

ABSTRACT
The herpesviral terminase complex is part of the intricate machinery that delivers a single viral genome into empty preformed capsids (encapsidation). The varicella-zoster virus (VZV) terminase components (pORF25, pORF30, and pORF45/42) have not been studied as extensively as those of herpes simplex virus 1 and human cytomegalovirus (HCMV). In this study, VZV bacterial artificial chromosomes (BACs) were generated with small (Δ30S), medium (Δ30M), and large (Δ30L) ORF30 internal deletions. In addition, we isolated recombinant viruses with specific alanine substitutions in the putative zinc finger motif (30-ZF3A) or in a conserved region (region IX) with predicted structural similarity to the human topoisomerase I core subdomains I and II (30-IXAla, 30-620A, and 30-622A). Recombinant viruses replicated in an ORF30-complementing cell line (ARPE30) but failed to replicate in noncomplementing ARPE19 and MeWo cells. Transmission electron microscopy of 30-IXAla-, 30-620A-, and 30-622A-infected ARPE19 cells revealed only empty VZV capsids. Southern analysis showed that cells infected with parental VZV (VZVLUC) or a repaired virus (30R) contained DNA termini, whereas cells infected with Δ30L, 30-IXAla, 30-620A, or 30-622A contained little or no processed viral DNA. These results demonstrated that pORF30, specifically amino acids 619 to 624 (region IX), was required for DNA encapsidation. A luciferase-based assay was employed to assess potential intermolecular complementation between the zinc finger domain and conserved region IX. Complementation between 30-ZF3A and 30-IXAla provided evidence that distinct pORF30 domains can function independently. The results suggest that pORF30 may exist as a multimer or participate in higher-order assemblies during viral DNA encapsidation.
IMPORTANCE Antivirals with novel mechanisms of action are sought as additional therapeutic options to treat human herpesvirus infections. Proteins involved in the viral DNA encapsidation process have become promising antiviral targets. For example, letermovir is a small-molecule drug targeting HCMV terminase that is currently in phase III clinical trials. It is important to define the structural and functional characteristics of proteins that make up viral terminase complexes to identify or design additional terminase-specific compounds. The VZV ORF30 mutants described in this study represent the first VZV terminase mutants reported to date. Targeted mutations confirmed the importance of a conserved zinc finger domain found in all herpesvirus ORF30 terminase homologs but also identified a novel, highly conserved region (region IX) essential for terminase function. Homology modeling suggested that the structure of region IX is present in all human herpesviruses and thus represents a potential structurally conserved antiviral target.
INTRODUCTION
During herpesvirus replication, large head-to-tail double-stranded DNA (dsDNA) concatemers accumulate in the infected-cell nucleus and are subsequently packaged into preformed viral capsids. The packaging process is complex and involves a group of proteins that recognize specific viral DNA sequences and simultaneously process and translocate unit-length genomes through the portal vertex of the procapsid (1). The protein complex that docks or interacts with the capsid portal protein consists of the heterotrimeric viral terminase. Terminases of DNA bacteriophages have been well described (2, 3), and those of several members of the Herpesviridae, including herpes simplex virus 1 (HSV-1) (4–14), human cytomegalovirus (HCMV) (15–21), Epstein-Barr virus (EBV) (22), and varicella-zoster virus (VZV) (23–25), are under investigation.
Members of the Alphaherpesvirinae, Betaherpesvirinae, and Gammaherpesvirinae share a core set of orthologous proteins required for DNA encapsidation. A heterotrimeric viral terminase complex consisting of three of these conserved core proteins was recently isolated from HSV-1-infected cells (4). The HSV-1 terminase consisted of a 1:1:1 equimolar complex of two different terminase protein subunits, pUL15 and pUL28, and a protein encoded by the UL33 gene family. The pUL33 component may act as an essential accessory protein involved in the formation and/or stability of the terminase complex (7). Homologous terminase components have also been identified for HCMV (pUL89, pUL56, and pUL51) (16–21, 26–28) and VZV (pORF45/42, pORF30, and pORF25) (23, 24). The pUL15, pUL89, and pORF45/42 homologs are encoded by spliced gene products (12, 24, 29) derived from two exons, and pUL51 and pORF25 are the HSV-1 pUL33 family homologs (5, 21, 23, 25, 30).
The functions of terminase complex components have been investigated via expression and purification of terminase proteins and/or analysis of herpesvirus mutants. For example, an essential role for HSV-1 pUL28 in viral replication was shown by use of temperature-sensitive and deletion mutants that were defective for cleavage and packaging of concatemeric viral DNA (31–33). Mutant UL28 viruses and/or coimmunoprecipitation experiments identified important functional regions of pUL28, including a potential zinc finger motif (4); interactions with other terminase components and viral capsids (6–11, 14, 34–37); and interaction with the viral portal protein (10, 38).
The VZV homolog, pORF30, has not been studied in the same detail. In transfected MeWo cells, pORF30 localized primarily to the cytoplasm, but in VZV-infected cells, nuclear localization was predominant (24). These results were consistent with those for the HSV-1 homolog, pUL28, which also localized to the cytoplasm of transfected cells and to the nuclei of HSV-1-infected cells (6, 10, 35). In agreement with studies on the HCMV pUL56 subunit (19), VZV pORF30 was shown to interact with the C terminus of the pORF45/42 terminase subunit (24). pORF30 was also shown to coprecipitate with pORF25, the UL33 gene family homolog, from virus-infected cells (23). Similarities between VZV pORF30 and HSV-1 pUL28 suggest that pORF30 may participate in a terminase complex analogous to that described for HSV-1.
Studies formally proving an essential role for the VZV terminase components have lagged behind those of HSV-1. Here we report the isolation of the first VZV ORF30 terminase mutants. VZV ORF30 was targeted for deletion to define its role in viral replication. Considering its homology to pUL28, pORF30 was predicted to be essential for DNA encapsidation. A human retinal pigmented epithelial cell line (ARPE19) stably expressing pORF30 (ARPE30) was isolated and used to complement a series of recombinant VZV ORF30 deletion and substitution constructs. A previously described VZV bacterial artificial chromosome (BAC) (39, 40) containing green fluorescent protein (GFP) and luciferase genes (VZVLUC) served as the backbone for deletion of ORF30 sequences (Δ30S, Δ30M, and Δ30L) via a galK selectable marker. A repaired Δ30L virus (30R) was isolated by replacing galK with the parental ORF30 gene. In addition, the Δ30 mutants were utilized to generate specific alanine substitutions in regions of pORF30 that are highly conserved in the Herpesviridae family. All recombinant ORF30 deletion and point mutant viruses were shown to be lethal, and pORF30 was shown to be essential for viral DNA cleavage and packaging. Allelic complementation by pORF30 mutants containing mutations in two different regions of ORF30 suggests that pORF30 forms higher-order structures with independently functioning domains.
MATERIALS AND METHODS
Cells and viruses.
Human retinal pigmented epithelial cells (ARPE19; ATCC CRL-2302), human melanoma cells (MeWo; ATCC HTB-65), and a stable ORF30 cell line (ARPE30) were maintained at 37°C in 5% CO2. ARPE19 and MeWo cells were grown in minimal essential medium (MEM) containing 5% and 10% fetal bovine serum, respectively, with 2 mM l-glutamine and antimicrobial supplements.
VZV stocks were prepared by resuspending trypsinized ARPE19 or ARPE30 infected-cell monolayers in 90% fetal bovine serum (FBS)–10% dimethyl sulfoxide (DMSO) and freezing them overnight at −80°C in a BioCision CoolCell unit (San Rafael, CA) prior to storage in liquid nitrogen vapor phase.
Isolation of a cell line stably expressing ORF30.
The ORF30 gene (2,313 bp) was cloned into the lentivirus vector pLenti-EF-1a-Rsv-Puro (GenTarget, San Diego, CA). ARPE19 cells were transduced with the ORF30 lentivirus in the presence of puromycin. Genomic DNA from the stable ARPE30 cell clone was validated via PCR with ORF30-specific primers (data not shown). ARPE30 cells were cultured as described above, with the addition of 0.625 μg/ml of puromycin (MP Biomedicals, LLC, Santa Ana, CA) for stable maintenance of lentivirus-ORF30 sequences (39).
Bioinformatics.
Terminase subunit sequences used for bioinformatic analyses were derived from HSV-1 (KOS) pUL28, HSV-2 (HG52) pUL28, VZV (Ellen) pORF30, EBV (B95-8) BALF3, HCMV (Towne) pUL56, human herpesvirus 6A (HHV-6A) (GS) pU40, HHV-6B (HST) pU40, HHV-7 (RK) pU40, and HHV-8 (GK18) pORF7. Primary amino acid sequences were submitted to the PHYRE2 server for structural homology modeling based on all currently submitted protein structures found in the Protein Data Bank (PDB) (41, 42). Clustal Omega was used to identify conserved regions of the primary amino acid sequences of human herpesvirus pORF30 homologs (43).
Recombineering of ORF30 deletion, ORF30 zinc finger, and ORF30 alanine substitution mutants.
All procedures using the VZVLUC BAC were performed as described previously (39, 40, 44), except that ORF30-specific primers were employed. Primer pairs (Table 1) were used to generate PCR products (Phusion DNA polymerase; New England BioLabs Inc., Ipswich, MA) containing homology arms to construct VZV BACs with the following three different internal ORF30 deletions: 1,918 bp (Δ30L), 1,637 bp (Δ30M), and 179 bp (Δ30S).
TABLE 1.
BACs, plasmids, primers, and strains used in this study
| Reagent | Description or purpose | Source, reference, or sequencea |
|---|---|---|
| BACs | ||
| VZVLUC | VZV pOKA with firefly luciferase and GFP | 40 |
| Δ30L | galK cassette replaced ORF30 bp 144–2063 in VZVLUC | This study |
| Δ30M | galK cassette replaced ORF30 bp 144–1782 in VZVLUC | This study |
| Δ30S | galK cassette replaced ORF30 bp 1080–1260 in VZVLUC | This study |
| 30R | Wild-type ORF30 replaced galK cassette in Δ30L BAC | This study |
| 30-620A | H-to-A mutation at ORF30 aa 620 | This study |
| 30-622A | K-to-A mutation at ORF30 aa 622 | This study |
| 30-IXAla | PHLKEE-to-AAAAAA mutation at ORF30 aa 619–624 | This study |
| 30-ZF3Ab | C-to-A, C-to-A, and H-to-A mutations at ORF30 aa 202, 225, and 227, respectively | This study |
| Plasmid | ||
| pgalK | Used to flank galK cassette with ORF30 homology arms | 44 |
| Bacterial strain | ||
| SW102 | Used to propagate/manipulate VZV BAC clones | 44 |
| Primersc | ||
| Δ30L-galK F | Used to generate ORF30 homology arm from bp 144 | GGGTGGCGTGTCGCTTTTTATATCGGTTAGCGGCTAACTGTTTGACAGTTcctgttgacaattaatcatcggca |
| Δ30L-galK R | Used to generate ORF30 homology arm from bp 2063 | CTTTAGGTTGAGACGTGCACCCGCGTGGATCCTTACCTAGACGGTCAACGtcagcactgtcctgctcctt |
| Δ30M-galK R | Used to generate ORF30 homology arm from bp 1782 | GTAAAAAATAAGGCGGTGTTAGGGGGTTGTGCAAAACGGTGTTCATCGTGtcagcactgtcctgctcctt |
| Δ30S-galK F | Used to generate ORF30 homology arm from bp 1080 | CACGCAGAACTTACAGCCGTAACGGTTGAGTTGGCGTTATTTGGAAAAACTcctgttgacaattaatcatcggca |
| Δ30S-galK R | Used to generate ORF30 homology arm from bp 1260 | TCATCCTCACACCCAACTCTTTCTAAAAGTTGGCGTAAGGCGGCTTCGTTtcagcactgtcctgctcctt |
| 30R F | Used to repair Δ30L and to make Ala mutations | ATGGAATTGGATATTAATCGAAC |
| 30R R | Used to repair Δ30L and to make Ala mutations | TTATGAAAACGCCGGGTCCGTTGAA |
| Δ30-620 F | Used with 30R R to generate H-to-A mutation at ORF30 aa 620 | CCGgcCTTAAAAGAGGAATTGGCAAAGTTTATG |
| Δ30-620 R | Used with 30R F to generate H-to-A mutation at ORF30 aa 620 | TTCCTCTTTTAAGgcCGGAAATAGGCCAACGTT |
| Δ30-622 F | Used with 30R R to generate K-to-A mutation at ORF30 aa 622 | CCGCACTTAgcAGAGGAATTGGCAAAGTTTATG |
| Δ30-622 R | Used with 30R F to generate K-to-A mutation at ORF30 aa 622 | TTCCTCTgcTAAGTGCGGAAATAGGCCAACGTT |
| Δ30-ALA F | Used with 30R R to generate ORF30 Ala substitutions at aa 619–624 | gctgccgcagcggctgccTTGGCAAAGTTTATG |
| Δ30-ALA R | Used with 30R F to generate ORF30 Ala substitutions at aa 619–624 | ggcagccgctgcggcagcAAATAGGCCAACGTT |
| 30-ZF F | Used with 30R R to generate C-to-A mutation at ORF30 aa 225 and H-to-A mutation at ORF30 aa 227 | AACCAAGGTGAGACCTTACATCGTAGATTATTAGGATGTATCgcCGATgcCGTTACT |
| 30-ZF Rb | Used with 30R F to generate C-to-A mutation at ORF30 aa 199 and C-to-A mutation at ORF30 aa 202 | GGTCTCACCTTGGTTAGCTGTTATACATAATTCTTCAAAAgcTATAGCAgcTGGATG |
| ARPE30 F | Used to validate ORF30 genomic integration | ATGGAATTGGATATTAATCG |
| ARPE30 R | Used to validate ORF30 genomic integration | TTATGAAAACGCCGGGTCCG |
| ORF29-3′ F | Used to identify genotypes present in complementation plaques | TTGCGTGTAGTCCTTACCCAT |
| ORF31-5′ R | Used to identify genotypes present in complementation plaques | GCTTGGAGAGACCGACACAA |
VZV sequences are shown in uppercase, VZV mutations in lowercase bold type, and galK sequences in lowercase lightface type.
30-ZF3A contains only 3 of the 4 targeted substitutions. The C-to-A substitution at position 199 was not observed in the sequence of the homology arm or the final BAC construct.
Primers are forward (F) or reverse (R) with respect to the genome map.
Δ30L was used as the backbone to generate a repaired virus, 30R, containing the intact full-length ORF30 gene. In addition, primer pairs encoding alanine substitutions within the zinc finger region or conserved region IX of ORF30 (Table 1) were used to synthesize overlapping DNA amplicons containing the mutations of interest. Equimolar amounts of purified PCR products were used in primer extension reaction mixtures with Q5 Hi Fidelity DNA polymerase (New England BioLabs Inc., Ipswich, MA) to obtain full-length mutant ORF30 genes for recombineering into Δ30L.
The ORF30 sequences of all VZV BAC DNA constructs were confirmed by DNA sequencing prior to transfection into ARPE19 or ARPE30 cells. Virus was reconstituted by transfecting 1 × 106 ARPE19 or ARPE30 cells with 10 μg of BAC DNA complexed with Lipofectamine LTX (Life Technologies, Carlsbad, CA). Plates were incubated for 5 days and observed for cytopathic effect and GFP-positive plaques.
DNA sequencing.
PCR products with nucleotide changes were cloned into pJet1.2 (Life Technologies, Carlsbad, CA) and sequenced (Eurofins WMG Operon, Louisville, KY) prior to recombineering. In addition, sequence analysis of recombinant VZV BAC DNA was performed to confirm the intended changes in ORF30 prior to transfection.
BAC DNA isolation and agarose gel electrophoresis.
BAC DNAs were isolated from Escherichia coli by using a NucleoBond Xtra BAC kit (Clontech Laboratories, Palo Alto, CA), digested with SapI, and analyzed in 0.65% agarose gels stained with ethidium bromide (EtBr) as described previously (39).
VZV replication kinetics.
Viral replication was determined via a previously described luciferase-based assay (39), with minor modifications. Three hundred PFU of cell-associated VZV stock was used to infect ∼3 × 105 ARPE19, ARPE30, or MeWo cells in 12-well monolayers in triplicate. Normalized data (based on 6-h relative luminescence units [RLU] readings to account for variations in initial inocula) were graphed as the averages ± standard deviations (SD) for triplicate samples.
DNA cleavage analysis.
Approximately 107 ARPE19 cells were infected with 1 × 104 PFU of VZVLUC, Δ30L, 30R, 30-IXAla, 30-620A, or 30-622A for 24 h. Infected-cell DNAs extracted at 24 h postinfection were digested with BamHI, separated in agarose gels, and subjected to Southern blotting with digoxigenin (DIG)-labeled probes specific for the short or long terminal repeats, as described previously (39).
TEM.
Transmission electron microscopy (TEM) was performed to confirm the phenotypes of selected ORF30 mutants. Samples were prepared and microscopy performed as described previously (39). Briefly, ARPE19 cell monolayers (∼106 cells) were infected with 104 PFU of VZV stock and harvested at 72 h postinfection. Samples were analyzed with a JEOL JEM-1230 transmission electron microscope (JEOL USA Inc., Peabody, MA) at 110 kV and imaged with an UltraScan 4000 charge-coupled device (CCD) camera (Gatan Inc., Pleasanton, CA).
Intermolecular complementation assay.
Coinfection with two different cell-free or cell-associated virus stocks was not efficient enough to perform complementation assays. This issue was overcome by preparing double mutant virus-infected cell stocks. ARPE30 cells in 100-mm tissue culture dishes were doubly infected with 1 × 104 PFU each of 30-IXAla and 30-ZF3A. Control stocks were prepared by infecting ARPE30 cells with 2 × 104 PFU of either 30-IXAla or 30-ZF3A. Infection mixtures were incubated at 37°C for 2 days, passaged 1:2, and incubated for an additional 2 days. Stocks were harvested and stored in liquid nitrogen as described above.
Twelve-well tissue culture plates were seeded with ARPE19 or ARPE30 cells to obtain 90% confluent monolayers (∼4 × 105 cells/well). Virus stocks were thawed, washed once in MEM, and resuspended at 1,000 PFU/ml in MEM-2% FBS. Approximately 1,000 PFU of each virus stock was plated in triplicate. Images of representative plaques were taken on day 3, using an Olympus IX51 inverted fluorescence microscope (Olympus America Inc., Center Valley, PA), an Olympus DP72 12.8-megapixel cooled digital color camera, and CellSens Entry 1.8 image acquisition software. On day 4, wells were washed with phosphate-buffered saline (PBS), lysed with 1× cell lysis buffer (Thermo Scientific-Pierce firefly luciferase glow assay kit; Life Technologies, Carlsbad, CA), and frozen at −80°C until assayed for luciferase activity.
On the day of infection, the titer of each virus stock was determined on ARPE30 cell monolayers to confirm that equal amounts of virus were added to the complementation assay wells.
Genotypic analysis of viral plaques.
ARPE19 monolayers were infected with dually ZF3A/IXAla-infected ARPE30 stocks as described for the complementation assay. Three well-isolated plaques were removed from the monolayer in 100 μl of PBS at 4 days postinfection. One hundred microliters of lysis buffer (10 mM Tris, pH 7.4, 10 mM EDTA, and 0.25% SDS) was added to each sample, followed by gentle mixing with an equal volume of chloroform. After centrifugation for 5 min at 16,000 × g, the upper layer was removed and sodium acetate (pH 5.2) added to 0.15 M. DNA was precipitated with 2 volumes of 100% ice-cold ethanol (EtOH) and pelleted at 16,000 × g for 15 min at 4°C. DNA pellets were washed with 70% EtOH, dried for 5 min, and resuspended in 20 μl of 10 mM Tris, pH 8.5. ORF30 products were amplified from each sample by using 1 μl of DNA as the template, Phusion DNA polymerase, and primers ORF29-3′F and ORF31-5′R (Table 1). Amplicons cloned into pJet1.2 were sequenced to identify VZVLUC or mutant ORF30 genotypes.
Statistical analysis.
P values for complementation data were calculated with GraphPad Prism 6.02 software (GraphPad Software, Inc., La Jolla, CA).
RESULTS
Relatedness of herpesvirus pORF30 terminase homologs.
The VZV pORF30 terminase subunit is well conserved with other Alphaherpesvirinae homologs, including pORF30 of cercopithecine herpesvirus 9 (simian varicella virus) (61%), pORF32 of equid herpesviruses 1 and 3 (∼50%), and pUL28 of HSV-1 and HSV-2 (∼47%). Aligning the pORF30 homologs of human alphaherpesviruses (HSV-1, HSV-2, and VZV) and betaherpesviruses (HCMV, HHV-6A, HHV-6B, and HHV-7) revealed 15 conserved regions (Fig. 1). Twelve of these overlapped those reported in a study on the HCMV homolog, pUL56 (15).
FIG 1.

Alignment of pORF30 terminase homologs of human alpha- and betaherpesviruses. Clustal Omega was used to align the pORF30 terminase subunit primary amino acid sequences for 3 human alphaherpesviruses (HSV-1, HSV-2, and VZV) and 4 human betaherpesviruses (HCMV, HHV-6A, HHV-6B, and HHV-7). The human gammaherpesviruses HHV-4 and HHV-8 were not included. Red sequences indicate the 12 conserved regions previously identified in HCMV pUL56 by Champier et al. (15). Green sequences represent the 15 conserved regions identified using the current alignment and are in close agreement with the previous analysis by Champier et al. (15). A highly conserved stretch of 11 amino acids found in the C terminus of region IX is indicated with x's. The conserved putative zinc finger motif, C-X2-C-X22-C-X-H (arrows mark the conserved cysteine/histidine residues), was found in region IV of all nine human herpesviruses. *, identical residues; colons, highly conserved residues; periods, conserved residues. Identity/conservation is indicated for alignment of the 3 alpha- and 4 betaherpesvirus pORF30 homologs; however, only the VZV and HCMV sequences are shown.
A conserved putative zinc finger motif, C-X2-C-X22-C-X-H, is present in pORF30 homologs of members of all three herpesvirus subfamilies (Fig. 1) (4, 15, 45). Mutagenesis of critical cysteine and/or histidine residues in HSV-1 (UL28) (4) and EBV (BALF3) (22) resulted in defective terminase activity. For HSV-1, recombinant viruses containing zinc finger amino acid substitutions were unable to effectively cleave and package viral DNA (4). We hypothesized that the analogous region in VZV pORF30 was essential for viral replication.
Relatedness of pORF30 and the human TopI core subdomains.
When the 770-amino-acid pORF30 sequence was submitted to the PHYRE2 protein fold recognition server, pORF30 residues 589 to 647 were modeled with >90.3% confidence to portions of human topoisomerase I (TopI) subdomains I and II (Fig. 2A). pORF30 region IX shared 24% identity and 61% similarity with sequences of the TopI core subdomains I and II. In addition, the longest continuous stretch of conserved amino acids in herpesvirus pORF30 homologs occurred in region IX (VZV pORF30 residues 617 to 627) (Fig. 1). Full-length sequences of the remaining pORF30 homologs were submitted to the PHYRE2 server. Sequences corresponding to region IX for all human herpesviruses were modeled with high confidence to TopI subdomains I and II (Fig. 2B). PHYRE2 did not model any other regions of pORF30 homologs to protein structures in the PDB.
FIG 2.
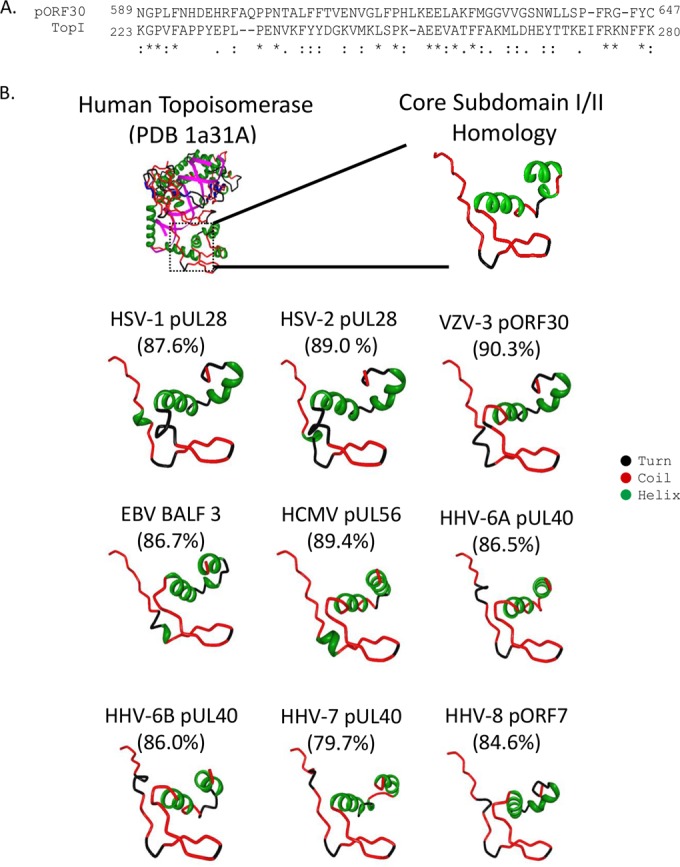
PHYRE2 modeling of pORF30 homologs. (A) The 770-amino-acid (full length) VZV strain Ellen pORF30 terminase sequence was submitted to the PHYRE2 Protein Fold Recognition Server (42). Sequences corresponding to region IX (pORF30 amino acid residues 589 to 647) contained structural homology to portions of human topoisomerase I (TopI) subdomains I and II (amino acids 223 to 280; includes the last 10 and first 48 amino acids of subdomains I and II, respectively [47]). The primary sequence alignment provided with the PHYRE2 model, showing amino acid identity (*) and similarity (periods and colons), is shown. (B) All nine human herpesvirus pORF30 homologs (HSV-1 pUL28, HSV-2 pUL28, VZV pORF30, EBV BALF3, HCMV pUL56, HHV-6A pUL40, HHV-6B pUL40, HHV-7 pUL40, and HHV-8 pORF7) were submitted to the PHYRE2 Protein Fold Recognition Server (42). Structural homology to portions of TopI subdomains I and II was identified. Models presented confidence levels between 79.7% (for HHV-7 pUL40) and 90.3% (for VZV pORF30). PHYRE2 did not model any other regions of pORF30 to known structures in the PDB. The structural features of each model are remarkably similar to each other and to the known structure of portions of human TopI domains I and II. The complete 765-amino-acid human TopI molecule associated with double-stranded DNA (fuchsia helix) is shown in the upper left corner (47) (PDB entry 1a31; human reconstituted DNA topoisomerase I in covalent complex with a 22-bp DNA duplex). Structural features of each model are coded as follows: green, helix; red, coil; and black, turn.
The results do not necessarily predict that pORF30 is a herpesvirus-encoded topoisomerase; however, the similarity to human TopI core subdomains (46, 47) suggests that pORF30 is likely to be involved in dsDNA “processing” (i.e., DNA clamping, cleavage, relaxation, and rotation). Regions or domains containing conserved sequences are likely to retain important functional or structural roles. Therefore, the zinc finger and the topoisomerase core-like domain (region IX) were selected for targeted mutagenesis studies.
Isolation of a stable cell line containing the VZV ORF30 gene.
As discussed earlier, HSV-1 pUL28 was shown to be essential for viral replication. Hence, the isolation of a stable ORF30 cell line was presumed to be necessary to complement VZV ORF30 mutant viruses. The 2,313-bp ORF30 gene was cloned downstream of the EF-1α promoter in a lentivirus expression vector. ARPE19 cells transduced with the ORF30 lentivirus were selected in the presence of puromycin. Genomic integration was confirmed by detection of a single PCR product, of ∼2.3 kb (data not shown), in a reaction mixture containing ARPE30 genomic DNA and ORF30-specific primers (Table 1). No gross morphological changes were noted for ARPE30 cells.
Construction of BAC ORF30 deletion mutants Δ30L, Δ30M, and Δ30S and BAC ORF30 zinc finger and region IX alanine substitution mutants via recombineering.
Analysis of the ORF30 region of the VZV genome sequence revealed that the 5′ coding region and promoter of VZV ORF31, an essential glycoprotein (gB) (48), overlap the 3′ coding region of ORF30 (Fig. 3A). The ARPE30 cell line might not complement a full-length deletion, because removal of the entire ORF30 coding region could affect the expression of gB. In addition, the 5′ region of ORF30 is in close proximity to another essential gene, the ORF29 gene, encoding a single-stranded DNA (ssDNA) binding protein (Fig. 3A) (49). Therefore, a strategy to generate small, medium, and large deletions within ORF30 was chosen to avoid unwanted effects on the flanking open reading frames (ORFs). Primer sets were used to prepare amplicons containing homology arms (Table 1) to generate 179-bp (Δ30S), 1,637-bp (Δ30M), and 1,918-bp (Δ30L) internal ORF30 deletions (Fig. 3B to D). A repaired BAC, BAC 30R, was isolated by recombining the wild-type ORF30 gene into BAC Δ30L (Fig. 3A). It was important to repair Δ30L because it provided the backbone for construction of the alanine substitution mutants.
FIG 3.
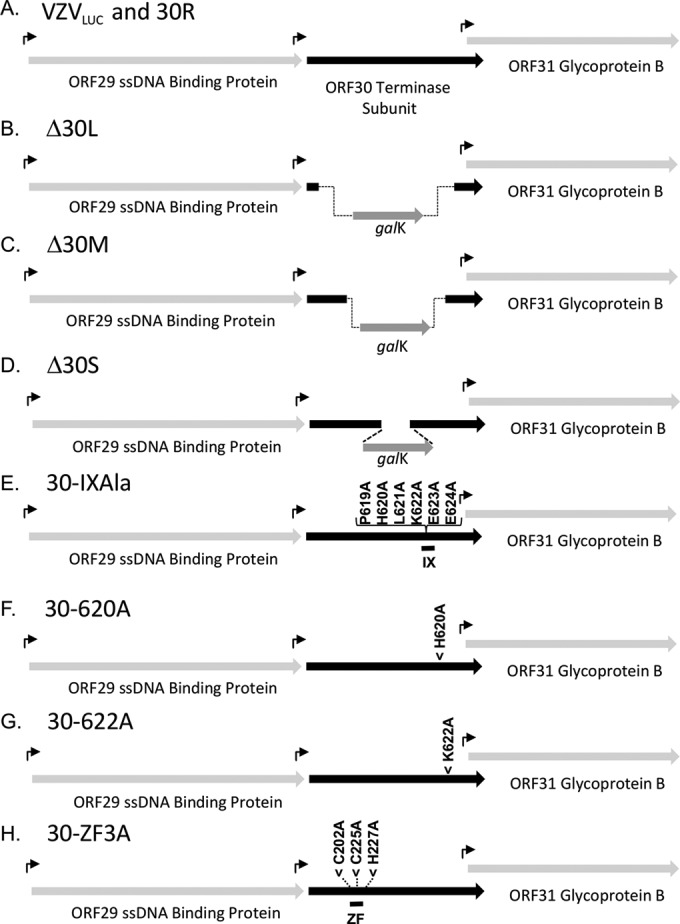
Predicted genome structures and features in the ORF30 region for parental and mutant viruses. (A) VZVLUC parental virus with shared coding regions for ORFs 30 and 31. The ORF31 promoter is predicted to fall within the 5′ coding sequence of the 2,310-bp ORF30 gene. (B) Δ30L (large ORF30 deletion) has an internal deletion of 1,918 bp. (C) Δ30M (medium ORF30 deletion) has an internal deletion of 1,337 bp. (D) Δ30S (small ORF30 deletion) has an internal deletion of 179 bp. (E to G) ORF30 sequences containing specific alanine substitutions in conserved terminase region IX. (H) ORF30 sequence containing three alanine substitutions in conserved terminase region IV, containing a putative zinc finger. The amino acid numbers are listed along with the amino acid changes. Promoters are indicated with arrows (galK promoter; for E. coli galactokinase).
Targeted mutagenesis of the ORF30 zinc finger motif and selected residues of the conserved stretch of amino acids in region IX was performed by using BAC Δ30L as the parent construct and recombining ORF30 fragments containing the indicated amino acid substitutions (Table 1; Fig. 3E to H).
The genomic structures of BACs VZVLUC, Δ30S, Δ30M, Δ30L, 30R, 30-IXAla, 30-620A, 30-622A, and 30-ZF3A were analyzed by restriction enzyme digestion with SapI (Fig. 4). The expected patterns were observed for BAC DNAs containing ORF30 deletions and reconstituted ORF30 (Fig. 4). Insertion of the galK gene, containing an internal SapI site, yielded the expected restriction fragments of 12.1 and 3.9 kb, 12.1 and 4.1 kb, and 13.0 and 4.7 kb for BAC Δ30L, BAC Δ30M, and BAC Δ30S, respectively. The genomic pattern for BAC 30R was identical to that for the parental virus VZVLUC. The expected patterns of DNA fragments were also observed for BACs digested with a second restriction enzyme, BssHII (data not shown).
FIG 4.

Analysis of VZV BAC genome structures. SapI restriction sites flanking the ORF30 gene and the resulting predicted fragment sizes are shown. Insertion of galK results in an additional SapI site. SapI-digested BAC DNAs were visualized after electrophoresis on a 0.65% agarose gel and EtBr staining. Mutants containing alanine substitutions are predicted to be identical to parental VZVLUC. All mutations were confirmed by sequence analysis. White arrows and dots mark the approximate locations of fragments in Δ30L, Δ30M, and Δ30S.
Replication of ORF30 mutants in ARPE19, ARPE30, and MeWo cells.
BAC DNAs were transfected into ARPE30 cells to prepare viral stocks. All three ORF30 deletion constructs were complemented in ARPE30 cells (data not shown). Mutant Δ30L was selected for further analysis and as the parent construct/virus for generation of zinc finger and region IX mutants. Using a previously described luciferase assay, the replication kinetics of VZVLUC, Δ30L, 30R, 30-IXAla, 30-620A, 30-622A, and 30-ZF3A were analyzed in the parental ARPE19, complementing ARPE30, and MeWo cell lines.
Monolayers were infected with 300 PFU of cell-associated virus stock and harvested at the indicated time points. VZVLUC and 30R replicated efficiently in ARPE19 (Fig. 5B), ARPE30 (Fig. 5C), and MeWo (Fig. 5A) cells. None of the ORF30 deletion or region IX alanine mutants formed plaques or replicated efficiently in ARPE19 (Fig. 5B) or MeWo (Fig. 5A) cells. Replication of Δ30L, 30-IXAla, 30-620A, and 30-622A in ARPE30 cells (Fig. 5C) suggested that providing pORF30 in trans complemented the loss of pORF30 function. However, none of the four mutants replicated to the same level as that of VZVLUC or 30R. There are several possibilities that might explain this observation: (i) expression of another VZV gene product was altered in the mutant viruses, (ii) pORF30 expression was not optimal for complete complementation in ARPE30 cells, or (iii) mutant pORF30s partially interfered with the function of wild-type pORF30 expressed in ARPE30 cells. The first explanation is unlikely, since single amino acid substitutions (i.e., in the 30-620A and 30-622A mutants) are not likely to affect expression of another VZV gene product. The fact that the deletion mutant and alanine substitution mutants replicated to approximately the same extent suggests that the second explanation is most plausible. However, since pORF30 is likely part of a terminase complex, mutant pORF30s could interfere with the normal function of ARPE30-expressed pORF30.
FIG 5.

Growth kinetics of region IX mutants. MeWo (A), ARPE19 (B), and ARPE30 (C) cell monolayers were infected in triplicate with VZVLUC, 30R, Δ30L, 30-IXAla, 30-620A, or 30-622A. Representative fields were photographed at 4 days postinfection. Cells were harvested in luciferase lysis buffer at the indicated time points. Firefly luciferase activity is shown in relative luminescence units (RLU). Each point is the average for three independent samples (the standard deviation is provided).
The replication kinetics of the zinc finger mutant 30-ZF3A were similar to those of the region IX mutants on noncomplementing cells. Monolayers were infected with 300 PFU of cell-associated virus stock and harvested at the indicated time points. VZVLUC and 30R replicated efficiently in ARPE19, ARPE30, and MeWo cells (Fig. 6). 30-ZF3A did not form plaques or replicate in ARPE19 or MeWo cells (Fig. 6). Replication of 30-ZF3A in ARPE30 cells (Fig. 6) suggested that providing pORF30 in trans complemented the loss of pORF30 function. These results are in agreement with previous studies showing that the zinc finger region of the HSV-1 homolog, pUL28, is essential for viral replication (4).
FIG 6.
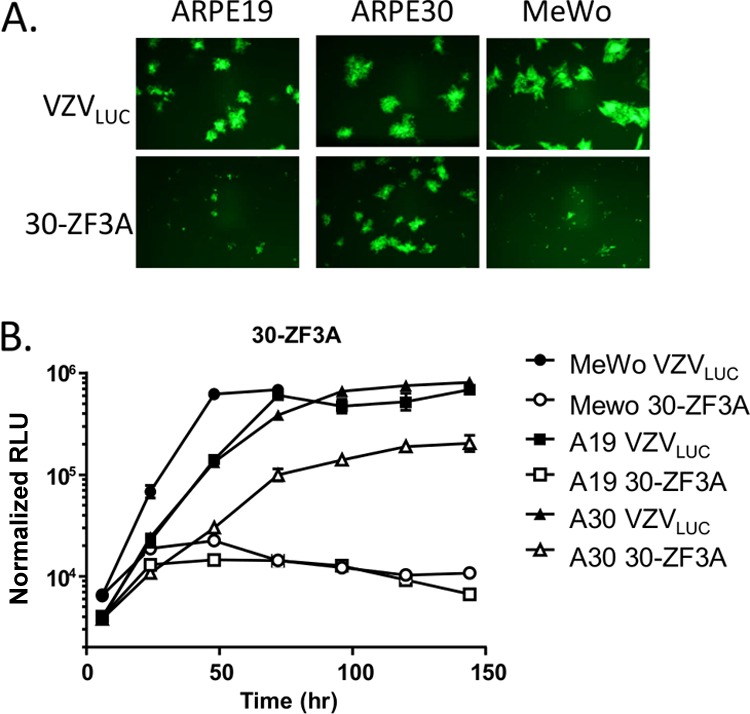
Growth kinetics of ORF30 zinc finger mutant 30-ZF3A. (A) ARPE19, ARPE30, and MeWo cells were infected with 30-ZF3A, and representative fields were photographed at 4 days postinfection. (B) Monolayers of ARPE19, ARPE30, and MeWo cells were infected in triplicate with VZVLUC or 30-ZF3A. Cells were harvested in luciferase lysis buffer at the indicated time points. Firefly luciferase activity is shown in RLU. Each point is the average for three independent samples (the standard deviation is provided).
Deletion of ORF30 and targeted mutations in region IX affect viral DNA processing in ARPE19 cells.
The absence of pORF30 or mutation of conserved functional or structural regions was hypothesized to result in defective DNA processing. ARPE19 cells were infected with VZVLUC, 30R, Δ30L, 30-IXAla, 30-620A, or 30-622A and harvested after 24 h. Total infected-cell DNA was digested with BamHI and analyzed via Southern blotting with a probe (TRL probe) specific for the VZV long terminal repeat region (Fig. 7A and B). The TRL probe detected a fragment of 0.9 kb representing viral DNA termini present only after cleavage of concatemeric viral DNA. The TRL probe also detected a 2.8-kb fragment representing uncleaved viral DNA. The 0.9-kb fragments observed for VZVLUC and the repaired virus, 30R, suggested that viral DNA processing occurred normally in infected ARPE19 cells. The amount of the 0.9-kb fragment was greatly reduced in ARPE19 cells infected with Δ30L, 30-IXAla, 30-620A, or 30-622A, suggesting that DNA processing was absent or inefficient.
FIG 7.
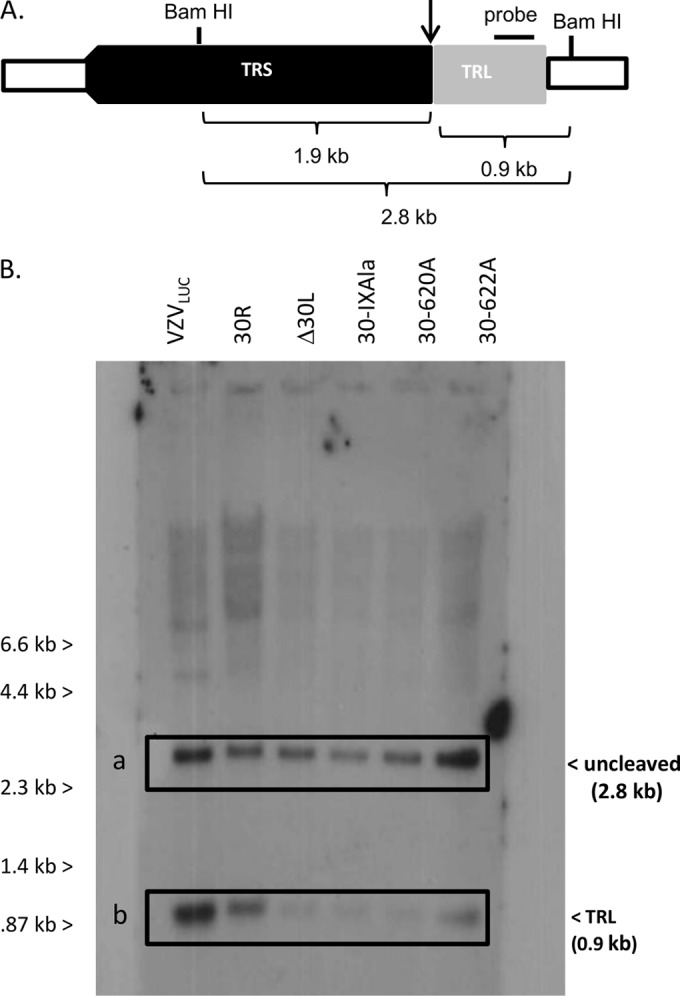
DNA cleavage assay. (A) Schematic of expected products from TRS-TRL junction, due to viral terminase cleavage (arrow), BamHI digestion, or both. (B) Monolayers of ARPE19 cells were infected with VZVLUC, 30R, Δ30L, 30-IXAla, 30-620A, or 30-622A. Cells were harvested at 24 h postinfection. Total infected cell DNA was digested with BamHI, fractionated by agarose gel electrophoresis, and analyzed by Southern blotting with a probe that specifically hybridized to the long terminal repeat (TRL). DNA fragments representing uncleaved DNA (2.8 kb) and the TRL fragment (0.9 kb) are indicated.
The results suggest that pORF30, and specifically the PHLKEE residues of region IX, are necessary for the efficient cleavage of viral genomic DNA. These results are consistent with those of previous studies showing that the HSV-1 homolog, pUL28, was required for proper viral DNA cleavage and packaging (33) and that a UL28 mutant with an in-frame linker insertion within region IX prevented DNA processing (7).
Absence of DNA-filled capsids in ORF30 deletion and region IX mutant-infected ARPE19 cells.
Viral DNA cleavage is tightly linked to genome packaging and requires an intact terminase complex to bind DNA, dock on the portal vertex, and deliver viral DNA into the empty preformed capsid. Therefore, ORF30 mutant-infected ARPE19 cells should contain only empty (unfilled) viral capsids if pORF30 is an integral component of the VZV terminase complex. Thin sections of 30R-, Δ30L-, 30-IXAla-, 30-620A-, and 30-622A-infected ARPE19 cells were examined by transmission electron microscopy. The repaired virus, 30R, was used as the positive control (Fig. 8). Three DNA-filled capsids with electron-dense staining cores could be seen in the same field. No DNA-filled capsids were observed in ARPE19 cells infected with Δ30L, 30-IXAla, 30-620A, or 30-622A (Fig. 8). Examination of multiple fields for each of the pORF30 mutants revealed crystalline-packed clusters of empty capsids (the 30-620A images provide a typical example).
FIG 8.
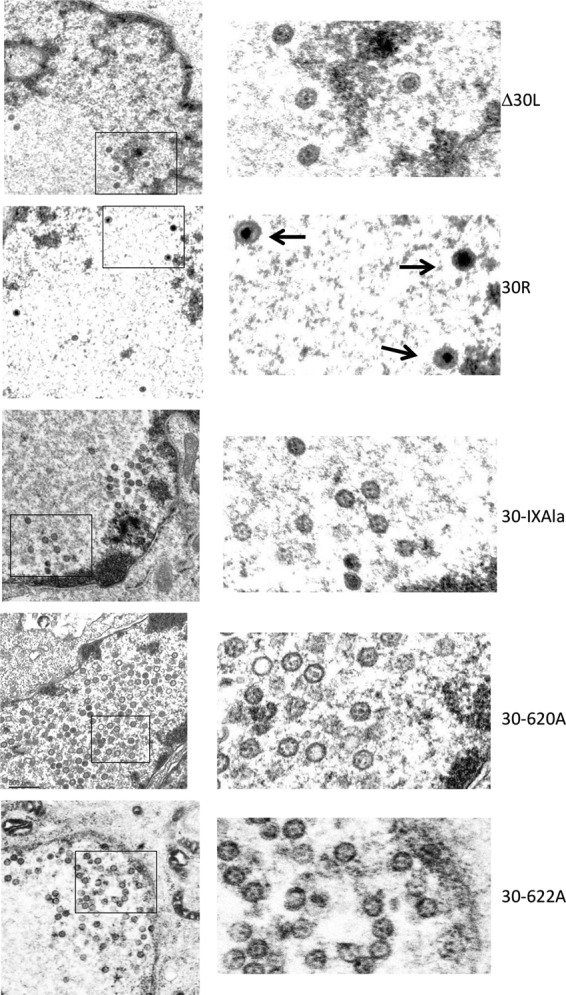
Electron microscopy of 30R, Δ30L, and region IX mutants. Monolayers of ARPE19 cells were infected with 30R-, Δ30L-, 30-IXAla-, 30-620A-, or 30-622A-infected cell stocks (multiplicity of infection of ∼0.001). Cells were harvested at 72 h postinfection, fixed, and examined via transmission electron microscopy. Boxed images in the left column are enlarged in the right column. Only 30R samples contained intranuclear DNA-filled capsids (dark arrows). All other capsids are unfilled and represent either A- or B-type capsids.
The results were consistent with previous studies showing that the HSV-1 and bovine herpesvirus 1 (BHV-1) homolog, pUL28, is required for DNA processing and packaging into preformed capsids (7, 31–33, 50).
Complementation assays define functionally distinct pORF30 domains.
The zinc finger and region IX sequences are approximately 400 amino acids apart with respect to the primary amino acid sequence. Their proximity in the pORF30 tertiary structure is not known, since the structures of pORF30 and pORF30 homologs have not been elucidated. The identification of 15 different conserved domains suggests that distinct regions of pORF30 may participate in different functions related to terminase activity, including DNA binding, DNA cleavage, and/or interaction with other encapsidation proteins. Figure 9A shows that when stocks of the 30-ZF3A/30-IXAla double mutant-infected ARPE30 cells were plated on ARPE19 cells, plaques similar to those of VZVLUC were observed. Only small or microplaques were seen for ARPE19 cells infected with individual 30-ZF3A or 30-IXAla mutant stocks. Single and double mutant stocks formed visible plaques in complementing ARPE30 cells, as expected. Luciferase activity in ARPE19 cells infected with the double mutant stock was significantly increased (∼10-fold; P < 0.002) compared to that in cells infected with the individual mutant stocks (Fig. 9B). The difference in luciferase activity between the individual mutant stocks was not significantly different (P > 0.8). The increased luciferase activity and identification of plaques in double mutant-infected ARPE19 cells suggested that intermolecular complementation occurred between defective ORF30 polypeptides and that pORF30 contains separate functional domains.
FIG 9.

Complementation assay. (A) Schematic representation depicting the association of wild-type and mutant heterotrimeric terminase complexes. Plaque phenotypes are shown for single mutant (30-IXAla or 30-ZF3A)- and double mutant (30-IX3A/30-ZF3A)-infected ARPE19 and ARPE30 cells (3 days postinfection). (B) Luciferase activities (RLU) of samples from single and double mutant stocks at 5 days postinfection. P values were calculated via one-way analysis of variance.
Analysis of viral plaques supports ORF30 allelic complementation.
The increased luciferase activity observed in the complementation assay could be due to reconstitution of ORF30 sequences (i.e., reversion to the parental genotype) that occurred during dual stock preparation. Therefore, PCR was performed on total DNAs extracted from three individual plaques derived from dual mutant virus-infected ARPE19 monolayers. Primers flanking the ORF30 gene (ORF29-3′F and ORF31-5′R) (Table 1) were used so that the resident ORF30 gene present in ARPE30 cells (inoculum) would not be amplified. Sequence analysis confirmed the presence of ORF30 mutant genotypes for 6/6 cloned amplicons (Table 2). ORF30 sequences were unchanged for DNA isolated from a VZVLUC plaque.
TABLE 2.
Genotypic analysis of plaques
| Plaque no. | PCR clone | Allele |
|
|---|---|---|---|
| 5′ end of ORF30 | 3′ end of ORF30 | ||
| 1 | A | ZF3A | No change |
| 2 | A | No change | IXAla |
| B | No change | IXAla | |
| 3 | A | ZF3A | No change |
| B | ZF3A | No change | |
| C | No change | IXAla | |
| VZVLUC | A | No change | No change |
Due to the cell-associated nature of the virus, cells transferred from a plaque resulting from complementation would be expected to form plaques on noncomplementing cells. Because we had shown that ORF30 mutant viruses replicated less efficiently that VZVLUC in complementing cells (Fig. 5C), it was unlikely that complementation would result in replication and plaque formation equal to those of VZVLUC. When individual plaques from the complementation assay were transferred to fresh ARPE19 monolayers, smaller, irregular plaques were formed (data not shown).
Based on the above-described evidence, coinfection with the 30-ZF3A and 30-IXAla mutant viruses resulted in true allelic complementation. These results were consistent with a previous study reporting allelic complementation between two HSV-1 pUL28 temperature-sensitive mutants containing lethal mutations in distant N- and C-terminal domains (32). A model is proposed in which a 1:1:1 heterotrimeric terminase complex participates in higher-order assemblies during DNA cleavage and packaging (Fig. 9A).
DISCUSSION
Nucleoside analogs that target the viral DNA polymerase have been the primary method for treating VZV, HSV, and HCMV infections (51). Although nucleoside analogs have proven effective, there are a number of problems associated with their use, including a limited antiviral spectrum and a requirement for treatment to begin early during the course of primary or recurrent infection (1). The limited number of approved drugs and the fact that they share the same antiviral target (DNA replication) are concerns with respect to drug-resistant infections. The development of resistance in immunosuppressed individuals has led to treatment failure for HSV-1 and -2, VZV, HCMV, and HHV-6 infections (51–55). Hence, there is a clinical need for new compounds directed against novel targets to treat both resistant infections and herpesviruses with limited therapeutic options (HCMV, EBV, and HHV-6, -7, and -8).
Previous research on pORF30 and the results presented here further support a role for pORF30 as a component of the VZV terminase complex. ORF30 deletion mutants replicated only in ORF30-complementing cell lines. In addition, deletion of ORF30 resulted in empty capsids in mutant-infected cells and in an absence of viral DNA processing. An ORF30 alanine substitution(s) in either the conserved zinc finger domain or pORF30 region IX was lethal, suggesting that these domains perform important enzymatic or structural functions. The zinc finger domain found in HSV pUL28 and EBV BALF3 was previously shown to be essential for viral DNA encapsidation and viral replication. The conserved region IX had not previously been investigated. The function of region IX remains unknown, but single alanine substitutions at H620 and K622 abrogated DNA cleavage and packaging. Region IX was modeled with high confidence to two subdomains that make up the human TopI core (46, 47). The precise functions of TopI subdomains I and II have not been determined. Studies suggest that subdomains I and II are not involved in DNA binding but instead transmit information related to DNA rotation and/or possible enzyme release (46). The related sequences in herpesvirus terminase homologs may transmit information about DNA rotation to terminase domains involved in binding and/or cleaving DNA during encapsidation. Additional studies on pORF30 may contribute to understanding the functions of herpesvirus and human DNA-metabolizing enzymes.
Bioinformatics revealed that the primary amino acid sequences of pORF30 homologs can be divided into 15 distinct, conserved regions. Complementation studies suggested that regions IV and IX function independently. It will be interesting to identify other terminase protein domains with separable enzymatic or structural functions. The results suggest that the pORF30 zinc finger and region IX domains are potential distinct antiviral targets. The other conserved pORF30 regions may serve as additional novel targets.
Viral proteins that play a role in DNA encapsidation have become promising drug targets. Small molecules that target the portal or terminase proteins of the DNA cleavage and packaging machinery of herpesviruses have been described (1, 39, 56–63). Several small-molecule compounds have been identified that specifically target HCMV pUL56 (the pORF30 homolog). 2-Bromo-5,6-dichloro-1-(β-d-ribofuranosyl) benzimidazole (BDCRB) and 2,5,6-trichloro-1-(β-d-ribofuranosyl) benzimidazole (TCRB) (benzimidazole ribonucleosides) inhibit HCMV replication by blocking viral DNA cleavage (45, 64). Letermovir (AIC246; C29H28F4N4O4) also targets pUL56 and is currently in clinical development (58, 62, 65). Letermovir has been used successfully to treat multidrug-resistant HCMV infections (66) and has been used prophylactically in hematopoietic cell (67) and kidney (68) transplantation recipients. These reports provide proof of principle that small-molecule inhibitors of terminase are viable antiviral options. A targeted screen against conserved domains found in terminase homologs could yield antiviral drug candidates active against one or more of the human herpesviruses.
ACKNOWLEDGMENTS
These studies were supported by a grant from the National Institutes of Health (grant 7R15AI062713-03) and by seed funding from the Mercer University School of Medicine.
We gratefully acknowledge the staff at Georgia Regents University for assistance with transmission electron microscopy and thank Hua Zhu, Rutgers New Jersey Medical School, for the parental VZVLUC BAC.
REFERENCES
- 1.Visalli RJ, van Zeijl M. 2003. DNA encapsidation as a target for anti-herpesvirus drug therapy. Antiviral Res 59:73–87. doi: 10.1016/S0166-3542(03)00108-6. [DOI] [PubMed] [Google Scholar]
- 2.Feiss M, Rao VB. 2012. The bacteriophage DNA packaging machine. Adv Exp Med Biol 726:489–509. doi: 10.1007/978-1-4614-0980-9_22. [DOI] [PubMed] [Google Scholar]
- 3.Rao VB, Feiss M. 2008. The bacteriophage DNA packaging motor. Annu Rev Genet 42:647–681. doi: 10.1146/annurev.genet.42.110807.091545. [DOI] [PubMed] [Google Scholar]
- 4.Heming JD, Huffman JB, Jones LM, Homa FL. 2014. Isolation and characterization of the herpes simplex virus 1 terminase complex. J Virol 88:225–236. doi: 10.1128/JVI.02632-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beilstein F, Higgs MR, Stow ND. 2009. Mutational analysis of the herpes simplex virus type 1 DNA packaging protein UL33. J Virol 83:8938–8945. doi: 10.1128/JVI.01048-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Higgs MR, Preston VG, Stow ND. 2008. The UL15 protein of herpes simplex virus type 1 is necessary for the localization of the UL28 and UL33 proteins to viral DNA replication centres. J Gen Virol 89:1709–1715. doi: 10.1099/vir.0.2008/000448-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jacobson JG, Yang K, Baines JD, Homa FL. 2006. Linker insertion mutations in the herpes simplex virus type 1 UL28 gene: effects on UL28 interaction with UL15 and UL33 and identification of a second-site mutation in the UL15 gene that suppresses a lethal UL28 mutation. J Virol 80:12312–12323. doi: 10.1128/JVI.01766-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sheaffer AK, Newcomb WW, Gao M, Yu D, Weller SK, Brown JC, Tenney DJ. 2001. Herpes simplex virus DNA cleavage and packaging proteins associate with the procapsid prior to its maturation. J Virol 75:687–698. doi: 10.1128/JVI.75.2.687-698.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang K, Baines JD. 2006. The putative terminase subunit of herpes simplex virus 1 encoded by UL28 is necessary and sufficient to mediate interaction between pUL15 and pUL33. J Virol 80:5733–5739. doi: 10.1128/JVI.00125-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang K, Homa F, Baines JD. 2007. Putative terminase subunits of herpes simplex virus 1 form a complex in the cytoplasm and interact with portal protein in the nucleus. J Virol 81:6419–6433. doi: 10.1128/JVI.00047-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu D, Weller SK. 1998. Herpes simplex virus type 1 cleavage and packaging proteins UL15 and UL28 are associated with B but not C capsids during packaging. J Virol 72:7428–7439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu D, Weller SK. 1998. Genetic analysis of the UL15 gene locus for the putative terminase of herpes simplex virus type 1. Virology 243:32–44. doi: 10.1006/viro.1998.9041. [DOI] [PubMed] [Google Scholar]
- 13.Selvarajan Sigamani S, Zhao H, Kamau YN, Baines JD, Tang L. 2013. The structure of the herpes simplex virus DNA-packaging terminase pUL15 nuclease domain suggests an evolutionary lineage among eukaryotic and prokaryotic viruses. J Virol 87:7140–7148. doi: 10.1128/JVI.00311-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beard PM, Taus NS, Baines JD. 2002. DNA cleavage and packaging proteins encoded by genes U(L)28, U(L)15, and U(L)33 of herpes simplex virus type 1 form a complex in infected cells. J Virol 76:4785–4791. doi: 10.1128/JVI.76.10.4785-4791.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Champier G, Couvreux A, Hantz S, Rametti A, Mazeron MC, Bouaziz S, Denis F, Alain S. 2008. Putative functional domains of human cytomegalovirus pUL56 involved in dimerization and benzimidazole d-ribonucleoside activity. Antivir Ther 13:643–654. [PubMed] [Google Scholar]
- 16.Giesen K, Radsak K, Bogner E. 2000. The potential terminase subunit of human cytomegalovirus, pUL56, is translocated into the nucleus by its own nuclear localization signal and interacts with importin alpha. J Gen Virol 81:2231–2244. [DOI] [PubMed] [Google Scholar]
- 17.Scheffczik H, Savva CG, Holzenburg A, Kolesnikova L, Bogner E. 2002. The terminase subunits pUL56 and pUL89 of human cytomegalovirus are DNA-metabolizing proteins with toroidal structure. Nucleic Acids Res 30:1695–1703. doi: 10.1093/nar/30.7.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scholz B, Rechter S, Drach JC, Townsend LB, Bogner E. 2003. Identification of the ATP-binding site in the terminase subunit pUL56 of human cytomegalovirus. Nucleic Acids Res 31:1426–1433. doi: 10.1093/nar/gkg229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thoma C, Borst E, Messerle M, Rieger M, Hwang JS, Bogner E. 2006. Identification of the interaction domain of the small terminase subunit pUL89 with the large subunit pUL56 of human cytomegalovirus. Biochemistry 45:8855–8863. doi: 10.1021/bi0600796. [DOI] [PubMed] [Google Scholar]
- 20.Wang JB, Zhu Y, McVoy MA, Parris DS. 2012. Changes in subcellular localization reveal interactions between human cytomegalovirus terminase subunits. Virol J 9:315. doi: 10.1186/1743-422X-9-315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Borst EM, Kleine-Albers J, Gabaev I, Babic M, Wagner K, Binz A, Degenhardt I, Kalesse M, Jonjic S, Bauerfeind R, Messerle M. 2013. The human cytomegalovirus UL51 protein is essential for viral genome cleavage-packaging and interacts with the terminase subunits pUL56 and pUL89. J Virol 87:1720–1732. doi: 10.1128/JVI.01955-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chiu SH, Wu MC, Wu CC, Chen YC, Lin SF, Hsu JT, Yang CS, Tsai CH, Takada K, Chen MR, Chen JY. 2014. Epstein-Barr virus BALF3 has nuclease activity and mediates mature virion production during the lytic cycle. J Virol 88:4962–4975. doi: 10.1128/JVI.00063-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Visalli RJ, Knepper J, Goshorn B, Vanover K, Burnside DM, Irven K, McGauley R, Visalli M. 2009. Characterization of the varicella-zoster virus ORF25 gene product: pORF25 interacts with multiple DNA encapsidation proteins. Virus Res 144:58–64. doi: 10.1016/j.virusres.2009.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Visalli RJ, Nicolosi DM, Irven KL, Goshorn B, Khan T, Visalli MA. 2007. The varicella-zoster virus DNA encapsidation genes: identification and characterization of the putative terminase subunits. Virus Res 129:200–211. doi: 10.1016/j.virusres.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vizoso Pinto MG, Pothineni VR, Haase R, Woidy M, Lotz-Havla AS, Gersting SW, Muntau AC, Haas J, Sommer M, Arvin AM, Baiker A. 2011. Varicella zoster virus ORF25 gene product: an essential hub protein linking encapsidation proteins and the nuclear egress complex. J Proteome Res 10:5374–5382. doi: 10.1021/pr200628s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bogner E, Radsak K, Stinski MF. 1998. The gene product of human cytomegalovirus open reading frame UL56 binds the pac motif and has specific nuclease activity. J Virol 72:2259–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bogner E, Reschke M, Reis B, Mockenhaupt T, Radsak K. 1993. Identification of the gene product encoded by ORF UL56 of the human cytomegalovirus genome. Virology 196:290–293. doi: 10.1006/viro.1993.1477. [DOI] [PubMed] [Google Scholar]
- 28.Giesen K, Radsak K, Bogner E. 2000. Targeting of the gene product encoded by ORF UL56 of human cytomegalovirus into viral replication centers. FEBS Lett 471:215–218. doi: 10.1016/S0014-5793(00)01407-1. [DOI] [PubMed] [Google Scholar]
- 29.Yu D, Sheaffer AK, Tenney DJ, Weller SK. 1997. Characterization of ICP6::lacZ insertion mutants of the UL15 gene of herpes simplex virus type 1 reveals the translation of two proteins. J Virol 71:2656–2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beard PM, Baines JD. 2004. The DNA cleavage and packaging protein encoded by the UL33 gene of herpes simplex virus 1 associates with capsids. Virology 324:475–482. doi: 10.1016/j.virol.2004.03.044. [DOI] [PubMed] [Google Scholar]
- 31.Addison C, Rixon FJ, Preston VG. 1990. Herpes simplex virus type 1 UL28 gene product is important for the formation of mature capsids. J Gen Virol 71:2377–2384. doi: 10.1099/0022-1317-71-10-2377. [DOI] [PubMed] [Google Scholar]
- 32.Cavalcoli JD, Baghian A, Homa FL, Kousoulas KG. 1993. Resolution of genotypic and phenotypic properties of herpes simplex virus type 1 temperature-sensitive mutant (KOS) tsZ47: evidence for allelic complementation in the UL28 gene. Virology 197:23–34. doi: 10.1006/viro.1993.1563. [DOI] [PubMed] [Google Scholar]
- 33.Tengelsen LA, Pederson NE, Shaver PR, Wathen MW, Homa FL. 1993. Herpes simplex virus type 1 DNA cleavage and encapsidation require the product of the UL28 gene: isolation and characterization of two UL28 deletion mutants. J Virol 67:3470–3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beard PM, Duffy C, Baines JD. 2004. Quantification of the DNA cleavage and packaging proteins U(L)15 and U(L)28 in A and B capsids of herpes simplex virus type 1. J Virol 78:1367–1374. doi: 10.1128/JVI.78.3.1367-1374.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koslowski KM, Shaver PR, Casey JT II, Wilson T, Yamanaka G, Sheaffer AK, Tenney DJ, Pederson NE. 1999. Physical and functional interactions between the herpes simplex virus UL15 and UL28 DNA cleavage and packaging proteins. J Virol 73:1704–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taus NS, Baines JD. 1998. Herpes simplex virus 1 DNA cleavage/packaging: the UL28 gene encodes a minor component of B capsids. Virology 252:443–449. doi: 10.1006/viro.1998.9475. [DOI] [PubMed] [Google Scholar]
- 37.Wills E, Scholtes L, Baines JD. 2006. Herpes simplex virus 1 DNA packaging proteins encoded by UL6, UL15, UL17, UL28, and UL33 are located on the external surface of the viral capsid. J Virol 80:10894–10899. doi: 10.1128/JVI.01364-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.White CA, Stow ND, Patel AH, Hughes M, Preston VG. 2003. Herpes simplex virus type 1 portal protein UL6 interacts with the putative terminase subunits UL15 and UL28. J Virol 77:6351–6358. doi: 10.1128/JVI.77.11.6351-6358.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Visalli MA, House BL, Selariu A, Zhu H, Visalli RJ. 2014. The varicella-zoster virus portal protein is essential for cleavage and packaging of viral DNA. J Virol 88:7973–7986. doi: 10.1128/JVI.00376-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Z, Rowe J, Wang W, Sommer M, Arvin A, Moffat J, Zhu H. 2007. Genetic analysis of varicella-zoster virus ORF0 to ORF4 by use of a novel luciferase bacterial artificial chromosome system. J Virol 81:9024–9033. doi: 10.1128/JVI.02666-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. 2000. The Protein Data Bank. Nucleic Acids Res 28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kelley LA, Sternberg MJ. 2009. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc 4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 43.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, Thompson JD, Higgins DG. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. 2005. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res 33:e36. doi: 10.1093/nar/gni035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krosky PM, Underwood MR, Turk SR, Feng KW, Jain RK, Ptak RG, Westerman AC, Biron KK, Townsend LB, Drach JC. 1998. Resistance of human cytomegalovirus to benzimidazole ribonucleosides maps to two open reading frames: UL89 and UL56. J Virol 72:4721–4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chillemi G, Fiorani P, Benedetti P, Desideri A. 2003. Protein concerted motions in the DNA-human topoisomerase I complex. Nucleic Acids Res 31:1525–1535. doi: 10.1093/nar/gkg242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Redinbo MR, Stewart L, Champoux JJ, Hol WG. 1999. Structural flexibility in human topoisomerase I revealed in multiple non-isomorphous crystal structures. J Mol Biol 292:685–696. doi: 10.1006/jmbi.1999.3065. [DOI] [PubMed] [Google Scholar]
- 48.Oliver SL, Sommer M, Zerboni L, Rajamani J, Grose C, Arvin AM. 2009. Mutagenesis of varicella-zoster virus glycoprotein B: putative fusion loop residues are essential for viral replication, and the furin cleavage motif contributes to pathogenesis in skin tissue in vivo. J Virol 83:7495–7506. doi: 10.1128/JVI.00400-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cohen JI, Krogmann T, Pesnicak L, Ali MA. 2007. Absence or overexpression of the varicella-zoster virus (VZV) ORF29 latency-associated protein impairs late gene expression and reduces VZV latency in a rodent model. J Virol 81:1586–1591. doi: 10.1128/JVI.01220-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Desloges N, Simard C. 2003. Role of the UL28 homologue of bovine herpesvirus 1 in viral DNA cleavage and packaging. Arch Virol 148:623–642. doi: 10.1007/s00705-002-0962-8. [DOI] [PubMed] [Google Scholar]
- 51.Agut H, Boutolleau D, Deback C, Bonnafous P, Gautheret-Dejean A. 2009. Testing the susceptibility of human herpesviruses to antivirals. Future Microbiol 4:1111–1123. doi: 10.2217/fmb.09.83. [DOI] [PubMed] [Google Scholar]
- 52.Piret J, Boivin G. 2011. Resistance of herpes simplex viruses to nucleoside analogues: mechanisms, prevalence, and management. Antimicrob Agents Chemother 55:459–472. doi: 10.1128/AAC.00615-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Piret J, Boivin G. 2014. Antiviral drug resistance in herpesviruses other than cytomegalovirus. Rev Med Virol 24:186–218. doi: 10.1002/rmv.1787. [DOI] [PubMed] [Google Scholar]
- 54.Komatsu TE, Pikis A, Naeger LK, Harrington PR. 2014. Resistance of human cytomegalovirus to ganciclovir/valganciclovir: a comprehensive review of putative resistance pathways. Antiviral Res 101:12–25. doi: 10.1016/j.antiviral.2013.10.011. [DOI] [PubMed] [Google Scholar]
- 55.Baldwin K. 2011. Ganciclovir-resistant human herpesvirus-6 encephalitis in a liver transplant patient: a case report. J Neurovirol 17:193–195. doi: 10.1007/s13365-011-0019-4. [DOI] [PubMed] [Google Scholar]
- 56.Visalli RJ, Fairhurst J, Srinivas S, Hu W, Feld B, DiGrandi M, Curran K, Ross A, Bloom JD, van Zeijl M, Jones TR, O'Connell J, Cohen JI. 2003. Identification of small molecule compounds that selectively inhibit varicella-zoster virus replication. J Virol 77:2349–2358. doi: 10.1128/JVI.77.4.2349-2358.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van Zeijl M, Fairhurst J, Jones TR, Vernon SK, Morin J, LaRocque J, Feld B, O'Hara B, Bloom JD, Johann SV. 2000. Novel class of thiourea compounds that inhibit herpes simplex virus type 1 DNA cleavage and encapsidation: resistance maps to the UL6 gene. J Virol 74:9054–9061. doi: 10.1128/JVI.74.19.9054-9061.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Goldner T, Hewlett G, Ettischer N, Ruebsamen-Schaeff H, Zimmermann H, Lischka P. 2011. The novel anticytomegalovirus compound AIC246 (letermovir) inhibits human cytomegalovirus replication through a specific antiviral mechanism that involves the viral terminase. J Virol 85:10884–10893. doi: 10.1128/JVI.05265-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hwang JS, Kregler O, Schilf R, Bannert N, Drach JC, Townsend LB, Bogner E. 2007. Identification of acetylated, tetrahalogenated benzimidazole d-ribonucleosides with enhanced activity against human cytomegalovirus. J Virol 81:11604–11611. doi: 10.1128/JVI.01130-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reefschlaeger J, Bender W, Hallenberger S, Weber O, Eckenberg P, Goldmann S, Haerter M, Buerger I, Trappe J, Herrington JA, Haebich D, Ruebsamen-Waigmann H. 2001. Novel non-nucleoside inhibitors of cytomegaloviruses (BAY 38-4766): in vitro and in vivo antiviral activity and mechanism of action. J Antimicrob Chemother 48:757–767. doi: 10.1093/jac/48.6.757. [DOI] [PubMed] [Google Scholar]
- 61.Underwood MR, Harvey RJ, Stanat SC, Hemphill ML, Miller T, Drach JC, Townsend LB, Biron KK. 1998. Inhibition of human cytomegalovirus DNA maturation by a benzimidazole ribonucleoside is mediated through the UL89 gene product. J Virol 72:717–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lischka P, Hewlett G, Wunberg T, Baumeister J, Paulsen D, Goldner T, Ruebsamen-Schaeff H, Zimmermann H. 2010. In vitro and in vivo activities of the novel anticytomegalovirus compound AIC246. Antimicrob Agents Chemother 54:1290–1297. doi: 10.1128/AAC.01596-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gable JE, Acker TM, Craik CS. 2 October 2014. Current and potential treatments for ubiquitous but neglected herpesvirus infections. Chem Rev doi: 10.1021/cr500255e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Komazin G, Townsend LB, Drach JC. 2004. Role of a mutation in human cytomegalovirus gene UL104 in resistance to benzimidazole ribonucleosides. J Virol 78:710–715. doi: 10.1128/JVI.78.2.710-715.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Verghese PS, Schleiss MR. 2013. Letermovir treatment of human cytomegalovirus infection antiinfective agent. Drugs Future 38:291–298. doi: 10.1358/dof.2013.038.05.1946425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kaul DR, Stoelben S, Cober E, Ojo T, Sandusky E, Lischka P, Zimmermann H, Rubsamen-Schaeff H. 2011. First report of successful treatment of multidrug-resistant cytomegalovirus disease with the novel anti-CMV compound AIC246. Am J Transplant 11:1079–1084. doi: 10.1111/j.1600-6143.2011.03530.x. [DOI] [PubMed] [Google Scholar]
- 67.Chemaly RF, Ullmann AJ, Stoelben S, Richard MP, Bornhauser M, Groth C, Einsele H, Silverman M, Mullane KM, Brown J, Nowak H, Kolling K, Stobernack HP, Lischka P, Zimmermann H, Rubsamen-Schaeff H, Champlin RE, Ehninger G, AIC246 Study Team . 2014. Letermovir for cytomegalovirus prophylaxis in hematopoietic-cell transplantation. N Engl J Med 370:1781–1789. doi: 10.1056/NEJMoa1309533. [DOI] [PubMed] [Google Scholar]
- 68.Stoelben S, Arns W, Renders L, Hummel J, Muhlfeld A, Stangl M, Fischereder M, Gwinner W, Suwelack B, Witzke O, Durr M, Beelen DW, Michel D, Lischka P, Zimmermann H, Rubsamen-Schaeff H, Budde K. 2014. Preemptive treatment of cytomegalovirus infection in kidney transplant recipients with letermovir: results of a phase 2a study. Transpl Int 27:77–86. doi: 10.1111/tri.12225. [DOI] [PubMed] [Google Scholar]