCycler data analysis software (version 1.5; Roche) using standard curves generated from known copy numbers of modified plasmid pCR 2.1 containing GP83 sequences. DNAemia was expressed as the total number of genome copies per milliliter of blood. The limit of detection was approximately 200 copies/ml. For the purpose of statistical comparisons, a level of 100 copies/ml was assigned to negative samples. Tissue viral loads were expressed as the number of genome copies per milligram of tissue. The limit of detection was approximately 2 genome copies/mg of tissue. For the purpose of statistical comparisons, a value of 1 copy/mg was assigned to negative samples. For the purpose of ascertaining congenital infection rates, any pup that had qPCR values falling below the limit of detection of the assay was considered uninfected.
Statistical analyses.
GraphPad Prism software (version 6.0) was used for statistical analyses. Pup mortality and transmission were compared using Fisher's exact test with one-sided comparisons. Pup weights in pregnancy/challenge studies were compared by use of the Kruskal-Wallis followed by Dunn's multiple-comparison test. Antibody titers were compared using the Mann-Whitney test. Parametric data included viral load measurements in blood and tissue and weight loss in cyclophosphamide challenge studies. These were compared using 2-way analysis of variance (ANOVA) followed by Bonferroni's multiple-comparison posttest.
RESULTS
Generation of Δ145 virus.
A gp145-null mutant was constructed using BAC mutagenesis in Escherichia coli. The BAC clone N13R10 contains the complete GPCMV genome derived from a tissue culture-passaged stock of GPCMV strain 22122 (39). A gp145-null mutant was constructed by insertion of a Kn marker cassette concomitant with deletion of the entire gp145 ORF using bacteriophage lambda Red-mediated recombination. Candidate clones were first screened by PCR and then confirmed by HindIII restriction fragment length polymorphism analyses, as indicated in Fig. 1B. The mutant BAC was predicted to lose the 8.8-kb HindIII-I fragment and to gain two novel HindIII fragments of 3.4 and 4.5 kb (Fig. 1B). A clone that demonstrated the expected HindIII pattern was selected and designated N13R10-Δ145 (Δ145). To determine if N13R10-Δ145 contained deletions or rearrangements elsewhere in the genome, N13R10-Δ145 was compared to N13R10 using three additional restriction enzymes. For each enzyme, the observed pattern changes were attributable to the mutation (Fig. 1B), suggesting that other regions of the genome were unaffected by deletions or rearrangements. Viruses designated N13R10 and Δ145 were reconstituted from the parental BAC N13R10 and mutant BAC N13R10-Δ145, respectively.
Evaluation of Δ145 genome structure and transcriptional analyses.
Large stocks of Δ145 were produced for in vivo studies (see below). To ensure that the Δ145 virus used in vivo retained the expected genome structure, the final stock was assessed by PCR analysis as described above. Two PCRs specific for regions that spanned the junctions between the Kn sequence and flanking GPCMV sequences to the left or right of the Kn insertion were performed. Both reactions produced products of the expected size when DNA from the Δ145 stock was amplified and produced no products from wild-type virus stocks (Fig. 1C). Amplification with GPCMV-specific primers spanning the Kn insertion in Δ145 or gp145 in wild-type virus produced the predicted ∼2.4-kb product from wild-type DNA and a predicted ∼1.5-kb product from Δ145 DNA, consistent with the smaller size of the Kn insertion than gp145 (Fig. 1C).
To confirm the knockout of gp145 at the transcriptional level, Northern blot studies were performed using probes designed to hybridize to the gp145 sequence. RNA was purified from Δ145- and N13R10-infected GPL cells at a late time point (72 h) after infection at an MOI of 5. A dsDNA probe corresponding to the gp145 ORF detected 2.2- and 2.5-kb RNAs in N13R10-infected GPL cells but not in Δ145-infected GPL cells (Fig. 2A, top). This result suggests that two distinct transcription start sites may drive gp145 transcription. As a control, infected cell RNAs were examined by Northern analysis using a probe specific for the gp147 ORF. Although the amount of the ∼8-kb gp147-specific transcript synthesized in cells infected with Δ145 was less than that in cells infected with N13R10 (Fig. 2A, bottom), compatible with the decreased replication efficiency of the Δ145 virus, the synthesis of the gp147 transcript in Δ145-infected cells confirmed that the absence of the gp145 transcript was not due to a generalized absence of viral gene expression.
To further compare the transcription of the N13R10 and Δ145 viruses and to evaluate whether the knockout of gp145 unexpectedly perturbed the synthesis of the adjacent gp144 and gp146 ORFs, RT-PCR was performed (Fig. 2B). Transcripts corresponding to gp144 and gp146 were readily detected in both viruses (Fig. 2B, middle and bottom), but a gp145-specific transcript could be identified only in cells infected with the N13R10 virus and not the Δ145 gp145-knockout virus (Fig. 2B, top). These results confirmed the knockout of the gp145 transcript and suggest that deletion of gp145 did not result in the alteration of transcription of nearby genes.
Characterization of Δ145 virus in cell culture.
The growth of the Δ145 virus was attenuated compared to that of parental virus N13R10, as seen in growth curves conducted at MOIs of 0.1 and 0.01 (Fig. 3A). Δ145 achieved peak titers roughly 100-fold lower than those of N13R10, and the times to the peak titer were delayed by 7 to 14 days (Fig. 3A). While titrating virus stocks, we observed that the rate of focus formation (assessed by the cytopathic effect) was substantially slower for Δ145 (data not shown). In order to utilize GFP to visualize viral spread, we used two additional recombinant viruses, N2 and N2-Δ145, that express GFP and either retain or lack gp145, respectively (Fig. 3B). Although the BAC origin in N13R10 includes a GFP expression cassette, when viruses are reconstituted from N13R10, it is necessary to use cre recombinase to excise the 8.8-kb LoxP-flanked BAC origin in order to prevent the overlong genome from spontaneously acquiring compensatory deletions (46). However, since BAC N2 has an 18-kb deletion in the HindIII-D region (39), viruses reconstituted from N2 can accommodate retention of the BAC origin (46). Thus, GFP-positive N2 BAC-derived viruses (reconstituted without cre-mediated excision of the BAC origin cassette) provided an expedient means to visualize the impact of deleting gp145.
To compare the ability of these two viruses to infect and spread within fibroblast cultures, GPL cells were infected with N2 or N2-Δ145 at an MOI of 0.001. One day after infection, six individual GFP-positive cells infected by each virus were identified, photographed, and marked. Daily thereafter the markings were used to locate and photograph the same cells or foci originating from those cells. All six of the N2-infected cells rapidly expanded to form large GFP-positive foci by 8 days postinfection. A series of micrographs showing focus expansion from one representative cell is shown in Fig. 3B. In contrast, only one of the six N2-Δ145-infected cells formed a focus, and its rate of formation was significantly lower than that of the N2 foci (Fig. 3B). Surprisingly, the remaining five N2-Δ145-infected cells that were initially GFP positive lost their GFP expression within a few days. These results suggest that the N2-Δ145 virus is far more likely than the N2 virus to enter an abortive pathway in which the cell initially expresses GFP (and, presumably, other viral proteins) but subsequently either dies or otherwise fails to progress to the production of viral progeny that spread to infect neighboring cells. Moreover, on the uncommon occasion that productive infection is achieved, the rate of N2-Δ145 spread to neighboring cells is low compared to that of the N2 virus.
Because gp145 was shown to bind to dsRNA and inhibit PKR in transfection assays (33), we evaluated its effect on the phosphorylation of eIF2α, the primary substrate of PKR, during infection. By 2 days postinfection, the abundance of phosphorylated eIF2α in cells infected with Δ145 was increased compared to that in cells that were mock infected and infected with the N13R10 virus (Fig. 3C). In this experiment, we pretreated the cells with interferon, but similar changes were also observed in the absence of interferon (data not shown). These results strengthen the hypothesis that antagonism of the PKR pathway by gp145 is necessary for efficient GPCMV replication.
Attenuation of Δ145 virus in immunocompromised guinea pigs.
Nonpregnant adult Hartley guinea pigs (n = 4/group) were treated with cyclophosphamide (100 mg/kg, i.p.) and 1 day later were challenged with 107 PFU of Δ145 or N13R10 inoculated s.c. or were sham inoculated with PBS. Animals were weighed every 3 to 5 days, and additional cyclophosphamide (100 mg/kg, i.p.) was administered on day 14 postchallenge. Patterns of weight gain (measured as the amount of weight gained over time as a percentage of the total body weight) were very similar in the Δ145-challenged animals and the sham (PBS)-inoculated group, but the patterns of weight gain were different in the N13R10-challenged group. Statistically significant differences in the patterns of weight gain were observed on days 6, 14, 17, and 20 postchallenge (Fig. 4). For example, on day 20 postchallenge, N13R10-challenged animals had a cumulative weight gain of 32%, whereas both the PBS group and the Δ145-challenged group had cumulative weight gains of 40% (P < 0.05 for the PBS group versus the N13R10 group and for the Δ145 group versus the N13R10 group; Fig. 4).
FIG 4.
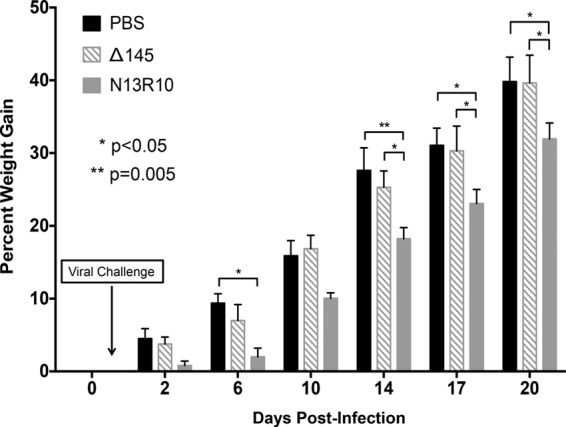
Deletion of gp145 results in attenuation of disease following challenge of cyclophosphamide-immunosuppressed guinea pigs. Adult Hartley guinea pigs were treated with cyclophosphamide 1 day before and 14 days after s.c. challenge with 107 PFU Δ145, 107 PFU N13R10, or PBS (n = 4 per treatment group). The timing of viral challenge (day 1) is indicated by an arrow. Animals were weighed on the indicated days after challenge, and the percent weight gain is plotted versus time; P values were determined by ANOVA.
Blood samples were collected on days 7 and 14 postinfection, and lung and spleen tissue samples were harvested upon termination of the experiment on day 35 postinfection. DNA PCR was used to measure viral loads in blood and end organs as a measure of viral dissemination. As expected, blood and tissue viral loads were below the limits of detection for all sham-infected animals (data not shown). Surprisingly, all blood and tissue viral loads for Δ145-challenged animals were also below the limits of detection. In contrast, all four N13R10-challenged animals had detectable DNAemia on day 7 postinfection, while half (2/4) remained DNAemic on day 14 postinfection. On day 7 the mean viral load in the N13R10 group was 6.3 × 103 genome copies/ml, whereas it was <100 genome copies/ml (the limit of detection) in Δ145-challenged animals (Table 1; P < 0.0001, 2-way ANOVA with Bonferroni's correction).
At necropsy, 4/4 N13R10-challenged animals had viral DNA present in liver homogenate and 2/4 had viral DNA present in spleen homogenate. In contrast, 0/4 cyclophosphamide-treated animals challenged with the Δ145 virus had any demonstrable viral DNA in organ homogenates (Table 1). Animals challenged with N13R10 had a mean viral load in the liver of 92 copies/mg. In contrast, the amount of viral DNA in Δ145-challenged animals was below the detection limit (1 genome copy/mg; P = 0.002, 2-way ANOVA with Tukey's posttest). Similarly, the spleens of N13R10-challenged animals had a significantly higher mean viral load of 430 copies/mg than the spleens of animals challenged with Δ145, in which the amount of viral DNA was below the limit of detection (<1 genome copy/mg; P = 0.03). Thus, even in highly immunosuppressed animals challenged with the Δ145 virus, no evidence of viremia or dissemination of virus to visceral organs was found, indicating that it was dramatically attenuated compared to the parental virus, N13R10.
Immune response to Δ145 vaccination.
The immunogenicity and efficacy of the Δ145 virus used as a live attenuated vaccine were next evaluated in immunocompetent guinea pigs. Young, weanling female guinea pigs were immunized using two regimens: the low-dose group (n = 8) received two s.c. injections of 105 PFU Δ145 1 month apart, while the high-dose group (n = 8) received two s.c. injections of 106 PFU Δ145 1 month apart. In contrast to cyclophosphamide-treated adult animals (see the previous section), some animals exhibited low-grade DNAemia at 7, 14, or 21 days after immunization with the first dose of the Δ145 vaccine. A total of 3/16 animals had DNAemia at day 7 postvaccination (mean viral load, 2 × 103 genomes/ml), 1/16 animals had DNAemia at day 14 (mean viral load, 1.8 × 103 genomes/ml), and 5/16 animals had DNAemia at day 21 (mean viral load, 3.2 × 103 genomes/ml). In total, 8/16 animals had at least one positive value by qPCR postimmunization. This biphasic pattern of low-grade DNAemia in immunized animals suggested that some limited but measurable viral replication took place in vivo.
Antibody responses to GPCMV antigens measured by ELISA and neutralizing responses in sera obtained 21 days after each dose revealed that both regimens were highly immunogenic. ELISA responses were higher in the group immunized with 106 PFU than the group immunized with 105 PFU following both doses, although the differences were statistically significant only when the titers following the second dose were compared (Fig. 5A). Surprisingly, these differences were not reflected in statistically significant differences in neutralizing antibody titers (Fig. 5B). In a separate experiment, the immune responses to vaccination were compared to the ELISA and neutralizing antibody responses in six naturally GPCMV-seropositive guinea pigs obtained from the same commercial vivarium that supplied the seronegative animals used in the vaccination study. Overall, the ELISA titers in the seropositive animals were significantly lower than the titers achieved after two doses of the Δ145 vaccine (P < 0.01 compared to the group immunized with 105 PFU and P < 0.001 compared to the group immunized with 106 PFU) and were comparable to those observed after a single dose of the Δ145 vaccine (Fig. 5A). These results indicate that one dose of Δ145 induced antibody responses comparable to those induced by natural infection, while two doses induced antibody responses superior to those induced by natural infection. The neutralizing responses following two doses of the Δ145 vaccine were statistically significantly equivalent to those induced by natural infection (Fig. 5B).
FIG 5.
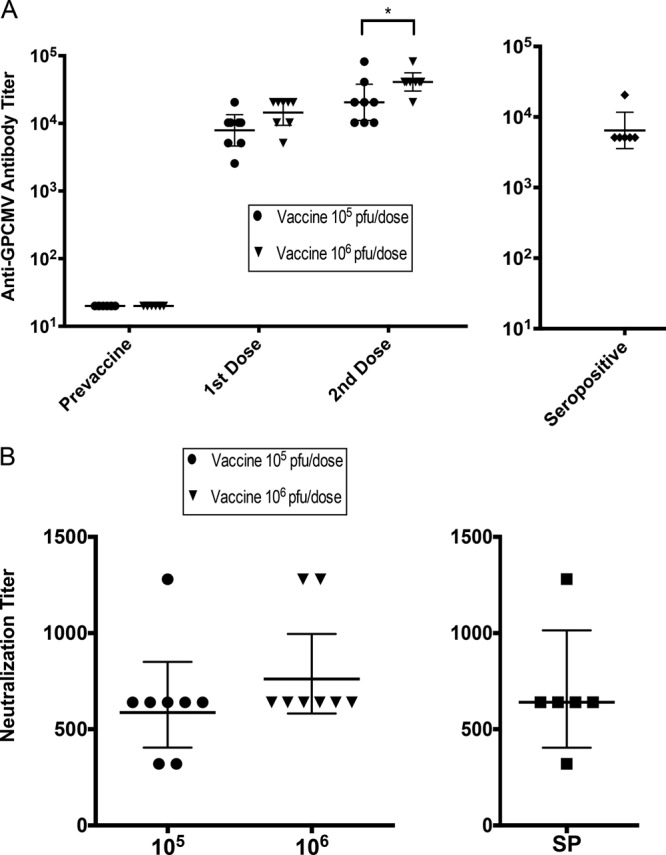
Immune responses to vaccination with Δ145 with comparisons to naturally seropositive animals. Young weanling female Hartley guinea pigs were vaccinated with two s.c. inoculations separated by 1 month, with each inoculum containing either 105 PFU or 106 PFU of Δ145 (n = 8 each group). (A) Anti-GPCMV antibody responses (geometric mean titers) were measured by ELISA using serum obtained 21 days after each inoculation (left). For comparison, the anti-GPCMV antibody titers for a group of six seropositive animals were determined in a separate experiment (right). Statistical analyses were conducted by the Kruskal-Wallis test, and the P values of the statistical significance of the differences between vaccine groups are provided in the Results. *, P < 0.01. (B) Neutralization titers were determined using serum obtained 21 days after the second vaccination, and neutralization assays were performed on guinea pig lung cells (as described in Materials and Methods) (left). The neutralization titers from a group of seropositive animals (SP; n = 6) were also determined in a separate experiment (right). Bars indicate the geometric mean titers ± 95% confidence intervals.
Pregnancy outcomes after challenge.
Of the animals described in the previous section, one animal in each vaccine group and two animals in the unvaccinated control group did not become pregnant. Hence, pup outcome data were available for 7 dams from each vaccine group and 6 unvaccinated controls. All pregnant dams were challenged with SG-passaged GPCMV in the third trimester of pregnancy. Vaccination resulted in a significant reduction in pup mortality from 26/31 (81%) pups in the unvaccinated control group to 4/29 (14%) pups in the group vaccinated with 105 PFU of Δ145 (P < 0.0001) and 1/24 (4%) pups in the group vaccinated with 106 PFU of Δ145 (P < 0.0001; Table 2). All 6 litters in the control group had at least one dead pup; in contrast, 1/7 litters in the group vaccinated with 106 PFU and 2/7 litters in the group vaccinated with 105 PFU had at least one dead pup (P < 0.005 for the vaccine groups combined versus the control group, Fisher's exact test).
TABLE 2.
Impact of Δ145 vaccination on pup mortality
| Group | Total no. of litters | Pup mortalitya | No. (%) of litters with dead pups |
|---|---|---|---|
| Control | 6 | 26/31 (81) | 6 (100) |
| Δ145 (105 PFU) | 7 | 4/29 (14)b | 2 (29) |
| Δ145 (106 PFU) | 7 | 1/24 (4)b | 1 (14) |
The data represent the number of dead pups/total number of pups delivered to the vaccine or control groups (percent).
P < 0.0001 versus the control group.
We also compared the weights of the pups in the vaccine and control groups as an indicator of the impact of vaccination on pup health. The weights of the pups from vaccinated dams were significantly improved, from a mean of 63.5 g for the control group to means of 96 g for the group vaccinated with 105 PFU and 95.6 g for the group vaccinated with 106 PFU (for both groups, P < 0.0001 versus the control group, Kruskal-Wallis and Dunn's multiple-comparison tests; Fig. 6).
FIG 6.
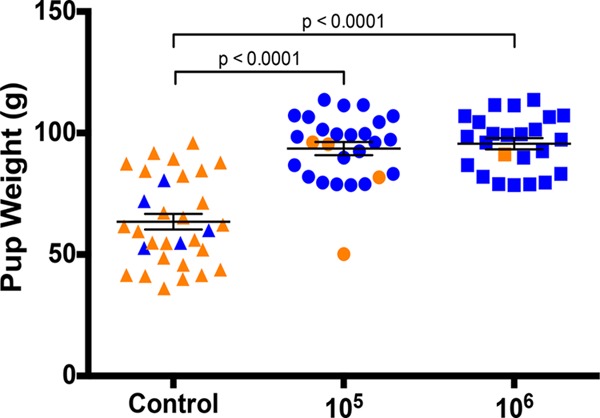
Vaccination with Δ145 improves pup weights after GPCMV challenge. Unvaccinated (control) guinea pigs or guinea pigs vaccinated with two inoculations of 105 PFU or 106 PFU of Δ145 were mated and challenged with SG-passaged GPCMV in the third trimester of pregnancy. The weights of pups born live (blue) or dead (orange) are shown; the P values were determined by the Kruskal-Wallis test and Dunn's multiple-comparison test.
Congenital infection and viral loads in dams and pups.
The viral loads in dams were examined by qPCR on days 7 and 14 after SG-passaged GPCMV challenge. Preconception vaccination had a significant impact on the magnitude and duration of DNAemia compared to those in the unvaccinated control dams (Fig. 7A). All dams in the control group and the group vaccinated with 105 PFU of Δ145 and 6/7 dams in the group vaccinated with 106 PFU of Δ145 were DNAemic on day 7. Three dams in the control group died after delivering pups but prior to the day 14 viremia time point (two at day 8 after SG-passaged virus challenge and one at day 10 postchallenge). On day 14, only 2/7 dams in the group vaccinated with 106 PFU were DNAemic, whereas 7/7 dams in the group vaccinated with 105 PFU and 3/3 surviving dams in the control group were DNAemic. On day 21, 0/7 dams in the group vaccinated with 106 PFU and 5/7 in the group vaccinated with 105 PFU remained DNAemic, while 3/3 surviving dams in the control group were DNAemic. The magnitude of DNAemia in dams vaccinated with the 105-PFU dose at both the day 14 and day 21 time points (1.2 × 107 and 3.3 × 105 genomes/ml, respectively) was statistically significantly lower than that in the unvaccinated controls (2.8 × 107 and 3.2 × 107 genomes/ml, respectively; P < 0.01). Vaccination with the 106-PFU dose similarly reduced the mean blood viral loads compared to those in the controls (P < 0.001) at the day 14 and 21 time points (Fig. 7A). There were also statistically significant differences in viral loads at days 14 and 21 when the groups vaccinated with 106 and 105 PFU were compared (P < 0.001).
FIG 7.
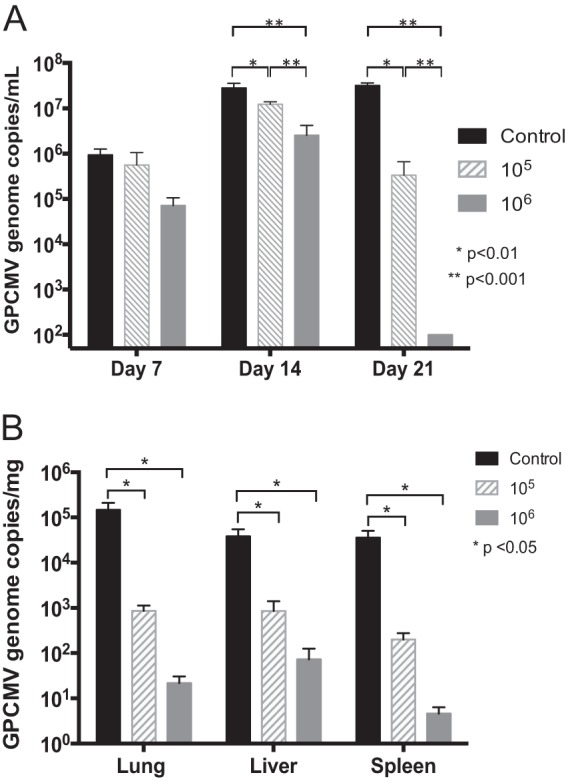
Vaccination with Δ145 reduces maternal and fetal viral loads. Unvaccinated (control) or Δ145-vaccinated guinea pigs were challenged during pregnancy, and qPCR was used to determine blood viral loads in dams and tissue viral loads in pups. (A) Blood viral loads in dams on days 7, 14, and 21 postchallenge. No dams in the group immunized with 106 PFU had detectable DNAemia at day 21, and the dams were therefore assigned a value of 100 genomes/ml (the threshold of detection of DNAemia by the qPCR assay). Significant reductions in DNAemia in both vaccine groups were noted at day 14, and significant differences were observed when the results for the groups vaccinated with 105 and 106 PFU at days 14 and 21 were compared; P values were determined by ANOVA. (B) Viral loads in pup tissues determined by qPCR. Negative samples were assigned a value of 1 genome/mg (the threshold of detection for the qPCR assay) for the purposes of statistical comparison. Comparisons were performed using ANOVA with Bonferroni's multiple comparisons; P values are indicated.
To compare the rates of congenital GPCMV transmission and the magnitude of the viral load in infected pups between the vaccine and control groups, real-time qPCR of liver, lung, and spleen tissue extracted from live-born and stillborn pups was performed. In total, 20/31 (65%) of pups from the control group had congenital GPCMV transmission, as evidenced by the fact that one or more samples were positive for GPCMV DNA by PCR. In comparison, there was a significant reduction in the rate of congenital transmission in pups born to dams from the group vaccinated with 106 PFU (8/24 [33%] pups were infected; P < 0.05 compared to the control group, Fisher's exact test). In the group vaccinated with 105 PFU, congenital transmission was observed in 19/29 (66%) pups (P was not significant when the results were compared to those for the controls; Table 3).
TABLE 3.
Impact of Δ145 vaccination on congenital GPCMV transmission
| Group | Transmissiona |
Overall transmission rateb | |
|---|---|---|---|
| Live born | Stillborn | ||
| Control | 3/5 | 17/26 | 20/31 (65) |
| Δ145 (105 PFU) | 17/25 | 2/4 | 19/29 (66) |
| Δ145 (106 PFU) | 8/23 | 0/1 | 8/24 (33)c |
The data represent the number of pups to which virus was transmitted/number of pups in the live-born and stillborn groups.
The data represent the number of pups to which virus was transmitted/total number of pups, whether stillborn or live-born (percent).
P < 0.05 versus the control group.
Viral loads in pup lung, liver, and spleen tissues were also reduced by vaccination. Viral loads in infected pups born to vaccinated dams that received the 106-PFU dose were reduced by 500- to 7,000-fold compared to those in pups born to the control dams (Fig. 7B). In the control group, mean viral loads were 1.5 × 105, 3.8 × 104, and 3.6 × 104 genome copies/mg tissue in lung, liver, and spleen tissue, respectively, while the mean viral loads in the 106-PFU-dose group were 22, 73, and 5 genome copies/mg tissue in lung, liver, and spleen tissue, respectively (P < 0.05 for all tissues compared to the control group). For the group vaccinated with 105 PFU of Δ145, the mean viral loads were 8.6 × 102, 8.6 × 102, and 2 × 102 genomes/mg tissue in lung, liver, and spleen tissue, respectively (P < 0.05 compared to the control group).
DISCUSSION
The strategy of attenuating live viral vaccines by deletion of PKR antagonists is founded on the idea that these viruses replicate poorly or not at all in vivo yet express a large set of viral proteins prior to the shutoff of protein synthesis that typically occurs at later stages of infection. Unchecked accumulation of dsRNA may also enhance the immune response through the activation of PKR and other dsRNA-sensing pathways. This approach has had success in the vaccinia virus (VACV) system, where a VACV lacking E3L (48, 49) provides a useful comparison. Similar to the PKR-inhibitory genes of cytomegaloviruses (26–33, 50), vaccinia virus E3L binds to and sequesters dsRNA and thereby prevents activation of type I interferon-induced PKR and oligoadenylate synthetase (51). In a primate study, an E3L gene deletion was engineered into the New York City Board of Health (NYCBH) strain of vaccinia, and the resulting virus (NYCBHΔE3L) was examined for its ability to protect cynomolgus macaques from heterologous challenge with monkeypox virus (35). Vaccination with the NYCBHΔE3L vaccine provided some protection of macaques from death, although it was unable to completely protect from the morbidity associated with monkeypox disease. It was proposed in this study that a minimum threshold of antigen was likely required to obtain an effective repertoire of neutralizing antibodies following NYCBHΔE3L immunization and that this antigen threshold may not have been met due to the severe attenuation for replication induced by the E3L mutation (35).
In the current report, we describe the construction of a recombinant GPCMV with a deletion of the PKR inhibitor gp145 for use as a live attenuated vaccine. GPCMVs lacking gp145 replicated poorly in cell culture, particularly when inoculated at a low MOI. Peak titers were reduced by more than 2 log units compared to those of the parental gp145-positive virus, and the time required to reach the peak titer was delayed by 1 to 2 weeks. Using GFP-tagged viruses, it was further revealed that deletion of gp145 results in a high incidence of abortive infection and a reduced rate of focus expansion. Infection of cells with Δ145 led to increased levels of eIF2α phosphate. These results are consistent with a presumed mechanism in which gp145-null viruses are unable to block the PKR-mediated shutdown of protein translation at later stages of infection. However, while gp145-null viruses retained some limited replication capability, human and murine cytomegaloviruses deleted of their known PKR antagonists are fully defective for replication in cell culture (27, 32). This difference raises the possibility that, like HCMV, GPCMV may encode a second, as yet unidentified PKR antagonist that enables Δ145 to replicate to a limited extent. It is also possible that guinea pig GPL cells have some inherent deficiency in the PKR pathway compared to cells used for studying the replication of human or mouse cytomegaloviruses.
In vivo the gp145-null virus Δ145 was highly attenuated for replication and dissemination in adult immunocompromised guinea pigs, as evidenced by improved weight gain and the absence of DNAemia or detectable viral DNA in end organ tissues, in marked contrast to the results for the parental N13R10 virus. Although there was no evidence of DNAemia in immunocompromised guinea pigs, there was evidence of transient DNAemia following the first vaccination of 8/16 young female weanling guinea pigs. The reason for the discrepancy is unclear, but it may be related to differences in the age or sex of the animals used in the two experiments. Irrespective of these differences, the Δ145 virus was clearly highly attenuated, although it was not completely disabled for replication in vivo following immunization of young female weanling guinea pigs.
Despite a high level of attenuation, Δ145 administered as a preconception vaccine was highly immunogenic, inducing robust ELISA and neutralizing antibody responses and resulting in improved pup outcomes following the third-trimester challenge of pregnant dams. Pups born to dams that received the 106-PFU-dose vaccine had substantially reduced mortality relative to that of the unvaccinated controls (4% and 81%, respectively), and the rate of congenital GPCMV transmission was reduced (33% and 65%, respectively). Viral loads were also substantially reduced in pups born to Δ145-immunized dams, particularly in the 106-PFU-dose group (reductions of 525-fold in liver tissue, 6,700-fold in lung tissue, and 7,700-fold in spleen tissue compared to the loads in the controls). However, there was clearly a dose dependence in the magnitude of protection, with the 106-PFU dose consistently being associated with significantly improved reductions in mortality, transmission, and viral load compared to the values obtained with the 105-PFU dose. The difference in efficacy was apparently not due to the higher neutralizing antibody responses conferred by the 106-PFU dose, although total antibody responses, measured by ELISA, were significantly higher after the second dose of vaccine in the group immunized with the 106-PFU dose than in the group immunized with the 105-PFU dose (Fig. 5A). It is possible that the 106-PFU dose resulted in a stronger antibody-dependent cytotoxicity response, a more robust cytokine response, or a better cell-mediated immune response.
The protection conferred following immunization with the Δ145 vaccine was comparable to that conferred by natural GPCMV seropositivity (23) or by preconception vaccination with adjuvanted, purified recombinant gB (22, 37), on the basis of comparisons of our results with those of other studies. Mortality data were previously evaluated in a study in pups born to naturally GPCMV-seropositive dams challenged with SG-passaged virus, using the same experimental design used in this study (23). In that study, five pregnant dams were challenged with the same dose of SG-passaged virus used in this study, and pup mortality was 29% (4/14), which is statistically significantly comparable to the mortality rates that we observed in Δ145-immunized dams. In studies from the CIDMTR laboratory of purified, recombinant gB vaccine adjuvanted with Freund's adjuvant, pup mortality rates have ranged from 14 to 36% and vertical transmission rates have ranged from 22 to 73% (22, 37). Thus, the metrics of protection conferred by the Δ145 vaccine compare very favorably to those associated with natural seropositivity or adjuvanted protein subunit immunization reported previously.
The results for the Δ145 vaccine also compare favorably with those for other live attenuated vaccine approaches tested in the GPCMV model. A study of a recombinant GPCMV vaccine deleted of three putative major histocompatibility complex class I homologs, 3DX (23), demonstrated that preconception vaccination conferred a reduction in pup mortality (22%) of approximately 2-fold compared to that for unimmunized controls (44%). Another study of a live attenuated GPCMV vaccine, v545 (engineered to have a deletion of a viral chemokine homolog of macrophage inflammatory protein 1α), demonstrated a reduction in the rate of pup mortality from 70% to 15% (41).
Both N13R10 and Δ145 have a 4-bp deletion that frameshifts the gp129 ORF (25) and renders these viruses unable to assemble a pentameric complex (PC) composed of gH, gL, gp129, gp131, and gp133 (42–44, 52). In this respect, they resemble the HCMV Towne strain variants that comprise the Towne vaccine, which also contains a mutation that disrupts expression of a PC subunit (17). This inability of the Towne virus to assemble a functional PC may contribute to the Towne vaccine's attenuation (53) and may limit its efficacy by impairing the vaccine's ability to induce epithelial entry-neutralizing antibodies directed against the PC (54). The potential to enhance the immunogenicity and protective efficacy of the Δ145 vaccine by restoring a wild-type gp129 sequence will require further study.
In summary, deletion of the GPCMV PKR antagonist gp145 resulted in a virus that was highly attenuated in nonpregnant immunosuppressed guinea pigs yet, when given as a preconception vaccine, protected against maternal viremia, congenital transmission, and pup mortality. In previous studies, live tissue culture attenuated GPCMV vaccines were able to cause congenital infection (23). Notably, the Δ145 virus demonstrated no evidence of end organ replication but nonetheless served as an effective vaccine in a dose-dependent fashion. This suggests that Δ145 is unlikely to be transmitted in utero, an important safety consideration for any analogously engineered live HCMV vaccine, particularly if inadvertent administration occurred in a pregnant woman. The antibody titers observed following Δ145 immunization were comparable to those observed following natural infection or immunization with other live attenuated GPCMV vaccine candidates (23, 41). Interestingly, we observed a dose-response effect on the magnitude of protection favoring the group vaccinated with 106 PFU, although neutralizing antibody titers were not statistically significantly different between the two groups. The magnitude of the cell-mediated immune response may have favored the group vaccinated with 106 PFU, and this will be an important area for future study. Although the Δ145 vaccine protected against GPCMV mortality in pups and reduced the viral load in both pups and dams postchallenge, sterilizing immunity was not observed. However, the inoculum of SG-passaged GPCMV used for this study was high (105 PFU) and delivered by a nonphysiologic route of challenge. Challenge by a more physiologic route, such as the intravaginal (55) or intranasal route, might demonstrate improved protection against maternal or fetal infection. Efforts to enhance the antibody response by employing Δ145 immunization in the context of a prime-boost approach that includes an adjuvanted recombinant glycoprotein(s) may help confer enhanced protection in the GPCMV model (56) and may be a strategy relevant to future human clinical trials. Although the gp145 mutant was able to replicate sufficiently to generate virus for vaccination, for ideal production strategies, it may be necessary for stocks to be propagated on complementing cells, particularly for an HCMV vaccine deleted of all PKR antagonists. Additionally, the fact that sterilizing immunity protecting against congenital GPCMV transmission was not conferred by the Δ145 vaccine may relate to the inability of the virus to form a functional PC, based on the known mutation in the gp129 gene in the parent N13R10 virus (25). Generation of a gp145 deletion against the background of a gp129-repaired BAC will help clarify the potential for the development and optimal design of HCMV vaccines based on knockout of PKR-inhibitory functions.
ACKNOWLEDGMENTS
Grant support from NIH awards HD044864 and HD082273 (to M.R.S.), HD068229 (supporting E.C.S.), and DE022732 (supporting C.J.B.) is acknowledged. We thank the University of Babylon for funds supporting a sabbatical opportunity for Z.A-M.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
REFERENCES
- 1.Griffiths P, Baraniak I, Reeves M. 2015. The pathogenesis of human cytomegalovirus. J Pathol 235:288–297. doi: 10.1002/path.4437. [DOI] [PubMed] [Google Scholar]
- 2.Boeckh M, Geballe AP. 2011. Cytomegalovirus: pathogen, paradigm, and puzzle. J Clin Invest 121:1673–1680. doi: 10.1172/JCI45449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arvin AM, Fast P, Myers M, Plotkin S, Rabinovich R. 2004. Vaccine development to prevent cytomegalovirus disease: report from the National Vaccine Advisory Committee. Clin Infect Dis 39:233–239. doi: 10.1086/421999. [DOI] [PubMed] [Google Scholar]
- 4.Khanna R, Diamond DJ. 2006. Human cytomegalovirus vaccine: time to look for alternative options. Trends Mol Med 12:26–33. doi: 10.1016/j.molmed.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 5.Sung H, Schleiss MR. 2010. Update on the current status of cytomegalovirus vaccines. Expert Rev Vaccines 9:1303–1314. doi: 10.1586/erv.10.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fu TM, An Z, Wang D. 2014. Progress on pursuit of human cytomegalovirus vaccines for prevention of congenital infection and disease. Vaccine 32:2525–2533. doi: 10.1016/j.vaccine.2014.03.057. [DOI] [PubMed] [Google Scholar]
- 7.McVoy MA. 2013. Cytomegalovirus vaccines. Clin Infect Dis 57(Suppl 4):S196–S199. doi: 10.1093/cid/cit587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pass RF, Zhang C, Evans A, Simpson T, Andrews W, Huang ML, Corey L, Hill J, Davis E, Flanigan C, Cloud G. 2009. Vaccine prevention of maternal cytomegalovirus infection. N Engl J Med 360:1191–1199. doi: 10.1056/NEJMoa0804749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kharfan-Dabaja MA, Boeckh M, Wilck MB, Langston AA, Chu AH, Wloch MK, Guterwill DF, Smith LR, Rolland AP, Kenney RT. 2012. A novel therapeutic cytomegalovirus DNA vaccine in allogeneic haemopoietic stem-cell transplantation: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Infect Dis 12:290–299. doi: 10.1016/S1473-3099(11)70344-9. [DOI] [PubMed] [Google Scholar]
- 10.Griffiths PD, Stanton A, McCarrell E, Smith C, Osman M, Harber M, Davenport A, Jones G, Wheeler DC, O'Beirne J, Thorburn D, Patch D, Atkinson CE, Pichon S, Sweny P, Lanzman M, Woodford E, Rothwell E, Old N, Kinyanjui R, Haque T, Atabani S, Luck S, Prideaux S, Milne RS, Emery VC, Burroughs AK. 2011. Cytomegalovirus glycoprotein-B vaccine with MF59 adjuvant in transplant recipients: a phase 2 randomised placebo-controlled trial. Lancet 377:1256–1263. doi: 10.1016/S0140-6736(11)60136-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Plotkin SA, Smiley ML, Friedman HM, Starr SE, Fleisher GR, Wlodaver C, Dafoe DC, Friedman AD, Grossman RA, Barker CF. 1984. Towne-vaccine-induced prevention of cytomegalovirus disease after renal transplants. Lancet i:528–530. [DOI] [PubMed] [Google Scholar]
- 12.Plotkin SA, Starr SE, Friedman HM, Brayman K, Harris S, Jackson S, Tustin NB, Grossman R, Dafoe D, Barker C. 1991. Effect of Towne live virus vaccine on cytomegalovirus disease after renal transplant. A controlled trial. Ann Intern Med 114:525–531. [DOI] [PubMed] [Google Scholar]
- 13.Adler SP, Starr SE, Plotkin SA, Hempfling SH, Buis J, Manning ML, Best AM. 1995. Immunity induced by primary human cytomegalovirus infection protects against secondary infection among women of childbearing age. J Infect Dis 171:26–32. doi: 10.1093/infdis/171.1.26. [DOI] [PubMed] [Google Scholar]
- 14.Heineman TC, Schleiss M, Bernstein DI, Spaete RR, Yan L, Duke G, Prichard M, Wang Z, Yan Q, Sharp MA, Klein N, Arvin AM, Kemble G. 2006. A phase 1 study of 4 live, recombinant human cytomegalovirus Towne/Toledo chimeric vaccines. J Infect Dis 193:1350–1360. doi: 10.1086/503365. [DOI] [PubMed] [Google Scholar]
- 15.Jacobson MA, Adler SP, Sinclair E, Black D, Smith A, Chu A, Moss RB, Wloch MK. 2009. A CMV DNA vaccine primes for memory immune responses to live-attenuated CMV (Towne strain). Vaccine 27:1540–1548. doi: 10.1016/j.vaccine.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 16.Jacobson MA, Sinclair E, Bredt B, Agrillo L, Black D, Epling CL, Carvidi A, Ho T, Bains R, Adler SP. 2006. Safety and immunogenicity of Towne cytomegalovirus vaccine with or without adjuvant recombinant interleukin-12. Vaccine 24:5311–5319. doi: 10.1016/j.vaccine.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 17.Dolan A, Cunningham C, Hector RD, Hassan-Walker AF, Lee L, Addison C, Dargan DJ, McGeoch DJ, Gatherer D, Emery VC, Griffiths PD, Sinzger C, McSharry BP, Wilkinson GW, Davison AJ. 2004. Genetic content of wild-type human cytomegalovirus. J Gen Virol 85:1301–1312. doi: 10.1099/vir.0.79888-0. [DOI] [PubMed] [Google Scholar]
- 18.Freed DC, Tang Q, Tang A, Li F, He X, Huang Z, Meng W, Xia L, Finnefrock AC, Durr E, Espeseth AS, Casimiro DR, Zhang N, Shiver JW, Wang D, An Z, Fu TM. 2013. Pentameric complex of viral glycoprotein H is the primary target for potent neutralization by a human cytomegalovirus vaccine. Proc Natl Acad Sci U S A 110:E4997–E5005. doi: 10.1073/pnas.1316517110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Söderberg-Nauclér C. 2006. Does cytomegalovirus play a causative role in the development of various inflammatory diseases and cancer? J Intern Med 259:219–246. doi: 10.1111/j.1365-2796.2006.01618.x. [DOI] [PubMed] [Google Scholar]
- 20.Söderberg-Nauclér C. 2012. Autoimmunity induced by human cytomegalovirus in patients with systemic lupus erythematosus. Arthritis Res Ther 14:101. doi: 10.1186/ar3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schleiss MR, Lacayo JC, Belkaid Y, McGregor A, Stroup G, Rayner J, Alterson K, Chulay JD, Smith JF. 2007. Preconceptual administration of an alphavirus replicon UL83 (pp65 homolog) vaccine induces humoral and cellular immunity and improves pregnancy outcome in the guinea pig model of congenital cytomegalovirus infection. J Infect Dis 195:789–798. doi: 10.1086/511982. [DOI] [PubMed] [Google Scholar]
- 22.Schleiss MR, Bourne N, Stroup G, Bravo FJ, Jensen NJ, Bernstein DI. 2004. Protection against congenital cytomegalovirus infection and disease in guinea pigs, conferred by a purified recombinant glycoprotein B vaccine. J Infect Dis 189:1374–1381. doi: 10.1086/382751. [DOI] [PubMed] [Google Scholar]
- 23.Crumpler MM, Choi KY, McVoy MA, Schleiss MR. 2009. A live guinea pig cytomegalovirus vaccine deleted of three putative immune evasion genes is highly attenuated but remains immunogenic in a vaccine/challenge model of congenital cytomegalovirus infection. Vaccine 27:4209–4218. doi: 10.1016/j.vaccine.2009.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mohr CA, Cicin-Sain L, Wagner M, Sacher T, Schnee M, Ruzsics Z, Koszinowski UH. 2008. Engineering of cytomegalovirus genomes for recombinant live herpesvirus vaccines. Int J Med Microbiol 298:115–125. doi: 10.1016/j.ijmm.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 25.Yang D, Alam Z, Cui X, Chen M, Sherrod CJ, McVoy MA, Schleiss MR, Dittmer DP. 2014. Complete genome sequence of cell culture-attenuated guinea pig cytomegalovirus cloned as an infectious bacterial artificial chromosome. Genome Announc 2(5):e00928-14. doi: 10.1128/genomeA.00928-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marshall EE, Geballe AP. 2009. Multifaceted evasion of the interferon response by cytomegalovirus. J Interferon Cytokine Res 29:609–619. doi: 10.1089/jir.2009.0064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marshall EE, Bierle CJ, Brune W, Geballe AP. 2009. Essential role for either TRS1 or IRS1 in human cytomegalovirus replication. J Virol 83:4112–4120. doi: 10.1128/JVI.02489-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hakki M, Marshall EE, De Niro KL, Geballe AP. 2006. Binding and nuclear relocalization of protein kinase R by human cytomegalovirus TRS1. J Virol 80:11817–11826. doi: 10.1128/JVI.00957-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Child SJ, Hanson LK, Brown CE, Janzen DM, Geballe AP. 2006. Double-stranded RNA binding by a heterodimeric complex of murine cytomegalovirus m142 and m143 proteins. J Virol 80:10173–10180. doi: 10.1128/JVI.00905-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Child SJ, Geballe AP. 2009. Binding and relocalization of protein kinase R by murine cytomegalovirus. J Virol 83:1790–1799. doi: 10.1128/JVI.01484-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Budt M, Niederstadt L, Valchanova RS, Jonjic S, Brune W. 2009. Specific inhibition of the PKR-mediated antiviral response by the murine cytomegalovirus proteins m142 and m143. J Virol 83:1260–1270. doi: 10.1128/JVI.01558-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Valchanova RS, Picard-Maureau M, Budt M, Brune W. 2006. Murine cytomegalovirus m142 and m143 are both required to block protein kinase R-mediated shutdown of protein synthesis. J Virol 80:10181–10190. doi: 10.1128/JVI.00908-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bierle CJ, Schleiss MR, Geballe AP. 2012. Antagonism of the protein kinase R pathway by the guinea pig cytomegalovirus US22-family gene gp145. Virology 433:157–166. doi: 10.1016/j.virol.2012.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chou J, Roizman B. 1992. The gamma 1(34.5) gene of herpes simplex virus 1 precludes neuroblastoma cells from triggering total shutoff of protein synthesis characteristic of programed cell death in neuronal cells. Proc Natl Acad Sci U S A 89:3266–3270. doi: 10.1073/pnas.89.8.3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Denzler KL, Babas T, Rippeon A, Huynh T, Fukushima N, Rhodes L, Silvera PM, Jacobs BL. 2011. Attenuated NYCBH vaccinia virus deleted for the E3L gene confers partial protection against lethal monkeypox virus disease in cynomolgus macaques. Vaccine 29:9684–9690. doi: 10.1016/j.vaccine.2011.09.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spector FC, Kern ER, Palmer J, Kaiwar R, Cha TA, Brown P, Spaete RR. 1998. Evaluation of a live attenuated recombinant virus RAV 9395 as a herpes simplex virus type 2 vaccine in guinea pigs. J Infect Dis 177:1143–1154. doi: 10.1086/515278. [DOI] [PubMed] [Google Scholar]
- 37.Schleiss MR, Choi KY, Anderson J, Mash JG, Wettendorff M, Mossman S, Van Damme M. 2014. Glycoprotein B (gB) vaccines adjuvanted with AS01 or AS02 protect female guinea pigs against cytomegalovirus (CMV) viremia and offspring mortality in a CMV-challenge model. Vaccine 32:2756–2762. doi: 10.1016/j.vaccine.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schleiss MR. 2008. Comparison of vaccine strategies against congenital CMV infection in the guinea pig model. J Clin Virol 41:224–230. doi: 10.1016/j.jcv.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 39.Cui X, McGregor A, Schleiss MR, McVoy MA. 2008. Cloning the complete guinea pig cytomegalovirus genome as an infectious bacterial artificial chromosome with excisable origin of replication. J Virol Methods 149:231–239. doi: 10.1016/j.jviromet.2008.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McGregor A, Schleiss MR. 2001. Molecular cloning of the guinea pig cytomegalovirus (GPCMV) genome as an infectious bacterial artificial chromosome (BAC) in Escherichia coli. Mol Genet Metab 72:15–26. doi: 10.1006/mgme.2000.3102. [DOI] [PubMed] [Google Scholar]
- 41.Leviton MP, Lacayo JC, Choi KY, Hernandez-Alvarado N, Wey A, Schleiss MR. 2013. An attenuated cytomegalovirus vaccine with a deletion of a viral chemokine gene is protective against congenital CMV transmission in a guinea pig model. Clin Dev Immunol 2013:906948. doi: 10.1155/2013/906948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nozawa N, Yamamoto Y, Fukui Y, Katano H, Tsutsui Y, Sato Y, Yamada S, Inami Y, Nakamura K, Yokoi M, Kurane I, Inoue N. 2008. Identification of a 1.6 kb genome locus of guinea pig cytomegalovirus required for efficient viral growth in animals but not in cell culture. Virology 379:45–54. doi: 10.1016/j.virol.2008.06.018. [DOI] [PubMed] [Google Scholar]
- 43.Gnanandarajah JS, Gillis PA, Hernandez-Alvarado N, Higgins L, Markowski TW, Sung H, Lumley S, Schleiss MR. 2014. Identification by mass spectrometry and immune response analysis of guinea pig cytomegalovirus (GPCMV) pentameric complex proteins GP129, 131 and 133. Viruses 6:727–751. doi: 10.3390/v6020727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Auerbach M, Yan D, Fouts A, Xu M, Estevez A, Austin CD, Bazan F, Feierbach B. 2013. Characterization of the guinea pig CMV gH/gL/GP129/GP131/GP133 complex in infection and spread. Virology 441:75–84. doi: 10.1016/j.virol.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 45.Wang JB, McVoy MA. 2008. Mutagenesis of the murine cytomegalovirus M56 terminase gene. J Gen Virol 89:2864–2868. doi: 10.1099/vir.0.2008/003137-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cui X, McGregor A, Schleiss MR, McVoy MA. 2009. The impact of genome length on replication and genome stability of the herpesvirus guinea pig cytomegalovirus. Virology 386:132–138. doi: 10.1016/j.virol.2008.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schleiss MR, Bernstein DI, McVoy MA, Stroup G, Bravo F, Creasy B, McGregor A, Henninger K, Hallenberger S. 2005. The non-nucleoside antiviral, BAY 38-4766, protects against cytomegalovirus (CMV) disease and mortality in immunocompromised guinea pigs. Antiviral Res 65:35–43. doi: 10.1016/j.antiviral.2004.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jentarra GM, Heck MC, Youn JW, Kibler K, Langland JO, Baskin CR, Ananieva O, Chang Y, Jacobs BL. 2008. Vaccinia viruses with mutations in the E3L gene as potential replication-competent, attenuated vaccines: scarification vaccination. Vaccine 26:2860–2872. doi: 10.1016/j.vaccine.2008.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vijaysri S, Jentarra G, Heck MC, Mercer AA, McInnes CJ, Jacobs BL. 2008. Vaccinia viruses with mutations in the E3L gene as potential replication-competent, attenuated vaccines: intra-nasal vaccination. Vaccine 26:664–676. doi: 10.1016/j.vaccine.2007.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang D, Tamburro K, Dittmer D, Cui X, McVoy MA, Hernandez-Alvarado N, Schleiss MR. 2013. Complete genome sequence of pathogenic guinea pig cytomegalovirus from salivary gland homogenates of infected animals. Genome Announc 1(2):e00054-13. doi: 10.1128/genomeA.00054-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Perdiguero B, Esteban M. 2009. The interferon system and vaccinia virus evasion mechanisms. J Interferon Cytokine Res 29:581–598. doi: 10.1089/jir.2009.0073. [DOI] [PubMed] [Google Scholar]
- 52.Yamada S, Fukuchi S, Hashimoto K, Fukui Y, Tsuda M, Kataoka M, Katano H, Inoue N. 2014. Guinea pig cytomegalovirus GP129/131/133, homologues of human cytomegalovirus UL128/130/131A, are necessary for infection of monocytes and macrophages. J Gen Virol 95:1376–1382. doi: 10.1099/vir.0.064527-0. [DOI] [PubMed] [Google Scholar]
- 53.Cui X, Adler SP, Davison AJ, Smith L, Habib E-SE, McVoy MA. 2012 Bacterial artificial chromosome clones of viruses comprising the Towne cytomegalovirus vaccine. J Biomed Biotechnol 2012:428498. doi: 10.1155/2012/428498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cui X, Meza BP, Adler SP, McVoy MA. 2008. Cytomegalovirus vaccines fail to induce epithelial entry neutralizing antibodies comparable to natural infection. Vaccine 26:5760–5766. doi: 10.1016/j.vaccine.2008.07.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Olejniczak MJ, Choi KY, McVoy MA, Cui X, Schleiss MR. 2011. Intravaginal cytomegalovirus (CMV) challenge elicits maternal viremia and results in congenital transmission in a guinea pig model. Virol J 8:89. doi: 10.1186/1743-422X-8-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schleiss MR. 2013. Developing a vaccine against congenital cytomegalovirus (CMV) infection: what have we learned from animal models? Where should we go next? Future Virol 8:1161–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]