

ABSTRACT
Assembly of hepatitis B virus (HBV) begins with packaging of the pregenomic RNA (pgRNA) into immature nucleocapsids (NC), which are converted to mature NCs containing the genomic relaxed circular (RC) DNA as a result of reverse transcription. Mature NCs have two alternative fates: (i) envelopment by viral envelope proteins, leading to secretion extracellularly as virions, or (ii) disassembly (uncoating) to deliver their RC DNA content into the host cell nucleus for conversion to the covalently closed circular (CCC) DNA, the template for viral transcription. How these two alternative fates are regulated remains to be better understood. The NC shell is composed of multiple copies of a single viral protein, the HBV core (HBc) protein. HBc mutations located on the surface of NC have been identified that allow NC maturation but block its envelopment. The potential effects of some of these mutations on NC uncoating and CCC DNA formation have been analyzed by transfecting HBV replication constructs into hepatoma cells. All envelopment-defective HBc mutations tested were competent for CCC DNA formation, indicating that core functions in envelopment and uncoating/nuclear delivery of RC DNA were genetically separable. Some of the envelopment-defective HBc mutations were found to alter specifically the integrity of mature, but not immature, NCs such that RC DNA became susceptible to nuclease digestion. Furthermore, CCC DNA formation could be enhanced by NC surface mutations that did or did not significantly affect mature NC integrity, indicating that the NC surface residues may be closely involved in NC uncoating and/or nuclear delivery of RC DNA.
IMPORTANCE Hepatitis B virus (HBV) infection is a major health issue worldwide. HBV assembly begins with the packaging into immature nucleocapsids (NCs) of a viral RNA pregenome, which is converted to the DNA genome in mature NCs. Mature NCs are then selected for envelopment and secretion as complete-virion particles or, alternatively, can deliver their DNA to the host cell nucleus to maintain the viral genome as nuclear episomes, which are the basis for virus persistence. Previous studies have identified mutations on the capsid surface that selectively block NC envelopment without affecting NC maturation. We have now discovered that some of the same mutations result in preferential alteration of mature NCs and increased viral nuclear episomes. These findings provide important new insights into the regulation of the two alternative fates of mature NCs and suggest new ways to perturb viral persistence by manipulating levels of viral nuclear episomes.
INTRODUCTION
The human pathogen hepatitis B virus (HBV) belongs to the family of Hepadnaviridae, a group of small, hepatotropic DNA viruses that also include closely related animal viruses, such as the duck hepatitis B virus (DHBV) (1). Hepadnaviruses contain a small (ca. 3-kb), partially double-stranded (ds-), relaxed circular (RC) DNA genome enclosed within an icosahedral capsid that is, in turn, formed by multiple copies (240 or 180) of the viral capsid or core protein (2, 3). All hepadnaviruses replicate their genomic DNA via an RNA intermediate, termed the pregenomic RNA (pgRNA), by reverse transcription (4, 5). Upon entering the host cells, the virion RC DNA is released into the nucleus for conversion into a covalently closed circular (CCC) DNA, which then serves as the viral transcriptional template for the synthesis of all viral RNAs, including pgRNA, by the host RNA polymerase II. After being packaged together with the viral reverse transcriptase (RT) protein into assembling immature nucleocapsid (NC) (6, 7), the pgRNA is converted by the multifunctional RT, first to a single-stranded DNA (ssDNA) and then to the characteristic RC DNA (4, 8). The mature (i.e., RC DNA-containing) NCs are then encapsulated by the viral envelop proteins and secreted extracellularly as virions, or they can deliver their RC DNA content to the nucleus to be converted to more CCC DNA via an intracellular CCC DNA amplification pathway (9–11).
The HBV core protein (HBc) consists of two separate domains: the N-terminal domain (NTD), which is sufficient to form the capsid shell, and the C-terminal domain (CTD), which is dispensable for capsid assembly but nevertheless essential for viral replication (12–14). The CTD is highly basic and dynamically phosphorylated, which is thought to be important for viral RNA packaging and DNA synthesis (14–24). The NTD has also been shown recently to play a role in viral DNA synthesis beyond its role in capsid assembly (25, 26).
The two alternative fates of mature NCs (i.e., envelopment versus CCC DNA amplification) are known to be regulated by the viral envelope proteins (9, 27–29). Since HBc forms the NC shell, it is also likely to play a key role in these processes (30, 31). Indeed, core mutants affecting NC envelopment have been identified (32, 33). In particular, substitutions of single residues within the N-terminal assembly domain, spatially located on the capsid surface, block envelopment of mature NC and thus secretion of complete HBV virions (34, 35). The effects of these mutations on CCC DNA are currently unknown. On the other hand, the HBc CTD harbors the nuclear localization signal (NLS) and thus is thought to play an important role in delivering the RC DNA in mature NCs to the nucleus for CCC DNA formation (15, 24, 36, 37). Since at least partial disassembly (uncoating) of the mature NCs is required to allow RC DNA release to the host cell nucleus for CCC DNA formation, NC stability or integrity likely plays a critical role in CCC DNA formation (9, 38). In established human hepatoma cells in culture, which have limited ability to support HBV CCC DNA formation, a processed form of RC DNA called protein-free (PF) or deproteinated (dp) RC DNA, also accumulates to high levels (9, 38). PF-RC DNA is derived from RC DNA, but the viral RT protein, which is used as a protein primer to initiate viral DNA synthesis (39, 40) and remains attached to RC DNA in mature NCs, has been removed. At least partial uncoating of the mature NCs is also thought to be required for the removal of RT from RC DNA and the generation of the PF-RC DNA (9, 37). Thus, regardless of whether PF-RC DNA is a true intermediate during the conversion of RC to CCC DNA (9, 37, 38), it may serve as a useful marker for the uncoating of mature NCs.
To explore the role of HBc in coordinating the two alternative fates of mature NCs—envelopment versus CCC DNA formation—we selected a number of HBc mutants that are defective in virion formation and tested their potential effects on CCC DNA formation. Our results indicate these events are genetically separable, and structural changes of mature NCs are likely involved in both of these processes.
MATERIALS AND METHODS
Plasmids.
DNA sequences encoding the wild-type (WT) and mutant HBc proteins were cloned from the pSVcore constructs (34) into pCMV-HBV/Env− (9). A 345-bp BglII-BspEII fragment encoding HBc amino acid residues 30 to 144 from pSVcore, containing the WT sequences, or the L60A, L95A, K96A, I126A substitutions was used to replace the corresponding sequences in pCMV-HBV/Env−. To transfer the WT and mutant core coding sequences to the envelope-WT HBV constructs, a 1.6-kb SnaBI-EcoRI fragment containing the cytomegalovirus (CMV) promoter plus the coding sequences for the 5′ third of pgRNA, encoding either the WT or mutant HBc proteins as described above, from pCMV-HBV/Env− was used to replace the CMV promoter and the 5′ HBV sequences (until the unique EcoRI site) in pCIdA-HBV (41). The resulting pCIdA-HBV/pgRNA constructs direct the expression of the HBV pgRNA, expressing either WT or mutant HBc, under the CMV promoter and expression of the WT envelope proteins from the native HBV promoters.
Transient transfection.
Transfection of HepG2 and Huh7 cells was done as previously described (42). Briefly, HepG2 cells in 60-mm dishes were transfected with 4 μg of plasmid using FuGENE6 (Promega). Huh7 cells seeded in 60-mm dishes were transfected with 10 μg of plasmid using the CalPhos mammalian transfection kit (Clontech). Cells were harvested on day 7 posttransfection for DNA analysis and the RNA packaging assay.
EPR.
NCs in 10 μl cytoplasmic lysate were pretreated with micrococcal nuclease (MNase [0.25 U/μl]) and CaCl2 (5 mM) at 37°C for 1 h. The MNase was then inactivated by adding EGTA to 10 mM. The treated lysates were used in the endogenous polymerase reaction (EPR) as previously described (30, 42). Briefly, the lysate was incubated with 100 μM each dATP, dGTP, dCTP, and TTP, an EDTA-free protease inhibitor cocktail (Roche), and the EPR buffer (50 mM Tris-HCl [pH 7.5], 10 mM MgCl2, 0.1% NP-40, 0.1% 2-mercaptoethanol) for 16 h at 37°C for EPR in a final volume of 20 μl, whereby the viral RT packaged within NCs synthesizes DNA using the endogenous viral RNA and DNA templates packaged within the NCs (4, 43). The DNA synthesized was then released by SDS-proteinase K digestion and detected by Southern blotting following agarose gel electrophoresis as previously described (9, 44).
Analysis of purified viral DNA.
HBV core DNA (i.e., NC-associated DNA) and PF DNAs were isolated as previously described (9), with minor modifications. Briefly, for isolation of core DNA, HepG2 or Huh7 cells were lysed in NP-40 lysis buffer. After removal of the nuclear pellet by brief centrifugation, the supernatant (cytoplasmic lysate) was incubated with MNase (150 U/ml) and CaCl2 (5 mM) at 37°C for 90 min, and proteinase K was then used to digest viral DNA-protein complexes after NCs were precipitated with polyethylene glycol and disrupted by SDS. Viral core DNA was then purified by phenol-chloroform extraction and ethanol precipitation. PF DNA was isolated by Hirt extraction (9, 45). Briefly, cells were lysed in SDS lysis buffer (50 mM Tris-HCl [pH 8.0], 10 mM EDTA, 150 mM NaCl, 1% SDS). The cell lysates were mixed with KCl and incubated at 4°C overnight with gentle rotation. The lysate was then spun at 14,000 × g for 20 min, and the supernatant was extracted three times with phenol and once with chloroform. The DNA was then recovered by ethanol precipitation. To isolate core DNA without MNase nuclease digestion, the MNase-CaCl2 treatment step was omitted, and DpnI digestion was used to remove plasmid DNA. DpnI digestion was also used to treat PF DNA to remove plasmid DNA (9). Confirmation of CCC DNA by heat denaturation and linearization by EcoRI digestion were performed as described before (9). Purified core or PF DNA was analyzed by agarose gel electrophoresis and Southern blotting, as previously described (9, 44). Various viral DNA species were quantified by phosphorimaging or densitometry, and the signal of a particular DNA species from the HBc mutants was normalized first to that of the corresponding DNA species from the WT analyzed on the same gel. The normalized values were then used to calculate the ratios of the different DNA species shown in Table 1.
TABLE 1.
Relative levels of HBV DNA species accumulated by WT and mutant NCs
| Envelope | Core | Relative level of DNA speciesa: |
|||||
|---|---|---|---|---|---|---|---|
| With MNase |
Without MNase |
||||||
| Core RC/SS | PF-RC/core RC | CCC/core RC | Core RC/SS | PF-RC/core RC | CCC/core RC | ||
| WT | WT | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 |
| L60A | − | High | High | ≤0.2b | ≥3.6 | ≥3.9 | |
| L95A | 0.8 | 1.8 | 0.9 | 1.2 | 1.1 | 0.8 | |
| K96A | 0.4 | 4.3 | 4.9 | 0.9 | 2.3 | 3.3 | |
| I126A | − | High | High | 0.4 | 4.2 | 12.8 | |
| Defective | WT | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 |
| L60A | − | High | High | ≤0.2b | ≥2.6 | ≥2.4 | |
| I126A | − | High | High | 0.3 | 3.7 | 7.8 | |
Reported as the ratio of the indicated viral DNA species normalized to that obtained with the WT NC, which is set as 1.0. Average values from multiple experiments are shown. The signal of a particular DNA species (e.g., core RC DNA) from the HBc mutants was normalized first to that of the corresponding DNA species from the WT analyzed on the same gel. The normalized values were then used to calculate the ratios of the different DNA species shown. Note that for the L60A and I126A HBc mutants, no RC DNA could be detected when MNase was used (indicated by “−”). Therefore, the exact ratios of PF-RC DNA or CCC DNA to core RC DNA in those cases could not be calculated but were much higher than those of the WT (“high”).
Due to the very small amounts of core RC DNA detected from the L60A mutant even in the absence of MNase digestion, this value might be an overrepresentation. Therefore, the ratios of PF-RC DNA or CCC DNA to core RC DNA in those cases represent only the minima.
Detection of NC-associated viral RNA (RNA packaging assay).
Viral RNA packaging was analyzed as previously described (21, 22, 42, 44). Briefly, intact NCs from MNase-digested cell lysate were analyzed by native agarose gel electrophoresis followed by detection using a 32P-labeled HBV plus-strand [(+)-strand]-specific riboprobe without the NaOH denaturation step prior to transfer to membrane. Under these conditions, we have shown previously that only pgRNA, but not (+)-strand DNA, in HBV NCs is detected (44, 46). The same membrane was subsequently probed with an anti-HBV core polyclonal antibody (Dako) to detect the core protein (9, 44).
RESULTS
Some but not all secretion-defective core NTD mutants accumulated little to no mature RC DNA.
Previously, a number of HBc NTD mutations were shown to block secretion of DNA-containing virions (34). These mutations still allowed the synthesis of mature HBV RC DNA in vitro in the endogenous polymerase assay. To analyze the potential effects of the HBc mutants on reverse transcription and CCC DNA synthesis inside the cells, we transfected HepG2 and Huh7 cells with HBV-replicating constructs harboring these NTD mutations. Two versions of these constructs were used, either with or without the expression of the viral envelope proteins, which are known to regulate CCC DNA formation (9, 27, 29). We found that in both cell lines, either with or without expression of the envelope proteins, the L95A and K96A mutants showed a core DNA (i.e., NC-associated DNA) pattern similar to that of the WT (Fig. 1 and Table 1). However, L60A and I126A mutants accumulated no detectable mature RC DNA. The minor form of mature DNA, the double-stranded linear (DSL) DNA (47), was affected the same way by the core mutations as RC DNA. All of these mutants made the ssDNA and immature dsDNA (Fig. 1). To further analyze the lengths of the viral minus-strand [(−)-strand] DNA and plus-strand [(+)-strand] DNA separately, the core DNA was first denatured before analysis. As expected from the analysis of the native core DNA, the WT and all HBc mutants made full-length (−)-strand DNA (Fig. 2). The level of full-length (−)-strand DNA [as well as the incomplete (−)-strand DNA] made by the L60A mutant in the HepG2 cells (in the Env− background) (Fig. 1A, lanes 7 and 15) or in Huh7 cells (in both the Env+ and Env− backgrounds) was less than the those of the others (Fig. 1B, lanes 2 and 7; Fig. 2B, lanes 2 and 12), due to the lower levels of pgRNA-containing capsids formed by L60A in that experiment (Fig. 3; see below). The L95A and K96A mutants showed a pattern of (+)-strand DNA similar to that of the WT, including the presence of full-length (+)-strand DNA (Fig. 2), in agreement with the detection of mature RC DNA in the native DNA (Fig. 1). As anticipated from the lack of mature RC DNA in the native DNA, the L60A and I126A mutants showed only incomplete (+)-strand DNA and no full-length (+) strands (Fig. 2).
FIG 1.
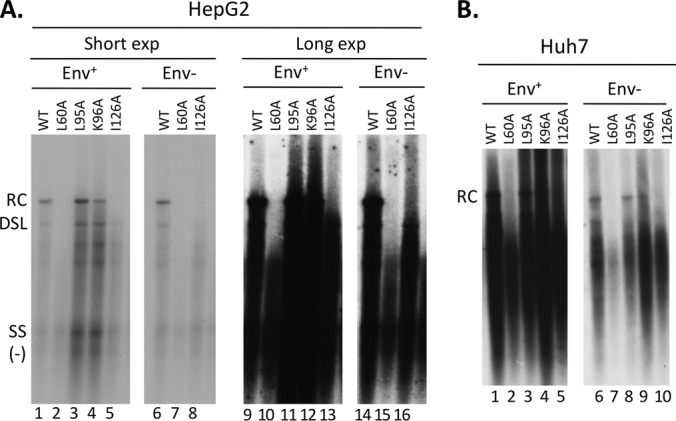
Analysis of NC-associated HBV DNA extracted following exogenous nuclease digestion. HBV-replicating plasmids expressing the WT or the indicated HBc mutants were transfected into HepG2 (A) or Huh7 (B) cells. HBV NC-associated DNA (core DNA) was isolated from cytoplasmic lysate of transfected cells following removal of the transfected plasmids by MNase digestion (see Materials and Methods) and analyzed by Southern blotting using a viral DNA probe. “Env+” or “Env−” denotes that the HBV envelope proteins were expressed or not expressed by the transfected plasmids. SS, single-stranded [(−)-strand] DNA. Lanes 9 to 16 in panel A represent a longer exposure (exp) of the same samples shown in lanes 1 to 8.
FIG 2.
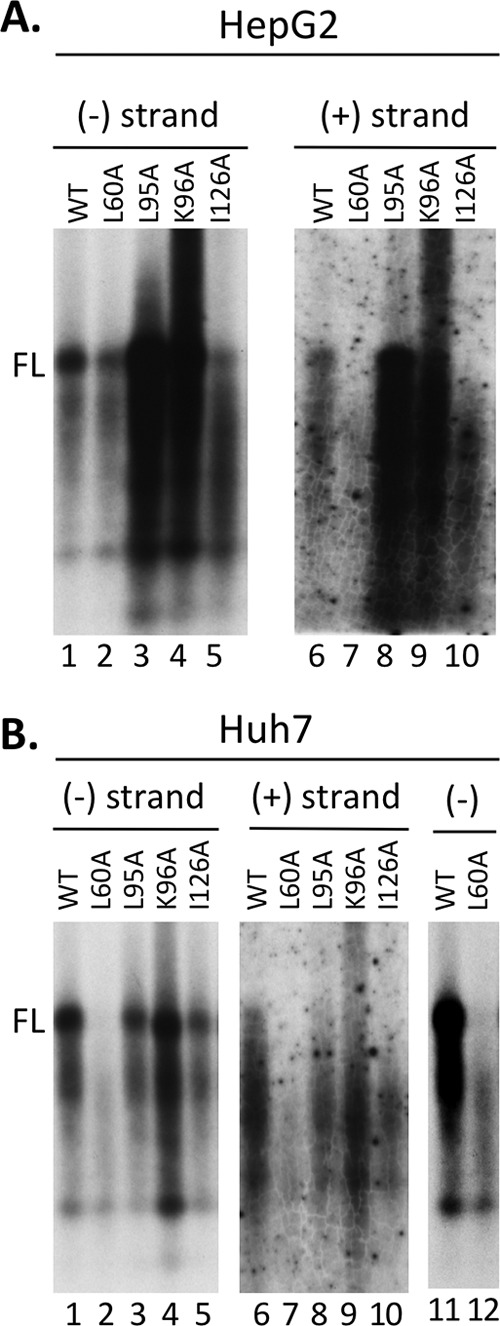
Strand-specific analysis of NC-associated HBV DNA extracted following exogenous nuclease digestion. The same HBV core DNA samples from Fig. 1—all from replication constructs expressing the envelope proteins (Env+) (HepG2 cells in Fig. 1A, lanes 1 to 5, and Huh7 cells in Fig. 1B, lanes 1 to 5)—were heat denatured first and then analyzed by Southern blotting using a strand-specific RNA probe to detect the (−)-strand (A, lanes 1 to 5; B, lanes 1 to 5, 11, and 12) or (+)-strand viral DNA (A and B, lanes 6 to 10). FL, full-length (−)- or (+)-strand DNA. Lanes 11 to 12 in panel B represent a longer exposure of the same samples shown in lanes 1 and 2.
FIG 3.
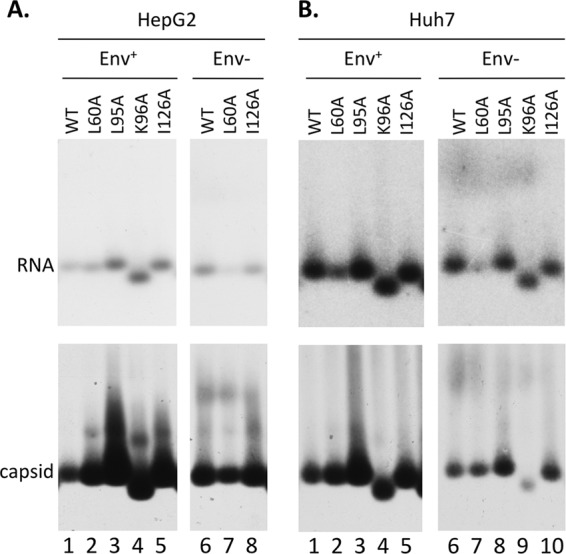
Analysis of HBV NC assembly and RNA packaging. The same HBV-replicating plasmids as in Fig. 1 were transfected into HepG2 (A) or Huh7 (B) cells. HBV NCs from cytoplasmic lysate of transfected cells were resolved by native agarose gel electrophoresis and transferred to nitrocellulose membrane. The NC-associated RNA was detected by an HBV-specific riboprobe (top), and the capsids were detected by an anti-HBc antibody (bottom).
As mentioned above, the levels of core DNA in the L60A mutant were significantly lower in some cases than in the WT and the other mutants. We thus measured the RNA packaging levels of the WT and mutant core proteins. All of the core mutants assembled similar or higher levels of pgRNA-containing capsids compared to the WT, but the L60A mutant showed 2- to 3-fold-lower levels of pgRNA-containing capsids in Huh7 cells (Fig. 3B, lanes 2 and 7) and when expressed from the Env− background in HepG2 cells (Fig. 3A, lane 7) in the experiment shown. This decrease in pgRNA packaging could therefore account for the lower levels of core DNA in the L60A mutant observed above (Fig. 1 and 2). The faster mobility of the K96A mutant (Fig. 3) was consistent with the loss of a positive charge contributed by K96 on the capsid surface, causing the mutant capsid to move faster toward the bottom (cationic) side of the agarose gel.
All secretion-defective HBc mutants tested accumulated high levels of PF DNA, including CCC DNA.
In contrast to the apparent defect in core DNA accumulation described above, all core mutants tested were able to accumulate PF-RC (and PF-DSL) and CCC DNA at levels similar to or higher than those of the WT in HepG2 cells (Fig. 4). The identity of the CCC DNA was verified by heat denaturation (Fig. 4B and C) and heat denaturation followed by linearization with the single-cutter restriction enzyme EcoRI (Fig. 4D), as we described earlier (9). In particular, even though the L60A and I126A mutants showed no RC DNA in NCs (Fig. 1), they showed abundant PF-RC DNA and CCC DNA. Since no core RC DNA, the precursor to both the PF-RC and CCC DNA, was detectable after the routine MNase digestion in the L60A and I126A mutants, the normalized PF-RC and CCC DNA levels in these two mutants would be infinitely higher than those in the WT (Table 1). However, as will be described in the next section below, we were able to detect low levels of core RC DNA in the I126A mutant when the MNase digestion was omitted during core DNA extraction. Upon normalization to the low but detectable levels of this nuclease-sensitive core RC DNA in the I126A mutant, the PF-RC and CCC DNA levels were 4.2- and 12.8-fold higher than those in the WT, respectively (Table 1). Similarly, very low levels of RC DNA were also detected in the L60A mutant NCs without the MNase digestion, which were even lower than those in the I126A mutant (Fig. 5 and Table 1). Upon normalization to the very small amounts of core RC DNA detected without the MNase digestion step, the PF-RC DNA and CCC DNA levels in L60A were at least 3.6- and 3.9-fold higher than those in the WT, respectively (Fig. 4A and B, lanes 2 and 7; Table 1). The K96A mutant, which accumulated mature RC DNA in contrast to the L60A and I126A mutants (Fig. 1), also showed higher levels (by 4- to 5-fold) of both PF-RC and CCC DNA than the WT (Table 1), whereas the L95A mutant had levels of PF-RC DNA and CCC DNA similar to those of the WT (Table 1). The presence or absence of the viral envelope proteins did not affect the pattern of the PF or core DNA from the L60A or I126A mutant (Fig. 1 and 4 and Table 1). The apparently somewhat lower level of PF-RC and CCC DNA from the L60A mutant (in the Env− background) compared to the WT (Fig. 4A to C, lane 7) could be accounted for by the smaller amount of pgRNA packaging and consequently lower core DNA levels (Fig. 1A, lanes 7 and 15, and 3A, lane 7).
FIG 4.
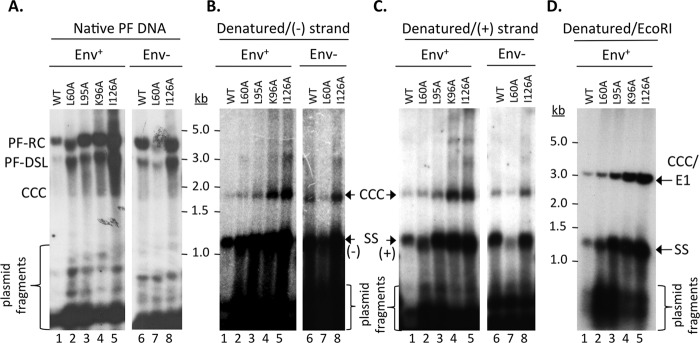
Analysis of HBV PF DNA. HBV-replicating plasmids as in Fig. 1 were transfected into HepG2 cells. PF DNA was extracted and analyzed by Southern blotting, using an HBV-specific DNA probe (A and D) or a (−)-strand-specific (B) or (+)-strand-specific (C) RNA probe. Plasmid DNA copurifying with the PF HBV DNA was degraded by DpnI digestion as described in Materials and Methods. The plasmid fragments are indicated (A). Following DpnI digestion, the DNA was further denatured by boiling before analysis (B and C) or boiling followed by EcoRI digestion (D). SS, single-stranded [full-length (−)- or (+)-strand] DNA derived from the denatured PF-RC and PF-DSL DNA; CCC/E1, CCC DNA linearized by EcoRI digestion.
FIG 5.
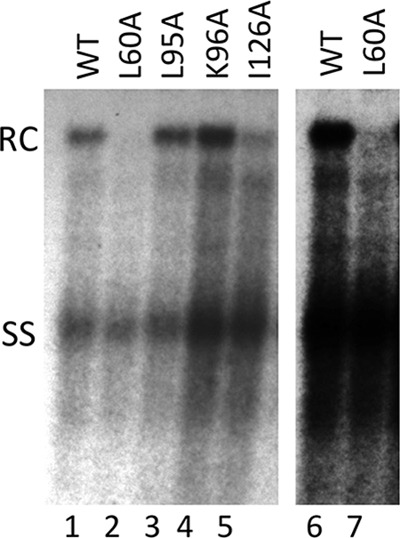
Analysis of NC-associated HBV DNA extracted without exogenous nuclease digestion. HBV-replicating plasmids (all in the Env− background) expressing the WT or the indicated HBc mutants were transfected into HepG2 cells. HBV NC-associated DNA (core DNA) was released from cytoplasmic lysate of transfected cells, without the MNase digestion step, by SDS-proteinase K digestion and analyzed by Southern blotting using an HBV DNA probe. Lanes 6 and 7 represent a longer exposure of lanes 1 and 2. SS, single-stranded [full-length (−)-strand] DNA.
The presence of full-length (+)-strand DNA, as well as full-length (−)-strand DNA, in the PF-RC DNA from the WT or all HBc mutants tested was further confirmed by its detection after denaturation of the PF-RC DNA (Fig. 4B and C), in contrast to the absence of full-length (+) strands in the core DNA from the L60A and I126A mutants (Fig. 2A and B, lanes 7 and 10). Also, essentially the same results were obtained in Huh7 cells, although the overall PF-RC and CCC DNA levels by the WT and core mutants were lower than those in the HepG2 cells (data not shown).
The HBc mutants failed to protect RC DNA.
The lack of RC DNA detection in NCs (Fig. 1) was apparently contradictory to the abundance of PF-RC and CCC DNAs (Fig. 3) for the L60A and I126A mutants, since the PF-RC and CCC DNAs are all derived from the core RC DNA following uncoating of mature NCs. One possibility that could account for this discrepancy was that these core mutants were in fact competent in making RC DNA, but mutant mature NCs containing the RC DNA were unstable and thus failed to accumulate. The instability of the mature NCs formed by the mutant core proteins, resulting in the disruption of the mutant mature NCs, could lead, on one hand, to enhanced uncoating resulting in a failure to accumulate mature NCs, as observed, and on the other, to rapid deproteination of RC DNA and CCC DNA formation, accounting for the abundant levels of these DNA species shown for these mutants (see Discussion). Since an exogenous nuclease (MNase in this study) treatment was used, as is routine in the field, to remove input plasmids during the core DNA extraction shown in Fig. 1 and 2 (see Materials and Methods), the putative unstable mature NCs formed by the mutant core proteins, which might have been accumulated in the cells, could have been eliminated as a result of the nuclease digestion. This possibility was supported by previous studies by us and others showing that certain capsid mutations of DHBV can indeed lead to preferential destabilization of mature NCs (21, 48, 49). To test this possibility, we extracted HBV core DNA in the absence of exogenous nuclease digestion of the cytoplasmic lysate. DpnI digestion was subsequently used to degrade the input plasmids (but not replicative viral DNA) from the purified core DNA.
As shown in Fig. 5, in the absence of MNase treatment, the levels of core RC DNA shown by the L95A and K96A core mutants were similar to those of the WT, just like the results obtained with core DNA extracted with MNase treatment (Fig. 1), indicating that these mutant capsids were able to protect the mature RC DNA like the WT capsids. Furthermore, significant amounts of core RC DNA were also detected in the I126A mutant, and low levels of RC DNA were detected in the L60A mutant (Fig. 5), despite the fact that no RC DNA was detected in cells transfected by these two core mutants when MNase was used to digest the lysate prior to viral DNA extraction. These results thus confirmed that at least some mature NCs were formed by the L60A and I126A mutants, but they failed to protect their RC DNA content. The failure of the mutant mature NC to protect its RC DNA content indicated a loss of structural integrity, which was consistent with its destabilization, but alternative interpretations remained possible (see Discussion). Upon normalization to the ssDNA levels, the levels of RC DNA in the I126A mutant were still ca. 3-fold lower than those of the WT, and those in the L60A mutant were at least 5-fold lower (Table 1). The fact that only very low levels of RC DNA were detected in the L60A mutant, even in the absence of MNase digestion, coupled with the detection of abundant levels of PF-RC DNA and CCC DNA in this mutant further indicated that mature L60A NCs might be even more unstable than the I126A mutant NCs and that the mutant failed to accumulate significant amounts of mature NCs due to rapid uncoating.
I126A mutant NCs matured in vitro by EPR also failed to protect their RC DNA content.
To test further the preferential instability induced by the I126A core mutation, we decided to make mature NCs in vitro using EPR. MNase digestion was used to eliminate all the mature NCs from the I126A mutant (and some WT mature NCs too) (30). The remaining (immature) NCs were then used for EPR to convert a portion of them to mature NCs, and the stability of the mature NCs made in vitro was tested by DNase I digestion (Fig. 6). The results showed that while a portion (ca. 30% or less) of mature WT NCs were unable to protect their RC DNA content, as observed before (30), all mature I126A mutant NCs formed in vitro failed to protect their RC DNA. Thus, the I126A mature NCs formed in vitro, like those formed in vivo, lost their integrity. In contrast, ssDNA inside immature NCs was protected from DNase digestion by either the WT or I126A mutant NCs, as shown above.
FIG 6.
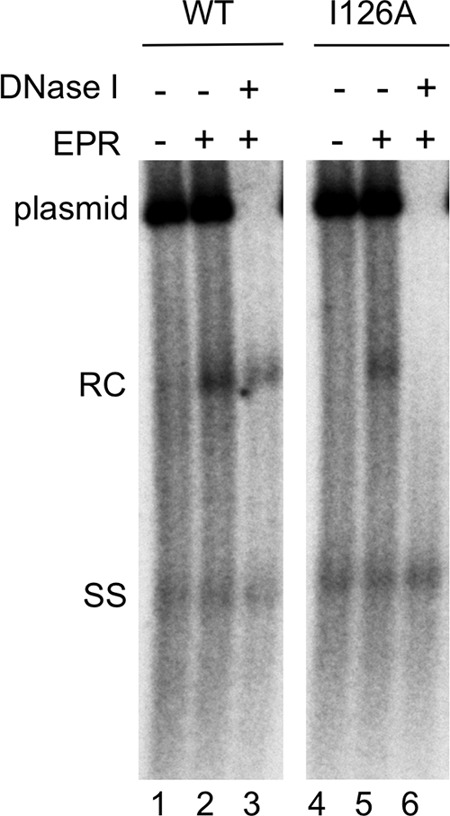
Analysis of HBV NCs by EPR. The HBV-replicating plasmid expressing the WT (lanes 1 to 3) or I126A mutant (lanes 4 to 6) HBc was transfected into HepG2 cells. Cytoplasmic lysate of transfected cells was treated with MNase and then used in EPR in vitro (lanes 2, 3, 5, and 6) or not (lanes 1 and 4). Following EPR, a portion of the reaction mixtures was treated with DNase I (1 mg/ml) (lanes 3 and 6). HBV core DNA was then released from NCs by SDS-proteinase K digestion and analyzed by Southern blotting using an HBV DNA probe. An HBV DNA-containing plasmid was included as a control to verify the efficacy of DNase digestion following EPR. SS, single-stranded [full-length (−)-strand] DNA.
DISCUSSION
We have shown recently that the mature HBV NCs are preferentially destabilized relative to immature ones (30). Here we have uncovered an important role for the HBc NTD in the integrity of mature NCs. We found that two NTD mutations, L60A and I126A, both located on the capsid surface and shown previously to block mature NC envelopment (34) (Fig. 7), failed to protect their RC DNA content from exogenous nuclease digestion. Analysis of NCs matured in vitro using EPR indicated that the loss of integrity of the I126A mature NCs was an intrinsic property conferred by the HBc mutation. A very recent report showed that mutations at another NTD site, V124, at the core-dimer interface (Fig. 7), close to I126, also led to a selective defect in mature RC DNA accumulation, although the defects of those mutants were attributed to RC DNA synthesis rather than destabilization (25). It is important to point out that the precise quantification of the synthesis versus degradation of the different DNA species was difficult, due to the dynamic nature of NC maturation and degradation, NC uncoating, RC DNA deproteination, and conversion to CCC DNA. It remains possible that some of the defect in accumulation of mature NCs by the L60A and I126A mutants was also due to decreased synthesis of mature RC DNA. Nevertheless, these results clearly demonstrate that HBV NCs are highly dynamic and undergo significant changes during maturation in vivo. In vitro, the core protein structure in the context of recombinant capsids is shown to be dynamic (i.e., capsid “breathing”) by hydrogen-deuterium exchange experiments (50, 51). In particular, consistent with our finding of preferential alteration of mature NCs in vivo, the HBV capsids assembled artificially in vitro in the presence of dsDNA, in contrast to those assembled in the presence of RNA or ssDNA, are reported to be in a spring-loaded, metastable structure (52). Furthermore, preferential destabilization of mature NCs is also caused by core mutations in DHBV (12, 49), and particularly by mutations mimicking constitutive CTD phosphorylation in the DHBV core protein (21). Thus, both the NTD and CTD of the core protein of hepadnaviruses are involved in regulating the dynamic stability of the mature NCs.
FIG 7.

Location of the HBc residues mapped onto the three-dimensional structure of an HBc dimer. The L60, L95, K96, and I126 positions analyzed here, as well as the V124 position alluded to in the Discussion section, are highlighted in dark gray. See the text for details.
It remains to be understood how mutations on the capsid surface such as L60A and I126A preferentially affect mature NC integrity. The role of the I126, together with that of the nearby V124 at the dimer interface (25) (Fig. 7), in mature NC formation/integrity suggests that maturation-associated NC structural changes, perhaps triggered by the altered interactions of HBc with the dsDNA in mature NCs versus the ssDNA or pgRNA in immature NCs (52), may be initiated and/or propagated through the dimer interface. Interestingly, a previous study indicated that the neighboring R127 is susceptible to protease digestion in the context of recombinant capsids as a consequence of dynamic local unfolding and refolding (53), consistent with an important role of this region in the structural dynamics of NC during maturation. L60 is located at the base of the spike on the capsid surface, and our results suggest that this location is also involved in regulating NC integrity associated with NC maturation. The exact nature of the NC alterations caused by the HBc mutations, which led to the selective nuclease sensitivity of mature NCs, remains to be determined. We have used the term “destabilization” or “loss of integrity” here fairly loosely to denote the fact that the mutant mature NCs failed to protect their RC DNA content from exogenous nuclease digestion during core DNA extraction, and they failed to accumulate in the cells, presumably due to their susceptibility to endogenous nucleases in the host cells.
We have recently shown that even with WT HBc, a fraction (ca. 30% or less) of mature NCs, so-called “M3” NCs, also fail to protect their RC DNA content (30). These M3 NCs further displayed slower sedimentation on sucrose gradients, slower migration on agarose gels, and increased susceptibility to protease degradation (30), which is consistent with a loosening up (or expansion) of the capsid shell allowing access of exogenous nuclease to the interior RC DNA. In the L60A and I126A mutants, it seems that all mature NCs, instead of only a small fraction, become destabilized, and their degree of destabilization may be even greater than that seen in the M3 subpopulation of the WT NCs as the I126A mutant mature NCs apparently could not survive the sucrose gradient ultracentrifugation (X.J.C. and J.H., unpublished results). It remains also possible that the thermodynamic stability of NCs during maturation is actually unaltered by these mutations but a barrier to active uncoating (NC disassembly) is lowered: e.g., by prematurely exposing a putative uncoating signal as a result of the mutations. Interestingly, the properties of the I126A mutant mature NCs are phenocopied by WT mature NCs formed in an immortalized mouse hepatocyte cell line, AML12HBV10, which also show a lack of accumulation of RC DNA and loss of integrity of mature NCs, accompanied by high levels of PF-RC DNA and CCC DNA (54). This suggests that NC integrity and uncoating are also subject to host regulation. As normal mouse hepatocytes are unable to form CCC DNA or significant amounts of PF-RC DNA (54, 55), possibly due to a deficiency in NC uncoating (54), and are resistant to HBV infection, even after receptor reconstitution (56, 57), these results further implicate NC disassembly or uncoating as a critical step in determining HBV species tropism.
Our results also demonstrate that the core NTD can significantly affect CCC DNA levels. The effect of L60A and I126A on mature NC integrity suggests that these mutations likely increased CCC DNA levels by enhancing NC uncoating and the release of RC DNA. On the other hand, the K96A mutation, which apparently did not significantly affect the integrity of the mature NCs, at least as measured here, still enhanced CCC DNA levels. Among other possibilities, K96A mutation may instead affect the intracellular trafficking and nuclear import of the mature NC and its RC DNA content, prerequisites for CCC DNA formation. Although only the core CTD is known to harbor nuclear localization signals (NLSs), it is possible that NTD could regulate the use of these NLSs in the CTD on the mature NC. Interestingly, the L95A mutation, right next to the K96A mutation, did not affect either NC integrity or CCC DNA formation (Fig. 7). Clearly, more work will be needed to elucidate further the mechanisms by which these NC surface mutations affect CCC DNA formation.
All of the core NTD mutations tested here are located on the exterior surface of NCs (Fig. 7) and are defective in forming complete virions, indicating that the residues affected by these mutations are involved in mediating the interaction of mature NCs with the viral envelope proteins during virion formation (34). These residues may form part of the actual binding site on mature NCs that directly interacts with the viral envelope proteins. Thus, the mutations at positions 95 and 96, which showed no significant effect on NC integrity as measured here, may be part of a site that is directly recognized by the envelope proteins (34, 58). Alternatively, the mutations at other sites (particularly L60 and I126) may affect NC-envelope interactions indirectly (e.g., by affecting the structural integrity of the mature NCs). In so doing, these mutations may interfere with the generation of the putative structural changes associated with NC maturation: i.e., with the emergence of the putative maturation signal that is recognized by the envelope proteins (4, 59). On the other hand, all of these mutants remain competent for delivering RC DNA to the host cell nucleus for CCC DNA formation. Thus, the determinants on the mature NC important for uncoating, nuclear import, and CCC DNA formation can be separated genetically from those for its envelopment.
The two alternative fates of the mature HBV NC, nuclear import for CCC DNA formation and envelopment for virion secretion, likely require that its stability be carefully regulated. If the mature NC is too stable, it will interfere with NC disassembly (uncoating). Thus, the preferential destabilization of mature NCs, as we reported recently (30), is most likely a reflection of the necessity to disassemble the mature NCs so their RC DNA content can be released into the host cell nucleus and is available for CCC DNA formation. On the other hand, if the NC is too unstable, as suggested here for the L60A and I126A mutants, it may interfere with the maturation-associated structural changes required to trigger envelopment and virion secretion. Therapeutic manipulation of NC stability/integrity can thus potentially block CCC DNA synthesis and/or virion secretion.
ACKNOWLEDGMENTS
We thank Jennifer Wentzel for technical assistance.
This work was supported by a Public Health Service grant (R01 AI074982 to J.H.) from the National Institutes of Health. V.B. was supported by the Deutsche Forschungsgemeinschaft (DFG), SFB402, Teilprojekt C2.
REFERENCES
- 1.Seeger C, Zoulim F, Mason WS. 2013. Hepadnaviruses, p 2185–2221. In Knipe DM, Howley PM (ed), Fields virology. Lippincott, Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Wynne SA, Crowther RA, Leslie AG. 1999. The crystal structure of the human hepatitis B virus capsid. Mol Cell 3:771–780. doi: 10.1016/S1097-2765(01)80009-5. [DOI] [PubMed] [Google Scholar]
- 3.Conway JF, Cheng N, Zlotnick A, Wingfield PT, Stahl SJ, Steven AC. 1997. Visualization of a 4-helix bundle in the hepatitis B virus capsid by cryo-electron microscopy. Nature 386:91–94. doi: 10.1038/386091a0. [DOI] [PubMed] [Google Scholar]
- 4.Summers J, Mason WS. 1982. Replication of the genome of a hepatitis B-like virus by reverse transcription of an RNA intermediate. Cell 29:403–415. doi: 10.1016/0092-8674(82)90157-X. [DOI] [PubMed] [Google Scholar]
- 5.Hu J, Seeger C. 2015. Hepadnavirus genome replication and persistence. Cold Spring Harb Perspect Med 5: a021386. doi: 10.1101/cshperspect.a021386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hirsch RC, Lavine JE, Chang LJ, Varmus HE, Ganem D. 1990. Polymerase gene products of hepatitis B viruses are required for genomic RNA packaging as well as for reverse transcription. Nature 344:552–555. doi: 10.1038/344552a0. [DOI] [PubMed] [Google Scholar]
- 7.Bartenschlager R, Schaller H. 1992. Hepadnaviral assembly is initiated by polymerase binding to the encapsidation signal in the viral RNA genome. EMBO J 11:3413–3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones SA, Hu J. 2013. Hepatitis B virus reverse transcriptase: diverse functions as classical and emerging targets for antiviral intervention. Emerg Microbes Infect 2:e56. doi: 10.1038/emi.2013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao W, Hu J. 2007. Formation of hepatitis B virus covalently closed circular DNA: removal of genome-linked protein. J Virol 81:6164–6174. doi: 10.1128/JVI.02721-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tuttleman JS, Pourcel C, Summers J. 1986. Formation of the pool of covalently closed circular viral DNA in hepadnavirus-infected cells. Cell 47:451–460. doi: 10.1016/0092-8674(86)90602-1. [DOI] [PubMed] [Google Scholar]
- 11.Wu TT, Coates L, Aldrich CE, Summers J, Mason WS. 1990. In hepatocytes infected with duck hepatitis B virus, the template for viral RNA synthesis is amplified by an intracellular pathway. Virology 175:255–261. doi: 10.1016/0042-6822(90)90206-7. [DOI] [PubMed] [Google Scholar]
- 12.Yu M, Summers J. 1991. A domain of the hepadnavirus capsid protein is specifically required for DNA maturation and virus assembly. J Virol 65:2511–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nassal M. 1992. The arginine-rich domain of the hepatitis B virus core protein is required for pregenome encapsidation and productive viral positive-strand DNA synthesis but not for virus assembly. J Virol 66:4107–4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schlicht HJ, Bartenschlager R, Schaller H. 1989. The duck hepatitis B virus core protein contains a highly phosphorylated C terminus that is essential for replication but not for RNA packaging. J Virol 63:2995–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liao W, Ou JH. 1995. Phosphorylation and nuclear localization of the hepatitis B virus core protein: significance of serine in the three repeated SPRRR motifs. J Virol 69:1025–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perlman DH, Berg EA, O'Connor PB, Costello CE, Hu J. 2005. Reverse transcription-associated dephosphorylation of hepadnavirus nucleocapsids. Proc Natl Acad Sci U S A 102:9020–9025. doi: 10.1073/pnas.0502138102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jung J, Hwang SG, Chwae YJ, Park S, Shin HJ, Kim K. 2014. Phosphoacceptors threonine 162 and serines 170 and 178 within the carboxyl-terminal RRRS/T motif of the hepatitis B virus core protein make multiple contributions to hepatitis B virus replication. J Virol 88:8754–8767. doi: 10.1128/JVI.01343-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lan YT, Li J, Liao W, Ou J. 1999. Roles of the three major phosphorylation sites of hepatitis B virus core protein in viral replication. Virology 259:342–348. doi: 10.1006/viro.1999.9798. [DOI] [PubMed] [Google Scholar]
- 19.Lewellyn EB, Loeb DD. 2011. Serine phosphoacceptor sites within the core protein of hepatitis B virus contribute to genome replication pleiotropically. PLoS One 6:e17202. doi: 10.1371/journal.pone.0017202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gazina EV, Fielding JE, Lin B, Anderson DA. 2000. Core protein phosphorylation modulates pregenomic RNA encapsidation to different extents in human and duck hepatitis B viruses. J Virol 74:4721–4728. doi: 10.1128/JVI.74.10.4721-4728.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Basagoudanavar SH, Perlman DH, Hu J. 2007. Regulation of hepadnavirus reverse transcription by dynamic nucleocapsid phosphorylation. J Virol 81:1641–1649. doi: 10.1128/JVI.01671-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu M, Summers J. 1994. Multiple functions of capsid protein phosphorylation in duck hepatitis B virus replication. J Virol 68:4341–4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ludgate L, Ning X, Nguyen DH, Adams C, Mentzer L, Hu J. 2012. Cyclin-dependent kinase 2 phosphorylates S/T-P sites in the hepadnavirus core protein C-terminal domain and is incorporated into viral capsids. J Virol 86:12237–12250. doi: 10.1128/JVI.01218-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu KC, Ludgate L, Yuan ZH, Hu J. 2015. Regulation of multiple stages of hepadnavirus replication by the carboxyl-terminal domain of viral core protein in trans. J Virol 89:2918–2930. doi: 10.1128/JVI.03116-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tan Z, Pionek K, Unchwaniwala N, Maguire ML, Loeb DD, Zlotnick A. 2015. The interface between HBV capsid proteins affects self-assembly, pgRNA packaging, and reverse transcription. J Virol 89:3275–3284. doi: 10.1128/JVI.03545-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tan Z, Maguire ML, Loeb DD, Zlotnick A. 2013. Genetically altering the thermodynamics and kinetics of hepatitis B virus capsid assembly has profound effects on virus replication in cell culture. J Virol 87:3208–3216. doi: 10.1128/JVI.03014-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Summers J, Smith PM, Horwich AL. 1990. Hepadnavirus envelope proteins regulate covalently closed circular DNA amplification. J Virol 64:2819–2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lenhoff RJ, Summers J. 1994. Coordinate regulation of replication and virus assembly by the large envelope protein of an avian hepadnavirus. J Virol 68:4565–4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lentz TB, Loeb DD. 2011. Roles of the envelope proteins in the amplification of covalently closed circular DNA and completion of synthesis of the plus-strand DNA in hepatitis B virus. J Virol 85:11916–11927. doi: 10.1128/JVI.05373-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cui X, Ludgate L, Ning X, Hu J. 2013. Maturation-associated destabilization of hepatitis B virus nucleocapsid. J Virol 87:11494–11503. doi: 10.1128/JVI.01912-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmitz A, Schwarz A, Foss M, Zhou L, Rabe B, Hoellenriegel J, Stoeber M, Pante N, Kann M. 2010. Nucleoporin 153 arrests the nuclear import of hepatitis B virus capsids in the nuclear basket. PLoS Pathog 6:e1000741. doi: 10.1371/journal.ppat.1000741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koschel M, Oed D, Gerelsaikhan T, Thomssen R, Bruss V. 2000. Hepatitis B virus core gene mutations which block nucleocapsid envelopment. J Virol 74:1–7. doi: 10.1128/JVI.74.1.1-7.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yuan TT, Sahu GK, Whitehead WE, Greenberg R, Shih C. 1999. The mechanism of an immature secretion phenotype of a highly frequent naturally occurring missense mutation at codon 97 of human hepatitis B virus core antigen. J Virol 73:5731–5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ponsel D, Bruss V. 2003. Mapping of amino acid side chains on the surface of hepatitis B virus capsids required for envelopment and virion formation. J Virol 77:416–422. doi: 10.1128/JVI.77.1.416-422.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pairan A, Bruss V. 2009. Functional surfaces of the hepatitis B virus capsid. J Virol 83:11616–11623. doi: 10.1128/JVI.01178-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rabe B, Vlachou A, Pante N, Helenius A, Kann M. 2003. Nuclear import of hepatitis B virus capsids and release of the viral genome. Proc Natl Acad Sci U S A 100:9849–9854. doi: 10.1073/pnas.1730940100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guo H, Mao R, Block TM, Guo JT. 2010. Production and function of the cytoplasmic deproteinized relaxed circular DNA of hepadnaviruses. J Virol 84:387–396. doi: 10.1128/JVI.01921-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo H, Jiang D, Zhou T, Cuconati A, Block TM, Guo JT. 2007. Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: an intermediate of covalently closed circular DNA formation. J Virol 81:12472–12484. doi: 10.1128/JVI.01123-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang GH, Seeger C. 1992. The reverse transcriptase of hepatitis B virus acts as a protein primer for viral DNA synthesis. Cell 71:663–670. doi: 10.1016/0092-8674(92)90599-8. [DOI] [PubMed] [Google Scholar]
- 40.Jones SA, Boregowda R, Spratt TE, Hu J. 2012. In vitro Epsilon RNA-dependent protein priming activity of human hepatitis B virus polymerase. J Virol 86:5134–5150. doi: 10.1128/JVI.07137-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nguyen DH, Hu J. 2008. Reverse transcriptase- and RNA packaging signal-dependent incorporation of APOBEC3G into hepatitis B virus nucleocapsids. J Virol 82:6852–6861. doi: 10.1128/JVI.00465-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nguyen DH, Gummuluru S, Hu J. 2007. Deamination-independent inhibition of hepatitis B virus reverse transcription by APOBEC3G. J Virol 81:4465–4472. doi: 10.1128/JVI.02510-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaplan PM, Greenman RL, Gerin JL, Purcell RH, Robinson WS. 1973. DNA polymerase associated with human hepatitis B antigen. J Virol 12:995–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu J, Flores D, Toft D, Wang X, Nguyen D. 2004. Requirement of heat shock protein 90 for human hepatitis B virus reverse transcriptase function. J Virol 78:13122–13131. doi: 10.1128/JVI.78.23.13122-13131.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hirt B. 1967. Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol 26:365–369. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- 46.Ning X, Nguyen D, Mentzer L, Adams C, Lee H, Ashley R, Hafenstein S, Hu J. 2011. Secretion of genome-free hepatitis B virus—single strand blocking model for virion morphogenesis of para-retrovirus. PLoS Pathog 7:e1002255. doi: 10.1371/journal.ppat.1002255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Staprans S, Loeb DD, Ganem D. 1991. Mutations affecting hepadnavirus plus-strand DNA synthesis dissociate primer cleavage from translocation and reveal the origin of linear viral DNA. J Virol 65:1255–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kock J, Kann M, Putz G, Blum HE, Von Weizsacker F. 2003. Central role of a serine phosphorylation site within duck hepatitis B virus core protein for capsid trafficking and genome release. J Biol Chem 278:28123–28129. doi: 10.1074/jbc.M300064200. [DOI] [PubMed] [Google Scholar]
- 49.Kock J, Wieland S, Blum HE, von Weizsacker F. 1998. Duck hepatitis B virus nucleocapsids formed by N-terminally extended or C-terminally truncated core proteins disintegrate during viral DNA maturation. J Virol 72:9116–9120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bereszczak JZ, Rose RJ, van Duijn E, Watts NR, Wingfield PT, Steven AC, Heck AJ. 2013. Epitope-distal effects accompany the binding of two distinct antibodies to hepatitis B virus capsids. J Am Chem Soc 135:6504–6512. doi: 10.1021/ja402023x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bereszczak JZ, Watts NR, Wingfield PT, Steven AC, Heck AJ. 2014. Assessment of differences in the conformational flexibility of hepatitis B virus core-antigen and e-antigen by hydrogen deuterium exchange-mass spectrometry. Protein Sci 23:884–896. doi: 10.1002/pro.2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dhason MS, Wang JC, Hagan MF, Zlotnick A. 2012. Differential assembly of hepatitis B virus core protein on single- and double-stranded nucleic acid suggest the dsDNA-filled core is spring-loaded. Virology 430:20–29. doi: 10.1016/j.virol.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hilmer JK, Zlotnick A, Bothner B. 2008. Conformational equilibria and rates of localized motion within hepatitis B virus capsids. J Mol Biol 375:581–594. doi: 10.1016/j.jmb.2007.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cui X, Guo JT, Hu J. 17 June 2015. Hepatitis B virus covalently closed circular DNA formation in immortalized mouse hepatocytes associated with nucleocapsid destabilization. J Virol doi: 10.1128/JVI.01261-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guidotti LG, Matzke B, Schaller H, Chisari FV. 1995. High-level hepatitis B virus replication in transgenic mice. J Virol 69:6158–6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ni Y, Lempp FA, Mehrle S, Nkongolo S, Kaufman C, Falth M, Stindt J, Koniger C, Nassal M, Kubitz R, Sultmann H, Urban S. 2014. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology 146:1070–1083. doi: 10.1053/j.gastro.2013.12.024. [DOI] [PubMed] [Google Scholar]
- 57.He W, Ren B, Mao F, Jing Z, Li Y, Liu Y, Peng B, Yan H, Qi Y, Sun Y, Guo JT, Sui J, Wang F, Li W. 2015. Hepatitis D virus infection of mice expressing human sodium taurocholate co-transporting polypeptide. PLoS Pathog 11:e1004840. doi: 10.1371/journal.ppat.1004840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Le Pogam S, Shih C. 2002. Influence of a putative intermolecular interaction between core and the pre-S1 domain of the large envelope protein on hepatitis B virus secretion. J Virol 76:6510–6517. doi: 10.1128/JVI.76.13.6510-6517.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Seeger C, Hu J. 1997. Why are hepadnaviruses DNA and not RNA viruses? Trends Microbiol 5:447–450. doi: 10.1016/S0966-842X(97)01141-4. [DOI] [PubMed] [Google Scholar]