

ABSTRACT
Measles virus (MeV) causes several unique syndromes, including transient immunosuppression. To clarify the cellular responses to MeV infection, we previously analyzed a MeV-infected epithelial cell line and a lymphoid cell line by microarray and showed that the expression of numerous genes was up- or downregulated in the epithelial cells. In particular, there was a characteristic comprehensive downregulation of housekeeping genes during late stage infection. To identify the mechanism underlying this phenomenon, we examined the phosphorylation status of transcription factors and kinase/phosphatase activities in epithelial cells after infection. MeV infection inactivated cellular protein phosphatase 5 (PP5) that consequently inactivated DNA-dependent protein kinase, which reduced Sp1 phosphorylation levels, and c-Myc degradation, both of which downregulated the expression of many housekeeping genes. In addition, intracellular accumulation of viral nucleocapsid inactivated PP5 and subsequent downstream responses. These findings demonstrate a novel strategy of MeV during infection, which causes the collapse of host cellular functions.
IMPORTANCE Measles virus (MeV) is one of the most important pathogens in humans. We previously showed that MeV infection induces the comprehensive downregulation of housekeeping genes in epithelial cells. By examining this phenomenon, we clarified the molecular mechanism underlying the constitutive expression of housekeeping genes in cells, which is maintained by cellular protein phosphatase 5 (PP5) and DNA-dependent protein kinase. We also demonstrated that MeV targets PP5 for downregulation in epithelial cells. This is the first report to show how MeV infection triggers a reduction in overall cellular functions of infected host cells. Our findings will help uncover unique pathogenicities caused by MeV.
INTRODUCTION
Measles virus (MeV) is one of the most important pathogens in humans and is a major cause of child mortality, particularly in developing countries (1). Therefore, MeV has been targeted for eradication by the World Health Organization. MeV infection causes several characteristic syndromes, including transient immunosuppression (1). MeV infection induces different immune responses in epithelial and lymphoid cells in vitro. For example, MeV infection induces the rapid activation of antiviral factors (2–4) and consequent production of various cytokines (3, 5, 6) in epithelial cells. In contrast, these cellular responses are suppressed in MeV-infected lymphoid cells (7–9).
To clarify the cell-type-specific responses to wild-type MeV infection, we previously performed a microarray analysis using two different cell types (10). Experiments were designed to assess the time course of gene expression patterns in human epithelial cells (HEK293 cell line that stably expresses the receptor SLAM; 293SLAM cells) and lymphoid cells (COBL-a cells), which are the cell types targeted by MeV during primary infection. Many genes were upregulated rapidly in 293SLAM cells, including genes involved in antiviral responses. In contrast, almost all gene regulation was suppressed in COBL-a cells. These results confirmed cell-type-specific responses described in previous reports. Furthermore, during the late stage of infection of 293SLAM cells, the transcription of numerous genes, especially housekeeping genes, was downregulated (10). Strikingly, this phenomenon was not previously reported for MeV. Based on these previous findings, we sought to investigate the mechanism that underlies the constitutive expression of housekeeping genes in cells and demonstrated how MeV causes the collapse of constitutive gene expression.
MATERIALS AND METHODS
Reagents.
Chemical inhibitors were purchased from Calbiochem and dissolved in dimethyl sulfoxide (DMSO) as a 1,000-fold-concentrated stock solution: PD98059 (50 μM), Ro-31-8220 (20 μM), DNA-dependent protein kinase (DNA-PK) inhibitor (100 μM), olomoucine (180 μM), casein kinase 2 inhibitor (10 μM), Gö 6976 (20 nM), ataxia-telangiectasia mutated kinase inhibitor (100 nM), Jun N-terminal kinase inhibitor II (500 nM), staurosporine (500 nM), and MG-132 (5 mM).
The primary antibodies were purchased from the following manufacturers: anti-Sp1 (rabbit polyclonal; Santa Cruz Biotechnology), anti-DNA-PKcs (mouse monoclonal, clone 18-2; GeneTex), anti-PP2A catalytic subunit (mouse monoclonal, clone 46; BD Biosciences), anti-PP5 (goat polyclonal; Santa Cruz Biotechnology), anti-PP6c (rabbit polyclonal; GeneTex), anti-Ku86 (mouse monoclonal, clone S10B1; Calbiochem), anti-glyceraldehyde phosphate dehydrogenase (GAPDH) (mouse monoclonal, clone MAB374; Chemicon), and anti-c-Myc (mouse monoclonal, clone 9E10; Clontech). Rabbit polyclonal antibody against MeV-N protein was prepared as described previously (11). Human universal type I interferon alpha (IFN-α) was purchased from PBL Biomedical Laboratories.
Cell culture and virus preparation.
COBL-a cells (12) were grown in RPMI 1640 medium with 100 U of penicillin/ml, 100 μg of streptomycin/ml, and 5% fetal calf serum (FCS) at 37°C in a 5% CO2 incubator. HEK293 cells and 293SLAM cells were grown in Dulbecco modified Eagle medium (DMEM) with 5% FCS, penicillin, and streptomycin at 37°C in a 5% CO2 incubator. The large-scale preparation and purification of the MeV-HL strain (13, 14) was performed as described previously (10).
Preparation of RNA samples for microarray analysis.
COBL-a and 293SLAM cells (3 × 107) were treated with 1,000 IU of IFN-α/ml. After 6 or 24 h, the medium was removed, and the cells were lysed with Isogen reagent (Nippon Gene). Total RNA was prepared from the Isogen lysates in accordance with the manufacturer's instructions. Poly(A)+ RNA was prepared from the total RNA with a MicroPoly(A)Purist kit (Ambion), according to the manufacturer's instructions.
Microarray preparation and expression profiles.
A DNA microarray analysis with a set of synthetic polynucleotides (80-mers) representing 22,272 human transcripts was performed as described previously (10). In accordance with “minimum information about a microarray experiment” (MIAME) guidelines, all of the the data were deposited in DNA Data Bank of Japan (DDBJ) via the Center for Information Biology gene EXpression database (CIBEX; http://cibex.nig.ac.jp/data/index.html) under accession number CBX32.
Electrophoretic mobility shift assay (EMSA).
Nuclear extracts were prepared with NE-PER nuclear protein extraction reagent (Pierce Biotechnology), according to the manufacturer's instructions. An oligonucleotide containing the Sp1-binding motif (5′-GAGGCGTGGTGAGGCGGAAT-3′) was labeled with [γ-32P]ATP (GE Healthcare) using T4 polynucleotide kinase (Toyobo). Unincorporated [γ-32P] was removed by centrifugation on a G-25 Sephadex column (GE Healthcare) according to the manufacturer's recommendations. The binding reactions were performed in 20-μl reaction volumes containing 15 mM Tris-HCl (pH 7.5), 75 mM NaCl, 1.5 mM EDTA, 0.3% Nonidet P-40, 7.5% glycerol, 1.5 mM dithiothreitol (DTT), 1 mg of bovine serum albumin (BSA)/ml, 50 μg of salmon sperm DNA/ml, and an aliquot of nuclear extract (∼5 μg). After each sample was preincubated on ice for 10 min, 32P-labeled probe DNA (40,000 cpm) was added to the reaction mixture, which was then incubated at room temperature for 20 min. The samples were subjected to electrophoresis on a native 6% polyacrylamide gel for 2 h at 150 V.
Immunoprecipitation of radiolabeled proteins.
293SLAM and COBL-a cells in cell culture dishes (3.5 cm in diameter) were infected with MeV at a multiplicity of infection (MOI) of 2, the same conditions as for microarray analysis (10), or were mock infected. At 24 h after infection, the medium was replaced with 1.5 ml of DMEM containing one-tenth the normal amount of methionine and cysteine, 5% FCS, and 150 μCi of [35S]EasyTag express protein labeling mix (Perkin-Elmer) or 1.5 ml of DMEM containing 1 mCi of [32PO4]phosphorous radionuclide (Perkin-Elmer). After 24 h, the cells were lysed with cell lysis buffer (0.5% Triton X-100, 0.5 mM EDTA, 50 mM NaF, 0.1 mM Na3VO4, and 2% [vol/vol] protease inhibitor cocktail [BD Bioscience] in phosphate-buffered saline [PBS]). The cell lysates were clarified by centrifugation at 16,000 × g for 10 min and subjected to an immunoprecipitation assay using anti-Sp1 antibody or anti-DNA-PKcs antibody. Each reaction mixture contained 20 μl of a protein A-Sepharose bead suspension (GE Healthcare). The samples were rocked at 4°C overnight. The beads were washed with PBS and subjected to SDS–10% PAGE. The immunoprecipitates were detected with a FLA-5000 imaging system (Fujifilm).
Construction and expression of GST-Sp1.
To create a plasmid expressing glutathione S-transferase–Sp1 (GST-Sp1), total RNA isolated from HEK293 cells with Isogen was reverse transcribed using a random primer (9-mer) and then PCR amplified with a specific primer pair corresponding to the Sp1 cDNA (5′-GAATTCCATGAGCGACCAAGATCACTC-3′ and 5′-GTCGACTCAGAAGCCATTGCCACTG-3′; the restriction sites are underlined). This PCR product was inserted into the EcoRI/SalI sites of pGEX-4T3 (GE Healthcare). Escherichia coli BL21(DE3), freshly transformed with the GST-Sp1 expression vector, was grown to mid-log phase, and protein expression was induced for 4 h with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside). The cells were harvested by centrifugation, lysed with E. coli lysis buffer (1% Triton X-100, 0.1× PBS), and then sonicated with a Sonifier 450 (Branson) for 5 min. The cell lysates were clarified by centrifugation at 16,000 × g for 10 min. GST-Sp1 was bound to glutathione-Sepharose beads (GE Healthcare) for 1 h at room temperature, and the unbound protein was removed by washing the beads with kinase buffer (20 mM Tris-HCl [pH 7.5], 0.5% Triton X-100, 10 mM MgCl2, 2 mM EGTA, 10 mM β-glycerophosphate, 0.1 mM Na3VO4, 50 mM NaF, 1 mM DTT, 2% [vol/vol] protease inhibitor cocktail).
In vitro kinase assay of GST-Sp1.
Cells were lysed with kinase buffer at 4°C for 2 h and clarified by centrifugation at 16,000 × g for 10 min. The GST-Sp1 bound to the glutathione-Sepharose beads was incubated with the cell lysate for 2 h at 4°C, and the beads were then washed with kinase buffer. The beads were resuspended in 20 μl of kinase buffer supplemented with 4 μCi of [γ-32P]ATP/μl (3,000 Ci/mmol) and incubated for 1 h at 30°C. The reaction was terminated by the addition of SDS sample buffer, and the phosphoproteins were analyzed with SDS-PAGE and autoradiography. For the kinase inhibitor assay, the cell lysates were supplemented with specific inhibitors, and then with GST-Sp1.
DNA-cellulose pulldown of DNA-PK and measurement of its kinase activity.
293SLAM cells (8 × 105 cells) were inoculated with MeV at an MOI of 2. After 24 h, the cells were lysed with kinase buffer and centrifuged at 16,000 × g for 10 min. The cell lysates were incubated with 20 μl of preswollen double-stranded DNA (dsDNA)–cellulose (GE Healthcare) for 30 min at 4°C. The DNA-cellulose was washed three times with DNA-PK reaction buffer (25 mM HEPES [pH 7.9], 50 mM KCl, 10 mM MgCl2, 10% [vol/vol] glycerol, 1 mM EDTA, 1 mM EGTA, 1 mM DTT). A SignaTECT DNA-dependent protein kinase assay system (Promega) was used to assess DNA-PK activity, with the following modifications. DNA-cellulose was resuspended in 20 μl of DNA-PK reaction buffer containing 100 μg of BSA/ml. The kinase reactions were conducted with 10-μl aliquots of the resuspended DNA-PK adsorbed to cellulose beads and were performed in the presence or absence of a biotinylated p53-derived DNA-PK substrate peptide and [γ-32P]ATP. No activator was used. The reactions were terminated, spotted onto a SAM2 biotin capture membrane (Promega), washed, and analyzed. Radioactivity was counted with a scintillation counter. The experiments were performed with at least three different extract preparations.
Reporter gene assay using luciferases.
A reporter plasmid carrying the c-Myc-binding motif (CACGTG; designated the “E box”) upstream from the firefly luciferase gene was constructed by inserting tandem repeats of the E box (CACGTG)5 into the SmaI site of pGL3-basic (Promega), and designated the “pGL3-Ebox.” 293SLAM cells in a 24-well plate were transfected with the reporter plasmid pGL3-Ebox or pGL3-basic (0.2 μg) and a reference plasmid phRG-B (Promega) encoding the Renilla luciferase gene (25 ng), using Lipofectamine 2000 transfection reagent (Life Technologies), according to the manufacturer's instructions. After 24 h, the cells were mock treated or infected with MeV at an MOI of 2. After 24 h, cells were lysed in passive lysis buffer (Promega), and the luciferase activities were measured with a Dual-Glo luciferase assay system (Promega) according to the manufacturer's instructions. Fluorescent intensity was detected by a Mini Lumat LB 9506 luminometer (Berthold). The experiments were performed in triplicate. The reporter activity was calculated as the ratio of firefly luciferase activity to Renilla luciferase activity.
In vitro phosphatase assay.
The cells were lysed in phosphatase lysis buffer (1% Nonidet P-40, 50 mM Tris-HCl [pH 7.5], 2 mM EDTA, 0.1% BSA, 2% [vol/vol] protease inhibitor cocktail). DNA-PK, protein phosphatase 2A (PP2A), PP5, or PP6 was immunoprecipitated with the appropriate antibody and protein A-Sepharose beads. The beads were then washed four times with phosphatase lysis buffer and four times with phosphatase assay buffer (50 mM Tris-HCl [pH 7.5], 2 mM EDTA, 2 mM EGTA). The phosphatase activity in the immunocomplexes was assayed with a Ser/Thr phosphatase assay kit 1 using KRpTIRR as the substrate peptide (Millipore), according to the manufacturer's instructions. All of the assays were performed with at least three different extract preparations.
siRNA knockdown of PP5.
Antisense small interfering RNAs (siRNAs) targeting PP5 mRNA and a mismatch negative-control siRNA were obtained from Sigma. 293SLAM cells in 24-well plates were transfected with 0.5 μg of the siRNA using X-tremeGENE siRNA transfection reagent (Roche), according to the manufacturer's instructions. After 2 days, the cells were subjected to immunoblotting, to EMSA, or to radiolabeling and immunoprecipitation, as described above, or else to quantitative real-time PCR (qPCR) as described below. The following siRNA sense oligonucleotide sequences were used: mismatch control, 5′-GGUAGAGCGAGAGGUAGGC-3′; PP5#1, 5′-AACAUAUUCGAGCUCAACGG-3′; and PP5#2, 5′-GAGGGUGAGGUGAAGGCCA-3′.
Relative quantification of host mRNAs.
Total RNAs were isolated with Isogen in accordance with the manufacturer's instructions. An aliquot of RNA was reverse transcribed with a 1:1 mixture of oligo(dT) primer and random primer and with PrimeScript reverse transcriptase (TaKaRa) according to the manufacturer's instructions. qPCR assays were conducted using specific primer pairs for 18S rRNA (RNA18S5), nonhousekeeping genes (HIST1H2AC, CASP3, JAK1, MAPK6, and IRF1) and housekeeping genes (NDUFB4, COX7C, RPL23, LAMA4, HSPA1L, and ATP5G3) described in the supplemental material. All qPCR assays were conducted on a Rotor-Gene Q cycler (Qiagen).
Minigenome assay.
A negative-strand minigenomic RNA, carrying both the 3′-leader and 5′-trailer sequences of the MeV-HL genome (nucleotide positions 1 to 107 and 15786 to 15894, respectively) at the ends of the firefly luciferase open reading frame, was synthesized, and the minigenome assay was performed as described previously (15). Briefly, HEK293 cells in 24-well plates were transfected with phRG-B (25 ng), together with N-, P-, and L-protein expression plasmids or empty plasmid, and mock or viral minigenomic RNA was transfected the following day. After 24, 36, or 48 h, the cells were lysed, and the luciferase activities were measured as described above. PP5 and PP2A assays, EMSA, and c-Myc immunoblotting were performed using cells in the same condition as the minigenome assay.
RESULTS
Overview of the microarray analysis.
First, we analyzed whether the significant alteration of gene expression in 293SLAM cells after MeV infection was caused by normal antiviral responses. 293SLAM cells and COBL-a cells were treated with IFN-α and then subjected to microarray analysis. The results of hierarchical clustering analysis were compared to those previously reported for MeV (10) and are summarized in Fig. 1. A limited number of genes, mainly IFN response genes, were upregulated after IFN-α treatment in both cell lines, and this upregulation did not increase with time. In particular, the comprehensive downregulation of housekeeping genes in 293SLAM cells in the later stages of infection with MeV (10) was not observed in the IFN-treated cells. This result indicates that the downregulation of gene expression induced by infection is an intrinsic feature of MeV infection and differs from the usual cellular antiviral response.
FIG 1.
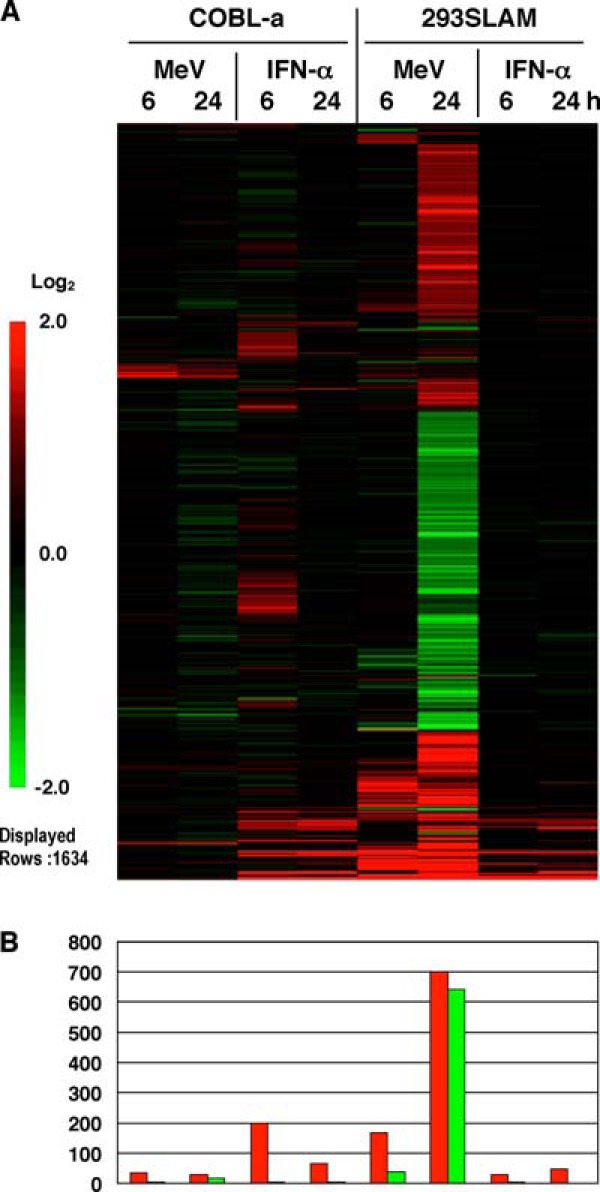
Gene expression profiles of cells inoculated with MeV or IFN-α. (A) Microarray gene expression profiles of 293SLAM and COBL-a cells treated with IFN-α, compared to those of cells inoculated with MeV. Genes that displayed a mean log ratio in all samples were extracted. Genes with a |mean log ratio| of ≥1 were further extracted, and the resulting 1,634 genes were assembled according to a hierarchical clustering analysis. (B) The numbers of genes extracted is shown on a graph.
MeV infection reduces the phosphorylation of Sp1 in 293SLAM cells.
To identify the signal that triggers the comprehensive downregulation of housekeeping genes during MeV infection, we focused on the transcription factor Sp1. The ubiquitously expressed Sp1 zinc finger protein regulates a large number of genes by binding to consensus sequences in the GC and GT boxes (16, 17) frequently found in the promoters and enhancers of ubiquitously expressed housekeeping genes. To confirm the activity of Sp1 after MeV infection, we performed an EMSA using nuclear extracts and an oligonucleotide containing the Sp1 consensus sequence. Sp1 binding activity was markedly reduced after MeV infection in 293SLAM cells (Fig. 2A). In contrast, Sp1 activity was not altered by MeV in COBL-a cells (Fig. 2A). This indicates that the downregulation of housekeeping genes after MeV infection correlates with reduced Sp1 activity.
FIG 2.
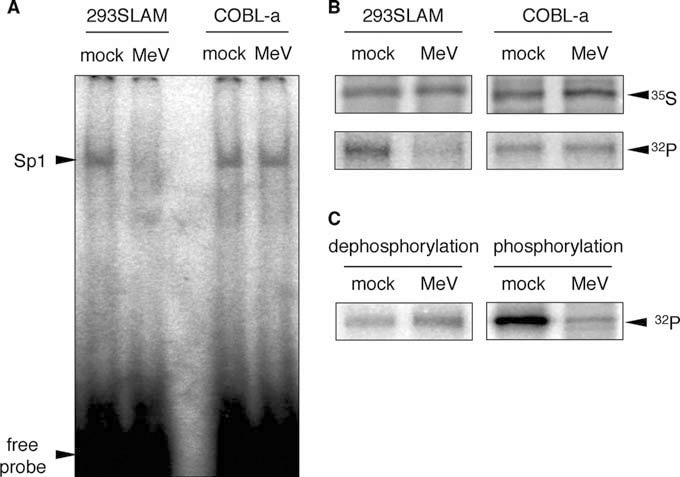
Binding activity and phosphorylation status of Sp1 after MeV infection. (A) Nuclear extracts were prepared from mock- or MeV-infected cells at 48 h postinfection, and the Sp1 DNA-binding activity was measured by EMSA using a radiolabeled probe containing the Sp1 consensus sequence. The position of the complex is indicated on the left. Specificity of binding was confirmed by competition with unlabeled probe (data not shown). (B) 293SLAM and COBL-a cells were mock infected or infected with MeV and then labeled with [35S]methionine-cysteine or [32PO4] for 24 h before harvest and extraction. Sp1 was immunoprecipitated from the cellular extracts, and the levels of 35S and 32P incorporation into the synthesized Sp1 were determined by SDS–10% PAGE and autoradiography. An arrowhead indicates Sp1. (C) 293SLAM cells were labeled with 32PO4 for 2 h before MeV infection (left: dephosphorylation) or 22 h after infection (right, phosphorylation) at an MOI of 2. After infection for 24 h, the cells were lysed, and Sp1 was immunoprecipitated.
Many studies have demonstrated that the phosphorylation of Sp1 occurs at various locations in the protein and positively and negatively regulates the expression of its target genes (18, 19). To determine the phosphorylation status of Sp1 during MeV infection, the expression levels and phosphorylation of Sp1 were examined by labeling equal numbers of cells with [35S] or [32PO4], followed by the immunoprecipitation of Sp1 (Fig. 2B). In MeV-infected 293SLAM cells, the incorporation of 32P into Sp1 decreased significantly. In contrast, in COBL-a cells, the phosphorylation of Sp1 was not altered by MeV, and the incorporation of 35S into the Sp1 protein was also unchanged (Fig. 2B). To determine whether the reduced phosphorylation of Sp1 in 293SLAM cells was caused by the suppression of phosphorylation or an increase in dephosphorylation, we labeled Sp1 with 32P in a pulse-chase experiment. The phosphorylation status of Sp1 observed before MeV infection was not altered by MeV infection (Fig. 2C, left panel), but the level of newly phosphorylated Sp1 was significantly reduced after infection (Fig. 2C, right panel). These results indicate that the inhibition of Sp1 phosphorylation induced by MeV infection caused the downregulated expression of housekeeping genes and strongly suggest that a specific kinase that phosphorylates Sp1 is inactivated during MeV infection.
Identification of Sp1-associated kinase activity.
Many different signals induce Sp1 phosphorylation, including viral infection, growth factors, certain drugs, cytokines, and mechanical stress, and this phosphorylation is catalyzed by different kinases (20). However, the kinase predominantly responsible for the constitutive phosphorylation of Sp1, maintaining its activity in cells in an unstimulated state, has not yet been identified. To identify the kinase activity involved in Sp1 phosphorylation and is affected by MeV infection, a bacterially expressed GST-Sp1 fusion protein was reacted with mock- or MeV-infected 293SLAM cell extracts and subjected to an in vitro kinase reaction. The kinase activity associated with GST-Sp1 was decreased markedly during MeV infection (Fig. 3A). Therefore, to identify the specific kinase that catalyzed the phosphorylation of Sp1 under unstimulated conditions, we performed an in vitro kinase assay using 293SLAM cell extracts that contained a specific kinase inhibitor corresponding to each kinase shown to act on Sp1 (summarized in Fig. 3B). Of these, a DNA-PK inhibitor caused a marked reduction in the phosphorylation of GST-Sp1 (Fig. 3C). These results suggest that DNA-PK acts as a Sp1 kinase in cells under unstimulated conditions, maintaining the constitutive transcriptional activity of Sp1.
FIG 3.
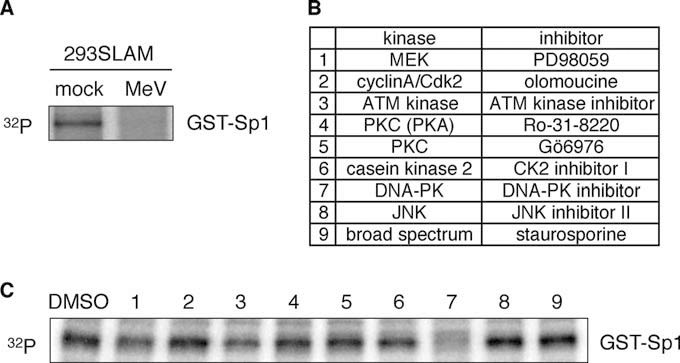
Identification of Sp1-associated kinase in cells under unstimulated conditions. (A) Phosphorylation of GST-Sp1 by Sp1-assocated kinase activity. GST-Sp1 was bound to glutathione-Sepharose beads and incubated with lysates from mock- or MeV-infected 293SLAM cells. After the beads were washed, the presence of kinase activity that could phosphorylate Sp1 was assessed by an in vitro kinase assay containing [γ-32P]ATP. The reactions were analyzed by SDS–10% PAGE and autoradiography. Arrowhead indicates phosphorylated GST-Sp1. (B) List of kinases reported to phosphorylate Sp1 and their corresponding specific inhibitors. (C) 293SLAM cell lysates treated with each kinase inhibitor and then subjected to an in vitro kinase assay.
Inactivation of DNA-PK by phosphorylation after MeV infection.
DNA-PK is composed of a catalytic subunit (DNA-PKcs) and a heterodimeric DNA-end-binding protein, Ku70/Ku86. DNA-PK plays a central role during the nonhomologous end-joining (NHEJ) pathway for the repair of dsDNA breaks generated by ionizing radiation and V(D)J recombination that occurs in developing lymphocytes (21). DNA-PK also acts as a serine/threonine protein kinase and phosphorylates a variety of protein substrates. However, the biological consequences of these activities are unclear (22, 23). To confirm that DNA-PK activity is affected by MeV infection, cell extracts were subjected to a DNA-PK assay, in which incorporation of 32P in DNA-PK-specific substrate peptides was measured. MeV infection markedly inhibited the activity of DNA-PK in 293SLAM cells (Fig. 4A).
FIG 4.
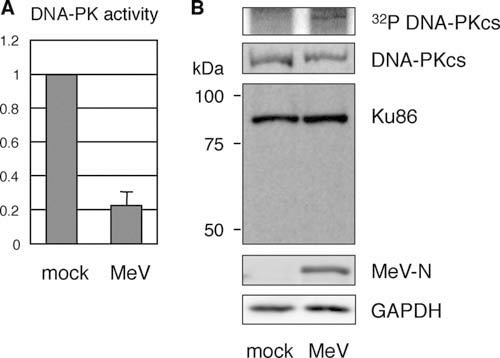
DNA-PK activity and modification of its subunits after MeV infection. (A) DNA-PK was partially purified with DNA-cellulose from cell lysates prepared from mock- or MeV-infected 293SLAM cells. Its kinase activity was assessed from the incorporation of 32P into peptide substrates. The results are expressed as values relative to the value of the mock-treated control sample. (B) Cells were labeled with 32PO4 after MeV infection for 24 h and subjected to immunoprecipitation with an anti-PKcs antibody (upper panel). Alternatively, the cell lysates were subjected to immunoblotting with anti-PKcs antibody, an anti-Ku86 monoclonal antibody that recognizes the N terminus of Ku86, anti-MeV-N antibody, and anti-GAPDH antibody.
Previous studies have shown that DNA-PK activity is regulated by its autophosphorylation (24). Several DNA-PK phosphorylation sites undergo autophosphorylation in vivo in response to DNA damage, and this event is crucial for correct NHEJ activity within a cell. In contrast, the autophosphorylation of the DNA-PK complex also results in the loss of its protein kinase activity (25). Therefore, we determined the phosphorylation status of DNA-PKcs after MeV infection by immunoprecipitation. In mock-infected cells, the 32P signal was below the level of detection, whereas phosphorylated DNA-PKcs was clearly detected after the MeV infection of the 293SLAM cells (Fig. 4B). No change in the amount of DNA-PKcs was detected after infection. This indicates that the phosphorylation of DNA-PKcs after MeV infection correlates with the reduction in DNA-PK activity.
The Ku86 subunit is the target of a cellular protease, and the degradation of Ku86 to a 69-kDa fragment to facilitate its removal from repaired DNA and the consequent inactivation of DNA-PK was previously shown in various cells (26). Therefore, we investigated whether Ku86 undergoes degradation during MeV infection, but this phenomenon was not detected (Fig. 4B). These results strongly suggest that the maintenance of the dephosphorylated state of DNA-PKcs is critical for its enzymatic activity, which in turn maintains the constitutive activity of Sp1.
Degradation of c-Myc by the inactivation of DNA-PK.
c-Myc is a well-studied oncogenic transcription factor that contributes to the genesis of many human cancers. In normal cells, c-Myc binds to the DNA consensus motif (CACGTG) known as the “Enhancer Box” (E box), and regulates the expression of a large number of genes involved in nucleotide metabolism, ribosome biogenesis, RNA processing, and DNA replication (27). Many of these genes were also identified in the downregulated gene cluster observed in our microarray analysis.
Interestingly, previous studies demonstrated that c-Myc interacts with DNA-PKcs (28) and that the inactivation of DNA-PKcs results in a marked reduction of c-Myc protein expression via the ubiquitin/proteasome pathway (29, 30). We examined whether MeV infection and the consequent DNA-PK inactivation also induce c-Myc degradation. As shown in Fig. 5A, c-Myc levels in 293SLAM cells were significantly reduced by MeV infection. To confirm the level of c-Myc activity after MeV infection, we used a reporter assay, with the E box in the promoter of the reporter construct. As expected, MeV infection significantly reduced the E-box-driven reporter activity (Fig. 5B), indicating that the reduction in c-Myc caused a reduction in transcription activity. To confirm that the reduction in c-Myc was dependent on the proteasomal pathway, MeV-infected cells were treated with the proteasome inhibitor MG132. The addition of MG132 restored c-Myc levels in a time-dependent manner (Fig. 5C), indicating that the reduction in c-Myc caused by MeV infection was mediated by the proteasomal pathway. We also confirmed that a DNA-PK inhibitor induced c-Myc degradation in 293SLAM cells, as reported previously (28) (Fig. 5D). These results indicate that DNA-PK inactivation, triggered by MeV infection, induces c-Myc degradation and the consequent comprehensive downregulation of housekeeping genes in 293SLAM cells, in combination with Sp1 inactivation.
FIG 5.
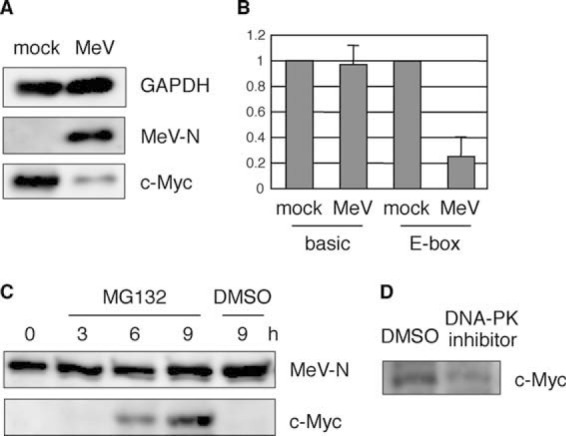
c-Myc degradation after MeV infection. (A) 293SLAM cells were infected with MeV for 24 h and then immunoblotted with anti-c-Myc, anti-MeV-N protein, and anti-GAPDH antibodies. (B) 293SLAM cells were transfected with a reporter plasmid, pGL3-basic or pGL3-Ebox, together with phRG-B as an internal reference. After 24 h, the cells were mock treated or infected with MeV for 24 h and then subjected to a dual-luciferase assay. The results are expressed as values relative to the value of the mock-treated control sample. (C) 293SLAM cells were infected with MeV for 24 h, treated with MG132 or the same volume of DMSO in fresh culture medium for the indicated times and then immunoblotted to detect c-Myc and MeV-N protein. (D) 293SLAM cells were treated with a DNA-PK inhibitor or the same volume of DMSO for 24 h and then immunoblotted to detect c-Myc.
Inactivation of PP5 by MeV infection.
Previous studies have shown that the inactivation of DNA-PK by autophosphorylation was regulated by a DNA-PK-associated PP2A-like protein phosphatase (31), and PP5 (32) and PP6 (33, 34) have been identified as binding partners of DNA-PKcs. These data suggest that a protein phosphatase regulates DNA-PKcs function in its active state. To determine the protein phosphatase activity associated with DNA-PK, cells were infected with MeV for 24 h and DNA-PKcs, immunoprecipitated, and the phosphatase activity in the immunocomplexes was determined. Figure 6A shows that the phosphatase activity correlated with the DNA-PK activity detected in Fig. 4A. To identify the corresponding phosphatase, PP2A, PP5, and PP6 were immunoprecipitated, and their activities were measured. The activities of PP2A and PP6 were negligibly affected by MeV infection, whereas the activity of PP5 was significantly reduced in MeV-infected 293SLAM cells under the same conditions (Fig. 6A). The level of PP5, determined by immunoblotting, did not change under these conditions (Fig. 6A).
FIG 6.

Inactivation of PP5 correlates with the suppression of DNA-PK activity. (A) 293SLAM cells were infected with MeV for 24 h and then immunoprecipitated with anti-DNA-PKcs, anti-PP2A, anti-PP5, or anti-PP6c antibody. Phosphatase activity in immunocomplexes was determined by an in vitro phosphatase assay. The results are expressed as values relative to the value of the mock-treated control sample (upper panel). The protein level of each protein was detected by immunoblotting (lower panel). (B) Knockdown of PP5 by RNA interference. 293SLAM cells were transfected with siRNA targeting PP5. For controls, cells were either mock treated or transfected with a nonspecific mismatched control siRNA (ctrl). After 48 h, the cells were lysed and subjected to immunoblotting or EMSA, or they were radiolabeled with 32PO4 and immunoprecipitated with anti-Sp1 antibody. (C) The cell lysates in panel B were subjected to immunoblotting with anti-c-Myc and anti-PP5 antibodies. (D) Total RNA prepared from cells transfected with ctrl siRNA or siRNA targeting PP5 described in panel B were then subjected to qPCR. The expression level in PP5 knock down cells was represented as a value relative to the that of ctrl siRNA-transfected cells. The specific primer pairs used are indicated by numbered columns as follows: 1, RNA18S5; 2, HIST1H2AC; 3, CASP3; 4, JAK1; 5, MAPK6; 6, IRF1; 7, NDUFB4; 8, COX7C; 9, RPL23; 10, LAMA4; 11, HSPA1L; and 12, ATP5G3. The official full names of these genes are described in the supplemental material.
To assess whether PP5 activity affected downstream Sp1 activity, we used specific siRNAs to knockdown PP5 expression in 293SLAM cells and performed radiolabeling with [32PO4] or EMSA. Sp1 showed reduced phosphorylation and binding activity (Fig. 6B). After PP5 knockdown, the intracellular c-Myc levels were also investigated with immunoblotting and, as expected, were significantly reduced (Fig. 6C).
To assess further the influence of PP5 knockdown on the expression of genes, we performed qPCR using specific primer pairs. PP5 knockdown had little effect on the expression of rRNA (RNA18S5) and nonhousekeeping genes, which showed no alteration or upregulation by MeV infection in the microarray analysis (Fig. 1A), including protein kinases (JAK1 and MAPK6), nuclear protein (HIST1H2AC), apoptosis (CASP3), and antiviral factor (IRF1) (Fig. 6D). In contrast, housekeeping genes, which were downregulated specifically in the microarray analysis and were characteristically categorized as electron transport system (NDUFB4 and COX7C), energy metabolism (ATP5G3), ribosomal protein (RPL23), cell structure (LAMA4), and chaperone protein (HSPA1L), showed a marked tendency to be suppressed by PP5 knockdown (Fig. 6D).
These results strongly suggest that PP5 maintains the dephosphorylation status of DNA-PKcs, which causes the constitutive phosphorylation of Sp1 and the levels of c-Myc protein in cells under unstimulated conditions and that MeV infection inactivates PP5, with the consequent downregulation of housekeeping gene expression in 293SLAM cells.
Accumulation of viral nucleocapsid inactivates PP5.
As seen by microarray analysis, a limited set of genes is upregulated in the early stage of infection in 293SLAM cells, and most are involved in antiviral responses, similar to those upregulated in IFN-α-treated cells (Fig. 1). In contrast, there was no significant change in the genes expressed in IFN-α-treated cells in the late stages of infection. These data strongly imply that the comprehensive downregulation of housekeeping genes is not a consequence of the usual cellular antiviral response but is attributable to the replication or accumulation of viral components. Therefore, we examined whether the inactivation of PP5 in 293SLAM cells was caused by the replication and accumulation of viral components. To do this, we tested the ability of viral nucleocapsid to inactivate PP5. To mimic viral genome replication and consequent intracellular accumulation of nucleocapsids, we used a minigenome assay (15). In brief, MeV minigenomic RNA with the MeV leader and trailer sequences flanking the luciferase gene was transfected into cells expressing MeV-N, -P, and -L proteins, and the replication of minigenomic RNA was evaluated as luciferase activity. As shown in Fig. 7A, when the minigenomic RNA was delivered alone, the PP5 activity was only slightly affected. Similarly, N, P and L expression showed no alteration of PP5 activity. However, progression of minigenomic RNA replication reduced the PP5 activity significantly (Fig. 7A). Under the same conditions, PP2A activity was not affected (Fig. 7B). To confirm whether reactions downstream from PP5 were affected by PP5 inactivation, we measured Sp1 activity with EMSA. N, P, and L expression induced no change, but the replication of minigenomic RNA caused a significant reduction in the binding activity of Sp1 (Fig. 7C). The intracellular c-Myc levels after minigenomic RNA replication were investigated by immunoblotting and, as expected, were significantly reduced (Fig. 7D).
FIG 7.

Accumulation of intracellular viral nucleocapsid inactivates PP5. (A) HEK293 cells were transfected with expression plasmids encoding MeV-N, -P, and -L genes (N, P, and L) or control empty plasmid, together with phRG-B as an internal reference. The following day, the cells were transfected with mock or negative-strand minigenomic RNA (minigenome). The cells were harvested at 24, 36, and 48 h after RNA transfection, and their lysates were subjected to luciferase activity analysis (upper panel), immunoblotting for MeV-N (middle panel), and an in vitro phosphatase assay with anti PP5 antibody (lower panel), as described above. (B) 293SLAM cells were transfected with N, P, and L expression plasmids and then transfected with mock or minigenomic RNA the following day. At 48 h after RNA transfection, cell lysates were immunoprecipitated with anti-PP5 or anti-PP2A antibody. Phosphatase activity in the immunocomplexes was determined as described above. (C) Nuclear extracts prepared from cells under the same conditions as panel B were subjected to EMSA. (D) The cell lysates from panel B and mock-treated cell lysates were immunoblotted to detect c-Myc, MeV-N, and GAPDH.
These results suggest that the usual rapid innate immune responses observed after infection are independent of the signal that downregulates the expression of housekeeping genes triggered by PP5 inactivation. The progression of viral RNA genome and resulting intracellular accumulation of viral nucleocapsid is required to induce this signal.
DISCUSSION
In the present study, we demonstrated that the intracellular accumulation of MeV nucleocapsid induced PP5 inactivation, which triggered the downregulation of housekeeping genes. PP5 is a multitasking regulator of the balance between phosphorylation and dephosphorylation. To date, PP5 has been identified in complexes containing many proteins that participate in signaling networks that initiate or regulate a variety of cellular events, including proliferation, migration, differentiation, electrolyte balance, apoptosis, and survival (35). This strongly implies that the inactivation of PP5 by MeV infection affects many other cellular signaling networks that were not identified in our microarray analysis, which contribute to the pathogenicity of MeV.
PP5 was first identified as a binding partner of DNA-PK (32), which contains many phosphorylation sites grouped into several clusters, that undergo complex autophosphorylation. Early studies demonstrated that autophosphorylation of purified DNA-PK resulted in kinase inactivation and the dissociation of DNA-PKcs from the DNA-end-bound Ku (25, 31). However, more recent work showed that the autophosphorylation of DNA-PKcs occurs at many sites and that phosphorylation at these different sites has distinct functional consequences (36–38). PP5 dephosphorylated threonine 2609 of DNA-PKcs (the phosphorylation of which is important for NHEJ progression) and serine 2056 (the autophosphorylation of which causes its inactivation) (32). In the present study, a marked phosphorylation of DNA-PKcs was detected after MeV infection, which correlated with a reduction in DNA-PK and PP5 activity (Fig. 4B). These results indicated that the kinase activity of DNA-PK is maintained by the continuous repression of its autophosphorylation by PP5.
The main function of DNA-PK is as a Ser/Thr protein kinase, and it has a key role in NHEJ, such as the repair of dsDNA breaks and V(D)J recombination. DNA-PK is also implicated in the phosphorylation of a range of other substrates, but its functions are poorly understood (23). In the present study, we demonstrated that DNA-PK induces the phosphorylation of Sp1 in cells under unstimulated conditions (Fig. 3). DNA-PK was first identified as a Sp1 kinase (39, 40), but the biological relevance of its phosphorylation was unclear. Our results indicate that the direct phosphorylation of Sp1 by DNA-PK is important for constitutive transcriptional activity.
Sp1 was originally identified as a transcription factor for the constitutive expression of thousands of genes, including housekeeping genes containing TATA-less GC-rich promoters. Sp1 undergoes various posttranslational modifications, and diverse cellular stimuli induce the phosphorylation of Sp1 by various kinases to alter its transcriptional activity both positively and negatively in a wide variety of physiological processes (20). Previous studies demonstrated that infection by several viruses, including simian virus 40 (39, 40), human immunodeficiency virus 1 (HIV-1) (41, 42), and herpes simplex virus 1 (43, 44), enhanced the phosphorylation of Sp1, but the signaling pathways that mediated this phosphorylation remain unclear. Interestingly, HIV-encoded Tat protein binds to Sp1 and DNA-PK in HIV-infected cells and these interactions enhance DNA-PK-mediated Sp1 phosphorylation and activate the HIV-1 promoter (41). These findings suggest that constitutive Sp1 phosphorylation and Tat-enhanced Sp1 phosphorylation have different consequences, although both are performed by DNA-PK.
We also demonstrated that PP5 inactivation and the consequent inactivation of DNA-PK induced c-Myc degradation. A number of recent genome-wide studies based on chromatin immunoprecipitation showed that c-Myc associates with a large fraction (>10 to 20%) of cellular genes in a variety of cell types (45–52). There have been few reports of c-Myc downregulation at the protein level induced by viral infection. Human papillomavirus oncoprotein E6-16 directly stimulates its ubiquitination resulting in the degradation of c-Myc (53). In the present study, we identified a novel mechanism whereby MeV infection induced c-Myc degradation via DNA-PK inactivation (Fig. 5).
Based on our results, we propose that the continuous dephosphorylation of DNA-PKcs by its associated PP5 induces the constitutive phosphorylation of Sp1 and the maintenance of c-Myc protein levels, both of which are crucial for the comprehensive expression of housekeeping genes in cells under nonstimulated steady-state conditions. We also showed that the accumulation of viral nucleocapsid triggered PP5 inactivation, reduced Sp1 activity, and caused the degradation of c-Myc (Fig. 8). These findings uncover a novel strategy of MeV during infection, which causes the collapse of host cellular functions.
FIG 8.
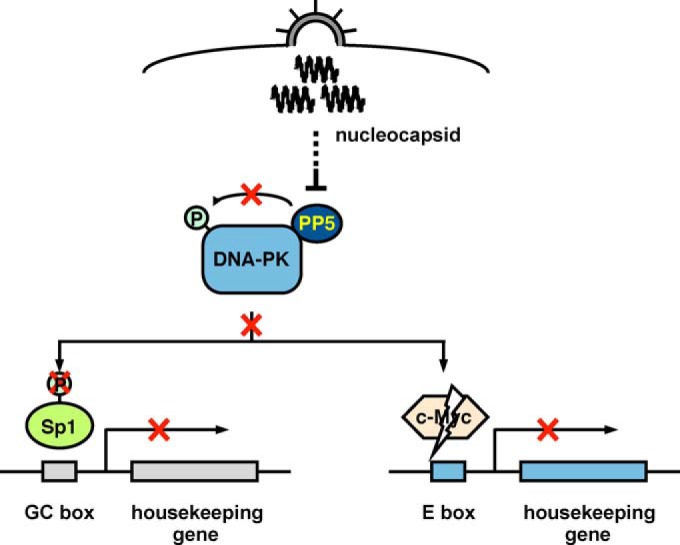
Model of the MeV-induced signal that triggers the comprehensive downregulation of cellular housekeeping gene expression. The intracellular accumulation of viral nucleocapsid causes the inactivation of PP5, autophosphorylation, and the resulting inactivation of DNA-PKcs and then the downstream reduction of Sp1 phosphorylation and degradation of c-Myc, both of which cause their inactivation and consequent downregulation of housekeeping genes in 293SLAM cells.
In contrast to 293SLAM cells, almost no up- or downregulated gene expression was observed in COBL-a cells (Fig. 1), strongly implying that signal transduction pathways induced by MeV infection are significantly suppressed in COBL-a cells. We have confirmed that the protein levels of PP5, DNA-PK and c-Myc in COBL-a cells are also comparable to those in 293SLAM cells (data not shown), suggesting that the cell-type-specific responses after MeV infection are caused by differences in posttranslational modifications. The mechanisms of this phenomenon and the related host factors have not been clarified to date. Further studies are required to identify the exact point in the downregulation signal at which MeV acts.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by Grants-in-Aid for Scientific Research (Kakenhi) from the Japan Society for the Promotion of Science, Japan, and a grant from the Program for the Promotion of Basic Research Activities for Innovative Biosciences (PROBRAIN), Japan.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00825-15.
REFERENCES
- 1.Griffin DE. 2008. Measles virus, p 1401–1441. In Knipe DM, Howley PM (ed), Fields virology, 4th ed Lippincott/The Williams & Wilkins Co, Philadelphia, PA. [Google Scholar]
- 2.Servant MJ, ten Oever B, LePage C, Conti L, Gessani S, Julkunen I, Lin R, Hiscott J. 2001. Identification of distinct signaling pathways leading to the phosphorylation of interferon regulatory factor 3. J Biol Chem 276:355–363. doi: 10.1074/jbc.M007790200. [DOI] [PubMed] [Google Scholar]
- 3.Helin E, Vainionpaa R, Hyypia T, Julkunen I, Matikainen S. 2001. Measles virus activates NF-κB and STAT transcription factors and production of IFN-α/β and IL-6 in the human lung epithelial cell line A549. Virology 290:1–10. doi: 10.1006/viro.2001.1174. [DOI] [PubMed] [Google Scholar]
- 4.tenOever BR, Servant MJ, Grandvaux N, Lin R, Hiscott J. 2002. Recognition of the measles virus nucleocapsid as a mechanism of IRF-3 activation. J Virol 76:3659–3669. doi: 10.1128/JVI.76.8.3659-3669.2002 (Erratum, J Virol 76:6413.). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sato H, Miura R, Kai C. 2005. Measles virus infection induces interleukin-8 release in human pulmonary epithelial cells: Comp Immunol Microbiol Infect Dis 28:311–320. [DOI] [PubMed] [Google Scholar]
- 6.Noe KH, Cenciarelli C, Moyer SA, Rota PA, Shin ML. 1999. Requirements for measles virus induction of RANTES chemokine in human astrocytoma-derived U373 cells. J Virol 73:3117–3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Naniche D, Yeh A, Eto D, Manchester M, Friedman RM, Oldstone MB. 2000. Evasion of host defenses by measles virus: wild-type measles virus infection interferes with induction of alpha/beta interferon production. J Virol 74:7478–7484. doi: 10.1128/JVI.74.16.7478-7484.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Obojes K, Andres O, Kim KS, Daubener W, Schneider-Schaulies J. 2005. Indoleamine 2,3-dioxygenase mediates cell type-specific anti-measles virus activity of gamma interferon. J Virol 79:7768–7776. doi: 10.1128/JVI.79.12.7768-7776.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Indoh T, Yokota S, Okabayashi T, Yokosawa N, Fujii N. 2007. Suppression of NF-κB and AP-1 activation in monocytic cells persistently infected with measles virus. Virology 361:294–303. doi: 10.1016/j.virol.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 10.Sato H, Honma R, Yoneda M, Miura R, Tsukiyama-Kohara K, Ikeda F, Seki T, Watanabe S, Kai C. 2008. Measles virus induces cell-type specific changes in gene expression. Virology 375:321–330. doi: 10.1016/j.virol.2008.02.015. [DOI] [PubMed] [Google Scholar]
- 11.Sugai A, Sato H, Yoneda M, Kai C. 2012. Phosphorylation of measles virus phosphoprotein at S86 and/or S151 downregulates viral transcriptional activity. FEBS Lett 586:3900–3907. doi: 10.1016/j.febslet.2012.09.021. [DOI] [PubMed] [Google Scholar]
- 12.Kobune F, Ami Y, Katayama M, Takahashi M, Tuul R, Korukluoglu G, Kiyohara T, Miura R, Sato H, Yoneda M, Kai C. 2007. A novel monolayer cell line derived from human umbilical cord blood cells shows high sensitivity to measles virus. J Gen Virol 88:1565–1567. doi: 10.1099/vir.0.82758-0. [DOI] [PubMed] [Google Scholar]
- 13.Yoneda M, Bandyopadhyay SK, Shiotani M, Fujita K, Nuntaprasert A, Miura R, Baron MD, Barrett T, Kai C. 2002. Rinderpest virus H protein: role in determining host range in rabbits. J Gen Virol 83:1457–1463. [DOI] [PubMed] [Google Scholar]
- 14.Yoneda M, Miura R, Barrett T, Tsukiyama-Kohara K, Kai C. 2004. Rinderpest virus phosphoprotein gene is a major determinant of species-specific pathogenicity. J Virol 78:6676–6681. doi: 10.1128/JVI.78.12.6676-6681.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hagiwara K, Sato H, Inoue Y, Watanabe A, Yoneda M, Ikeda F, Fujita K, Fukuda H, Takamura C, Kozuka-Hata H, Oyama M, Sugano S, Ohmi S, Kai C. 2008. Phosphorylation of measles virus nucleoprotein upregulates the transcriptional activity of minigenomic RNA. Proteomics 8:1871–1879. doi: 10.1002/pmic.200701051. [DOI] [PubMed] [Google Scholar]
- 16.Briggs MR, Kadonaga JT, Bell SP, Tjian R. 1986. Purification and biochemical characterization of the promoter-specific transcription factor, Sp1. Science 234:47–52. doi: 10.1126/science.3529394. [DOI] [PubMed] [Google Scholar]
- 17.Kadonaga JT, Tjian R. 1986. Affinity purification of sequence-specific DNA binding proteins. Proc Natl Acad Sci U S A 83:5889–5893. doi: 10.1073/pnas.83.16.5889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chu S, Ferro TJ. 2005. Sp1: regulation of gene expression by phosphorylation. Gene 348:1–11. doi: 10.1016/j.gene.2005.01.013. [DOI] [PubMed] [Google Scholar]
- 19.Tan NY, Khachigian LM. 2009. Sp1 phosphorylation and its regulation of gene transcription. Mol Cell Biol 29:2483–2488. doi: 10.1128/MCB.01828-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wierstra I. 2008. Sp1: emerging roles–beyond constitutive activation of TATA-less housekeeping genes. Biochem Biophys Res Commun 372:1–13. doi: 10.1016/j.bbrc.2008.03.074. [DOI] [PubMed] [Google Scholar]
- 21.Meek K, Gupta S, Ramsden DA, Lees-Miller SP. 2004. The DNA-dependent protein kinase: the director at the end. Immunological Rev 200:132–141. doi: 10.1111/j.0105-2896.2004.00162.x. [DOI] [PubMed] [Google Scholar]
- 22.Burma S, Chen DJ. 2004. Role of DNA-PK in the cellular response to DNA double-strand breaks. DNA Repair 3:909–918. doi: 10.1016/j.dnarep.2004.03.021. [DOI] [PubMed] [Google Scholar]
- 23.Collis SJ, DeWeese TL, Jeggo PA, Parker AR. 2005. The life and death of DNA-PK. Oncogene 24:949–961. doi: 10.1038/sj.onc.1208332. [DOI] [PubMed] [Google Scholar]
- 24.Meek T, Dang V, Lees-Miller SP. 2009. DNA-PK: the means to justify the ends? Adv Immunol 99:33–58. [DOI] [PubMed] [Google Scholar]
- 25.Chan DW, Lees-Miller SP. 1996. The DNA-dependent protein kinase is inactivated by autophosphorylation of the catalytic subunit. J Biol Chem 271:8936–8941. doi: 10.1074/jbc.271.15.8936. [DOI] [PubMed] [Google Scholar]
- 26.Gullo CA, Ge F, Cow G, Teoh G. 2008. Ku86 exists as both a full-length and a protease-sensitive natural variant in multiple myeloma cells. Cancer Cell Int 8:4. doi: 10.1186/1475-2867-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perna D, Faga G, Verrecchia A, Gorski MM, Barozzi I, Narang V, Khng J, Lim KC, Sung WK, Sanges R, Stupka E, Oskarsson T, Trumpp A, Wei CL, Muller H, Amati B. 2012. Genome-wide mapping of Myc binding and gene regulation in serum-stimulated fibroblasts. Oncogene 31:1695–1709. doi: 10.1038/onc.2011.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.An J, Xu QZ, Sui JL, Bai B, Zhou PK. 2005. Downregulation of c-myc protein by siRNA-mediated silencing of DNA-PKcs in HeLa cells. Int J Cancer 117:531–537. doi: 10.1002/ijc.21093. [DOI] [PubMed] [Google Scholar]
- 29.An J, Yang DY, Xu QZ, Zhang SM, Huo YY, Shang ZF, Wang Y, Wu DC, Zhou PK. 2008. DNA-dependent protein kinase catalytic subunit modulates the stability of c-Myc oncoprotein. Mol Cancer 7:32. doi: 10.1186/1476-4598-7-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yan YQ, Zhang B, Wang L, Xie YH, Peng T, Bai B, Zhou PK. 2007. Induction of apoptosis and autophagic cell death by the vanillin derivative 6-bromine-5-hydroxy-4-methoxybenzaldehyde is accompanied by the cleavage of DNA-PKcs and rapid destruction of c-Myc oncoprotein in HepG2 cells. Cancer Lett 252:280–289. doi: 10.1016/j.canlet.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 31.Douglas P, Moorhead GB, Ye R, Lees-Miller SP. 2001. Protein phosphatases regulate DNA-dependent protein kinase activity. J Biol Chem 276:18992–18998. doi: 10.1074/jbc.M011703200. [DOI] [PubMed] [Google Scholar]
- 32.Wechsler T, Chen BP, Harper R, Morotomi-Yano K, Huang BC, Meek K, Cleaver JE, Chen DJ, Wabl M. 2004. DNA-PKcs function regulated specifically by protein phosphatase 5. Proc Natl Acad Sci U S A 101:1247–1252. doi: 10.1073/pnas.0307765100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Douglas P, Zhong J, Ye R, Moorhead GB, Xu X, Lees-Miller SP. 2010. Protein phosphatase 6 interacts with the DNA-dependent protein kinase catalytic subunit and dephosphorylates γ-H2AX. Mol Cell Biol 30:1368–1381. doi: 10.1128/MCB.00741-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hosing AS, Valerie NC, Dziegielewski J, Brautigan DL, Larner JM. 2012. PP6 regulatory subunit R1 is bidentate anchor for targeting protein phosphatase-6 to DNA-dependent protein kinase. J Biol Chem 287:9230–9239. doi: 10.1074/jbc.M111.333708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Golden T, Swingle M, Honkanen RE. 2008. The role of serine/threonine protein phosphatase type 5 (PP5) in the regulation of stress-induced signaling networks and cancer. Cancer Metastasis Rev 27:169–178. doi: 10.1007/s10555-008-9125-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cui X, Yu Y, Gupta S, Cho YM, Lees-Miller SP, Meek K. 2005. Autophosphorylation of DNA-dependent protein kinase regulates DNA end processing and may also alter double-strand break repair pathway choice. Mol Cell Biol 25:10842–10852. doi: 10.1128/MCB.25.24.10842-10852.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Douglas P, Cui X, Block WD, Yu Y, Gupta S, Ding Q, Ye R, Morrice N, Lees-Miller SP, Meek K. 2007. The DNA-dependent protein kinase catalytic subunit is phosphorylated in vivo on threonine 3950, a highly conserved amino acid in the protein kinase domain. Mol Cell Biol 27:1581–1591. doi: 10.1128/MCB.01962-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meek K, Douglas P, Cui X, Ding Q, Lees-Miller SP. 2007. trans Autophosphorylation at DNA-dependent protein kinase's two major autophosphorylation site clusters facilitates end processing but not end joining. Mol Cell Biol 27:3881–3890. doi: 10.1128/MCB.02366-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jackson SP, MacDonald JJ, Lees-Miller S, Tjian R. 1990. GC box binding induces phosphorylation of Sp1 by a DNA-dependent protein kinase. Cell 63:155–165. doi: 10.1016/0092-8674(90)90296-Q. [DOI] [PubMed] [Google Scholar]
- 40.Saffer JD, Jackson SP, Thurston SJ. 1990. SV40 stimulates expression of the transacting factor Sp1 at the mRNA level. Genes Dev 4:659–666. doi: 10.1101/gad.4.4.659. [DOI] [PubMed] [Google Scholar]
- 41.Chun RF, Semmes OJ, Neuveut C, Jeang KT. 1998. Modulation of Sp1 phosphorylation by human immunodeficiency virus type 1 Tat. J Virol 72:2615–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jeang KT, Chun R, Lin NH, Gatignol A, Glabe CG, Fan H. 1993. In vitro and in vivo binding of human immunodeficiency virus type 1 Tat protein and Sp1 transcription factor. J Virol 67:6224–6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iwahori S, Shirata N, Kawaguchi Y, Weller SK, Sato Y, Kudoh A, Nakayama S, Isomura H, Tsurumi T. 2007. Enhanced phosphorylation of transcription factor sp1 in response to herpes simplex virus type 1 infection is dependent on the ataxia telangiectasia-mutated protein. J Virol 81:9653–9664. doi: 10.1128/JVI.00568-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim DB, DeLuca NA. 2002. Phosphorylation of transcription factor Sp1 during herpes simplex virus type 1 infection. J Virol 76:6473–6479. doi: 10.1128/JVI.76.13.6473-6479.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, Loh YH, Yeo HC, Yeo ZX, Narang V, Govindarajan KR, Leong B, Shahab A, Ruan Y, Bourque G, Sung WK, Clarke ND, Wei CL, Ng HH. 2008. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 133:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- 46.Eilers M, Eisenman RN. 2008. Myc's broad reach. Genes Dev 22:2755–2766. doi: 10.1101/gad.1712408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fernandez PC, Frank SR, Wang L, Schroeder M, Liu S, Greene J, Cocito A, Amati B. 2003. Genomic targets of the human c-Myc protein. Genes Dev 17:1115–1129. doi: 10.1101/gad.1067003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kidder BL, Yang J, Palmer S. 2008. Stat3 and c-Myc genome-wide promoter occupancy in embryonic stem cells. PLoS One 3:e3932. doi: 10.1371/journal.pone.0003932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Z, Van Calcar S, Qu C, Cavenee WK, Zhang MQ, Ren B. 2003. A global transcriptional regulatory role for c-Myc in Burkitt's lymphoma cells. Proc Natl Acad Sci U S A 100:8164–8169. doi: 10.1073/pnas.1332764100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim J, Chu J, Shen X, Wang J, Orkin SH. 2008. An extended transcriptional network for pluripotency of embryonic stem cells. Cell 132:1049–1061. doi: 10.1016/j.cell.2008.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Seitz V, Butzhammer P, Hirsch B, Hecht J, Gutgemann I, Ehlers A, Lenze D, Oker E, Sommerfeld A, von der Wall E, Konig C, Zinser C, Spang R, Hummel M. 2011. Deep sequencing of MYC DNA-binding sites in Burkitt lymphoma. PLoS One 6:e26837. doi: 10.1371/journal.pone.0026837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zeller KI, Zhao X, Lee CW, Chiu KP, Yao F, Yustein JT, Ooi HS, Orlov YL, Shahab A, Yong HC, Fu Y, Weng Z, Kuznetsov VA, Sung WK, Ruan Y, Dang CV, Wei CL. 2006. Global mapping of c-Myc binding sites and target gene networks in human B cells. Proc Natl Acad Sci U S A 103:17834–17839. doi: 10.1073/pnas.0604129103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gross-Mesilaty S, Reinstein E, Bercovich B, Tobias KE, Schwartz AL, Kahana C, Ciechanover A. 1998. Basal and human papillomavirus E6 oncoprotein-induced degradation of Myc proteins by the ubiquitin pathway. Proc Natl Acad Sci U S A 95:8058–8063. doi: 10.1073/pnas.95.14.8058. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.