

ABSTRACT
Cytomegalovirus (CMV) is a ubiquitous beta-herpesvirus whose reactivation from latency is a major cause of morbidity and mortality in immunocompromised hosts. Mouse CMV (MCMV) is a well-established model virus to study virus-host interactions. We showed in this study that the CD8-independent antiviral function of myeloid dendritic cells (mDC) is biologically relevant for the inhibition of MCMV replication in vivo and in vitro. In vivo ablation of CD11c+ DC resulted in higher viral titers and increased susceptibility to MCMV infection in the first 3 days postinfection. We developed in vitro coculture systems in which we cocultivated MCMV-infected endothelial cells or fibroblasts with T cell subsets and/or dendritic cells. While CD8 T cells failed to control MCMV replication, bone marrow-derived mDC reduced viral titers by a factor of up to 10,000. Contact of mDC with the infected endothelial cells was crucial for their antiviral activity. Soluble factors secreted by the mDC blocked MCMV replication at the level of immediate early (IE) gene expression, yet the viral lytic cycle reinitiated once the mDC were removed from the cells. On the other hand, the mDC did not impair MCMV replication in cells deficient for the interferon (IFN) alpha/beta receptor (IFNAR), arguing that type I interferons were critical for viral control by mDC. In light of our recent observation that type I IFN is sufficient for the induction of latency immediately upon infection, our results imply that IFN secreted by mDC may play an important role in the establishment of CMV latency.
IMPORTANCE Numerous studies have focused on the infection of DC with cytomegaloviruses and on the establishment of latency within them. However, almost all of these studies have relied on the infection of DC monocultures in vitro, whereas DC are just one among many cell types present in an infection site in vivo. To mimic this aspect of the in vivo situation, we cocultured DC with infected endothelial cells or fibroblasts. Our data suggest that direct contact with virus-infected endothelial cells activates CD11c+ DC, which leads to reversible suppression of MCMV replication at the level of IE gene expression by a mechanism that depends on type I IFN. The effect matches the formal definition of viral latency. Therefore, our data argue that the interplay of dendritic cells and infected neighboring cells might play an important role in the establishment of viral latency.
INTRODUCTION
Human cytomegalovirus (HCMV) is a betaherpesvirus which establishes a lifelong latent infection in immunocompetent hosts. Latent HCMV is present in the majority of people worldwide, but the primary infection is usually asymptomatic. The primary infection is well contained by the immune cells, such as natural killer (NK) cells and T cells, which also prevent viral reactivation from latency (1, 2).Their activation depends on cross talk with dendritic cells (DC) (3, 4), and this interaction plays an important role in CMV control (5–7). The direct effect of DC on viral replication remains, however, unclear.
In immunocompromised hosts, like AIDS patients or people undergoing transplantation, the virus cannot be contained, and its reactivation from latency has been associated with severe disease (8). Therefore, to develop new therapeutic approaches against CMV disease, it is exceedingly important to understand the immune mechanisms that drive the virus into latency.
Murine cytomegalovirus (MCMV) is a natural pathogen of the mouse. It shows numerous analogies in latency and reactivation to the human virus, and its genome displays substantial similarity to the HCMV one (9). Therefore, MCMV is a widely used model for CMV infection and immunity (10–12).
During primary infection, MCMV infects various different cell types, such as macrophages and DC but also nonhematopoietic cells, including endothelial and epithelial cells (13). On the other hand, the establishment of latency appears to be restricted to certain cell types. Latent HCMV was found in blood monocytes and in progenitor cells of the myeloid lineage (14–19), whereas liver sinusoidal endothelial cells (LSEC) were shown to be a site of MCMV latency and reactivation (20, 21), although myeloid cells might also present a latent reservoir in the mouse (22, 23).
DC are heterogeneous mononuclear phagocytes which can be classified in different subsets due to their ontogeny, surface markers, and functions (24). Murine plasmacytoid DC (pDC) are described as CD11b− CD11cint SiglecH+ and are the major source of interferon alpha/beta (IFN-α/β) in response to MCMV infection (25–27). We and colleagues showed recently that pDC mount high type I interferon (IFN-I) responses after MCMV infection (28), even though the cells do not appear to be productively infected. In these cells, the sensing of MCMV is completely dependent on myeloid differentiation primary response gene 88 (MyD88), with a major contribution of the Toll-like receptor 9 (TLR9) (7, 29, 30). Type I IFNs play a prominent role in the protection against CMV infection, and we have recently shown that IFN-β is able to block MCMV immediate early (IE) gene expression in a reversible manner, which is consistent with the formal definition of viral latency (21).
In contrast to pDC, which morphologically resemble plasma cells, conventional myeloid DC (mDC) and macrophages (Mϕ) are derived from a common myeloid progenitor cell (31). The nonplasmacytoid DC may also secrete IFN-I upon infection with RNA viruses, such as lymphoid choriomeningitic virus or NS1-deficient influenza mutants (32). The susceptibility of mDC and Mϕ to CMV was addressed in different studies. MCMV can infect and replicate in both cell types, but the replication is suboptimal in macrophages and even less efficient in mDC (28). They secrete IFN-β and a small amount of IFN-α in response to MCMV infection, but uninfected bystander mDC produce just minor amounts of type I IFN (28). However, all of these studies were performed with monocultures of DC infected at a high multiplicity of infection (MOI), which might not reflect the in vivo situation, where most of the infected cells are epithelial and endothelial cells, which show higher permissiveness for MCMV than DC. Hence, it is still unclear if DC restrict or contribute to viral replication and spread (24).
While the role of mDC in activating CD8 T cells or NK cells and their cytokine release after direct infection was investigated in various studies, the direct effect of mDC on MCMV replication has remained unexplored. Here we show that CD11c+ mDC are sufficient for the inhibition of MCMV replication immediately after infection in vivo and in vitro, in a manner that is independent of their function as antigen-presenting cells (APC). While the coculture of mDC with infected embryonic fibroblasts (MEF) or endothelial cells activated mDC to release IFN and reversibly inhibit IE1 gene expression, direct MCMV infection of mDC in monoculture did not show this effect. Therefore, infection experiments with cocultures of permissive target cells and mDC revealed a novel mechanism by which mDC-secreted IFN may contribute to the early establishment of virus latency.
MATERIALS AND METHODS
Mice.
All animal experiments were handled in compliance with the German regulations for animal experimentation (Animal Welfare Act) and approved by the responsible state office (Lower Saxony State Office of Consumer Protection and Food Safety) under permit numbers 33.9-42502-04-11/0426 and 33.19-42502-04-14/1710. C57BL/6 mice for bone marrow (BM) isolation were purchased from Janvier (Le Genest St Isle, France) at 4 weeks of age. B6.FVB-TG (Itgax-DTR/EGFP)57Lan/J mice and C57BL/6 littermates were purchased from The Jackson Laboratory (Bar Harbor, ME) at 5 to 6 weeks of age. gBT-I mice (33) were housed under specific-pathogen-free (SPF) conditions and kindly provided by Frank Carbone (University of Melbourne) and Georg Behrens (Medical School Hanover).
Cells.
M2-10B4 and NIH 3T3 cells were maintained in Dulbecco's modified Eagle medium supplemented with 10% fetal calf serum, 1% glutamine, and 1% penicillin-streptomycin. Primary C57BL/6 and IFNAR−/− MEF were prepared and maintained as described previously (11). LSEC were isolated from in-house-bred C57BL/6 mice, and the LSECB6 line was generated by conditional immortalization and cultivation as previously described (34).
Viruses.
The bacterial artificial chromosome (BAC)-derived wild-type MCMV (MCMV WT) (35) and MCMVr strains (34) have been described previously. To generate the MCMVIE2SL-MIEP mutant, the sequence encoding the SSIEFARL peptide was inserted at the 3′ end of the ie2 gene, as described before (36), whereupon the full-length major immediate early promoter (MIEP) sequence was inserted into this mutant using a previously described construct and insertion site (34). Viral growth of the new mutant was assessed by infecting NIH 3T3 cells with MCMVIE2SL-MIEP or MCMV WT at an MOI of 0.1 as described elsewhere (34), and no growth impairment was observed. All MCMV clones were grown on M2-10B4 cells and purified as described before (37). Virus infectious titers were determined by plaque assay on MEF as described previously (38).
Diphtheria toxin (DT) treatment and in vivo infection.
Eight- to 14-week-old Itgax-DTR/EGFP and C57BL/6 mice were intraperitoneally (i.p.) injected with DT (Sigma-Aldrich, St. Louis, MO) diluted in phosphate-buffered saline (PBS) at a dose of 9 ng per g of body weight. Control groups were injected with the same volume of PBS. Infection with 106 PFU of purified, tissue culture-derived MCMV WT was performed intraperitoneally 12 to 24 h after DT treatment. Mice were monitored for signs of illness and loss of body weight throughout the experiment. On day 3 postinfection, mice were sacrificed by CO2 administration. Spleen, lungs, and weighed parts of the liver were harvested under sterile conditions and stored at −70°C until titration was performed. Organ homogenates were titrated as described previously (38).
Isolation and sorting of CD11c+ myeloid dendritic cells.
BM cells were isolated by flushing the femur and tibia of C57BL/6 mice with RPMI medium supplemented with 10% fetal calf serum, 1% glutamine, and 1% penicillin-streptomycin.
After red blood cell lysis with ammonium chloride-potassium (ACK) lysing buffer, cells were washed and resuspended at a density of 1 × 106/ml in medium supplemented with 100 ng/ml of granulocyte-macrophage colony-stimulating factor (GM-CSF). Cells were seeded in T175 flasks and incubated at 37°C, 5% CO2, and 5% O2 for 6 days. Medium was changed on day 3 or 4, depending on the status of the cultures, by centrifuging the cells (1,200 rpm for 5 min) and replacing the supernatant with fresh GM-CSF-supplemented medium. On day 6 nonadherent cells were harvested and sorted. Cells were stained for 20 min at 4°C with CD11c-phycoerythrin (PE) antibody (Biolegend, San Diego, CA), washed with PBS–2.5% fetal bovine serum (FBS) to remove unbound primary antibody, and afterwards stained with 20 μl of anti-PE MicroBeads (Miltenyi Biotec) per 107 total cells for 15 min at 4°C. After the washing step, cells were resuspended in 2 ml of PBS–2.5% FBS and purified by magnetic sorting.
Isolation and sorting of CD4− CD19− CD8+ T cells from gBT-I mice.
gBT-I mice were used as donors for CD4− CD19− CD8+ T cells. In brief, spleens were homogenized and erythrocytes were lysed with ACK buffer. Cells were washed and stained with anti-CD8b-PE-Cy7, anti-CD19-PerCP/Cy5.5, and anti-CD4-Pacific blue (Biolegend). Cells were washed and sorted for CD8+ CD19− CD4− using a FACS Aria II (BD Bioscience) cell sorter, and purity was >98% in all experiments.
Isolation and sorting of NK cells.
NK cells were isolated from naive C57BL/6 mice. In brief, spleens were isolated and homogenized, and erythrocytes were lysed with ACK buffer. Cells were stained with anti-CD3-PE and anti-NKp46-eFluor660 (Bioscience). Cells were washed and prepared for sorting. For this, cells were transferred to 15-ml conical centrifugation tubes. The cell suspension was further filtered through CellTrics filters to remove cell debris. The cells were acquired to gate on CD3− Nkp46+ using a FACS Aria II (BD Bioscience) cell sorter. The marked cells were sorted into 15-ml conical centrifugation tubes containing RPMI medium. The sorted cells were counted and directly used for the coculture experiments.
In vitro infection and coculture protocols.
Confluent monolayers of noncycling LSEC, C57BL/6 MEF, IFNAR−/− MEF, or CD11c+ mDC were infected in 96-well or 48-well plates with MCMVr or MCMVIE2SL-MIEP at the desired MOIs. After 1 h, the infectious supernatant was replaced either with fresh RPMI 1640 or with CD11c+ mDC and/or CD8+ CD19− CD4− cells at the desired ratios. To test if CD8+ T cells sorted from gBT-I mice are activated by LSEC infected with MCMVIE2SL-MIEP (MOI of 1), IFN-γ released in the supernatants was measured at 1 day postinfection; more than a 1,000-fold induction could be observed.
Two-chamber experiments.
Collagen-coated 0.4-μm-pore-size Transwell chambers suitable for mounting on 24-well plates were purchased from Corning.
LSEC were seeded in the lower chamber and infected with MCMVr at an MOI of 0.1. LSEC or mDC were infected (MOI of 0.1) alone or in coculture in the upper chamber. Infectious virus titers were determined in supernatants from the lower chambers at day 7 postinfection.
Supernatant experiments.
LSEC or CD11c+ DC were seeded in T75 flasks and infected with MCMVr. The virus was washed from LSEC cultures after 1 h of infection, and RPMI 1640 or CD11c+ DC were added. This wash step was omitted in CD11c+ DC cultures to increase virus infection in the low-permissiveness cell type. Supernatants were harvested after 5 to 7 days, filtered through a 0.1-μm filter (Pall Newquay, Cornwall, United Kingdom), and added to cultures of LSEC infected with MCMVr. Seven days later, titers of infectious virus in supernatants from secondary cultures were determined. Where indicated, the supernatant was replaced with normal medium at day 5 postinfection, upon which the medium or supernatants were exchanged with fresh batches every 2 days up to day 12 postinfection. Infected cells were monitored via fluorescence microscopy for enhanced yellow fluorescent protein (EYFP) gene expression, and wells with one single positive cell were classified as productively infected. Medium was taken at day 5, day 7, day 9, and day 12 to determine virus titers before and after the medium exchange.
RESULTS
In vivo ablation of CD11c+ mDC increases susceptibility to MCMV infection.
The in vivo relevance of numerous immune subsets in the control of MCMV infection can be studied by targeted depletion strategies. Still, the role of mDC remains unclear. To determine the effect of CD11c+ myeloid dendritic cell depletion during early murine cytomegalovirus infection, we took advantage of the well-described ltgax-DTR-GFP transgenic mouse model (39), which allows for inducible depletion of CD11c+ dendritic cells by a single treatment with diphtheria toxin (DT). Eighteen hours after DT application all DC are depleted, and they return to normal levels 5 days later (40).
We treated Itgax-DTR-Tg mice or BL/6 littermates with DT or a PBS vehicle control, 12 to 24 h prior to MCMV infection. DT-treated Itgax-DTR-Tg animals showed signs of illness, like lethargy or ruffled fur (data not shown), and lost, on average, 16.5% of their total body weight in the first 3 days after the infection, whereas BL/6 littermates or Itgax-DTR-Tg mice with the PBS vehicle control lost about 3 to 4% on day 1 postinfection but then stabilized (Fig. 1A). At day 3 postinfection, weight was significantly reduced (P < 0.001) in MCMV-infected Itgax-DTR-Tg mice treated with DT compared to that in all control groups. Control Itgax-DTR-Tg mice, which were treated with DT but not infected with MCMV, did not lose weight (data not shown), which is in line with published data (39, 41).
FIG 1.
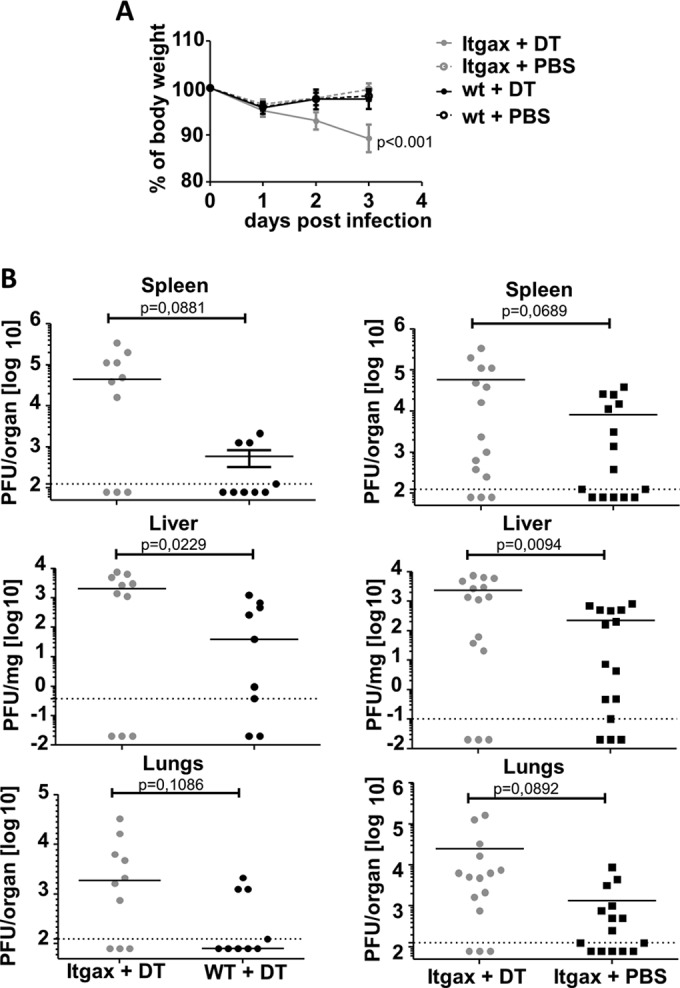
Impact of depletion of CD11c+ dendritic cells on body weight loss and viral titers after MCMV infection. Itgax-DTR-TG mice or WT littermates were treated with DT (9 ng/g of body weight) or a PBS vehicle as control 1 day prior to infection with 106 PFU of MCMV WT. (A) Mice were weighed on infection and daily thereafter, and average group weights are shown. Errors bars indicate the standard errors of the means (SEM). Weight loss over time in Itgax-DTR-Tg mice treated with DT was compared with all control groups by repeated-measures two-way analysis of variance (ANOVA) followed by Bonferroni postanalysis. (B) Lung, spleen, and liver homogenates were plaque assayed for infectious MCMV titers at day 3 postinfection. Each symbol represents one mouse, and horizontal lines indicate group medians. The left and right sides show independent sets of two (left graphs) or three (right graphs) experiments. Statistics were performed using the Student t test with Welch correction.
Three days after MCMV infection, plaque assay revealed a >10-fold median increase in infectious MCMV titers in livers, lungs, and spleens of DT-treated Itgax-DTR-Tg mice in comparison to those in DT-treated BL/6 mice, Itgax-DTR-Tg littermates with an intact DC population (Fig. 1B), and other control groups (data not shown). On the other hand, the MCMV titers observed in organs of DC-depleted mice overlapped in part with those from the control groups, which was reflected by P values ranging from 0.01 to 0.1 in the tested organs and below 0.05 only in the liver. The experiment was performed twice (for DT-treated Itgax-DTR-Tg mice versus DT-treated BL/6 mice) or three times (DT-treated Itgax-DTR-Tg mice versus PBS-treated DTR-Tg mice), and data were pooled. Therefore, our data indicated that CD11c+ DC might play an important role in limiting MCMV replication early after infection.
Myeloid dendritic cells repress MCMV immediate early gene expression in vitro independently of CD8+ T cell and NK cell response.
Since we observed a clear effect of DC ablation on weight loss and MCMV growth at 3 days postinfection, which is before detectable CD8 responses to MCMV may be measured in the blood or lymphatic organs (data not shown), we surmised that these effects might be CD8+ T cell independent. On the other hand, DC ablation by DT administration diminishes CD8+ CTL function after herpes simplex virus 1 (HSV-1) infection (41). Thus, we wondered if DC impaired MCMV replication by activating CD8+ T cells or by direct effects of CD11c+ DC on virus replication.
To study this question directly, we derived mDC from bone marrow cultures by culturing them with GM-CSF and sorted the CD11c+ subset. CD11c+ DC were cocultured with syngeneic MCMV-infected LSEC or with MCMV-infected LSEC and antigen-specific CD8+ T cells. We used the new LSEC line LSECB6 and generated a recombinant MCMV (MCMVIE2SL-MIEP) expressing two fluorescent proteins under the control of the full-length major immediate early promoter-enhancer, akin to the previously published MCMVr (34). The mutant also encoded the Kb-restricted peptide SSIEFARL as a C-terminal tag on the IE2 viral gene, because this expression context results in robust SSIEFARL-specific CD8 responses upon infection with recombinant MCMV (36). More importantly, this allowed us to use SSIEFARL-specific CD8+ T cells derived from T cell receptor (TCR)-transgenic gBT-I mice (33), recognizing virus-infected cells in coculture experiments.
In line with published data (42), we observed that MCMV-infected LSEC are able to activate CD8+ T cells, which results in IFN-γ production (data not shown). Cocultures of CD8+ T cells and infected cells moderately impaired virus replication over the control group (Fig. 2A). However, viral titers were substantially decreased when infected LSEC were cocultivated with CD11c+-sorted mDC from naive mice. Identical titers were observed when DC and infected LSEC were cocultured with virus-specific CD8 T cells, arguing that mDC have a direct antiviral function in vitro, which is independent of their function as APC.
FIG 2.

Myeloid dendritic cells are able to repress immediate early gene expression in coculture with LSEC or MEF. MEF or LSEC were infected with recombinant MCMVs and cocultured with antigen-specific CD8+ T cells or mDC. (A) LSEC infected with MCMVIE2SL-MIEP (MOI of 0.001). Where indicated, LSEC were cocultured with CD4− CD19− CD8+ sorted cells from gBT-I (ratio of 1:1) mice, CD11c+ mDC (ratio of 1:1), or both mDC and CD8 T cells. Virus titer in supernatants on day 5 postinfection was assessed in triplicate, and average values ± SEM are shown. (B) LSEC infected with MCMVr (MOI of 0.01) were cocultured with CD3− Nkp46+ sorted NK cells (ratio of 4:1), CD11c+ mDC (ratio of 4:1), or both NK cells and DC and compared to LSEC monocultures as control. Virus titer in supernatants on day 6 postinfection was assessed in triplicate, and average values ± SEM are shown. (C) LSEC were infected with MCMVr at an MOI of 0.1 and cocultured with increasing proportions of CD11c+ mDC. Each condition was performed in biological triplicates in two independent experiments, and average virus titers in supernatants at day 6 postinfection are shown. Errors bars indicate SEM. (D) MEF were infected with MCMVr at the indicated MOIs and cocultured with CD11c+ mDC at a ratio of 1:4. Supernatants from biological triplicates at the indicated days were titrated on MEF, and graphs show average values ± SEM. (E) Representative bright-field and fluorescence microscopy pictures of MEF infected at an MOI of 0.1 in the presence or absence of CD11c+ mDC. Pictures were taken at day 6 postinfection. DL, detection limit.
Nevertheless, besides their function in activating T cells, DC contribute to the early control of MCMV infection by activating NK cells (43). The reduced control of MCMV after the depletion of mDC in vivo might therefore result from inefficient priming of NK cells. To test if mDC and NK cells would synergize to control MCMV replication in LSEC, a coculture with mDC and NK cells was set up. LSECB6 were infected with MCMVr and cultured alone or in the presence of CD11c+ mDC or CD3− NKp46+ NK cells, or together with both. The addition of mDC to MCMV-infected LSECB6 resulted in a marked decrease of viral titers, consistent with previous results, whereas coculture with NK cells did not efficiently diminish viral titers (Fig. 2B). The inability of NK cells to diminish MCMV replication in vitro is in line with recently published data (44). Coculture with both NK cells and DC did not result in a further decrease of MCMV titers in comparison to the culture with mDC only (Fig. 2B), arguing that the in vivo effect might be independent of a synergy between DC and NK cells.
To test the effector-target ratio (ETR) required for the abrogation of viral replication, we cocultured infected LSEC with increasing proportions of mDC in cell culture (from 1:10 to 10:1). We observed an inhibition of viral replication starting at an ETR of 1:1 and complete abrogation at an ETR of 10:1 (Fig. 2C). Hence, the number of DC was relevant for the suppression of virus titers (Fig. 2C).
To test if CD11c+ DC control MCMV replication only in an endothelial cell line and at defined MOIs, or in a variety of infection conditions, we infected MEF with different MOIs of MCMV and monitored the kinetic of viral replication in the presence or absence of DC. Virus replication appeared similar to that in the control group until day 2 postinfection but was impaired thereafter in all tested conditions (Fig. 2D). To further test the ability of DC to control virus replication in primary cells, we cocultured CD11c+ mDC with infected primary LSEC and observed a similar reduction in virus replication (data not shown).
To clarify if DC blocked MCMV growth by killing infected cells, or by impairing its dissemination, we infected MEF with MCMVr, a recently described reporter MCMV which expresses two fluorescent proteins under the control of the major immediate early promoter (MIEP) of MCMV (21, 34). MCMVr infection of MEF resulted in strong EYFP expression (Fig. 2E). In contrast, hardly any signal was detected in the coculture of MCMVr infected MEF and CD11c+ mDC at 6 days postinfection with an MOI of 0.1. The MEF monolayer was maintained, with a few single infected cells within it, indicating that the infection was not spreading to bystander cells in the presence of DC but also that infected cells were not directly killed (Fig. 2E).
In summary, these experiments provided evidence for direct antiviral function of CD11c+ sorted mDC obtained from the bone marrow of naive mice in a new coculture assay with infected fibroblasts or endothelial cells.
Infected neighbor cells are necessary for activation of mDC and the release of an antiviral factor.
Dendritic cells are known as professional APC with high endocytotic properties but also capable of secreting cytokines and chemokines (3). To determine whether MCMV replication was blocked by a factor that was secreted by the mDC and if this factor is secreted during contact with virus particles or infected cells, we performed two-chamber experiments. We used commercially available systems with membranes carrying pores of 0.4 μm in diameter, which completely block the migration of cells from one chamber to the other but still allow the virus to move between chambers. LSEC were seeded in the lower chamber and infected with MCMV. In the upper chamber, we seeded CD11c+ mDC, cocultures of infected LSEC with CD11c+ mDC. or just infected LSEC, as reference control. At day 7 postinfection, the virus titer in the lower chamber showed no differences from the reference control in the groups where mDC were seeded alone in the upper chamber, whereas cocultures of LSEC and mDC in the upper well reduced viral titers in the lower well by a factor of 10, as normalized to values from the control group (Fig. 3A).
FIG 3.
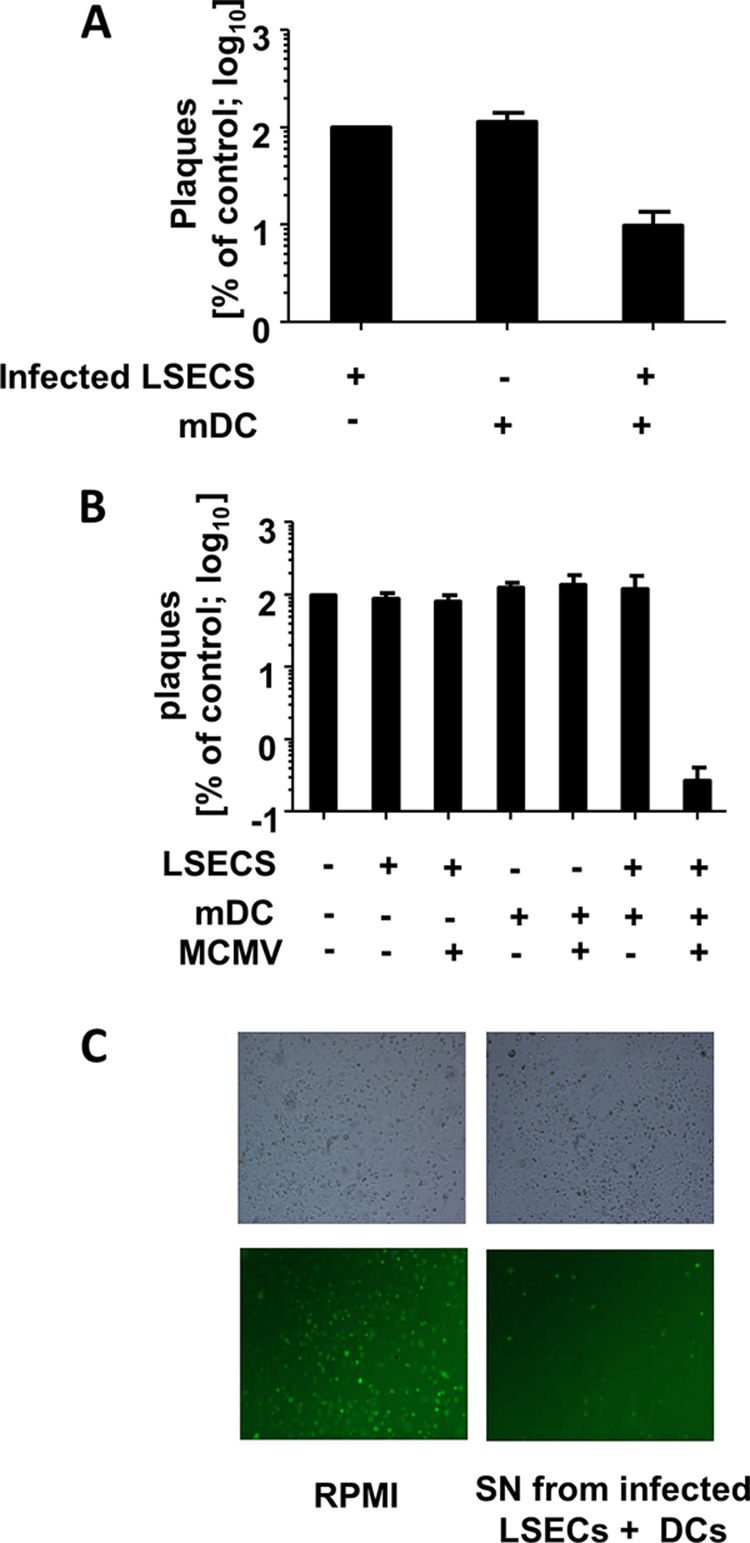
Direct contact between CD11c+ mDC and infected target cells is required for secretion of a factor, which can block mCMV replication. (A) Virus growth in a two-chamber assay. LSEC were seeded in triplicates in the lower chamber and infected with MCMVr (MOI 0.1), while the upper chamber was seeded with mDC, infected LSEC (MOI 0.1), or mDC and infected LSEC. Virus titers were determined by plaque assay in supernatants from the lower chamber and normalized to values from the control group (no mDC in the upper chamber). Shown is the growth as a percentage of the control. Cells in the upper chambers are indicated below the x axis. Experiments were performed twice, and data were pooled. (B) Single cultures or cocultures of LSEC and mDC were infected (MOIs of 0.01 for LSEC and 1 for DC) or left uninfected. Supernatants were taken at day 6 or 7, filtered to prevent virus carryover, and transferred to fresh LSEC cultures infected with MCMVr (MOI of 0.1). Virus titers at day 6 postinfection were normalized to LSEC infected in the presence of standard cell culture medium (RPMI) and are shown on the y axis. Keys below the x axis indicate SN used as medium during second infection. Data from three independent experiments were pooled and are shown. (C) Representative bright-field and fluorescence microscopy pictures of LSEC infected at an MOI of 1 with MCMVr cultured in SN taken from coculture or medium control. Pictures were taken at 20 h postinfection.
These data suggested that mDC secreted one or more soluble factors that controlled MCMV growth in LSEC in the lower chamber, but also that mDC inhibited virus replication only if they were in direct contact with LSEC in the upper chamber. However, it remained unclear if the mDC were activated by contact with infected LSEC or if contact with uninfected LSEC would suffice for their activation. Furthermore, since the pores between chambers could not prevent MCMV from moving between chambers, we could not exclude that MCMV disseminated into the upper chambers and activated the mDC directly. To determine which conditions were required to release the soluble factors that controlled MCMV replication, we harvested conditioned supernatants from MCMV-infected or uninfected LSEC, CD11c+ mDC, or their combinations. Supernatants were filtered through 0.1-μm filters to prevent virus carryover and transferred to newly infected LSEC to investigate the antiviral properties of the different supernatants. Interestingly, only the supernatants gathered from the coculture of infected LSEC and mDC impaired viral growth. Under these conditions, the viral titer was 100-fold reduced in comparison to that in reference controls (Fig. 3B). In contrast, direct infection of mDC or the coculture of uninfected endothelial cells with mDC failed to activate DCs to secrete antiviral cytokines. Therefore, we conclude that the antiviral properties of mDCs are mainly due to the direct interaction with the infected target cell, whereas direct activation of DCs by viral particles and/or virus-derived pathogen-associated molecular patterns (PAMPs) play an indistinguishable role in their activation. We considered that the antiviral action may be induced by gap junction transfer of cGAMP (45) from LSEC into mDC. In this scenario, cGAMP would be transferred from the infected endothelial cells via gap junctions to the mDC where cGAMP is sensed by STING, which leads to induction of IFN-I. However, our preliminary data argued that this is not the case, because STING-deficient mDC were also able to control MCMV replication in LSEC (data not shown).
The same supernatant strongly inhibited the expression of a reporter gene expressed with immediate early kinetics in LSEC, and this inhibition was evident even at 20 h postinfection (Fig. 3C). On the other hand, the supernatants obtained from single cultures of LSEC or DC or the coculture of uninfected LSEC and mDC did not impair immediate early gene expression (data not shown). Therefore, the antiviral substance released by mDC appeared to act at the level of immediate early expression or prior to it.
CD11c+ DC secrete IFN type I to block virus replication in a reversible manner.
The inhibition of viral replication at the immediate early level was reminiscent of our recent observation that IFN-β suppresses MCMV transcription at this step of the viral replication cycle (21). Importantly, we had shown that IFN-β silences gene expression and viral replication in a reversible manner, consistent with the induction of latency (21). To test if the inhibition of MCMV replication by mDC is due to a transient inhibition of viral gene expression or is a reflection of permanent virus clearance, infected LSEC were monitored over time. We seeded the cells in dodecuplicate wells of a 96-well plate, infected them with MCMVr, and added filtered supernatants from cocultures of infected LSEC and CD11c+ mDC. Control LSEC were infected in the presence of standard tissue culture medium. Wells were scanned for fluorescent gene expression, and if even one single EYFP-positive cell was observed within 5 days of infection (Fig. 4B, left), the whole well was classified as positive for lytic infection (21). We observed a complete abrogation of viral gene expression in approximately 75% of the wells that received conditioned supernatants (SNs) from LSEC-mDC cocultures, while viral replication occurred in almost all of the control wells. To test if the suppression of viral gene expression is reversible, we replaced the supernatants at day 5 postinfection with standard medium and monitored the cells for additional 7 days. Remarkably, MCMV IE gene expression initiated in all of the wells where the supernatants were replaced but only in a few of those kept with the original SNs from LSEC-mDC cocultures (Fig. 4). Similarly, no or very low infectious viral titers were observed in the supernatants of the wells cultured with the conditioned supernatants, but the exchange of the medium resulted in a sharp increase in infectious MCMV titers (Fig. 4B, right). Since MCMV latency is defined as the persistence of viral genomes in the absence of lytic viral replication and gene expression, and by the viral ability to reinitiate the lytic cycle from this state, the reversible inhibition of viral replication in our model implied that supernatants from LSEC-DC cocultures may be sufficient to drive the virus into latency, similar to effects that we recently described for type I IFN (21).
FIG 4.

Soluble factor secreted from CD11c+ myeloid dendritic cells impairs MCMVr replication in a reversible manner. LSEC were infected with MCMVr at an MOI of 0.0001 in dodecuplicates and cultured afterwards with medium control or filtered SN from coculture of infected LSEC plus CD11c+ mDC. Wells were analyzed at multiple time points, and those with even one infected cell were classified as positive. At dpi 5, SN was removed in selected wells (+/−) and wells were further cultured with RPMI medium. (A) Representative EYFP fluorescence and bright-field microscopy of medium control or LSEC cultured in filtered SN from coculture 1 and 7 days after SN retraction. (B) Percent of wells showing any viral gene expression at the indicated time points (right panel). Group averages ± SEM from three independent experiments are shown. Supernatant was collected at indicated time points and titrated on MEF (left panel). To monitor for virus reactivation, only wells which showed no viral replication on day 5 were monitored.
It is well known that type I interferons have antiviral potential (46). While pDC are generally accepted as main producers of IFN-I early after MCMV infection (25, 47), nonplasmacytoid CD11chigh DC may also act as interferon-producing cells (32). Thus, we considered that mDC might have been controlling MCMV by secreting IFN-I in our coculture studies. To test this, we compared viral growth in fibroblasts deficient for the IFN-α/β receptor (IFNAR−/− MEF) to IFNAR−/− MEF cocultured with CD11c+ mDC. Additional control groups included fibroblasts derived from wild-type mice (C57BL/6 MEF) infected with MCMV in monocultures or in the presence of mDC. While the coculture of fibroblasts that respond to IFN-I resulted in a 100-fold decrease of virus titers after 5 days (Fig. 5A, black bars), the addition of CD11c+ mDC to IFNAR−/− MEF showed only minor differences in virus titers in comparison to the control (Fig. 5A, gray bars).
FIG 5.
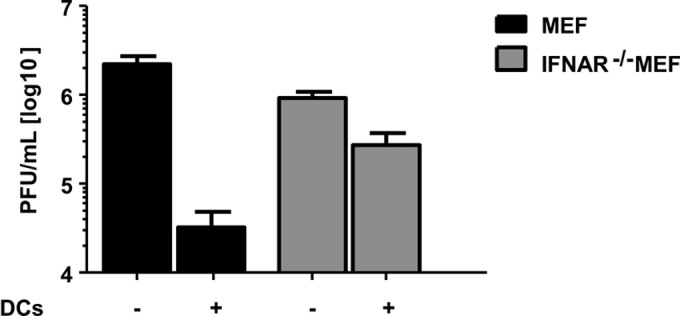
Type I IFN secreted from CD11c+ myeloid dendritic cells impairs MCMVr replication. MEF or IFNAR−/− MEF were infected with MCMVr at an MOI of 0.1 and cocultured in the absence or presence of CD11c+ mDC (ratio of 1:4). Supernatants were collected in triplicates at day 5, and titration was performed on MEF. Graphs show average values ± SEM from three independent experiments.
In summary, our results argue that CD11c+ mDC are necessary for the block of virus replication early after infection in vivo and sufficient in vitro, and this is independent of their function as APC. CD11c+ mDC suppressed MCMV replication in a reversible manner through a mechanism requiring IFN-I signaling and thus may play a critical role in the onset of CMV latency.
DISCUSSION
Numerous studies have focused on mDC permissiveness for infection and on their role in the immune control of CMV infection, but all of them depended on the infection of monocultures of mDC with MCMV (14, 26, 27, 48–50). To the best of our knowledge, this is the first study aiming to address the effect of mDC cocultured with cells that are fully permissive for MCMV infection. This allowed us to observe that only direct contact with infected fibroblasts or endothelial cells provides activating signals to mDC that induce potent antiviral cytokines. Since mDC are naturally in contact with such cells in vivo, our data highlight the limitation of in vitro modeling based on cells kept in monoculture and strongly argue that coculture of immune cells and permissive target cells is a strategy that may allow the identification of hitherto-unknown principles of high biological relevance.
We showed recently that IFN-β is sufficient for the establishment of latency in murine liver endothelial cells in vitro and in vivo (21), but the in vivo source of IFN remained unclear. pDC, rather than mDC, have been considered to play a critical role in the induction of IFN responses that control MCMV in vivo (26, 51). On the other hand, other viral infections were shown to induce potent IFN responses from the mDC subset (32), and a recent study has shown that methods that were used to in vivo deplete pDC by targeting SIGLEC-H also deplete macrophages and mDC (52). Therefore, the relative contribution of different DC subsets to early MCMV control has remained unclear, especially since both mDC and pDC may respond to CMV infection by secreting IFN (28). Our in vivo data argue that CD11c+ DC play an important role in the control of MCMV replication immediately upon infection, and our in vitro data showed that this effect occurs in the absence of CD8 T cells and, thus, that it may be independent of their function as professional APC. On the other hand, our data do not exclude the possibility that pDC also play a crucial role in antiviral control, where a concerted action of both DC subsets is necessary to contain the MCMV replication and spread. Furthermore, we do not exclude the possibility that antigen cross-presentation by mDC and cross talk to other immune cells, such as the previously described cross talk with NK cells (43), contribute to their antiviral function in vivo. In line with this, we cannot rule out the possibility that mDC limit MCMV replication in vivo in an IFN-I-independent manner. While our results showed a strong effect of DC depletion on weight loss upon MCMV infection, a statistically significant (P < 0.05) effect of DC depletion on viral titers was observed only in the liver and not in the spleen or lungs of infected animals. One should be cautious in assuming organ-specific effects, because a similar trend was observed in all organs and in all of the experiments, and the P values in the liver groups, while formally satisfying the significance criterion, were not much lower than those in other organs. Therefore, more experimental data are necessary to corroborate and clarify our in vivo observations.
Interestingly, coculture with antigen-specific CD8 T cells did not impair MCMV replication in LSEC as efficiently as coculture with mDC, although MCMV-infected LSEC may activate antigen-specific CD8 T cells (42). Our results are consistent with in vivo observations that the killing of MCMV-infected cells and viral control by CD8 T cells may be a relatively inefficient process due to MCMV immune-evasive genes (53, 54). While direct infection of mDC with MCMV induces IFN-β and IFN-α responses, and MCMV gene M27 may impair mDC function and IFN-α secretion in response to MCMV infection (28), we showed here that direct contact with infected LSEC (or MEF) activates mDC much more efficiently than virus infection of mDC (and, by extension, more efficiently than direct contact of uninfected mDC with the infected ones). Future studies will elucidate if this cross talk between infected endothelial cells and mDC applies also for the HCMV situation, as well as the exact mechanisms of this activation axis.
MCMV latency in the mouse model of infection is clearly documented in LSEC (20), and IFN-β contains lytic MCMV gene expression in this cell type (21). In contrast, latent HCMV has been described in precursor cells of the myeloid lineage (14), and their ex vivo differentiation into mature mDC results in chromatin remodeling and initiation of lytic gene expression (55). The data presented here may indicate that IFN released by myeloid cells plays an important role in the regulation of CMV latency. Therefore, it is tempting to speculate that the in vitro models of HCMV latency in myeloid cells and their precursors (56) may also depend on robust IFN-I responses. However, such claims would need to be validated by direct evidence in HCMV models of latency.
Virus kinetics in coculture experiments of mDC with infected MEFs showed impaired replication from 3 days postinfection (dpi) onwards. This may appear counterintuitive if one considers that cytokines, especially interferons, are induced very rapidly. However, since the mDC were added to cocultures only after the MEF were infected and the viral inoculum was removed (to avoid direct infection of DCs in the experiment), the first replication cycle necessarily initiated before mDC were able to sense the infection. Furthermore, once MCMV was internalized in LSECs, the activation of DCs could occur only once the virus would reemerge form the infected cell or the infected cell was altered in a manner that was sensed by the nearby DCs. It is important to note that the supernatant derived from the coculture of infected LSEC and mDC had an immediate effect on virus replication at the immediate early level. Taken together, our data argued that the delay in DC activation and buildup of cytokines in the SN likely accounts for the observed delay in the control of virus replication.
It is very intriguing that MCMV replication in LSEC was blocked by adding mDC after the infection (Fig. 2). Since the antiviral activity of mDC depended on IFN signaling (Fig. 5), this implies that the IFN, which is released after virus entry into LSEC, controlled MCMV replication. This in stern contrast to our protocols based on the addition of IFN-β to MCMV-infected LSEC cultures, where IFN-β had to be added at least 8 h prior to infection in order to upregulate host genes that prevent MCMV infection (28, 57). Taken together, these observations lead to the conclusion that additional factors released from mDC in the presence of infected LSEC are required to complement and/or synergize with the antiviral activity of IFN. If one was to identify these factors, novel antiviral immunotherapeutic strategies may become available. This study paves the way toward such efforts.
ACKNOWLEDGMENTS
This work was supported the Helmholtz Association (Helmholtz Virtual Institute VH-VI-424), the German Center for Infection Research (DZIF, TTU 07/804) and the German Scientific Foundation (DFG) through the Collaborative Research Center 900, subproject B2 to L.C.-S. J.K.H. was supported by a stipend from the HZI Graduate School.
We thank Frank Carbone (University of Melbourne) and Georg Behrens (MHH) for providing the gBT-I mice. We thank Ayse Barut, Jennifer Wolf, Thomas Marandu, and Jennifer Oduro for technical assistance.
Tobias May has filed a patent which covers the technology for establishing conditionally immortalized cell lines.
REFERENCES
- 1.Miletic A, Krmpotic A, Jonjic S. 2013. The evolutionary arms race between NK cells and viruses: who gets the short end of the stick? Eur J Immunol 43:867–877. doi: 10.1002/eji.201243101. [DOI] [PubMed] [Google Scholar]
- 2.Lathbury LJ, Allan JE, Shellam GR, Scalzo AA. 1996. Effect of host genotype in determining the relative roles of natural killer cells and T cells in mediating protection against murine cytomegalovirus infection. J Gen Virol 77(Part 10):2605–2613. [DOI] [PubMed] [Google Scholar]
- 3.Banchereau J, Steinman RM. 1998. Dendritic cells and the control of immunity. Nature 392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 4.Tyznik AJ, Verma S, Wang Q, Kronenberg M, Benedict CA. 2014. Distinct requirements for activation of NKT and NK cells during viral infection. J Immunol 192:3676–3685. doi: 10.4049/jimmunol.1300837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andrews DM, Scalzo AA, Yokoyama WM, Smyth MJ, Degli-Esposti MA. 2003. Functional interactions between dendritic cells and NK cells during viral infection. Nat Immunol 4:175–181. doi: 10.1038/ni880. [DOI] [PubMed] [Google Scholar]
- 6.Mandaric S, Walton SM, Rulicke T, Richter K, Girard-Madoux MJ, Clausen BE, Zurunic A, Kamanaka M, Flavell RA, Jonjic S, Oxenius A. 2012. IL-10 suppression of NK/DC crosstalk leads to poor priming of MCMV-specific CD4 T cells and prolonged MCMV persistence. PLoS Pathog 8:e1002846. doi: 10.1371/journal.ppat.1002846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krug A, French AR, Barchet W, Fischer JA, Dzionek A, Pingel JT, Orihuela MM, Akira S, Yokoyama WM, Colonna M. 2004. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity 21:107–119. doi: 10.1016/j.immuni.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 8.Razonable RR, Paya CV. 2002. The impact of human herpesvirus-6 and -7 infection on the outcome of liver transplantation. Liver Transplant 8:651–658. doi: 10.1053/jlts.2002.34966. [DOI] [PubMed] [Google Scholar]
- 9.Rawlinson WD, Farrell HE, Barrell BG. 1996. Analysis of the complete DNA sequence of murine cytomegalovirus. J Virol 70:8833–8849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reddehase MJ, Simon CO, Seckert CK, Lemmermann N, Grzimek NK. 2008. Murine model of cytomegalovirus latency and reactivation. Curr Top Microbiol Immunol 325:315–331. [DOI] [PubMed] [Google Scholar]
- 11.Reddehase MJ, Podlech J, Grzimek NK. 2002. Mouse models of cytomegalovirus latency: overview. J Clin Virol 25(Suppl 2):S23–S36. [DOI] [PubMed] [Google Scholar]
- 12.Hummel M, Abecassis MM. 2002. A model for reactivation of CMV from latency. J Clin Virol 25(Suppl 2):S123–136. [DOI] [PubMed] [Google Scholar]
- 13.Krmpotic A, Bubic I, Polic B, Lucin P, Jonjic S. 2003. Pathogenesis of murine cytomegalovirus infection. Microbes Infect 5:1263–1277. doi: 10.1016/j.micinf.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 14.Hahn G, Jores R, Mocarski ES. 1998. Cytomegalovirus remains latent in a common precursor of dendritic and myeloid cells. Proc Natl Acad Sci U S A 95:3937–3942. doi: 10.1073/pnas.95.7.3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reeves M, Sinclair J. 2008. Aspects of human cytomegalovirus latency and reactivation. Curr Top Microbiol Immunol 325:297–313. [DOI] [PubMed] [Google Scholar]
- 16.Kondo K, Xu J, Mocarski ES. 1996. Human cytomegalovirus latent gene expression in granulocyte-macrophage progenitors in culture and in seropositive individuals. Proc Natl Acad Sci U S A 93:11137–11142. doi: 10.1073/pnas.93.20.11137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bevan IS, Daw RA, Day PJ, Ala FA, Walker MR. 1991. Polymerase chain reaction for detection of human cytomegalovirus infection in a blood donor population. Br J Haematol 78:94–99. doi: 10.1111/j.1365-2141.1991.tb04388.x. [DOI] [PubMed] [Google Scholar]
- 18.Kondo K, Kaneshima H, Mocarski ES. 1994. Human cytomegalovirus latent infection of granulocyte-macrophage progenitors. Proc Natl Acad Sci U S A 91:11879–11883. doi: 10.1073/pnas.91.25.11879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Söderberg-Nauclér C, Fish KN, Nelson JA. 1997. Reactivation of latent human cytomegalovirus by allogeneic stimulation of blood cells from healthy donors. Cell 91:119–126. doi: 10.1016/S0092-8674(01)80014-3. [DOI] [PubMed] [Google Scholar]
- 20.Seckert CK, Renzaho A, Tervo HM, Krause C, Deegen P, Kuhnapfel B, Reddehase MJ, Grzimek NK. 2009. Liver sinusoidal endothelial cells are a site of murine cytomegalovirus latency and reactivation. J Virol 83:8869–8884. doi: 10.1128/JVI.00870-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dag F, Dolken L, Holzki J, Drabig A, Weingartner A, Schwerk J, Lienenklaus S, Conte I, Geffers R, Davenport C, Rand U, Koster M, Weiss S, Adler B, Wirth D, Messerle M, Hauser H, Cicin-Sain L. 2014. Reversible silencing of cytomegalovirus genomes by type I interferon governs virus latency. PLoS Pathog 10:e1003962. doi: 10.1371/journal.ppat.1003962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brautigam AR, Dutko FJ, Olding LB, Oldstone MB. 1979. Pathogenesis of murine cytomegalovirus infection: the macrophage as a permissive cell for cytomegalovirus infection, replication and latency. J Gen Virol 44:349–359. doi: 10.1099/0022-1317-44-2-349. [DOI] [PubMed] [Google Scholar]
- 23.Pollock JL, Presti RM, Paetzold S, Virgin HWt. 1997. Latent murine cytomegalovirus infection in macrophages. Virology 227:168–179. doi: 10.1006/viro.1996.8303. [DOI] [PubMed] [Google Scholar]
- 24.Alexandre YO, Cocita CD, Ghilas S, Dalod M. 2014. Deciphering the role of DC subsets in MCMV infection to better understand immune protection against viral infections. Front Microbiol 5:378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dalod M, Salazar-Mather TP, Malmgaard L, Lewis C, Asselin-Paturel C, Briere F, Trinchieri G, Biron CA. 2002. Interferon alpha/beta and interleukin 12 responses to viral infections: pathways regulating dendritic cell cytokine expression in vivo. J Exp Med 195:517–528. doi: 10.1084/jem.20011672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dalod M, Hamilton T, Salomon R, Salazar-Mather TP, Henry SC, Hamilton JD, Biron CA. 2003. Dendritic cell responses to early murine cytomegalovirus infection: subset functional specialization and differential regulation by interferon alpha/beta. J Exp Med 197:885–898. doi: 10.1084/jem.20021522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scheu S, Dresing P, Locksley RM. 2008. Visualization of IFNbeta production by plasmacytoid versus conventional dendritic cells under specific stimulation conditions in vivo. Proc Natl Acad Sci U S A 105:20416–20421. doi: 10.1073/pnas.0808537105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doring M, Lessin I, Frenz T, Spanier J, Kessler A, Tegtmeyer P, Dag F, Thiel N, Trilling M, Lienenklaus S, Weiss S, Scheu S, Messerle M, Cicin-Sain L, Hengel H, Kalinke U. 17 September 2014. M27 expressed by cytomegalovirus counteracts effective type I IFN induction of myeloid cells, but not of plasmacytoid dendritic cells. J Virol doi: 10.1128/JVI.00216-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tabeta K, Georgel P, Janssen E, Du X, Hoebe K, Crozat K, Mudd S, Shamel L, Sovath S, Goode J, Alexopoulou L, Flavell RA, Beutler B. 2004. Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc Natl Acad Sci U S A 101:3516–3521. doi: 10.1073/pnas.0400525101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Delale T, Paquin A, Asselin-Paturel C, Dalod M, Brizard G, Bates EE, Kastner P, Chan S, Akira S, Vicari A, Biron CA, Trinchieri G, Briere F. 2005. MyD88-dependent and -independent murine cytomegalovirus sensing for IFN-alpha release and initiation of immune responses in vivo. J Immunol 175:6723–6732. doi: 10.4049/jimmunol.175.10.6723. [DOI] [PubMed] [Google Scholar]
- 31.Merad M, Sathe P, Helft J, Miller J, Mortha A. 2013. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol 31:563–604. doi: 10.1146/annurev-immunol-020711-074950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Diebold SS, Montoya M, Unger H, Alexopoulou L, Roy P, Haswell LE, Al-Shamkhani A, Flavell R, Borrow P, Reis e Sousa C. 2003. Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. Nature 424:324–328. doi: 10.1038/nature01783. [DOI] [PubMed] [Google Scholar]
- 33.Mueller SN, Heath W, McLain JD, Carbone FR, Jones CM. 2002. Characterization of two TCR transgenic mouse lines specific for herpes simplex virus. Immunol Cell Biol 80:156–163. doi: 10.1046/j.1440-1711.2002.01071.x. [DOI] [PubMed] [Google Scholar]
- 34.Dag F, Weingartner A, Butueva M, Conte I, Holzki J, May T, Adler B, Wirth D, Čičin-Šain L. 2013. A new reporter mouse cytomegalovirus reveals maintained immediate-early gene expression but poor virus replication in cycling liver sinusoidal endothelial cells. Virol J 10:197. doi: 10.1186/1743-422X-10-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jordan SJK, Prager A, Mitrovic M, Jonjic S, Koszinowski UH, Adler B. 2011. Virus progeny of murine cytomegalovirus bacterial artificial chromosome pSM3fr show reduced growth in salivary glands due to a fixed mutation of MCK-2. J Virol 85:10346–10353. doi: 10.1128/JVI.00545-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dekhtiarenko I, Jarvis MA, Ruzsics Z, Cicin-Sain L. 2013. The context of gene expression defines the immunodominance hierarchy of cytomegalovirus antigens. J Immunol 190:3399–3409. doi: 10.4049/jimmunol.1203173. [DOI] [PubMed] [Google Scholar]
- 37.Reddehase MJ, Keil GM, Koszinowski UH. 1984. The cytolytic T lymphocyte response to the murine cytomegalovirus. II. Detection of virus replication stage-specific antigens by separate populations of in vivo active cytolytic T lymphocyte precursors. Eur J Immunol 14:56–61. [DOI] [PubMed] [Google Scholar]
- 38.Cicin-Sain L, Podlech J, Messerle M, Reddehase MJ, Koszinowski UH. 2005. Frequent coinfection of cells explains functional in vivo complementation between cytomegalovirus variants in the multiply infected host. J Virol 79:9492–9502. doi: 10.1128/JVI.79.15.9492-9502.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jung S, Unutmaz D, Wong P, Sano G, De los Santos K, Sparwasser T, Wu S, Vuthoori S, Ko K, Zavala F, Pamer EG, Littman DR, Lang RA. 2002. In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens. Immunity 17:211–220. doi: 10.1016/S1074-7613(02)00365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Probst HC, Tschannen K, Odermatt B, Schwendener R, Zinkernagel RM, Van Den Broek M. 2005. Histological analysis of CD11c-DTR/GFP mice after in vivo depletion of dendritic cells. Clin Exp Immunol 141:398–404. doi: 10.1111/j.1365-2249.2005.02868.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kassim SH, Rajasagi NK, Zhao X, Chervenak R, Jennings SR. 2006. In vivo ablation of CD11c-positive dendritic cells increases susceptibility to herpes simplex virus type 1 infection and diminishes NK and T-cell responses. J Virol 80:3985–3993. doi: 10.1128/JVI.80.8.3985-3993.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kern M, Popov A, Scholz K, Schumak B, Djandji D, Limmer A, Eggle D, Sacher T, Zawatzky R, Holtappels R, Reddehase MJ, Hartmann G, Debey-Pascher S, Diehl L, Kalinke U, Koszinowski U, Schultze J, Knolle PA. 2010. Virally infected mouse liver endothelial cells trigger CD8+ T-cell immunity. Gastroenterology 138:336–346. doi: 10.1053/j.gastro.2009.08.057. [DOI] [PubMed] [Google Scholar]
- 43.Andoniou CE, van Dommelen SL, Voigt V, Andrews DM, Brizard G, Asselin-Paturel C, Delale T, Stacey KJ, Trinchieri G, Degli-Esposti MA. 2005. Interaction between conventional dendritic cells and natural killer cells is integral to the activation of effective antiviral immunity. Nat Immunol 6:1011–1019. doi: 10.1038/ni1244. [DOI] [PubMed] [Google Scholar]
- 44.Riese P, Trittel S, May T, Cicin-Sain L, Chambers BJ, Guzman CA. 2015. Activated NKT cells imprint NK-cell differentiation, functionality and education. Eur J Immunol 45:1794–1807. doi: 10.1002/eji.201445209. [DOI] [PubMed] [Google Scholar]
- 45.Ablasser A, Schmid-Burgk JL, Hemmerling I, Horvath GL, Schmidt T, Latz E, Hornung V. 2013. Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature 503:530–534. doi: 10.1038/nature12640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Müller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. 1994. Functional role of type I and type II interferons in antiviral defense. Science 264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 47.Asselin-Paturel C, Boonstra A, Dalod M, Durand I, Yessaad N, Dezutter-Dambuyant C, Vicari A, O'Garra A, Biron C, Briere F, Trinchieri G. 2001. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat Immunol 2:1144–1150. doi: 10.1038/ni736. [DOI] [PubMed] [Google Scholar]
- 48.Andrews DM, Andoniou CE, Granucci F, Ricciardi-Castagnoli P, Degli-Esposti MA. 2001. Infection of dendritic cells by murine cytomegalovirus induces functional paralysis. Nat Immunol 2:1077–1084. doi: 10.1038/ni724. [DOI] [PubMed] [Google Scholar]
- 49.Benedict CA, Loewendorf A, Garcia Z, Blazar BR, Janssen EM. 2008. Dendritic cell programming by cytomegalovirus stunts naive T cell responses via the PD-L1/PD-1 pathway. J Immunol 180:4836–4847. doi: 10.4049/jimmunol.180.7.4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reeves MB, Lehner PJ, Sissons JG, Sinclair JH. 2005. An in vitro model for the regulation of human cytomegalovirus latency and reactivation in dendritic cells by chromatin remodelling. J Gen Virol 86:2949–2954. doi: 10.1099/vir.0.81161-0. [DOI] [PubMed] [Google Scholar]
- 51.Swiecki M, Gilfillan S, Vermi W, Wang Y, Colonna M. 2010. Plasmacytoid dendritic cell ablation impacts early interferon responses and antiviral NK and CD8(+) T cell accrual. Immunity 33:955–966. doi: 10.1016/j.immuni.2010.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Swiecki M, Wang Y, Riboldi E, Kim AH, Dzutsev A, Gilfillan S, Vermi W, Ruedl C, Trinchieri G, Colonna M. 2014. Cell depletion in mice that express diphtheria toxin receptor under the control of SiglecH encompasses more than plasmacytoid dendritic cells. J Immunol 192:4409–4416. doi: 10.4049/jimmunol.1303135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Holtappels R, Podlech J, Pahl-Seibert MF, Julch M, Thomas D, Simon CO, Wagner M, Reddehase MJ. 2004. Cytomegalovirus misleads its host by priming of CD8 T cells specific for an epitope not presented in infected tissues. J Exp Med 199:131–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stahl FR, Heller K, Halle S, Keyser KA, Busche A, Marquardt A, Wagner K, Boelter J, Bischoff Y, Kremmer E, Arens R, Messerle M, Förster R. 2013. Nodular inflammatory foci are sites of T cell priming and control of murine cytomegalovirus infection in the neonatal lung. PLoS Pathog 9:e1003828. doi: 10.1371/journal.ppat.1003828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reeves MB, MacAry PA, Lehner PJ, Sissons JG, Sinclair JH. 2005. Latency, chromatin remodeling, and reactivation of human cytomegalovirus in the dendritic cells of healthy carriers. Proc Natl Acad Sci U S A 102:4140–4145. doi: 10.1073/pnas.0408994102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goodrum FD, Jordan CT, High K, Shenk T. 2002. Human cytomegalovirus gene expression during infection of primary hematopoietic progenitor cells: a model for latency. Proc Natl Acad Sci U S A 99:16255–16260. doi: 10.1073/pnas.252630899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brinkmann MM, Dag F, Hengel H, Messerle M, Kalinke U, Cicin-Sain L. 2015. Cytomegalovirus immune evasion of myeloid lineage cells. Medical Microbiol Immunol 204:367–382. doi: 10.1007/s00430-015-0403-4. [DOI] [PubMed] [Google Scholar]