

ABSTRACT
Viruses that spread systemically from a peripheral site of infection cause morbidity and mortality in the human population. Innate myeloid cells, including monocytes, macrophages, monocyte-derived dendritic cells (mo-DC), and dendritic cells (DC), respond early during viral infection to control viral replication, reducing virus spread from the peripheral site. Ectromelia virus (ECTV), an orthopoxvirus that naturally infects the mouse, spreads systemically from the peripheral site of infection and results in death of susceptible mice. While phagocytic cells have a requisite role in the response to ECTV, the requirement for individual myeloid cell populations during acute immune responses to peripheral viral infection is unclear. In this study, a variety of myeloid-specific depletion methods were used to dissect the roles of individual myeloid cell subsets in the survival of ECTV infection. We showed that DC are the primary producers of type I interferons (T1-IFN), requisite cytokines for survival, following ECTV infection. DC, but not macrophages, monocytes, or granulocytes, were required for control of the virus and survival of mice following ECTV infection. Depletion of either plasmacytoid DC (pDC) alone or the lymphoid-resident DC subset (CD8α+ DC) alone did not confer lethal susceptibility to ECTV. However, the function of at least one of the pDC or CD8α+ DC subsets is required for survival of ECTV infection, as mice depleted of both populations were susceptible to ECTV challenge. The presence of at least one of these DC subsets is sufficient for cytokine production that reduces ECTV replication and virus spread, facilitating survival following infection.
IMPORTANCE Prior to the eradication of variola virus, the orthopoxvirus that causes smallpox, one-third of infected people succumbed to the disease. Following successful eradication of smallpox, vaccination rates with the smallpox vaccine have significantly dropped. There is now an increasing incidence of zoonotic orthopoxvirus infections for which there are no effective treatments. Moreover, the safety of the smallpox vaccine is of great concern, as complications may arise, resulting in morbidity. Like many viruses that cause significant human diseases, orthopoxviruses spread from a peripheral site of infection to become systemic. This study elucidates the early requirement for innate immune cells in controlling a peripheral infection with ECTV, the causative agent of mousepox. We report that there is redundancy in the function of two innate immune cell subsets in controlling virus spread early during infection. The viral control mediated by these cell subsets presents a potential target for therapies and rational vaccine design.
INTRODUCTION
Prior to the induction of the adaptive immune response, cells of the innate immune system are deployed early during viral infection to control the pathogen and limit its spread from the site of infection. Effector cells of the innate immune system include myeloid cell subsets: granulocytes (neutrophils, eosinophils, and basophils), monocytes, macrophages, and dendritic cells (DC). It has become clear that each of these myeloid cell populations consists of many subpopulations that are phenotypically and functionally specialized, even in the steady state (1). During infection, the complexity of the myeloid cell subsets is increased, with cells such as inflammatory monocytes and monocyte-derived DC (mo-DC) increasing in number compared to the steady state (2, 3). The innate response is required to both activate the adaptive immune response and allow time for recruitment of the activated T and B cells. The majority of previous studies have examined the abilities of various myeloid cells to activate naive lymphocytes, particularly CD4+ (TCD4+) and CD8+ (TCD8+) T cells. Monocytes, macrophages, mo-DC, and DC can all present the antigen required for induction of T cell responses (4). Furthermore, during viral infection, neutrophils may also be capable of presenting to TCD8+ (5). Besides their well-defined role in antigen presentation, myeloid cell populations can also produce cytokines in response to viral infection. These cytokines may be directly antiviral, such as type I interferons (T1-IFN) (6), or may create an inflammatory environment, thereby facilitating the recruitment of other effector immune cell populations (7). Here, we examine the identities of myeloid cell populations required for induction of inflammation following infection with a natural mouse pathogen via its natural route.
Upon infection of mice with ectromelia virus (ECTV), an orthopoxvirus that causes mousepox, there is an early expansion of myeloid cells in naturally resistant mice (8). ECTV infection is characterized by lymphohematogenous spread of the virus from the peripheral site of infection (the footpad), and susceptible mice succumb to systemic disease (9). Survival following ECTV challenge requires recruitment of natural killer (NK) cells (10–12), the induction of TCD8+ (13, 14) and TCD4+ (15), and production of antibody by virus-specific B cells (16, 17).
The requirement for specific, individual populations of myeloid cells for an effective immune response to ECTV is currently unknown. Phagocytic cells are critical for survival following ECTV infection, as resistant mice injected with clodronate liposomes (CLL), which deplete phagocytes, succumb to infection (13). CLL deplete macrophages (18), monocytes (19), and some splenic DC (20), but neutrophils do not appear to be affected (18). Multiple populations of phagocytic cells, including macrophages and DC, are infected by ECTV. These myeloid cell subsets present antigen for activation of the required TCD8+ response (4). Plasmacytoid DC (pDC) are typically thought to be the primary producers of T1-IFN, including interferon alpha (IFN-α), during viral infection (21). IFN-α is required for the survival of mice following ECTV infection and is important for mediating control of the virus (22). Virus infection induces changes in the myeloid cell compartment, including changes in cell numbers, cellular phenotype, and cytokine production. The cytokines produced by myeloid cells, including pDC and non-pDC populations, following ECTV infection have not been examined. In the current study, we assessed whether myeloid cell populations produce T1-IFN following ECTV infection. Moreover, we determined the in vivo roles of myeloid cell populations in immunity to ECTV infection by utilizing multiple complementary depletion models. This allowed us to define the requisite role(s) of specific phagocytic myeloid cell subsets during a peripheral viral infection. We observed that granulocytes, inflammatory monocytes, mo-DC, and macrophages alone are not required for survival following ECTV infection. However, DC are required for survival following ECTV infection. Specifically, our results indicate that both pDC and lymphoid-resident CD8α+ DC produce cytokines following exposure to ECTV. Thus, pDC and CD8α+ DC exhibit a redundant function in protective immune responses necessary for control of the virus and ultimately for survival of ECTV challenge.
MATERIALS AND METHODS
Mice.
CD11c:cre (catalog no. 008068) (23), inducible diphtheria toxin receptor (iDTR) (catalog no. 007900) (24), CCR2 knockout (CCR2KO) (catalog no. 004999) (25), and Batf3KO (catalog no. 013755) (26) mice were purchased from the Jackson Laboratory and bred at the Penn State College of Medicine. All mouse strains were on the C57BL/6 background. CD11cwt/cre × iDTR+/− mice (CD11c:cre × iDTR) were generated by breeding CD11cwt/cre mice with iDTR+/+ mice, and the genotype was verified by PCR. The mice were maintained in the specific-pathogen-free facility at the Penn State College of Medicine. All animal experiments were approved by the Penn State College of Medicine IACUC.
Virus infection.
For infections, 6- to 12-week-old mice were used. All experiments used wild-type (WT) ECTV (Moscow strain). Mice were injected with 3 × 103 PFU of ECTV in one hind footpad or mock infected with Hanks balanced salt solution (HBSS)-0.1% bovine serum albumin (BSA). The mice were monitored daily for morbidity and mortality. ECTV was inactivated (27) by the addition of psoralen (final concentration, 10 μg/ml) and subsequent treatment under a UVC lamp for 2 h. This treatment ablated virus replication and measurable early gene expression (not shown). When indicated, mice were treated with 100 mg cidofovir (Mylan International)/kg of body weight intraperitoneally (i.p.) in phosphate-buffered saline (PBS) on days 3 and 6 postinfection (p.i).
CLL-based depletion.
CLL from Boehringer Mannheim GmbH (Mannheim, Germany) were injected intravenously (i.v.) in a total volume of 200 μl at 1 day p.i. for depletion. Control mice were injected with 200 μl PBS i.v. at 1 day p.i.
DT-based depletion.
CD11c:cre × iDTR mice were injected with 20 ng/g diphtheria toxin (DT) (Sigma) i.p. at 1 day p.i. unless otherwise indicated. This dose provided optimal depletion with minimal toxicity over the period of challenge. Control mice received PBS i.p. The efficacy of depletion is outlined below (see Fig. 3A and B).
FIG 3.
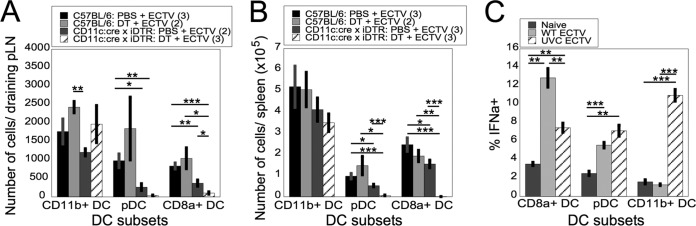
CD8α+ DC and pDC are depleted in susceptible DT-injected CD11c:cre × iDTR mice and produce IFN-α in response to live ECTV. (A and B) WT or CD11c:cre × iDTR mice were infected with ECTV on day 0 and injected i.p. with DT or PBS at 1 day p.i. D-LN (A) or spleens (B) were harvested at 2 days p.i., and the cells were stained for flow cytometry for CD11b+ DCs, pDCs, and CD8α+ DCs as in Fig. 1A. The data are representative of two independent experiments (means ± SEM). (C) Spleens from WT mice were harvested 10 to 14 days after administration of Flt3L. Purified DC were left untreated (naive) or treated with live WT ECTV (WT ECTV) or UVC-inactivated WT ECTV (UVC ECTV) at an MOI of 10 for 6 h. The cells were stained for flow cytometry and gated on pDC, CD8α+ DC, or CD11b+ DC producing IFN-α. The data are representative of four independent experiments with experimental triplicates (means ± SEM). *, P < 0.05; **, P < 0.01; ***, P < 0.001; Student's unpaired t test.
Antibody-based depletion.
For depletion of Ly6G+ cells, mice were injected i.v. on day −1 and then every third day from day −1 with 0.7 mg anti-Ly6G (1A8; BioXcell) in 500 μl PBS. Control mice received 0.7 mg rat IgG2a isotype control (2A3; BioXcell) in 500 μl PBS following the same schedule. The efficacy of depletion is outlined in Fig. 1D.
FIG 1.

Survival of ECTV infection does not require inflammatory monocytes or mo-DC, granulocytes, or macrophages. (A) Representative gating of myeloid cell populations from ECTV-infected mice. All gates were defined by fluorescence minus one controls, and the numbers represent percentages of cells. Large arrows indicate the flow of the analyses, with each population being a subset of the previous one. (B) Quantification of myeloid cell populations from WT mice infected with ECTV and injected i.v. with either CLL or PBS at 1 day p.i. Spleens were harvested at 2 days p.i. (C) WT or CCR2KO mice were infected with ECTV and monitored for survival until 21 days p.i. (D and E) WT mice were injected i.v. with 0.5 ml of 1.4 mg/ml anti-Ly6G depleting antibody (1A8) or with an IgG2a control antibody (2A3) in PBS on day −1 and then every third day and infected with ECTV on day 0. Depletion of Ly6C+ Ly6G+ cells in the spleen at 21 days p.i. (D) and survival (E) were measured. (F and G) WT mice were injected i.p. with 3 mg/mouse anti-CD115 depleting antibody or vehicle control every other day starting at day −5 and infected with ECTV. Depletion of splenic macrophage populations (F) and survival (G) were monitored until 22 days p.i. (B to G) Data were pooled from two independent experiments (means ± SEM). ***, P < 0.001; Student's unpaired t test.
For depletion of CD115+ cells, mice were injected i.p. every other day starting at day −5 with 3 mg AFS98 antibody (anti-CD115 depleting antibody) in 200 μl 0.1% BSA- HBSS. Control mice were injected i.p. with 200 μl vehicle following the same schedule. The efficacy of depletion is outlined in Fig. 1F.
For depletion of pDC, mice were injected i.p. on day −2, day −1, and then every third day from day −1 throughout the experiment with 500 μg BX444 anti-pDC antibody (BioXCell) in 500 μl PBS. Control mice were injected i.p. with 500 μg isotype control rat IgG1 anti-horseradish peroxidase (HRPN) antibody (BioXCell) in 500 μl PBS following the same schedule.
Viral titers.
Infected footpads, draining lymph nodes (D-LN), spleens, and livers were harvested at 5 days p.i. and frozen in HBSS-0.1% BSA at −80°C. Titers were determined by plaque assay. Briefly, tissues were homogenized using a TissueLyser II (Qiagen). For weighed tissues, the number of PFU per gram tissue was calculated.
Flow cytometry.
D-LN or spleens were harvested and digested using 1 mg/ml collagenase D (Roche) for 30 min at 37°C. Cells were blocked and stained on ice in 2.4G2 supernatant-10% mouse serum (Sigma). For myeloid cell analysis, unless otherwise noted, all antibodies were purchased from eBioscience. They included biotin-conjugated CD19 (1D3), NK1.1 (PK136), and CD90.2 (53-2.1). Phycoerythrin (PE)-Cy7-streptavidin was used to reveal staining with biotin-conjugated antibodies. Other antibodies included CD45 (30-F11; BD Bioscience), CD11b (M1/70; BD Bioscience), B220 (RA3-6B2; BD Bioscience), CD8α (53-6.7; BD Bioscience), CD11c (N418; Biolegend), and CD64 (X54-5/7.1; Biolegend). Sample acquisition was performed with an LSRII flow cytometer (BD Bioscience), and data were analyzed using FlowJo software (TreeStar). All myeloid cells were initially gated as live cells, singlets, CD45+, and CD19− CD90.2− NK1.1− (Fig. 1A), and myeloid cells were identified as follows: mo-DC (CD11c+ CD11b+ CD64+); CD11b+ DC (CD11c+ CD11b+ CD8α− CD64−); pDC (CD11c+ CD11b− B220+); CD8α+ DC (CD11c+ CD11b− B220− CD8α+); granulocytes, including both neutrophils and eosinophils (CD11c− CD11bhi); and monocytes (CD11c− CD11blo).
For NK cell and T cell analyses, cells were stained in 2.4G2 with mouse serum and 50 μl/well BD Horizon Brilliant Stain Buffer (BD Bioscience). Unless otherwise noted, all antibodies were from BD Bioscience. The antibodies included CD45 (30-F11; Biolegend), CD8α (53-6.7; Biolegend), CD11b (M1/70; Biolegend), CD69 (H1.2F3; eBioscience), CD4 (RM4-5), CD3e (145-2C11), NK1.1 (PK136), CD27 (LG.3A10), CD44 (1M7), and CD62L (MEL-14). All cells were initially gated as live cells, singlets, and CD45+ populations. T cells were gated as CD3e+ NK1.1−, with TCD8+ gated as CD8α+ CD4− (total TCD8+) and TCD4+ gated as CD4+ CD8α− (total TCD4+). TCD8+ and TCD4+ were then gated as naive (CD44lo CD62Lhi), effector (CD44hi CD62Llo), and memory (CD44hi CD62Lhi) populations and CD69+ populations. NK cells were gated as NK1.1+ CD3e− cells (total NK cells). The NK cells were then gated into subsets as CD62L+, CD69+, immature (CD11b− CD27+), intermediate (CD11b+ CD27+), and mature (CD11b+ CD27−).
Intracellular cytokine staining.
For ex vivo analyses, mice were infected with 3 × 103 PFU in the footpad, and the D-LN were harvested and stained with antibodies to exclude T, B, and NK cell populations. The cells were then stained with anti-CD11c and anti-CD11b as shown. For in vitro analyses, spleens were harvested from mice treated with FMS-like tyrosine kinase 3 ligand (Flt3L) and digested using 1 mg/ml collagenase D (Roche), and DC were sorted using panDC microbeads (Miltenyi Biotec). DC were left untreated or were treated with wild-type ECTV (crude preparation) or UVC-inactivated WT ECTV (crude preparation) at a multiplicity of infection (MOI) of 10 in Dulbecco's modified Eagle's medium (DMEM) containing 5% fetal bovine serum (FBS) for 6 h. The cells were incubated in 2.4G2 cell supernatant-10% mouse serum and stained using CD11c (N418), CD11b (M1/70), B220 (RA3-6B2), and CD8α (53-6.7), all from BD Biosciences.
For both ex vivo and in vitro analyses, cells were incubated in 10 μg/ml brefeldin A (Sigma) for a minimum of 4 h to allow accumulation of T1-IFN. The cells were then stained to identify myeloid cell populations as outlined above, fixed in 1% paraformaldehyde for 10 min at room temperature, and then washed using PBS. To reveal T1-IFN staining, the cells were permeabilized with 0.5% saponin and stained with fluorescein isothiocyanate (FITC)-labeled anti-IFN-α (clone RMMA-1; R&D Systems). Samples were then washed and analyzed as outlined above. All acquisition was performed on the same day as tissue and cell harvest to prevent an increase in T1-IFN staining background following overnight storage of fixed samples.
Immunofluorescence microscopy.
Mice were infected in the footpad with 1 × 106 PFU ECTV expressing green fluorescent protein (GFP). Twelve hours postinfection, D-LN were harvested, mounted in OCT, and frozen; 20-μm sections were cut and incubated with Fab donkey anti-mouse IgG (Jackson ImmunoResearch) and then stained with directly labeled Alexa 647-conjugated anti-CD11c (N418) (eBioscience). Staining was visualized using an Olympus 1X81 DSU Spinning Disk microscope and Slidebook 5.0 software.
Data display and statistical analysis.
Statistical analyses were performed using an unpaired, two-tailed Student t test as applicable. P values of <0.05 were considered significant. When applicable, data are displayed as means ± standard errors of the mean (SEM).
RESULTS
Depletion of myeloid cell subsets by CLL.
Phagocytic cells are required for survival of mice following ECTV infection, as depletion of phagocytes using CLL led to the death of mice at approximately 8.5 days p.i. (13). Phagocytes comprise a heterogeneous population of cells; therefore, one or more cell subsets required for a protective immune response were depleted by CLL treatment. Following internalization of CLL by phagocytic cells, the cells die via apoptotic mechanisms, resulting in the depletion of macrophages (18), monocytes (19), and some splenic DC subsets (20). Depletion of macrophage subpopulations is dependent on the route of CLL injection (28). However, during virus infection, the plasticity of the myeloid cell compartment likely produces novel populations that may also be susceptible to CLL-induced death. To examine the myeloid cell subset(s) required for survival of mice following ECTV infection, we carefully analyzed the myeloid cell subsets depleted by CLL injection during ECTV infection. Myeloid cell subsets were defined as shown in Fig. 1A, with gates defined by “fluorescence minus one” controls. Mice were infected with ECTV and injected i.v. with CLL at 1 day p.i. CLL treatment depleted multiple myeloid cell subsets in the spleen, including mo-DC, CD11b+ DC, pDC, CD8α+ DC, and monocytes (Fig. 1B). Granulocytes, however, were not depleted by CLL treatment (Fig. 1B). We found depletion of similar myeloid cell subsets by CLL during infection of mice with another orthopoxvirus, vaccinia virus (VACV) (M. L. Davies, C. Soni, L. W. Kaminsky, I. E. Reider, M. A. Fischer, N. Van Rooijen, Z. S. M. Rahman, and C. C. Norbury, unpublished data).
Involvement of monocytes, mo-DC, granulocytes, and macrophages in survival following ECTV infection.
We next sought to examine the requirement for individual myeloid cell populations depleted by CLL in survival of mice after lethal infection with ECTV. To isolate the requirement for inflammatory monocytes and mo-DC, we challenged CCR2KO mice with ECTV. CCR2 is a chemokine receptor that is necessary for inflammatory monocytes to exit the bone marrow and enter the vasculature (29). In CCR2KO mice, inflammatory monocytes are unable to traffic to the site of infection, and mo-DC are then unable to develop from inflammatory monocytes at the site of infection or inflammation (25, 30). CCR2KO mice survived ECTV infection (Fig. 1C), indicating that neither inflammatory monocytes nor mo-DC alone are required for survival of ECTV infection. Although CLL does not appear to significantly deplete neutrophils in our system (Fig. 1B), there is a single report indicating that neutrophils may play a role in immunity to ECTV (31). To examine the role of neutrophils in ECTV infection, we depleted mice of Ly6G+ cells with 1A8 anti-Ly6G or isotype control (Fig. 1D). A small number of mice treated with anti-Ly6G died compared to treatment with the isotype control, but the treatment failed to produce greater than 75% lethality in multiple experiments (Fig. 1E), indicating that neutrophils likely do not play a requisite role in protection following ECTV infection. CLL can also deplete tissue-resident macrophages (28), so we next investigated the role of tissue-resident macrophage populations in mediating survival of ECTV infection. We administered the anti-CD115 (CSF-1R)-blocking antibody AFS98, a monoclonal antibody commonly used to target tissue-resident cells of the monocyte-macrophage lineage, with high availability throughout the body (32–34). As expected, macrophages were partially, but not fully, depleted in the spleens of AFS98-injected mice, as not all macrophage populations within secondary lymphoid organs are sensitive to anti-CD115 treatment (Fig. 1F). Neither mice treated with anti-CD115 nor mice treated with vehicle control died upon ECTV challenge (Fig. 1G), indicating that tissue-resident macrophage populations were not required for anti-ECTV immunity. From these data, we can conclude that neutrophils, tissue-resident macrophage populations, inflammatory monocytes, and mo-DC were not required for mice to survive lethal ECTV challenge.
Cytokine production by myeloid cell populations.
Although macrophage populations did not appear to be essential for the survival of mice after ECTV challenge, DC subsets were also depleted by CLL (Fig. 1B), indicating a potential role for these DC subsets in mediating survival following ECTV infection. Infection of mice with recombinant ECTV expressing green fluorescent protein (GFP) revealed colocalization of GFP and CD11c, an integrin that is primarily expressed by DC, within 12 h of infection in the D-LN (Fig. 2A) (4). We know that production of T1-IFN, particularly IFN-α, is required for a protective response during ECTV infection (22, 35). Therefore, we examined the ability of myeloid cell populations to produce IFN-α following ECTV infection. Initially we examined IFN-α production by CD11b+ CD11c− cells (likely macrophages and granulocytes), CD11b+ CD11c+ cells (likely conventional DC), and CD11b− CD11c+ cells (likely lymphoid-resident DC and pDC) in the D-LN at 3 days p.i. Intracellular cytokine staining for IFN-α is technically challenging, but our analyses revealed that it was primarily CD11b− CD11c+ cells that produced IFN-α following ECTV infection (Fig. 2B and C).
FIG 2.

CD11c+ cells control viral replication to mediate survival of ECTV infection. (A) Mice were infected in the footpad with 1 × 106 PFU ECTV expressing NP-S-EGFP (green). Twelve hours postinfection, D-LN were harvested and then stained with anti-CD11c-Alexa 647 (red). The data are representative of the results of 7 independent experiments. (B and C) Mice were infected with 3 × 103 PFU ECTV in the footpad, and at 2 days p.i., D-LN were harvested and stained for flow cytometry. The cells were gated on live cells, singlets, and NK cell- and T cell-negative status and then on CD11b+ CD11c+ (conventional DC), CD11b+ CD11c− (macrophages), and CD11b− CD11c+ (pDC and CD8α+ DC). Representative histograms of IFN-α expression are shown (B), and the percentages of IFN-α+ cells were quantified (C). “Uninfected” represents IFN-α production by CD11b− CD11c+ cells in the absence of ECTV infection. The data are representative of two independent experiments. The error bars indicate SEM. (D) CD11c:cre × iDTR mice were either (i) injected i.p. with DT on day 1 and mock infected in the left hind footpad on day 0; (ii) injected i.p. with PBS on day 1 and then infected with ECTV on day 0; or (iii) infected with ECTV on day 0 and then injected i.p. with DT on day −1 or day 1, 3, or 5 postinfection. Survival was monitored until 15 days p.i. The data from four independent experiments were combined. (E to H) WT or CD11c:cre × iDTR mice were injected i.p. with DT or PBS on day 1 p.i. All the mice were infected with ECTV as for panel D, and infected footpads (E), D-LN (F), livers (G), and spleens (H) were harvested at 5 days p.i. The tissues were processed and plated in serial dilutions on cell monolayers for viral titer analysis. The data from two independent experiments were combined. (I) CD11c:cre × iDTR mice were infected with ECTV as for panel D and injected i.p. with DT on day 1 p.i. The mice received 100 mg/kg cidofovir or PBS i.p. at 3 days p.i. and 6 days p.i. Survival was monitored to 21 days p.i. The data from two individual experiments were combined. *, P < 0.05; Student's unpaired t test.
Role of DC in survival of ECTV infection and control of ECTV replication.
To investigate the requirement for DC in protective immunity to ECTV, we used CD11c:cre × iDTR mice. In CD11c:cre × iDTR mice, Cre recombinase expression is driven by the CD11c promoter (23). Expression of Cre recombinase leads to the removal of a loxP-flanked stop cassette, resulting in the expression of the diphtheria toxin receptor (DTR) (24). We depleted CD11c+ cells by injecting DT at different time points during ECTV infection. Control C57BL/6 WT mice injected with DT all survived ECTV infection (data not shown), as did CD11c:cre × iDTR mice infected with ECTV but not treated with DT (Fig. 2D). Mice depleted of DC at day −1 or day 3 or 5 postinfection had 45% survival, 50% survival, and 50% survival, respectively (Fig. 2D). However, of the mice treated with only a single injection of DT at 1 day p.i., greater than 90% died from ECTV infection (Fig. 2D), indicating that DC are required early to mediate survival following ECTV infection.
To examine whether DC-depleted mice were dying from uncontrolled viral replication or from a damaging aberrant immune response, mice were treated with DT on 1 day p.i., and various tissues were harvested at 5 days p.i., the peak of viral replication in the spleens of WT mice. There was no difference between the viral titers from the infected footpad (Fig. 2E) or D-LN (Fig. 2F) in DC-depleted mice and those from vehicle-treated CD11c:cre × iDTR mice, DT-treated mice, or vehicle-treated mice. However, there was an ∼2-log-unit increase in the levels of ECTV in the spleens of DC-depleted mice compared to vehicle-injected C57BL/6 WT mice (Fig. 2H). There was also a higher virus titer in the livers of DC-depleted mice than in those of control mice that survived ECTV infection (Fig. 2G). The higher viral titers in the spleens and livers of DC-depleted mice indicated that the depleted mice are unable to control the spread of the virus following peripheral infection. To examine whether increased ECTV replication was responsible for the death of DC-depleted mice, we attempted to rescue these mice by pharmacologically controlling virus replication. We treated infected DC-depleted mice or controls with cidofovir, a nucleotide analog that inhibits the viral DNA polymerase and terminates the growing viral DNA strand. Cidofovir has been shown to be effective against ECTV (15, 36). Mice depleted of DC at 1 day p.i. died from ECTV infection, but mice that were subsequently treated with cidofovir on day 3 and day 6 postinfection survived ECTV infection (Fig. 2I). Therefore, the death of ECTV-infected DC-depleted mice was likely mediated by a lack of control of viral infection, but it may also result from a damaging immune response triggered by enhanced virus replication in the absence of DC.
Roles of DC subpopulations in protective anti-ECTV immunity.
DC are a complex, heterogeneous population of cells consisting of subpopulations that are phenotypically and functionally specialized. We dissected which DC subpopulations were depleted in the ECTV-infected CD11c:cre × iDTR mice that were susceptible to death, i.e., those that were treated with a single dose of DT at 1 day p.i. We found that there was no significant change in the number of CD11b+ “conventional DC” (cDC) in either the D-LN (Fig. 3A) or spleen (Fig. 3B) 24 h after DT treatment. However, DT treatment of CD11c:cre × iDTR mice led to a marked reduction of both pDC and lymphoid-resident CD8α+ DC in the D-LN (Fig. 3A) and spleen (Fig. 3B). As there was a clear differential ability of DT to deplete certain DC subpopulations, we examined the potential function of each DC population in response to ECTV.
As outlined above, T1-IFN (particularly IFN-α) are required for survival of mice infected with ECTV (22, 35). Our preliminary investigations indicated that CD11b− CD11c+ cells were the major producers of T1-IFN upon ECTV infection (Fig. 2B and C), so we sought to examine if there was a link between the cells producing T1-IFN in response to exposure to ECTV and those depleted by DT treatment of CD11c:cre × iDTR mice. Mice were treated with Flt3L to expand DC populations, DC were purified, and DC subpopulations were examined for the ability to produce IFN-α in response to either live WT ECTV or UVC/psoralen-inactivated replication-incompetent ECTV (UVC ECTV). When exposed to live WT ECTV, CD11b+ DC failed to produce levels of T1-IFN above background (Fig. 3C). In contrast, both pDC and CD8α+ DC produced T1-IFN in response to live ECTV (Fig. 3C). Both pDC and CD8α+ DC also produced T1-IFN in response to UVC ECTV, although the production of T1-IFN by CD8α+ DC after stimulation with UVC ECTV was not as robust as that of live ECTV (Fig. 3C). Notably, CD11b+ DC produced significantly large quantities of T1-IFN upon exposure to UVC-treated ECTV (Fig. 3C). Therefore, it appears that the DC populations producing T1-IFN are similar to those that are depleted in CD11c:cre × iDTR mice treated with a single dose of DT, and these populations may be required for resistance to ECTV following lethal challenge.
To examine the requirement for the T1-IFN-producing DC subpopulations, pDC and CD8α+ DC, in mediating survival following ECTV infection, we utilized experimental systems in which CD8α+ DC, pDC, or both populations were ablated or depleted. Batf3KO mice lack a transcription factor necessary for development of CD8α+ DC (26). Although some inflammatory conditions may induce production of CD8α+ DC in Batf3KO mice (37), we found that Batf3KO mice that were infected with ECTV lacked these cells (Fig. 4D). When challenged with ECTV, over 80% of Batf3KO mice survived infection (Fig. 4A), indicating that CD8α+ DC alone are not required for survival following ECTV infection. To examine the role of pDC, we treated mice with anti-PDCA-1 depleting antibody, which depleted pDC in ECTV-infected mice to the maximal extent we were able to achieve using this methodology (Fig. 4D). When anti-PDCA-1-treated mice were challenged with ECTV, again, over 80% of the mice survived (Fig. 4B), indicating that pDC alone are not required for survival of ECTV infection. As both pDC and CD8α+ DC produce T1-IFN in response to ECTV (Fig. 3C), it is possible that redundancy between the populations may allow survival of ECTV challenge in the absence of one DC population. To examine the requirement for both pDC and CD8α+ DC subsets, we depleted pDC in Batf3KO mice (Fig. 4D). ECTV-infected pDC-depleted Batf3KO mice died from infection at ∼8.25 days p.i. (Fig. 4C). Therefore, it appears that alone, neither pDC nor CD8α+ DC are required for survival of ECTV-infected mice but that at least one of the DC populations is required for survival of mice following ECTV infection.
FIG 4.

CD8α+ DC or pDC are required for survival of mice following ECTV infection. (A) WT or Batf3KO mice were mock infected or infected with ECTV, and survival was monitored to 21 days p.i. The data from three independent experiments were pooled. (B) WT mice were injected i.p. with pDC-depleting or isotype control antibody and infected with ECTV. Survival was monitored to 14 days p.i. The data from three independent experiments were combined. (C) Batf3KO mice were injected i.p. with pDC-depleting antibody or control antibody, and survival was monitored to 14 days p.i. The data from three independent experiments were combined. (D) WT or Batf3KO mice were injected i.p. with pDC-depleting or control antibody and infected with ECTV. Spleens were harvested at 3 days p.i., and cells were stained for flow cytometry for pDC and CD8α+ DC as for Fig. 1A. The data are representative of two independent experiments (means ± SEM). *, P < 0.05; **, P < 0.01; ***, P < 0.001; Student's unpaired t test.
To examine whether depletion of pDC, CD8α+ DC, or both populations leads to uncontrolled ECTV replication, which would be similar to the cause of the death of DT-treated CD11c:cre × iDTR mice, we determined the titers of ECTV from multiple tissues of isotype-treated, pDC-depleted Batf3KO or pDC-depleted Batf3KO mice. There was no difference between the viral titers in the infected footpads of pDC-depleted Batf3KO mice and those of mice lacking either pDC or CD8α+ DC alone or WT mice (Fig. 5A). There was a greater viral titer in the D-LN of the pDC-depleted Batf3KO mice than in those of WT mice and pDC-depleted mice (Fig. 5B). However, there was no difference between the viral titers in the D-LN of susceptible pDC-depleted Batf3KO mice and those of resistant Batf3KO mice (Fig. 5B). This suggested that the increased viral titer in the D-LN does not support an inability to control the virus in the D-LN of pDC-depleted Batf3KO mice. However, pDC-depleted Batf3KO mice had a 1-log-unit increase in the ECTV titer in the spleen compared to either pDC-depleted mice or Batf3KO mice and a 2-log-unit increase over WT mice treated with isotype control antibody (Fig. 5C). By depleting both pDC and CD8α+ DC, localized virus was able to disseminate systemically to higher virus loads. To ascertain whether uncontrolled ECTV replication in pDC-depleted Batf3KO mice was responsible for the death of challenged mice, we treated mice with the antiviral cidofovir as described above. pDC-depleted Batf3KO mice that were treated with cidofovir on day 3 and day 6 postinfection survived ECTV infection (Fig. 5D), demonstrating the requirement for either pDC or CD8α+ DC in control of virus replication and spread.
FIG 5.

CD8α+ DC or pDC are required to control ECTV infection. (A, B, and C) WT or Batf3KO mice were injected i.p. with pDC-depleting or control antibody and infected with ECTV. Infected footpads (A), D-LN (B), and spleens (C) were harvested at 5 days p.i. The tissues were processed, and the titer of virus was determined. The data from two independent experiments were combined. (D) Batf3KO mice were injected i.p. with pDC-depleting antibody and infected with ECTV as for panel A. The mice received 100 mg/kg cidofovir or PBS i.p. at 3 and 6 days p.i. Survival was monitored to 17 days p.i. The data from two independent experiments were combined. *, P < 0.05; **, P < 0.01; ***, P < 0.001; Student's unpaired t test.
Although we have demonstrated production of antiviral T1-IFN by both pDC and CD8α+ DC, other major functions of DC include recruitment and activation of NK cells and initiation of the adaptive T cell response, both of which are required for protective immunity following ECTV challenge (10–14). To ascertain whether the antiviral activity of pDC and CD8α+ DC is direct or whether it is mediated primarily through bystander cell activation, we examined the numbers and activation status of NK cells, TCD8+, or TCD4+ at 4 days p.i. of WT or Batf3KO mice treated with anti-PDCA-1 or istoype control antibodies. Although there was a decline in total, immature, intermediate, mature, and CD69+ NK cells in susceptible pDC-depleted Batf3KO mice, the reduction was not statistically significant compared to resistant pDC-depleted WT mice (Fig. 6A). Therefore, the mild reduction in NK cells following anti-PDCA-1 treatment is unlikely to result in lethal susceptibility to ECTV challenge. Under some circumstances, pDC have been implicated in antigen presentation to TCD4+ (38, 39), but there was no reduction in numbers of total, naive, effector, memory, or CD69+ TCD4+ in susceptible pDC-depleted Batf3KO mice compared to resistant control mice (Fig. 6B). Finally, CD8α+ DC have been implicated in cross presentation of ECTV antigens to TCD8+ (4). During ECTV infection, the TCD8+ response to a recombinant antigen, or to the immunodominant ECTV-encoded peptide, was unaltered in Batf3KO mice (4). To broaden our analyses to bulk TCD8+ and to analyze the response in pDC-depleted Batf3KO mice, we examined the numbers of total, naive, effector, memory, and recently activated (CD69+) TCD8+ in mice 4 days after ECTV infection. Although there was a minimal reduction of the total numbers of TCD8+ in pDC-depleted Batf3KO compared to either pDC-depleted or Batf3KO mice, it was not statistically significant in multiple experiments (Fig. 6C). In addition, there was no significant difference in populations of naive, effector, memory, or recently activated TCD8+ in pDC-depleted Batf3KO compared to resistant control mice (Fig. 6C). Therefore, there are no large differences in the lymphocyte responses to ECTV following depletion of both pDC and CD8α+ DC that confer susceptibility to lethal ECTV challenge.
FIG 6.
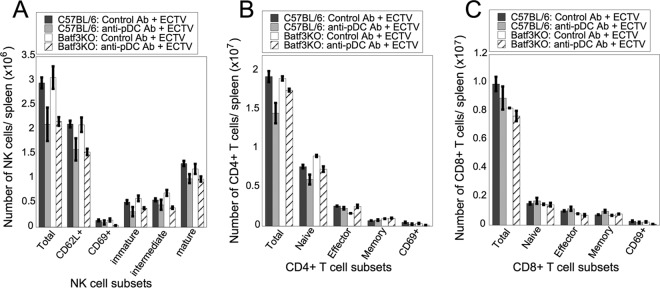
The absence of both CD8α+ DC and pDC does not result in reduced numbers of NK cells, TCD4+, or TCD8+. WT or Batf3KO mice were injected i.p. with pDC-depleting or control antibody and infected with ECTV. Spleens were harvested at 4 days p.i., processed, and stained for NK cell subsets (A), TCD4+ subsets (B), or TCD8+ subsets (C). The data are representative of two independent experiments (n = 2 or 3). Student's unpaired t test was used to compare the pDC-depleted Batf3KO group to either the pDC-depleted WT group or the control antibody Batf3KO group. Statistics would have been shown only if both of these comparisons had a significant P value (P < 0.05).
DISCUSSION
During a primary infection with ECTV, the peripheral infection spreads through the lymphohematogenous route through the D-LN to the spleen and liver, establishing a fulminant systemic infection that can cause death in susceptible mice. Although the virus spreads to all organs in both susceptible and resistant mice, the levels of ECTV found in the spleen and liver are higher in susceptible mice, establishing the D-LN as an organ that can control throughput of virus and subsequent resistance or susceptibility (40). Recruitment of NK cells and subsequent lytic activity are vital for an effective response (11, 12), but we show here that either pDC or CD8α+ DC play a vital role in reducing virus replication and allow mice to survive following lethal challenge.
Previous studies have used CLL to deplete phagocyte populations and concluded that macrophages were essential to survive ECTV challenge (13). However, we have found that during infection with ECTV or a related poxvirus, VACV (Davies et al., unpublished), CLL depletes multiple populations of phagocytic cells, including macrophages, mo-DC, CD11b+ DC, pDC, and CD8α+ DC, complicating the previous interpretation. By using a variety of complementary strategies, we showed that neither tissue-resident macrophage nor recruited monocyte or mo-DC populations were required for protective immunity to ECTV challenge. The original investigations of phagocyte function used either carrageenan (41) or CLL (13) depletion, neither of which is completely specific for macrophage/monocyte populations. Macrophage-derived nitric oxide was thought to be essential for survival of ECTV challenge (42), but other cells, such as pDC, can also produce nitric oxide during virus infection (43). Macrophages are infected with ECTV (4) and likely serve as reservoirs for spread of the virus following productive replication (44–46). Therefore, depletion of macrophages may actually reduce ECTV replication in vivo, potentially confounding any study of their importance in natural resistance. The ability of ECTV to replicate efficiently in macrophage populations starkly contrasts with the inability of the related poxvirus VACV to replicate within macrophages (47), likely due to expression of the host factor SAMHD1 in these cells (48). SAMHD1 depletes intracellular deoxynucleoside triphosphate (dNTP) pools, limiting the replication of DNA viruses (48). Macrophage populations are essential in preventing systemic spread of VACV (Davies et al., unpublished), likely due in part to the filtration of blood and lymph by macrophages that then become infected by VACV but do not produce progeny. ECTV, in contrast, productively infects macrophages, and this may result in the massively different pathogeneses of VACV and ECTV in the mouse system.
Our observation that macrophages alone were not required for protective immunity to ECTV led us to ask which other innate cells were required to mediate protection against ECTV. Given that a previous report implicated Ly6G+ granulocytes in protective immunity to ECTV challenge (31), we first examined the requirement for neutrophils. Despite our ability to produce a marked depletion of neutrophils upon administration of anti-Ly6G antibody, this treatment did not confer susceptibility to ECTV infection. Upon closer examination, we discovered that the previous study had used an anti-Gr-1 antibody (clone RB6-8C5) to deplete granulocytes (31). Gr-1 is a shared epitope present on the Ly6C and Ly6G molecules (49), so administration of this antibody would deplete multiple populations of cells expressing Ly6C, including monocytes and macrophages (50), bone marrow cells (51), thymocytes (52), B cells (53), and subsets of TCD8+ (54) and TCD4+ (52). Our study targeted Ly6G specifically using a different antibody (clone 1A8) that does not recognize significant populations of activated T or B lymphocytes (55). Gr-1-expressing cells, but not Ly6G+ cells, have previously been implicated in protection against herpes simplex virus 1 (HSV-1) infection (56, 57), indicating a clear difference in the antiviral roles of these different populations of cells.
Multiple populations of DC are infected in the D-LN of ECTV-infected mice, and these infected DC can present antigen to naive TCD8+ (4). Depletion of CD11c+ cells conferred susceptibility to lethal ECTV challenge, demonstrating that CD11c+ cells are required for resistance. However, CD11c is an integrin that is also expressed by subsets of NK cells and activated TCD8+, both of which are required for protective immunity (11, 13). Therefore, we needed to further refine our methodology to investigate the role of DC subsets in protective immunity to ECTV. DT treatment of CD11c:cre × iDTR mice primarily depleted pDC and CD8α+ DC during ECTV infection. Tahiliani et al., using a similar antibody-mediated depletion model, found that pDC-depleted mice died from ECTV infection (31). However, we were unable to reproduce this result. This could have been for a number of reasons. (i) Tahiliani et al. used an antibody dose of 1 mg/mouse every other day, while we used our optimized dose of 0.5 mg/mouse every third day. An increased dose of antibody did not increase the efficacy of pDC depletion but may have increased the side effects of administration. (ii) Tahiliani et al. used 25% weight loss as a cutoff during a survival challenge, and all mice losing greater than 25% body weight were euthanized and counted as nonsurvivors. However, weight loss has been demonstrated to be a poor marker of disease progression following lethal ECTV infection (58) or infection with other poxviruses (59–61). We have observed survival of mice challenged with either ECTV or VACV that have lost significantly more than 25% of their total body weight on many occasions. Therefore, we feel it is likely that, when examining the role of pDC in protective anti-ECTV immunity, using a weight loss cutoff can produce potentially confounding data. (iii) It is possible that the use of different substrains of C57BL/6 WT mice or different animal housing conditions in our study and that of Tahiliani et al. could have altered the susceptibility of pDC-depleted mice. It is known that the endogenous microbiota differ widely between animal facilities and can significantly alter the protective immune response to virus infection (62–64). Therefore, it may not be possible to completely replicate the conditions used in the Tahiliani et al. publication.
We found that either pDC or CD8α+ DC could confer resistance to lethal ECTV challenge. ECTV challenge of either CD11c:cre × iDTR or pDC-depleted Batf3KO mice consistently led to death within 10 days of infection. However, major histocompatibility complex (MHC) class II-deficient or TCD4+-depleted mice do not die until significantly after this point (13). Therefore, it is unlikely that a deficit in antigen presentation to TCD4+ in CD11c:cre × iDTR or pDC-depleted Batf3KO mice accounted for susceptibility. Indeed, we found no deficit in the numbers of TCD4+ following ECTV infection. TCD8+-depleted mice succumb to lethal ECTV challenge much sooner than TCD4+-deficient mice (∼8.5 days p.i.) (13). This time frame was similar to what we observed following infection of DT-depleted CD11c:cre × iDTR or pDC-depleted Batf3KO mice. We have previously shown that ECTV-infected pDC and CD8α+ DC can both present MHC class I-peptide complexes from virus-encoded antigen to naive TCD8+ (4). In addition, uninfected CD8α+ DC can also cross-present antigen acquired from ECTV-infected cells (4). However, numerous other antigen-presenting cell types, including macrophages, CD11b+ DC, and B cells, can present MHC class I-peptide complexes upon ECTV infection (4). It is likely that there is significant functional redundancy that would allow the full generation of a protective anti-ECTV TCD8+ response. Indeed, the TCD8+ response to ECTV appears unaltered in pDC-depleted Batf3KO mice, indicating that, while MHC class I-restricted antigen presentation could be diminished in the absence of pDC and CD8α+ DC, there is still significant presentation occurring to generate protective TCD8+ responses.
One of the initial indicators of the importance of pDC and CD8α+ DC during our study was the observation that CD11c+ CD11b− populations from the D-LN produce IFN-α upon ECTV infection. Mice deficient in the T1-IFN receptor, but not IFN-β, are acutely sensitive to ECTV infection, indicating a requirement for IFN-α (22). Indeed, ECTV encodes a soluble protein that binds to IFN-α, but not IFN-β, and is essential for virulence (35). pDC were originally characterized as natural interferon-producing cells, as they are known to produce large quantities of T1-IFN in response to various pathogens (65). However, injection of the TLR9 ligand CpG-B can induce the production of large quantities of T1-IFN by CD8α+ DC (66). Moreover, virus infection can stimulate non-pDC to become potent T1-IFN producers (67). Mice deficient in Toll-like receptor 9 (TLR9) are very susceptible to lethal infection with ECTV (68), indicating a critical role for sensing of ECTV by this pattern recognition receptor. Therefore, it is likely that TLR9-mediated recognition of ECTV induces production of T1-IFN by both pDC and CD8α+ DC and that either subpopulation of DC can produce enough T1-IFN to protect mice from lethal infection. Unfortunately, the nature of the multiple IFN-α genes in the genome currently makes study of the requirement for production by individual cell types very challenging.
We observed that both pDC and CD8α+ DC can produce T1-IFN in response to both live and inactivated ECTV but that CD11b+ DC can recognize only inactivated ECTV. This implies either that CD11b+ DC cannot detect live ECTV or, more likely, that ECTV can block the innate sensing mechanism used by CD11b+ DC, which is distinct from that used by pDC and CD8α+ DC. It is not clear whether pDC and CD8α+ DC use the same mechanism to sense ECTV infection and produce T1-IFN, but it is clear that sensing of ECTV by one of these cell populations is essential to survive ECTV challenge. Our data do not address the roles of other combined populations of innate effector cells in protective immunity to ECTV. For example, inflammatory monocytes and CD11b+ DC may have redundant function required for survival following ECTV challenge, and depletion of these cells following clodronate administration may be the main reason for the death of clodronate-treated animals. In addition, our data do not address the role of CD11b+ DC in resistance to ECTV infection, as these cells, or subpopulations of the cells, are not efficiently depleted in susceptible DT-depleted CD11c:cre × iDTR mice. However, it is clear that there is a distinct function mediated by either pDC or CD8α+ DC that CD11b+ DC, or other innate effector populations, cannot replace following ECTV infection. To date, the majority of publications have focused upon the specialization of DC populations to present antigen during a virus infection. The data presented here show that pDC or CD8α+ DC can fulfill a role that other myeloid cell populations, including monocytes, macrophages, mo-DC, granulocytes, and CD11b+ DC, cannot perform during ECTV infection. Therefore, the targeted activation of these cells (and the equivalent populations in humans) may be an important step in therapeutic control of a fulminant systemic virus infection.
ACKNOWLEDGMENTS
We thank Nate Sheaffer, Jade Vogel, and Joe Bednarcyk in the Hershey Medical Center flow cytometry core facility; Melanie Epler, Phyllis Rupp, and Karen Reigle of central laboratory resources; and Karen Briar, Robin Goshorn, Jeanette Mohl, Linley Eberhart, Heidi Piper, and Tiffany Whitcomb for animal handling and veterinary support.
This work was supported by NIH grants AI083008, AI056094, AI070537, AI097787, and AI115230 to C.C.N. and by training grant 5 T32 CA60395-15.
REFERENCES
- 1.Malissen B, Tamoutounour S, Henri S. 2014. The origins and functions of dendritic cells and macrophages in the skin. Nat Rev Immunol 14:417–428. doi: 10.1038/nri3683. [DOI] [PubMed] [Google Scholar]
- 2.Shortman K, Naik SH. 2007. Steady-state and inflammatory dendritic-cell development. Nat Rev Immunol 7:19–30. doi: 10.1038/nri1996. [DOI] [PubMed] [Google Scholar]
- 3.Naik SH, Metcalf D, van Nieuwenhuijze A, Wicks I, Wu L, O'Keeffe M, Shortman K. 2006. Intrasplenic steady-state dendritic cell precursors that are distinct from monocytes. Nat Immunol 7:663–671. doi: 10.1038/ni1340. [DOI] [PubMed] [Google Scholar]
- 4.Sei JJ, Haskett S, Kaminsky LW, Lin E, Truckenmiller ME, Bellone CJ, Buller RM, Norbury CC. 2015. Peptide-MHC-I from endogenous antigen outnumber those from exogenous antigen, irrespective of APC phenotype or activation. PLoS Pathog 11:e1004941. doi: 10.1371/journal.ppat.1004941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hufford MM, Richardson G, Zhou H, Manicassamy B, Garcia-Sastre A, Enelow RI, Braciale TJ. 2012. Influenza-infected neutrophils within the infected lungs act as antigen presenting cells for anti-viral CD8(+) T cells. PLoS One 7:e46581. doi: 10.1371/journal.pone.0046581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Y, Swiecki M, McCartney SA, Colonna M. 2011. dsRNA sensors and plasmacytoid dendritic cells in host defense and autoimmunity. Immunol Rev 243:74–90. doi: 10.1111/j.1600-065X.2011.01049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schuster S, Hurrell B, Tacchini-Cottier F. 2013. Crosstalk between neutrophils and dendritic cells: a context-dependent process. J Leukoc Biol 94:671–675. doi: 10.1189/jlb.1012540. [DOI] [PubMed] [Google Scholar]
- 8.Buller RM, Palumbo GJ. 1991. Poxvirus pathogenesis. Microbiol Rev 55:80–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fenner F. 1948. The pathogenesis of the acute exanthems; an interpretation based on experimental investigations with mousepox; infectious ectromelia of mice. Lancet ii:915–920. [DOI] [PubMed] [Google Scholar]
- 10.Parker AK, Parker S, Yokoyama WM, Corbett JA, Buller RM. 2007. Induction of natural killer cell responses by ectromelia virus controls infection. J Virol 81:4070–4079. doi: 10.1128/JVI.02061-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fang M, Lanier LL, Sigal LJ. 2008. A role for NKG2D in NK cell-mediated resistance to poxvirus disease. PLoS Pathog 4:e30. doi: 10.1371/journal.ppat.0040030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fang M, Orr MT, Spee P, Egebjerg T, Lanier LL, Sigal LJ. 2011. CD94 is essential for NK cell-mediated resistance to a lethal viral disease. Immunity 34:579–589. doi: 10.1016/j.immuni.2011.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karupiah G, Buller RM, Van Rooijen N, Duarte CJ, Chen J. 1996. Different roles for CD4+ and CD8+ T lymphocytes and macrophage subsets in the control of a generalized virus infection. J Virol 70:8301–8309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fang M, Sigal LJ. 2006. Direct CD28 costimulation is required for CD8+ T cell-mediated resistance to an acute viral disease in a natural host. J Immunol 177:8027–8036. doi: 10.4049/jimmunol.177.11.8027. [DOI] [PubMed] [Google Scholar]
- 15.Fang M, Siciliano NA, Hersperger AR, Roscoe F, Hu A, Ma X, Shamsedeen AR, Eisenlohr LC, Sigal LJ. 2012. Perforin-dependent CD4+ T-cell cytotoxicity contributes to control a murine poxvirus infection. Proc Natl Acad Sci U S A 109:9983–9988. doi: 10.1073/pnas.1202143109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fang M, Sigal LJ. 2005. Antibodies and CD8+ T cells are complementary and essential for natural resistance to a highly lethal cytopathic virus. J Immunol 175:6829–6836. doi: 10.4049/jimmunol.175.10.6829. [DOI] [PubMed] [Google Scholar]
- 17.Chaudhri G, Panchanathan V, Bluethmann H, Karupiah G. 2006. Obligatory requirement for antibody in recovery from a primary poxvirus infection. J Virol 80:6339–6344. doi: 10.1128/JVI.00116-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qian Q, Jutila MA, Van Rooijen N, Cutler JE. 1994. Elimination of mouse splenic macrophages correlates with increased susceptibility to experimental disseminated candidiasis. J Immunol 152:5000–5008. [PubMed] [Google Scholar]
- 19.Sunderkotter C, Nikolic T, Dillon MJ, Van Rooijen N, Stehling M, Drevets DA, Leenen PJ. 2004. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J Immunol 172:4410–4417. doi: 10.4049/jimmunol.172.7.4410. [DOI] [PubMed] [Google Scholar]
- 20.Leenen PJ, Radosevic K, Voerman JS, Salomon B, van Rooijen N, Klatzmann D, van Ewijk W. 1998. Heterogeneity of mouse spleen dendritic cells: in vivo phagocytic activity, expression of macrophage markers, and subpopulation turnover. 160:2166–2173. [PubMed] [Google Scholar]
- 21.Asselin-Paturel C, Boonstra A, Dalod M, Durand I, Yessaad N, Dezutter-Dambuyant C, Vicari A, O'Garra A, Biron C, Briere F, Trinchieri G. 2001. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat Immunol 2:1144–1150. doi: 10.1038/ni736. [DOI] [PubMed] [Google Scholar]
- 22.Rubio D, Xu RH, Remakus S, Krouse TE, Truckenmiller ME, Thapa RJ, Balachandran S, Alcami A, Norbury CC, Sigal LJ. 2013. Cross-talk between the type I interferon and nuclear factor kappa B pathways confers resistance to a lethal virus infection. Cell Host Microbe 13:701–710. doi: 10.1016/j.chom.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Caton ML, Smith-Raska MR, Reizis B. 2007. Notch-RBP-J signaling controls the homeostasis of CD8- dendritic cells in the spleen. J Exp Med 204:1653–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buch T, Heppner FL, Tertilt C, Heinen TJ, Kremer M, Wunderlich FT, Jung S, Waisman A. 2005. A Cre-inducible diphtheria toxin receptor mediates cell lineage ablation after toxin administration. Nat Methods 2:419–426. doi: 10.1038/nmeth762. [DOI] [PubMed] [Google Scholar]
- 25.Boring L, Gosling J, Cleary M, Charo IF. 1998. Decreased lesion formation in CCR2-/- mice reveals a role for chemokines in the initiation of atherosclerosis. Nature 394:894–897. doi: 10.1038/29788. [DOI] [PubMed] [Google Scholar]
- 26.Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, Schreiber RD, Murphy TL, Murphy KM. 2008. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 322:1097–1100. doi: 10.1126/science.1164206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fischer MA, Tscharke DC, Donohue KB, Truckenmiller ME, Norbury CC. 2007. Reduction of vector gene expression increases foreign antigen-specific CD8+ T-cell priming. J Gen Virol 88:2378–2386. doi: 10.1099/vir.0.83107-0. [DOI] [PubMed] [Google Scholar]
- 28.van Rooijen N, Hendrikx E. 2010. Liposomes for specific depletion of macrophages from organs and tissues. Methods Mol Biol 605:189–203. doi: 10.1007/978-1-60327-360-2_13. [DOI] [PubMed] [Google Scholar]
- 29.Willenborg S, Lucas T, van Loo G, Knipper JA, Krieg T, Haase I, Brachvogel B, Hammerschmidt M, Nagy A, Ferrara N, Pasparakis M, Eming SA. 2012. CCR2 recruits an inflammatory macrophage subpopulation critical for angiogenesis in tissue repair. Blood 120:613–625. doi: 10.1182/blood-2012-01-403386. [DOI] [PubMed] [Google Scholar]
- 30.Lin KL, Suzuki Y, Nakano H, Ramsburg E, Gunn MD. 2008. CCR2+ monocyte-derived dendritic cells and exudate macrophages produce influenza-induced pulmonary immune pathology and mortality. J Immunol 180:2562–2572. doi: 10.4049/jimmunol.180.4.2562. [DOI] [PubMed] [Google Scholar]
- 31.Tahiliani V, Chaudhri G, Eldi P, Karupiah G. 2013. The orchestrated functions of innate leukocytes and T cell subsets contribute to humoral immunity, virus control, and recovery from secondary poxvirus challenge. J Virol 87:3852–3861. doi: 10.1128/JVI.03038-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hume DA, MacDonald KP. 2012. Therapeutic applications of macrophage colony-stimulating factor-1 (CSF-1) and antagonists of CSF-1 receptor (CSF-1R) signaling. Blood 119:1810–1820. doi: 10.1182/blood-2011-09-379214. [DOI] [PubMed] [Google Scholar]
- 33.Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, Becker CD, See P, Price J, Lucas D, Greter M, Mortha A, Boyer SW, Forsberg EC, Tanaka M, van Rooijen N, Garcia-Sastre A, Stanley ER, Ginhoux F, Frenette PS, Merad M. 2013. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 38:792–804. doi: 10.1016/j.immuni.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fend L, Accart N, Kintz J, Cochin S, Reymann C, Le Pogam F, Marchand JB, Menguy T, Slos P, Rooke R, Fournel S, Bonnefoy JY, Preville X, Haegel H. 2013. Therapeutic effects of anti-CD115 monoclonal antibody in mouse cancer models through dual inhibition of tumor-associated macrophages and osteoclasts. PLoS One 8:e73310. doi: 10.1371/journal.pone.0073310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu RH, Rubio D, Roscoe F, Krouse TE, Truckenmiller ME, Norbury CC, Hudson PN, Damon IK, Alcami A, Sigal LJ. 2012. Antibody inhibition of a viral type 1 interferon decoy receptor cures a viral disease by restoring interferon signaling in the liver. PLoS Pathog 8:e1002475. doi: 10.1371/journal.ppat.1002475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buller RM, Owens G, Schriewer J, Melman L, Beadle JR, Hostetler KY. 2004. Efficacy of oral active ether lipid analogs of cidofovir in a lethal mousepox model. Virology 318:474–481. doi: 10.1016/j.virol.2003.11.015. [DOI] [PubMed] [Google Scholar]
- 37.Tussiwand R, Lee WL, Murphy TL, Mashayekhi M, Wumesh KC, Albring JC, Satpathy AT, Rotondo JA, Edelson BT, Kretzer NM, Wu X, Weiss LA, Glasmacher E, Li P, Liao W, Behnke M, Lam SS, Aurthur CT, Leonard WJ, Singh H, Stallings CL, Sibley LD, Schreiber RD, Murphy KM. 2012. Compensatory dendritic cell development mediated by BATF-IRF interactions. Nature 490:502–507. doi: 10.1038/nature11531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sapoznikov A, Fischer JA, Zaft T, Krauthgamer R, Dzionek A, Jung S. 2007. Organ-dependent in vivo priming of naive CD4+, but not CD8+, T cells by plasmacytoid dendritic cells. J Exp Med 204:1923–1933. doi: 10.1084/jem.20062373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Villadangos JA, Young L. 2008. Antigen-presentation properties of plasmacytoid dendritic cells. Immunity 29:352–361. doi: 10.1016/j.immuni.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 40.Xu RH, Fang M, Klein-Szanto A, Sigal LJ. 2007. Memory CD8+ T cells are gatekeepers of the lymph node draining the site of viral infection. Proc Natl Acad Sci U S A 104:10992–10997. doi: 10.1073/pnas.0701822104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsuru S, Kitani H, Seno M, Abe M, Zinnaka Y, Nomoto K. 1983. Mechanism of protection during the early phase of a generalized viral infection. I. Contribution of phagocytes to protection against ectromelia virus. J Gen Virol 64:2021–2026. [DOI] [PubMed] [Google Scholar]
- 42.Karupiah G, Fredrickson TN, Holmes KL, Khairallah LH, Buller RM. 1993. Importance of interferons in recovery from mousepox. J Virol 67:4214–4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Giordano D, Li C, Suthar MS, Draves KE, Ma DY, Gale M Jr, Clark EA. 2011. Nitric oxide controls an inflammatory-like Ly6C(hi)PDCA1+ DC subset that regulates Th1 immune responses. J Leukoc Biol 89:443–455. doi: 10.1189/jlb.0610329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seamer J. 1965. Mouse macrophages as host cells for murine viruses. Arch Gesamte Virusforsch 17:654–663. doi: 10.1007/BF01262241. [DOI] [PubMed] [Google Scholar]
- 45.Senkevich TG, Wolffe EJ, Buller RM. 1995. Ectromelia virus RING finger protein is localized in virus factories and is required for virus replication in macrophages. J Virol 69:4103–4111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Senkevich TG, Koonin EV, Buller RM. 1994. A poxvirus protein with a RING zinc finger motif is of crucial importance for virulence. Virology 198:118–128. doi: 10.1006/viro.1994.1014. [DOI] [PubMed] [Google Scholar]
- 47.Broder CC, Kennedy PE, Michaels F, Berger EA. 1994. Expression of foreign genes in cultured human primary macrophages using recombinant vaccinia virus vectors. Gene 142:167–174. doi: 10.1016/0378-1119(94)90257-7. [DOI] [PubMed] [Google Scholar]
- 48.Hollenbaugh JA, Gee P, Baker J, Daly MB, Amie SM, Tate J, Kasai N, Kanemura Y, Kim DH, Ward BM, Koyanagi Y, Kim B. 2013. Host factor SAMHD1 restricts DNA viruses in non-dividing myeloid cells. PLoS Pathog 9:e1003481. doi: 10.1371/journal.ppat.1003481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dunay IR, Damatta RA, Fux B, Presti R, Greco S, Colonna M, Sibley LD. 2008. Gr1(+) inflammatory monocytes are required for mucosal resistance to the pathogen Toxoplasma gondii. Immunity 29:306–317. doi: 10.1016/j.immuni.2008.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jutila MA, Kroese FG, Jutila KL, Stall AM, Fiering S, Herzenberg LA, Berg EL, Butcher EC. 1988. Ly-6C is a monocyte/macrophage and endothelial cell differentiation antigen regulated by interferon-gamma. Eur J Immunol 18:1819–1826. doi: 10.1002/eji.1830181125. [DOI] [PubMed] [Google Scholar]
- 51.Philbrick WM, Maher SE, Bridgett MM, Bothwell AL. 1990. A recombination event in the 5′ flanking region of the Ly-6C gene correlates with impaired expression in the NOD, NZB and ST strains of mice. EMBO J 9:2485–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McHeyzer-Williams LJ, McHeyzer-Williams MG. 2004. Developmentally distinct Th cells control plasma cell production in vivo. Immunity 20:231–242. doi: 10.1016/S1074-7613(04)00028-7. [DOI] [PubMed] [Google Scholar]
- 53.Wrammert J, Kallberg E, Agace WW, Leanderson T. 2002. Ly6C expression differentiates plasma cells from other B cell subsets in mice. Eur J Immunol 32:97–103. doi:. [DOI] [PubMed] [Google Scholar]
- 54.Jaakkola I, Merinen M, Jalkanen S, Hanninen A. 2003. Ly6C induces clustering of LFA-1 (CD11a/CD18) and is involved in subtype-specific adhesion of CD8 T cells. J Immunol 170:1283–1290. doi: 10.4049/jimmunol.170.3.1283. [DOI] [PubMed] [Google Scholar]
- 55.Fleming TJ, Fleming ML, Malek TR. 1993. Selective expression of Ly-6G on myeloid lineage cells in mouse bone marrow. RB6-8C5 mAb to granulocyte-differentiation antigen (Gr-1) detects members of the Ly-6 family. J Immunol 151:2399–2408. [PubMed] [Google Scholar]
- 56.Wojtasiak M, Pickett DL, Tate MD, Bedoui S, Job ER, Whitney PG, Brooks AG, Reading PC. 2010. Gr-1(+) cells, but not neutrophils, limit virus replication and lesion development following flank infection of mice with herpes simplex virus type-1. Virology 407:143–151. doi: 10.1016/j.virol.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 57.Wojtasiak M, Pickett DL, Tate MD, Londrigan SL, Bedoui S, Brooks AG, Reading PC. 2010. Depletion of Gr-1+, but not Ly6G+, immune cells exacerbates virus replication and disease in an intranasal model of herpes simplex virus type 1 infection. J Gen Virol 91:2158–2166. doi: 10.1099/vir.0.021915-0. [DOI] [PubMed] [Google Scholar]
- 58.Parker AK, Yokoyama WM, Corbett JA, Chen N, Buller RM. 2008. Primary naive and interleukin-2-activated natural killer cells do not support efficient ectromelia virus replication. J Gen Virol 89:751–759. doi: 10.1099/vir.0.83205-0. [DOI] [PubMed] [Google Scholar]
- 59.Meyer VS, Kastenmuller W, Gasteiger G, Franz-Wachtel M, Lamkemeyer T, Rammensee HG, Stevanovic S, Sigurdardottir D, Drexler I. 2008. Long-term immunity against actual poxviral HLA ligands as identified by differential stable isotope labeling. J Immunol 181:6371–6383. doi: 10.4049/jimmunol.181.9.6371. [DOI] [PubMed] [Google Scholar]
- 60.Zaitseva M, Kapnick SM, Scott J, King LR, Manischewitz J, Sirota L, Kodihalli S, Golding H. 2009. Application of bioluminescence imaging to the prediction of lethality in vaccinia virus-infected mice. J Virol 83:10437–10447. doi: 10.1128/JVI.01296-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Clark KM, Johnson JB, Kock ND, Mizel SB, Parks GD. 2011. Parainfluenza virus 5-based vaccine vectors expressing vaccinia virus (VACV) antigens provide long-term protection in mice from lethal intranasal VACV challenge. Virology 419:97–106. doi: 10.1016/j.virol.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Osborne LC, Monticelli LA, Nice TJ, Sutherland TE, Siracusa MC, Hepworth MR, Tomov VT, Kobuley D, Tran SV, Bittinger K, Bailey AG, Laughlin AL, Boucher JL, Wherry EJ, Bushman FD, Allen JE, Virgin HW, Artis D. 2014. Coinfection. Virus-helminth coinfection reveals a microbiota-independent mechanism of immunomodulation. Science 345:578–582. doi: 10.1126/science.1256942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gopinath S, Iwasaki A. 2015. Cervicovaginal microbiota: simple is better. Immunity 42:790–791. doi: 10.1016/j.immuni.2015.05.006. [DOI] [PubMed] [Google Scholar]
- 64.Ichinohe T, Pang IK, Kumamoto Y, Peaper DR, Ho JH, Murray TS, Iwasaki A. 2011. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc Natl Acad Sci U S A 108:5354–5359. doi: 10.1073/pnas.1019378108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nakano H, Yanagita M, Gunn MD. 2001. CD11c(+)B220(+)Gr-1(+) cells in mouse lymph nodes and spleen display characteristics of plasmacytoid dendritic cells. J Exp Med 194:1171–1178. doi: 10.1084/jem.194.8.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hochrein H, Shortman K, Vremec D, Scott B, Hertzog P, O'Keeffe M. 2001. Differential production of IL-12, IFN-alpha, and IFN-gamma by mouse dendritic cell subsets. J Immunol 166:5448–5455. doi: 10.4049/jimmunol.166.9.5448. [DOI] [PubMed] [Google Scholar]
- 67.Diebold SS, Montoya M, Unger H, Alexopoulou L, Roy P, Haswell LE, Al-Shamkhani A, Flavell R, Borrow P, Reis e Sousa C. 2003. Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. Nature 424:324–328. doi: 10.1038/nature01783. [DOI] [PubMed] [Google Scholar]
- 68.Samuelsson C, Hausmann J, Lauterbach H, Schmidt M, Akira S, Wagner H, Chaplin P, Suter M, O'Keeffe M, Hochrein H. 2008. Survival of lethal poxvirus infection in mice depends on TLR9, and therapeutic vaccination provides protection. J Clin Invest 118:1776–1784. doi: 10.1172/JCI33940. [DOI] [PMC free article] [PubMed] [Google Scholar]