

ABSTRACT
Despite the validation of direct-acting antivirals for hepatitis C treatment, the discovery of new compounds with different modes of action may still be of importance for the treatment of special patient populations. We recently identified a natural molecule, epigallocatechin-3-gallate (EGCG), as an inhibitor of hepatitis C virus (HCV) targeting the viral particle. The aim of this work was to discover new natural compounds with higher anti-HCV activity than that of EGCG and determine their mode of action. Eight natural molecules with structure similarity to EGCG were selected. HCV JFH1 in cell culture and HCV pseudoparticle systems were used to determine the antiviral activity and mechanism of action of the compounds. We identified delphinidin, a polyphenol belonging to the anthocyanidin family, as a new inhibitor of HCV entry. Delphinidin inhibits HCV entry in a pangenotypic manner by acting directly on the viral particle and impairing its attachment to the cell surface. Importantly, it is also active against HCV in primary human hepatocytes, with no apparent cytotoxicity and in combination with interferon and boceprevir in cell culture. Different approaches showed that neither aggregation nor destruction of the particle occurred. Cryo-transmission electron microscopy observations of HCV pseudoparticles treated with delphinidin or EGCG showed a bulge on particles that was not observed under control conditions. In conclusion, EGCG and delphinidin inhibit HCV entry by a new mechanism, i.e., alteration of the viral particle structure that impairs its attachment to the cell surface.
IMPORTANCE In this article, we identify a new inhibitor of hepatitis C virus (HCV) infection, delphinidin, that prevents HCV entry. This natural compound, a plant pigment responsible for the blue-purple color of flowers and berries, belongs to the flavonoid family, like the catechin EGCG, the major component present in green tea extract, which is also an inhibitor of HCV entry. We studied the mode of action of these two compounds against HCV and demonstrated that they both act directly on the virus, inducing a bulging of the viral envelope. This deformation might be responsible for the observed inhibition of virus attachment to the cell surface. The discovery of such HCV inhibitors with an unusual mode of action is important to better characterize the mechanism of HCV entry into hepatocytes and to help develop a new class of HCV entry inhibitors.
INTRODUCTION
Hepatitis C is a major health problem, with as many as 130 million people chronically infected worldwide (1). It is caused by hepatitis C virus (HCV), which has a high propensity to establish a persistent infection. Chronic hepatitis C can lead to the development of cirrhosis and hepatocellular carcinoma. Recent improvements in the standard of care, with direct-acting antivirals (DAAs) that inhibit HCV NS3/4A protease, NS5B polymerase, or NS5A, and now without interferon (IFN), which causes severe adverse effects, have raised the hope that HCV infection can be managed efficiently in countries with adequate medical infrastructures (2), although the cost of this therapy is extremely high. To reduce the risk of selecting viral escape mutants, a combination of DAAs is needed that, ideally, should include inhibitors targeting different steps of the HCV infectious cycle, entry, replication, and assembly/secretion, and should be efficient against all HCV genotypes. It was shown recently that the addition of entry inhibitors to other DAAs induces a synergy in the efficacy of the antiviral effect (3). In developed countries, identification of efficient HCV entry inhibitors is still needed for patients undergoing liver transplantation. Graft reinfection with HCV after transplantation occurs in nearly all cases, and long-term outcomes are unsatisfactory (4). Prevention of donor liver reinfection by inhibiting viral entry into hepatocytes might be achieved by using DAAs targeting entry.
HCV entry is a very complex process that involves a series of host entry factors (5). On the viral particle, HCV envelope glycoproteins E1 and E2 play a major role during entry. The viral particle probably initially binds to glycosaminoglycans on the surface of the target cell. It has been proposed that interactions between the low-density lipoprotein receptor and apolipoproteins associated with the viral particle might also participate in initial binding to the cell surface. Following these rather nonspecific initial binding events, several host factors are specifically involved in the virus entry process (6). Recently, we and others (7–9) have independently identified a natural molecule of plant origin, epigallocatechin-3-gallate (EGCG), as a new inhibitor of HCV infection. We demonstrated that EGCG inhibits entry by acting directly on the viral particle and preventing its attachment to the cell surface. This was confirmed recently by Colpitts and Schang (10), who showed that EGCG inhibits HCV binding by competing with heparan sulfates. Furthermore, EGCG acts in a pangenotypic manner and was also shown to have antiviral activities against a number of unrelated viruses (11). This molecule is the most abundant flavonoid in the subclass of catechins present in green tea extract. Consumption of up to 800 mg of EGCG increases the concentration of EGCG detected in the plasma (12), but this concentration is 5 to 10 times lower than the 50% inhibitory concentration (IC50) required for HCV (7).
The use of natural molecules in the treatment of hepatitis C might be a way to reduce this major health burden in developing countries, where the prevalence of HCV infection is by far the highest (13). Indeed, the improvement of hepatitis C treatment efficacy by the use of costly DAAs is predicted to have a low impact on the reduction of hepatitis C disease in these countries (13, 14). In the last few years, reports on the identification of natural molecules of plant origin with anti-HCV activities have emerged (15, 16). The most-studied plant extract is silymarin, an extract of the milk thistle plant that was used for years to treat liver diseases and has been proved to inhibit HCV infection at different steps of the viral life cycle and to display a hepatoprotective capacity (17). Silymarin and its derived compound silibinin have been tested in liver transplant patients and nonresponder patients with a substantial positive effect (18–20). Moreover, some other purified molecules, like EGCG, ladanein, cucurmin, and saikosaponin b2, seem to be good candidates for use as HCV entry inhibitors (7, 9, 15, 21, 22).
In the present study, our objectives were to start from the EGCG scaffold to identify new natural molecules with enhanced anti-HCV capacity and to determine their mode of action. We identified delphinidin, an anthocyanidin present in plant pigment, as a new HCV entry inhibitor with enhanced activity compared to that of EGCG. Delphinidin inhibits HCV attachment to the cell surface, and it is active in primary human hepatocytes (PHH), one of the most relevant preclinical models of HCV infection. Furthermore, our results showed that EGCG and delphinidin both act on the viral particle's structure, leading to a loss of shape that might impair its attachment to the cell surface.
MATERIALS AND METHODS
Chemicals.
Dulbecco's modified Eagle's medium (DMEM), phosphate-buffered saline (PBS), GlutaMAX-I, and goat and fetal bovine sera were purchased from Invitrogen (Carlsbad, CA). 4′,6-Diamidino-2-phenylindole (DAPI) was from Molecular Probes (Invitrogen). EGCG was from Calbiochem (Merck Chemicals, Darmstadt, Germany). Gallocatechin-3-gallate (GCG) was from Sigma (St. Louis, MO). Delphinidin chloride, cyanidin chloride, myrtillin chloride, pelargonidin chloride, tricetinidin chloride, myricetin, and petunidin chloride were from Extrasynthèse (Lyon, France) and were >96% pure. Stocks were resuspended in dimethyl sulfoxide (DMSO) at 0.5 M. Boceprevir was kindly provided by Philippe Halfon (Hôpital Européen, Laboratoire Alphabio, Marseille, France). IFN and other chemicals were from Sigma (St. Louis, MO).
Antibodies.
Mouse anti-E1 monoclonal antibody (MAb) A4 (23) was produced in vitro. Cy3-conjugated goat anti-mouse IgG was from Jackson ImmunoResearch (West Grove, PA).
Cells and culture conditions.
Huh-7 (24) and HEK 293T (ATCC CRL-11268) cells were grown in DMEM supplemented with GlutaMAX-I and 10% fetal bovine serum. PHH (Biopredic, Rennes, France) were isolated from normal-appearing liver tissue obtained from adult patients undergoing hepatectomy for the therapy of metastases who were seronegative for HCV, hepatitis B virus, and human immunodeficiency virus. Hepatocytes were seeded at high density into collagen-coated plates (in order to achieve full confluence, i.e., 1.5 × 105 cells/cm2) and maintained in primary culture as described previously (25).
Viruses and infectivity assays. (i) HCVcc.
We used a modified JFH1 virus containing titer-enhancing mutations and in which the A4 epitope of HCV glycoprotein E1 of genotype 1a was reconstituted (26). Virus purification was performed as described previously (7). For infection assays, Huh-7 cells were inoculated with HCV grown in Huh-7 cell culture (HCVcc) at a multiplicity of infection (MOI) of 0.8 for 2 h at 37°C. The inoculum was removed, and cells were further incubated in complete culture medium for 28 h at 37°C and then fixed.
(ii) HCVpp.
The luciferase-based pseudotyped retroviral particle infection assay was performed as previously described (27, 28). Pseudoparticles pseudotyped with either HCV E1E2 envelope proteins (HCVpp) of different genotypes or the G envelope glycoprotein of vesicular stomatitis virus (VSV-Gpp) were produced, as well as pseudoparticles with no viral envelope protein (NoEnvpp). Pseudoparticles were used to inoculate Huh-7 cells into the wells of 24-well plates for 3 h of incubation in the presence of the molecules. The inoculum was removed, and cells were further incubated with culture medium for 45 h.
(iii) HCV grown in primary culture (HCVpc).
For HCV infection, PHH were inoculated 3 days postseeding at an MOI of 5 with a nonmodified JFH1 virus (HCVcc) as previously described (25). After a 6-h incubation at 37°C, the inoculum was removed and monolayers were washed three times with PBS. Supernatants containing infectious HCVpc were harvested 2 days later, and infectious titers were evaluated by a focus-forming assay on Huh-7 cells as previously described (29).
(iv) Indirect immunofluorescence microscopy for HCV infection quantification in 96-well plates.
The day before infection, Huh-7 cells were plated into the wells of 96-well plates (6 ×103 cells/well). Cells were infected for 2 h with HCVcc at an MOI of 0.8. The inoculum was then removed, and the cells were overlaid with fresh medium. After 28 h, the cells were fixed with ice-cold methanol and processed for immunofluorescent detection of E1 as previously described (30). Nuclei were stained with 1 μg/ml DAPI. Confocal images were recorded on an automated confocal microscope (IN Cell Analyzer 6000; GE Healthcare Life Sciences) with a 20× objective with exposure parameters of 405/450 and 561/610 nm. Six fields per well were recorded. Each image was then processed with the Columbus image analysis software (PerkinElmer). Nuclei were first segmented, and the cytoplasm region was extrapolated on the basis of DAPI staining. A cell was considered “infected” when a high ratio of the intensity of E1 staining in the cytoplasm region relative to that in the nucleus was found. The quantity of cells per well and MOI were calculated to obtain 30 to 40% infected cells at 30 h postinfection, allowing automated quantification. Data were then normalized to a DMSO control, which was expressed as 100% infection.
Cellular integrity assays. (i) Viability assay.
The viability of Huh-7 cells grown in culture medium supplemented with the molecules was determined with an MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxy-methoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt]-based viability assay as described previously (7).
(ii) Cytotoxicity assay.
To assess the potential cytotoxicity of delphinidin in PHH, the activity of lactate dehydrogenase released into culture supernatants was measured in triplicate with the CytoTox 96R Non-Radioactive Cytotoxicity Assay (Promega), and the ratio of lactate dehydrogenase leakage relative to a carrier control was calculated.
Spectrophotometric quantification of delphinidin conversion.
A thermoregulated spectrophotometer was used to quantify delphinidin conversion. The absorbance spectrum of a solution of 50 μM delphinidin in HEPES (1 M, pH 7.4) was recorded every 5 min. Delphinidin conversion was monitored by the decrease of the 596-nm peak over time.
Quantitative binding assay.
HCV RNA bound to Huh-7 cells was quantified as described previously (7).
Luciferase infection assays.
A JFH1-Luc plasmid containing a Renilla luciferase reporter gene, JFH1-ΔE1E2-Luc, has been described previously (26, 31).
Quantification of HCV core protein.
HCV core antigen expressed within cells or secreted into the supernatant was quantified as described previously (32).
RNase protection assays.
Two milliliters of HCVcc stock diluted in DMEM at 1.5 × 105 focus-forming units (FFU)/ml was incubated with 50 μM EGCG, 50 μM delphinidin, 50 μM petunidin, or 0.5% Triton X-100 in the presence of 50 μg/ml RNase A for 1 h at 37°C. Treated particles were then deposited on top of 10 to 40% continuous iodixanol equilibrium gradients and ultracentrifuged for 16 h at 100,000 × g at 4°C. Twelve fractions were collected from the top of the gradients. Quantitative real-time PCR (qRT-PCR)-based viral RNA measurement (33), infectious titer determination, and buoyant density determination were performed for each fraction.
Viral aggregation analysis by velocity gradients.
HCVcc purified virions (106 FFU) were incubated with DMSO or 250 μM EGCG or delphinidin in a final volume of 1 ml in PBS for 1 h at 37°C. Treated particles were ultracentrifuged at 4°C for 90 min at 100,000 × g in a 5 to 20% iodixanol gradient. Twelve fractions were collected, and the viral RNA quantity in each fraction was determined.
Cryo-TEM of pseudoparticles.
Pseudoparticles were concentrated 100-fold by centrifugation through a 25% sucrose cushion at 100,000 × g for 120 min at 4°C and then resuspended in PBS. Pseudoparticles were then incubated for 1 h at 37°C with DMSO, 50 μM EGCG, 50 μM delphinidin, or 50 μM petunidin. Cryo-transmission electron microscopy (cryo-TEM) lacey carbon copper grids (Ted Pella Inc.) were then prepared with an EM GP plunge freezer (Leica). Five microliters of sample was deposited onto the grid placed in the chamber maintained at 15°C and 70% relative humidity. The excess of sample was removed with filter paper, and the grid was plunged into a liquid ethane bath cooled at −183°C. Specimens were maintained at a temperature of approximately −170°C with a cryo-transfer holder (Gatan, Inc.) and observed with an FEI Tecnai F20 electron microscope operating at 200 kV at a nominal magnification of ×29,000 under low-dose conditions. Images were recorded with a USC 1000 slow-scan charge-coupled device camera (2,048 by 2,048 pixels; Gatan).
Statistical analysis.
The data were analyzed with Prism statistical software (Graph Pad Inc.) by using the Mann-Whitney nonparametric test with a confidence interval of 95%. Each treated group was compared with an untreated group (DMSO control). We report two-tailed P values. Differences between treated and control groups were considered significant for any P value of <0.05. P values are indicated whenever significant differences were observed. The results are presented as a mean value ± the standard error of the mean (SEM) of triplicate experiments.
RESULTS
Delphinidin inhibits HCV infection more efficiently than EGCG does.
We recently demonstrated that EGCG inhibits HCV entry (7). In order to better understand the activity of the molecule and find potentially more potent anti-HCV molecules, we tested the antiviral capacities of different natural polyphenol molecules structurally related to EGCG. Our previous study of different catechins highlighted the importance of the galloyl group at R3 and of the hydroxyl group at R5′ in conferring on EGCG the ability to efficiently inhibit HCV entry (7). In order to determine the role of the residues at other positions, gallocatechin gallate (GCG, epimer of EGCG), tricetinidin chloride, cyanidin chloride, pelargonidin chloride, myricetin, and delphinidin were compared (Fig. 1A). Huh-7 cells were inoculated with HCVcc in the presence of the different molecules, and then the inoculum was removed and replaced with culture medium without molecules. The number of infected cells was quantified by immunofluorescence labeling of the E1 envelope protein at 28 h postinoculation. GCG exhibited infection inhibition similar to that of its epimer EGCG, showing that the spatial orientation of the trihydroxyphenyl group is not crucial for HCV inhibition (Fig. 1B). Tricetinidin chloride, cyanidin chloride, and myricetin led to only moderate inhibition of HCV infection, and no inhibition was observed with pelargonidin chloride. However, delphinidin exhibited strong inhibition of infection, showing that the trihydroxyphenyl group at R2 and the hydroxyl group at R3 are necessary to confer anti-HCV activity. In a second set of experiments, molecules derived from delphinidin were tested. Myrtillin chloride, a glycosylated delphinidin, was completely inactive against HCV infection, and petunidin, a methylated delphinidin, reduced infection by 50%, highlighting the requirement of free hydroxyl groups at R3 and R5′ to confer antiviral activity on delphinidin. Taken together, these results showed that the anthocyanidin delphinidin is a new inhibitor of HCV infection.
FIG 1.

Delphinidin inhibits HCVcc infection in a dose-dependent manner. (A) Schematic representation of the different molecules tested. (B) Huh-7 cells were inoculated with HCVcc in the presence of each molecule separately at 50 μM and incubated for 2 h. The inoculum was removed and replaced with fresh medium without the compounds. DMSO was used as a control. (C) Huh-7 cells were infected with HCVcc in the presence of the concentrations of delphinidin or EGCG shown. Numbers of infected cells were quantified by immunofluorescence assay. Data are normalized to the DMSO control, which is expressed as 100% infection. Error bars represent the SEM of three independent experiments performed in triplicate.
The IC50 of delphinidin was calculated to be 3.7 ± 0.8 μM, which is about one-third of the IC50 of EGCG calculated under the same conditions (10.6 ± 2.9 μM) (Fig. 1C).
Delphinidin and its conversion product both inhibit HCV infection.
To further characterize delphinidin, its cellular toxicity was determined. Our results showed that delphinidin is not toxic at concentrations of up to 100 μM, even when present in the medium for 72 h (Fig. 2A). Delphinidin is a plant pigment that gives a blue-purple color to flowers and berries. It is worth noting that the blue-purple color of delphinidin disappeared within approximately 2 h when it was added to the cell culture medium at 37°C, indicating a potential chemical conversion of the molecule in the medium. This was confirmed by measuring optical density at 596 nm in a time course experiment at 37°C. As shown in Fig. 2B, a time-dependent decrease of the absorbance peak of delphinidin was observed, with the molecule becoming faintly detectable after 1 h and undetectable at 2 h. To test if the conversion product is still active as an antiviral molecule, delphinidin was incubated for 1 h at 37°C in cell culture medium prior to its addition to HCVcc. As shown in Fig. 2C, the conversion product generated in this way also inhibited HCV infection in a dose-dependent manner with an IC50 of 18.7 μM. Thus, despite the disappearance of the blue-purple color of delphinidin, the colorless conversion product present in the medium is also an active anti-HCV molecule.
FIG 2.

Delphinidin and its conversion products are all active against HCVcc. (A) Toxicity of delphinidin was quantified in Huh-7 cells. Huh-7 cells were cultured in the presence of the delphinidin concentrations shown. Their viability was monitored with an MTS-based viability assay after 24, 48, and 72 h. Data are expressed as a ratio with respect to the control, i.e., the condition without delphinidin. Error bars represent the SEM from three independent experiments performed in triplicate. (B) Delphinidin at 50 μM in HEPES buffer was incubated at 37°C, and the optical density at 596 nm was determined every 5 min. (C) Delphinidin was preincubated or not for 1 h at 37°C in culture medium. Huh-7 cells were infected with the virus in the presence of increasing concentrations of freshly dissolved (circles) or preincubated (squares) delphinidin. Infected cells were quantified by immunofluorescence assay. Data are normalized to the DMSO control, which is expressed as 100% infection. Data are the mean value ± SEM obtained in three independent experiments performed in triplicate.
Delphinidin inhibits HCV entry.
To characterize the step of the HCV infectious cycle inhibited by delphinidin, the molecule was added to the culture medium for 2 h of incubation before, during, or after the inoculation step. As shown in Fig. 3A, delphinidin was only active against HCV when added during the inoculation step. No effect was observed when delphinidin was added to the cells prior to or after inoculation, suggesting direct activity on the virus at an early step of the virus infectious cycle, the entry step. We confirmed that delphinidin indeed acts on HCV entry, by the use of HCVpp harboring E1E2 envelope glycoproteins of different genotypes. Our results showed that delphinidin inhibits HCVpp entry regardless of the genotype (Fig. 3B).
FIG 3.

Delphinidin inhibits HCV entry (A) Delphinidin was added for 2 h of incubation before (cell pretreatment), during (inoculation), and/or after inoculation (postinoculation), as indicated. Infected cells were quantified by immunofluorescence assay. (B) Huh-7 cells were infected with HCVpp of the indicated genotypes or with VSV-Gpp for 2 h in the presence of either DMSO or delphinidin at 3.5 or 35 μM. Luciferase activity was quantified at 46 h postinfection. Data are normalized to the DMSO control, which is expressed as 100% infection. Error bars represent the SEM from three independent experiments performed in triplicate. (C) JFH1-ΔE1E2-Luc RNA was electroporated into Huh-7 cells. At 4 h postelectroporation, molecules (50 μM delphinidin or 2 IU/ml IFN) were added to the culture medium. Cells were lysed at the postelectroporation time points indicated, and luciferase activity was measured. (D) Cells were inoculated with HCVcc for 2 h and cultured in the absence or presence of 50 μM delphinidin for 70 h. Supernatants were collected, cells were lysed, and amounts of intracellular and extracellular core protein were measured. Data are the mean value ± SEM of three independent experiments performed in triplicate. Statistical analyses were performed with the Mann-Whitney nonparametric test. *, P < 0.05; *** P < 0.005; ns, not significant.
To test its impact on postentry steps, JFH1-ΔE1E2-Luc RNA was electroporated into Huh-7 cells and 50 μM delphinidin or 2 UI/ml IFN was added to the cells at 4 h postelectroporation. Luciferase activity was measured at different time points. Our data indicate that delphinidin did not affect luciferase activity, whereas a significant decrease in luciferase activity was observed with IFN treatment at 24, 48, and 72 h postelectroporation (Fig. 3C). These results indicate that delphinidin has no effect on HCV replication. Furthermore, by quantifying intracellular or extracellular HCV core protein, no effect of delphinidin on the secretion/assembly step was detected (Fig. 3D).
Delphinidin acts directly on HCV particles, inhibiting entry at the binding step.
As shown above, delphinidin inhibits HCV entry and seems to have no effect when added to cells prior to inoculation suggesting that it acts directly on the viral particle (Fig. 3A). To test this hypothesis, HCV was preincubated with 50 μM delphinidin for 1 h before inoculation and then a mixture of the virus and the molecule was diluted 10 times before being used for the inoculation period (at a final concentration of 5 μM). Importantly, the MOI was kept constant under all of the conditions. As shown in Fig. 4A, the inhibitory effect of delphinidin was higher when the virus was preincubated with the molecule prior to inoculation, demonstrating that delphinidin acts directly on the HCV particle. Delphinidin, by acting directly on the viral particle, might affect the attachment of the virion to the cell surface. To test this hypothesis, HCV binding assays were performed by quantifying HCV RNA attached to the cell surface after incubation of purified virions with Huh-7 cells in the presence or absence of delphinidin. Heparin and EGCG, both known to inhibit HCV binding (7), were added as positive controls. As shown in Fig. 4B, delphinidin decreased the amount of HCV RNA bound to the cell surface in the same range as EGCG, revealing an inhibition of HCV binding. Taken together, these results show that delphinidin inhibits HCV entry at the attachment step.
FIG 4.
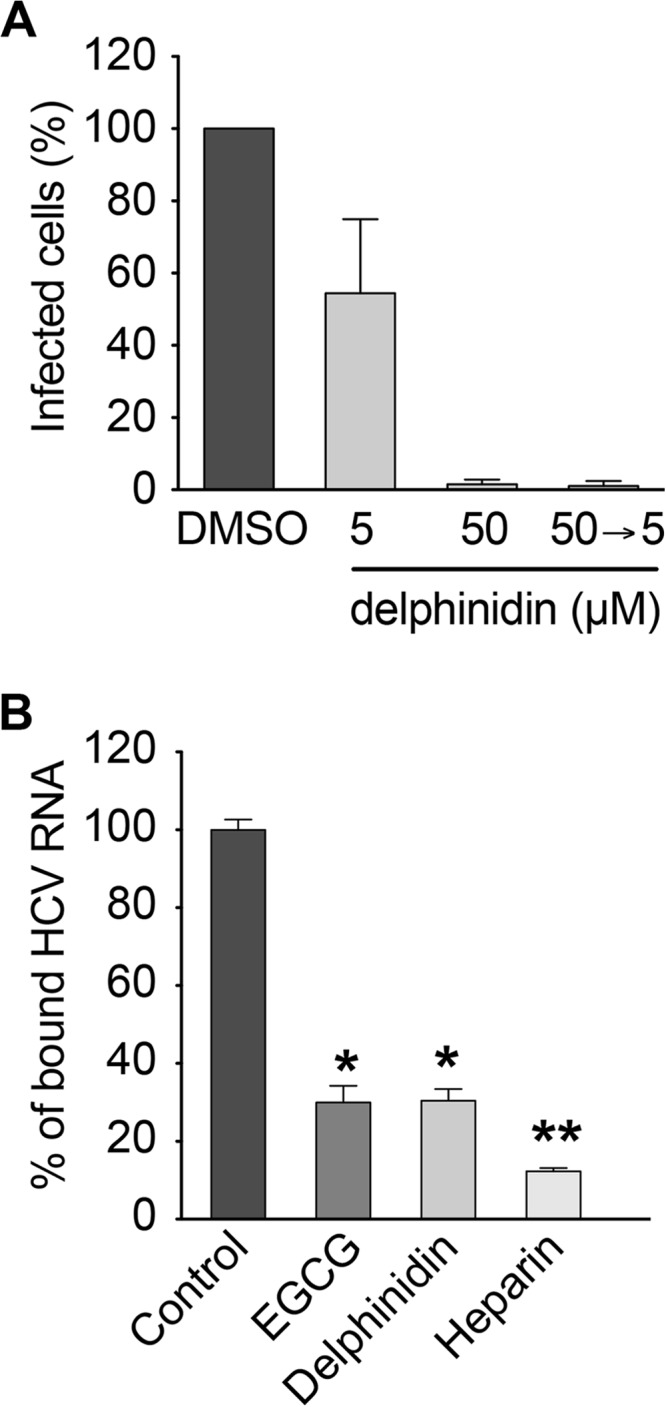
Delphinidin acts directly on the viral particle and inhibits HCV binding to the cell surface. (A) HCVcc were pretreated with 50 μM delphinidin prior to inoculation and used to infect cells after 10-fold dilution of the inoculum (50→5). Infection with untreated virus was performed in parallel in the presence of the delphinidin concentrations indicated. (B) Cells were inoculated with HCVcc at 4°C in the presence of 50 μM EGCG, 50 μM delphinidin, or 500 μg/ml heparin. Bound HCV virions were measured by quantification of HCV RNA. Data are the mean ± SEM of three experiments performed in triplicate. Statistical analyses were performed with the Mann-Whitney nonparametric test. *, P < 0.05; ** P < 0.01; ns, not significant.
Delphinidin is effective in combination with IFN or boceprevir.
Newly identified DAAs are intended to be used in combination with either IFN-α or ribavirin, the standard therapy, or with other DAAs, like protease or polymerase inhibitors. The idea is to decrease the dose of the molecule administered to diminish side effects, to target different steps of the virus infectious cycle, and to reduce the risk of viral resistance. As proof of the concept that delphinidin can be used in combination with other antivirals, Huh-7 cells were inoculated with HCVcc in the presence of different concentrations of delphinidin, and then the virus was removed and the cell culture medium was supplemented with boceprevir, a viral protease inhibitor, or IFN-α at different concentrations. As shown in Fig. 5A and B, delphinidin in combination with either IFN-α or boceprevir effectively inhibited HCV infection. The addition of delphinidin at 5 μM led to a 5-fold decrease in the IC50 of boceprevir, from 0.15 μM (boceprevir alone) to 0.03 μM, and to a 10-fold decrease of the IC50 of IFN-α, from 6.30 IU/ml (IFN-α alone) to 0.59 IU/ml. Neither synergy nor antagonism was observed when using the calculation method of Prichard and Shipman (data not shown) (34, 35). Taken together, these results show that delphinidin could be used in combination with boceprevir or IFN-α with increased efficiency, demonstrating its potential as a new anti-HCV agent.
FIG 5.

Delphinidin is effective in combination with IFN and boceprevir and in primary human hepatocytes. (A, B) Huh-7 cells were inoculated with HCVcc in the presence of delphinidin at 5 or 10 μM and then overlaid with fresh medium containing increasing concentrations of either IFN-α (A) or boceprevir (B). Numbers of infected cells were determined by immunofluorescence assay. (C, D) PHH were inoculated with HCVcc in the presence of the delphinidin concentrations indicated. (C) Infectious virus titers in culture supernatants are expressed as ratios with respect to the carrier control. (D) Cytotoxicity is expressed as a ratio with respect to the carrier control. Data are the mean ± SEM of three independent experiments performed in triplicate.
Delphinidin inhibits HCV entry into primary human hepatocytes.
To confirm and reinforce our data obtained with Huh-7 cells, the antiviral activity of delphinidin was determined in PHH. Under the culture conditions used in this study, PHH retain gene expression and physiological functions specific to normal differentiated hepatocytes, thus providing the closest in vitro model for the natural host cell of HCV and the most physiologically relevant cell culture system to confirm antiviral activities (25). PHH were inoculated with HCVcc in the presence of increasing concentrations of delphinidin, and culture supernatants were collected 2 days later to measure infectious virus titers and evaluate cytotoxicity. As shown in Fig. 5C and D, delphinidin was able to inhibit HCV infection in PHH in a dose-dependent manner with an increased IC50 of 25 ± 1.2 μM and no apparent toxicity. Of note, a higher IC50 in PHH than in hepatoma cell lines was previously observed with the HCV entry inhibitors EGCG and ladanein (9, 21).
EGCG and delphinidin modify the morphology of HCVpp.
To further characterize the mechanism of action of both EGCG and delphinidin, their impact on particle integrity was determined. RNase protection assays were performed with purified HCVcc particles. HCVcc were incubated with EGCG, delphinidin, petunidin, DMSO, or Triton X-100 in the presence of RNase A. Treated particles were then separated on iodixanol gradients (Fig. 6A). Under the control condition (DMSO), viral RNA was readily detected at a density of 1.11 g/ml, showing that the particles were not destroyed, as confirmed by the high infectious virus titers observed in the same fractions. Conversely, as expected, no RNA was detected in Triton-treated HCVcc, showing that the viral envelope was destroyed by the detergent. For both EGCG- and delphinidin-treated particles, no significant decrease in the HCV RNA level compared to that in the control was observed, showing that the particles were not destroyed by the treatment. However, the infectious virus titers were about 100 times lower than those of the control, confirming the antiviral activity of the molecules in this experiment. The petunidin-treated HCVcc showed results similar to those of the control. Taken together, these results show that neither EGCG nor delphinidin destroys the envelope of viral particles.
FIG 6.

EGCG and delphinidin do not alter HCVcc integrity and do not aggregate HCVcc. (A) HCVcc was incubated with 50 μM EGCG, 50 μM delphinidin, 50 μM petunidin, DMSO, or 0.5% Triton X-100 in the presence of RNase A at 50 μg/ml. Treated particles were then separated on 10 to 40% iodixanol equilibrium gradients. Viral RNA quantity, infectious titer, and buoyant density were determined in each fraction. Data are the mean ± SEM of three experiments performed in triplicate. (B) After incubation with DMSO or 250 μM EGCG or delphinidin, HCVcc virions were ultracentrifuged in a 5 to 20% iodixanol velocity gradient. HCV RNA was quantified by qRT-PCR. Data are representative of three experiments.
To test if EGCG or delphinidin aggregates HCVcc particles, iodixanol velocity gradient analyses were performed. After incubation with EGCG or delphinidin, purified HCVcc virions were ultracentrifuged in 5 to 20% iodixanol gradients (Fig. 6B). No difference in HCVcc mobility in the gradient was observed between treated and untreated particles, showing that neither EGCG nor delphinidin aggregates HCVcc particles.
Finally, the impact of EGCG and delphinidin on HCV morphology was determined. Because of difficulties in visualizing HCVcc by electron microscopy, the experiments were performed with HCVpp, for which we previously published the visualization of the particles by cryo-TEM (36). HCVpp of genotype 2a were incubated with EGCG or delphinidin and visualized by cryo-TEM. We observed that the addition of EGCG or delphinidin deformed the viral envelope shape of HCVpp, inducing a bulging of the particle (Fig. 7). The bulging effect was more prominent with delphinidin than with EGCG. No deformation of HCVpp was observed with DMSO (data not shown). Petunidin, which has no strong inhibitory effect on HCV infection (Fig. 1B), was used as a control, and unlike with EGCG or delphinidin, no deformation of the particles was observed (Fig. 7). These results indicate that HCVpp bulging is specifically induced by EGCG and delphinidin. The quantification of the number of deformed particles observed under each condition is presented in Table 1 and confirmed the impact of EGCG and delphinidin on HCVpp morphology, with a stronger effect of delphinidin than EGCG. To determine whether this envelope deformation was specifically related to the presence of HCV E1E2 glycoproteins, NoEnvpp and VSV-Gpp were incubated with those molecules. None of the pseudoparticles exhibited any membrane deformation (Fig. 7). Taken together, these results show that the observed bulging effect of EGCG and delphinidin on HCVpp is specifically related to the presence of HCV E1E2 glycoproteins at the surface of the particles. Neither destruction nor aggregation of HCVpp in the presence of EGCG or delphinidin was observed by cryo-TEM, confirming the results presented in Fig. 6. Taken together, these data confirm the direct effect of EGCG and delphinidin on HCV particles by an unexpected mechanism of action.
FIG 7.

EGCG and delphinidin induce HCVpp deformation. HCVpp of genotype 2a, NoEnvpp, and VSV-Gpp were incubated for 1 h at 37°C in the presence of 50 μM EGCG, delphinidin, or petunidin and processed for cryo-TEM. Cryo-TEM images representative of HCVpp, NoEnvpp, and VSV-Gpp treated with EGCG, delphinidin, and petunidin are presented. The arrows indicate membrane deformations on HCVpp incubated with EGCG or delphinidin. Bars, 50 nm.
TABLE 1.
Quantification of the numbers of deformed HCVpp observed by cryoTEM
| Treatment | No. of HCVpp counted | No. of deformed HCVpp | % of deformed HCVpp |
|---|---|---|---|
| DMSO | 51 | 1 | 2 |
| EGCG | 77 | 30 | 39 |
| Delphinidin | 91 | 61 | 67 |
| Petunidin | 74 | 3 | 4 |
DISCUSSION
In this article, we identify a new inhibitor of HCV entry, delphinidin, that might have some applications in hepatitis C therapy. This natural compound belongs to the flavonoid family, like the catechin EGCG identified by us and others as a new anti-HCV agent (7–9). Our results show that the hydroxyl groups at R3′, R5′, and R3 are important in conferring antiviral activity on delphinidin, whereas a free hydroxyl group at R3 is not required in the case of EGCG (7). Nevertheless, we show here that the mechanisms of action of EGCG and delphinidin against HCV seem to be similar, suggesting that the nature of the ester group at R3 is not important for the activity of the molecules.
We demonstrated that delphinidin, like EGCG, acts at an early step of entry, probably by inhibiting docking of the virus to the cell surface. Furthermore, our results show that delphinidin acts on the virion itself and is active against all HCV genotypes. Since delphinidin acts on the viral particle and inhibits both HCVpp and HCVcc infections, it is reasonable to think that it might interfere with E1E2 envelope glycoprotein function. This was confirmed by studying the mechanism of action of EGCG and delphinidin. Cryo-TEM observations of HCVpp treated with the molecules show that both EGCG and delphinidin induce a change in the shape of the particles that is dependent on the presence of the E1E2 envelope glycoproteins. Neither aggregation nor destruction of the particle was observed by different techniques. This uncommon mechanism of action has, to our knowledge, never been reported before. Membrane deformation is more important after incubation with delphinidin than after incubation with EGCG, suggesting that delphinidin could be more prone to interact with E1E2 glycoproteins. It is worth noting that delphinidin possesses a positive charge that is not present on EGCG and might favor its interaction with E1E2 envelopes. We hypothesize that the mechanism of the interaction of EGCG or delphinidin with E1E2 underlying the deformation of the particles impairs their capacity to interact with the cell surface. It would be interesting to observe HCVcc particles treated with EGCG and delphinidin by cryo-TEM, but this technique is not sensitive enough to observe such a phenomenon with HCVcc because of difficulties in purifying and visualizing the virus. A morphological effect of EGCG on herpes simplex virus particles was previously described (37) in which destruction of the viral envelope was observed, probably because of the interaction of EGCG with viral envelope glycoproteins gB and gD. Selection of resistant viruses could help to better understand the mechanism of action of EGCG or delphinidin on inhibition of HCV entry. Unfortunately, our efforts to generate such viruses resistant to either EGCG or delphinidin were unsuccessful. This is likely due to the mode of action of the molecules that target the virus before its attachment to the cell. This could also be due to the fact that these molecules might be able to interact with proteins at multiple binding sites.
We showed that delphinidin could be used in combination with antiviral drugs like IFN-α or boceprevir. Furthermore, the capacity of delphinidin to inhibit HCV infection in PHH, the most physiologically relevant in vitro model for preclinical validation of drugs, suggests that it could be efficient in the context of liver physiology. The latter two observations reinforce the potential use of delphinidin in hepatitis C treatment.
HCV entry is a crucial step for the initiation, spread, and maintenance of virus infection and represents an interesting target for antiviral therapy (3), especially in the case of liver transplantation. Infection with HCV is the main indication for liver transplantation in developed countries. Unfortunately, reinfection of the transplanted liver with HCV is universal and greatly compromises both patient and graft survival (4). The use of an HCV entry inhibitor to prevent graft reinfection might be needed in future combined therapies. Indeed, the compound can be administered before liver transplantation and graft reperfusion, and this should limit potential toxicity to the new donor liver.
Many studies report the health benefit of the consumption of anthocyanidin-rich food or beverages, more precisely in the case of cancers (38), cardiovascular diseases (39), and Alzheimer's disease (40). This could be explained by their anti-inflammatory and antioxidative properties and their capacity to improve lipid profiles and to activate specific regulatory pathways. A key issue is to correlate the observed benefit of anthocyanidin consumption with a precise mechanism of action. According to the United States Department of Agriculture database for the flavonoid content of selected foods (release 3.1, May 2014), delphinidin is very abundant in bilberries (97.59 mg/100 g) and black currants (87.86 mg/100 g) and even more in their concentrated juice (up to 201.28 mg/100 g) and, very interestingly, is also highly abundant in a cowpea cultivar with black seeds (94.60 mg/100 g). This leguminous plant (Vigna unguiculata) is one of the most important food legume crops in different regions of Africa, Asia, Southern Europe, and Central and South America, covering many countries where the HCV infection prevalence is very high. Further investigation is needed to determine the routes of delivery of delphinidin after the consumption of berries or black cowpeas, its concentration in plasma, and its safety and bioavailability in humans.
Low bioavailability of anthocyanidins in humans has been reported (41, 42). However, a very recent study by Czank et al. (43) performed with humans and radiolabeled cyanidin-3-glucoside showed that, with a bioavailability of 12%, anthocyanidins might be as available as other flavonoids. The peak concentration of the anthocyanidin was 0.14 μmol/liter. Interestingly, those authors also reported the detection of 24 conversion or degradation products after the ingestion of cyanidin-3-glucoside. This finding might explain the disappearance of delphinidin in cell culture that we observed. We tried to characterize the colorless conversion product(s) but were unsuccessful, probably because of the conversion or degradation of delphinidin in many different products. As already demonstrated for EGCG with herpes simplex virus (44), the pH might alter the antiviral activity of delphinidin. Hence, the fact that both delphinidin and its colorless conversion product are active against HCV might be a strong argument for the use of this molecule in humans. Curcumin is another example of a dietary polyphenol with multiple positive effects on several human diseases. However, this compound is highly unstable and its bioavailability is very low. It is supposed that, like the degradation products of delphinidin, those of curcumin are also active (45). Experiments need to be performed to better understand the metabolism and degradation pathways of these polyphenols and determine their modes of action. To overcome the low bioavailability of plant polyphenols like anthocyanidins or catechins, different studies report the use of nanoparticles that are able to encapsulate the molecules and potentiate their activity (46).
If the low toxicity of delphinidin is confirmed in vivo, the activity of this molecule on HCV entry and its pangenotypic action represent major assets of this molecule. Furthermore, delphinidin is readily available and cheap, which could limit the cost of therapy. For all of these reasons, it would be interesting to further evaluate the antiviral activity of delphinidin in a humanized mouse model and, if positive results are obtained, in HCV-infected patients.
ACKNOWLEDGMENTS
This work was supported by the French Agence Nationale de Recherche sur le Sida et les Hépatites Virales (ANRS) and by the Agence Nationale de Recherche (ANR-10-EQPX-04-01) and the Feder (12001407 [D-AL] Equipex Imaginex BioMed). A.A.M. is the recipient of a Ph.D. fellowship provided by the Egyptian Government. P.M. is supported by a concerted research action grant (01G01712) from Ghent University.
We thank Takaji Wakita for providing essential reagents. We thank François Helle, Stéphane Lobbens, and Cécile Lecoeur for helpful discussions and Marion Giard for technical assistance. We are grateful to Sophana Ung for his assistance with the illustrations.
REFERENCES
- 1.Lavanchy D. 2011. Evolving epidemiology of hepatitis C virus. Clin Microbiol Infect 17:107–115. doi: 10.1111/j.1469-0691.2010.03432.x. [DOI] [PubMed] [Google Scholar]
- 2.Pawlotsky J, Feld J, Zeuzem S, Hoofnagle J. 2015. From non-A, non-B hepatitis to hepatitis C virus cure. J Hepatol 62:S87–99. doi: 10.1016/j.jhep.2015.02.006. [DOI] [PubMed] [Google Scholar]
- 3.Xiao F, Fofana I, Thumann C, Mailly L, Alles R, Robinet E, Meyer N, Schaeffer M, Habersetzer F, Doffoel M, Leyssen P, Neyts J, Zeisel MB, Baumert TF. 2015. Synergy of entry inhibitors with direct-acting antivirals uncovers novel combinations for prevention and treatment of hepatitis C. Gut 64:483–494. doi: 10.1136/gutjnl-2013-306155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crespo G, Marino Z, Navasa M, Forns X. 2012. Viral hepatitis in liver transplantation. Gastroenterology 142:1373–1383 e1371. doi: 10.1053/j.gastro.2012.02.011. [DOI] [PubMed] [Google Scholar]
- 5.Belouzard S, Cocquerel L, Dubuisson J. 2011. Hepatitis C virus entry into the hepatocyte. Cent Eur J Biol 6:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meredith LW, Wilson GK, Fletcher NF, McKeating JA. 2012. Hepatitis C virus entry: beyond receptors. Rev Med Virol 22:182–193. doi: 10.1002/rmv.723. [DOI] [PubMed] [Google Scholar]
- 7.Calland N, Albecka A, Belouzard S, Wychowski C, Duverlie G, Descamps V, Hober D, Dubuisson J, Rouillé Y, Séron K. 2012. (−)-Epigallocatechin-3-gallate is a new inhibitor of hepatitis C virus entry. Hepatology 55:720–729. doi: 10.1002/hep.24803. [DOI] [PubMed] [Google Scholar]
- 8.Chen C, Qiu H, Gong J, Liu Q, Xiao H, Chen XW, Sun BL, Yang RG. 2012. (−)-Epigallocatechin-3-gallate inhibits the replication cycle of hepatitis C virus. Arch Virol 157:1301–1312. doi: 10.1007/s00705-012-1304-0. [DOI] [PubMed] [Google Scholar]
- 9.Ciesek S, von Hahn T, Colpitts CC, Schang LM, Friesland M, Steinmann J, Manns MP, Ott M, Wedemeyer H, Meuleman P, Pietschmann T, Steinmann E. 2011. The green tea polyphenol, epigallocatechin-3-gallate, inhibits hepatitis C virus entry. Hepatology 54:1947–1955. doi: 10.1002/hep.24610. [DOI] [PubMed] [Google Scholar]
- 10.Colpitts CC, Schang LM. 2014. A small molecule inhibits virion attachment to heparan sulfate- or sialic acid-containing glycans. J Virol 88:7806–7817. doi: 10.1128/JVI.00896-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steinmann J, Buer J, Pietschmann T, Steinmann E. 2013. Anti-infective properties of epigallocatechin-3-gallate (EGCG), a component of green tea. Br J Pharmacol 168:1059–1073. doi: 10.1111/bph.12009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chow HH, Cai Y, Hakim IA, Crowell JA, Shahi F, Brooks CA, Dorr RT, Hara Y, Alberts DS. 2003. Pharmacokinetics and safety of green tea polyphenols after multiple-dose administration of epigallocatechin gallate and polyphenon E in healthy individuals. Clin Cancer Res 9:3312–3319. [PubMed] [Google Scholar]
- 13.Hajarizadeh B, Grebely J, Dore GJ. 2013. Epidemiology and natural history of HCV infection. Nat Rev Gastroenterol Hepatol 10:553–562. doi: 10.1038/nrgastro.2013.107. [DOI] [PubMed] [Google Scholar]
- 14.Thomas DL. 2010. Curing hepatitis C with pills: a step toward global control. Lancet 376:1441–1442. doi: 10.1016/S0140-6736(10)61497-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anggakusuma, Colpitts CC, Schang LM, Rachmawati H, Frentzen A, Pfaender S, Behrendt P, Brown RJ, Bankwitz D, Steinmann J, Ott M, Meuleman P, Rice CM, Ploss A, Pietschmann T, Steinmann E. 2014. Turmeric curcumin inhibits entry of all hepatitis C virus genotypes into human liver cells. Gut 63:1137–1149. doi: 10.1136/gutjnl-2012-304299. [DOI] [PubMed] [Google Scholar]
- 16.Calland N, Dubuisson J, Rouillé Y, Séron K. 2012. Hepatitis C virus and natural compounds: a new antiviral approach? Viruses 4:2197–2217. doi: 10.3390/v4102197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Polyak S, Oberlies N, Pécheur E, Dahari H, Ferenci P, Pawlotsky J. 2013. Silymarin for HCV infection. Antivir Ther 18:141–147. doi: 10.3851/IMP2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beinhardt S, Rasoul-Rockenschaub S, Scherzer T, Ferenci P. 2011. Silibinin monotherapy prevents graft infection after orthotopic liver transplantation in a patient with chronic hepatitis C. J Hepatol 54:591–592. doi: 10.1016/j.jhep.2010.09.009. [DOI] [PubMed] [Google Scholar]
- 19.Ferenci P, Scherzer T, Kerschner H, Rutter K, Beinhardt S, Hofer H, Schöniger-Hekele M, Holzmann H, Steindl-Munda P. 2008. Silibinin is a potent antiviral agent in patients with chronic hepatitis C not responding to pegylated interferon/ribavirin therapy. Gastroenterology 135:1561–1567. doi: 10.1053/j.gastro.2008.07.072. [DOI] [PubMed] [Google Scholar]
- 20.Neumann U, Biermer M, Eurich D, Neuhaus P, Berg T. 2010. Successful prevention of hepatitis C virus (HCV) liver graft reinfection by silibinin mono-therapy. J Hepatol 52:951–952. doi: 10.1016/j.jhep.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 21.Haid S, Novodomska A, Gentzsch J, Grethe C, Geuenich S, Bankwitz D, Chhatwal P, Jannack B, Hennebelle T, Bailleul F, Keppler OT, Poenisch M, Bartenschlager R, Hernandez C, Lemasson M, Rosenberg AR, Wong-Staal F, Davioud-Charvet E, Pietschmann T. 2012. A plant-derived flavonoid inhibits entry of all HCV genotypes into human hepatocytes. Gastroenterology 143:213–222 e215. doi: 10.1053/j.gastro.2012.03.036. [DOI] [PubMed] [Google Scholar]
- 22.Lin LT, Chung CY, Hsu WC, Chang SP, Hung TC, Shields J, Russell RS, Lin CC, Li CF, Yen MH, Tyrrell DL, Richardson CD. 2015. Saikosaponin b2 is a naturally occurring terpenoid that efficiently inhibits hepatitis C virus entry. J Hepatol 62:541–548. doi: 10.1016/j.jhep.2014.10.040. [DOI] [PubMed] [Google Scholar]
- 23.Dubuisson J, Hsu HH, Cheung RC, Greenberg HB, Russell DG, Rice CM. 1994. Formation and intracellular localization of hepatitis C virus envelope glycoprotein complexes expressed by recombinant vaccinia and Sindbis viruses. J Virol 68:6147–6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakabayashi H, Taketa K, Miyano K, Yamane T, Sato J. 1982. Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res 42:3858–3863. [PubMed] [Google Scholar]
- 25.Podevin P, Carpentier A, Pène V, Aoudjehane L, Carriere M, Zaidi S, Hernandez C, Calle V, Meritet JF, Scatton O, Dreux M, Cosset FL, Wakita T, Bartenschlager R, Demignot S, Conti F, Rosenberg AR, Calmus Y. 2010. Production of infectious hepatitis C virus in primary cultures of human adult hepatocytes. Gastroenterology 139:1355–1364. doi: 10.1053/j.gastro.2010.06.058. [DOI] [PubMed] [Google Scholar]
- 26.Goueslain L, Alsaleh K, Horellou P, Roingeard P, Descamps V, Duverlie G, Ciczora Y, Wychowski C, Dubuisson J, Rouillé Y. 2010. Identification of GBF1 as a cellular factor required for hepatitis C virus RNA replication. J Virol 84:773–787. doi: 10.1128/JVI.01190-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bartosch B, Dubuisson J, Cosset FL. 2003. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J Exp Med 197:633–642. doi: 10.1084/jem.20021756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Op De Beeck A, Voisset C, Bartosch B, Ciczora Y, Cocquerel L, Keck Z, Foung S, Cosset FL, Dubuisson J. 2004. Characterization of functional hepatitis C virus envelope glycoproteins. J Virol 78:2994–3002. doi: 10.1128/JVI.78.6.2994-3002.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pène V, Hernandez C, Vauloup-Fellous C, Garaud-Aunis J, Rosenberg AR. 2009. Sequential processing of hepatitis C virus core protein by host cell signal peptidase and signal peptide peptidase: a reassessment. J Viral Hepat 16:705–715. doi: 10.1111/j.1365-2893.2009.01118.x. [DOI] [PubMed] [Google Scholar]
- 30.Rouillé Y, Helle F, Delgrange D, Roingeard P, Voisset C, Blanchard E, Belouzard S, McKeating J, Patel AH, Maertens G, Wakita T, Wychowski C, Dubuisson J. 2006. Subcellular localization of hepatitis C virus structural proteins in a cell culture system that efficiently replicates the virus. J Virol 80:2832–2841. doi: 10.1128/JVI.80.6.2832-2841.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rocha-Perugini V, Montpellier C, Delgrange D, Wychowski C, Helle F, Pillez A, Drobecq H, Le Naour F, Charrin S, Levy S, Rubinstein E, Dubuisson J, Cocquerel L. 2008. The CD81 partner EWI-2wint inhibits hepatitis C virus entry. PLoS One 3:e1866. doi: 10.1371/journal.pone.0001866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alsaleh K, Delavalle PY, Pillez A, Duverlie G, Descamps V, Rouillé Y, Dubuisson J, Wychowski C. 2010. Identification of basic amino acids at the N-terminal end of the core protein that are crucial for hepatitis C virus infectivity. J Virol 84:12515–12528. doi: 10.1128/JVI.01393-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Castelain S, Descamps V, Thibault V, Francois C, Bonte D, Morel V, Izopet J, Capron D, Zawadzki P, Duverlie G. 2004. TaqMan amplification system with an internal positive control for HCV RNA quantitation. J Clin Virol 31:227–234. doi: 10.1016/j.jcv.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 34.Prichard MN, Shipman C Jr. 1990. A three-dimensional model to analyze drug-drug interactions. Antiviral Res 14:181–205. doi: 10.1016/0166-3542(90)90001-N. [DOI] [PubMed] [Google Scholar]
- 35.Prichard MN, Shipman C Jr. 1996. Analysis of combinations of antiviral drugs and design of effective multidrug therapies. Antivir Ther 1:9–20. [PubMed] [Google Scholar]
- 36.Bonnafous P, Perrault M, Le Bihan O, Bartosch B, Lavillette D, Penin F, Lambert O, Pecheur EI. 2010. Characterization of hepatitis C virus pseudoparticles by cryo-transmission electron microscopy using functionalized magnetic nanobeads. J Gen Virol 91:1919–1930. doi: 10.1099/vir.0.021071-0. [DOI] [PubMed] [Google Scholar]
- 37.Isaacs CE, Wen GY, Xu W, Jia JH, Rohan L, Corbo C, Di Maggio V, Jenkins EC Jr, Hillier S. 2008. Epigallocatechin gallate inactivates clinical isolates of herpes simplex virus. Antimicrob Agents Chemother 52:962–970. doi: 10.1128/AAC.00825-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Del Rio D, Borges G, Crozier A. 2010. Berry flavonoids and phenolics: bioavailability and evidence of protective effects. Br J Nutr 104(Suppl 3):S67–S90. doi: 10.1017/S0007114510003958. [DOI] [PubMed] [Google Scholar]
- 39.Wallace TC. 2011. Anthocyanins in cardiovascular disease. Adv Nutr 2:1–7. doi: 10.3945/an.110.000042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ho L, Ferruzzi MG, Janle EM, Wang J, Gong B, Chen TY, Lobo J, Cooper B, Wu QL, Talcott ST, Percival SS, Simon JE, Pasinetti GM. 2013. Identification of brain-targeted bioactive dietary quercetin-3-O-glucuronide as a novel intervention for Alzheimer's disease. FASEB J 27:769–781. doi: 10.1096/fj.12-212118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Manach C, Williamson G, Morand C, Scalbert A, Remesy C. 2005. Bioavailability and bioefficacy of polyphenols in humans. I. Review of 97 bioavailability studies. Am J Clin Nutr 81(1 Suppl):230S–242S. [DOI] [PubMed] [Google Scholar]
- 42.Williamson G, Manach C. 2005. Bioavailability and bioefficacy of polyphenols in humans. II. Review of 93 intervention studies. Am J Clin Nutr 81(1 Suppl):243S–255S. [DOI] [PubMed] [Google Scholar]
- 43.Czank C, Cassidy A, Zhang Q, Morrison DJ, Preston T, Kroon PA, Botting NP, Kay CD. 2013. Human metabolism and elimination of the anthocyanin, cyanidin-3-glucoside: a (13)C-tracer study. Am J Clin Nutr 97:995–1003. doi: 10.3945/ajcn.112.049247. [DOI] [PubMed] [Google Scholar]
- 44.Isaacs CE, Xu W, Merz G, Hillier S, Rohan L, Wen GY. 2011. Digallate dimers of (−)-epigallocatechin gallate inactivate herpes simplex virus. Antimicrob Agents Chemother 55:5646–5653. doi: 10.1128/AAC.05531-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schneider C, Gordon O, Edwards R, Luis P. 30 March 2015. Degradation of curcumin: from mechanism to biological implication. J Agric Food Chem doi: 10.1021/acs.jafc.5b00244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Munin A, Edwards-Levy F. 2011. Encapsulation of natural polyphenolic compounds; a review. Pharmaceutics 3:793–829. doi: 10.3390/pharmaceutics3040793. [DOI] [PMC free article] [PubMed] [Google Scholar]