

ABSTRACT
The accessory HIV protein Vpu inhibits a number of cellular pathways that trigger host innate restriction mechanisms. HIV Vpu-mediated degradation of tetherin allows efficient particle release and hampers the activation of the NF-κB pathway thereby limiting the expression of proinflammatory genes. In addition, Vpu reduces cell surface expression of several cellular molecules such as newly synthesized CD4. However, the role of HIV Vpu in regulating the type 1 interferon response to viral infection by degradation of the interferon regulatory factor 3 (IRF3) has been subject of conflicting reports. We therefore systematically investigated the expression of IRF3 in primary CD4+ T cells and macrophages infected with HIV at different time points. In addition, we also tested the ability of Vpu to interfere with innate immune signaling pathways such as the NF-κB and the IRF3 pathways. We report here that HIV Vpu failed to degrade IRF3 in infected primary cells. Moreover, we observed that HIV NL4.3 Vpu had no effect on IRF3-dependent gene expression in reporter assays. On the other hand, HIV NL4.3 Vpu downmodulated NF-κB-dependent transcription. Mutation of two serines (positions 52 and 56) involved in the binding of NL4.3 Vpu to the βTrCP ubiquitin ligase abolishes its ability to inhibit NF-κB activity. Taken together, these results suggest that HIV Vpu regulates antiviral innate response in primary human cells by acting specifically on the NF-κB pathway.
IMPORTANCE HIV Vpu plays a pivotal role in enhancing HIV infection by counteraction of Tetherin. However, Vpu also regulates host response to HIV infection by hampering the type 1 interferon response. The molecular mechanism by which Vpu inhibits the interferon response is still controversial. Here we report that Vpu affects interferon expression by inhibiting NF-κB activity without affecting IRF3 levels or activity. These data suggest that Vpu facilitates HIV infection by regulating NF-κB transcription to levels sufficient for viral transcription while limiting cellular responses to infection.
INTRODUCTION
The success of the immediate innate immune response relies on the recognition of conserved pathogen structures, termed pathogen-associated molecular patterns (PAMPs; reviewed in reference 1). PAMPs induce intracellular signaling events, such as activation of the NF-κB and interferon (IFN) regulatory factor (IRF) pathways (reviewed in reference 2). The potent, but short-lived, activation of these innate response pathways triggers the induction of cytokines and interferons, which restrict replication of the pathogen (3). In addition, induction of the innate immune system is required for activation of long-lived adaptive immune responses (reviewed in reference 4). Many viruses have adapted to the presence of an innate immune system by specifically counteracting critical components of these pathways (reviewed in reference 5). Our understanding on how HIV efficiently evades immune recognition remains incomplete, despite recent findings describing how HIV can induce activation of the innate immune response in humans (reviewed in reference 6).
The accessory HIV protein Vpu antagonizes a number of different host restriction factors (reviewed in reference 7). It counteracts the inhibitory effect of Tetherin on particle release, but it also limits the expression of proinflammatory genes by hampering the activation of the NF-κB pathway (8–10). NF-κB inhibition is achieved by degradation of tetherin and sequestration of βTrCP (10–12). In addition, Vpu reduces the cell surface expression of several cellular molecules, such as the newly synthesized CD4, and the NK T cell and NK cell activating proteins, CD1d and NTB-A (13–15).
Reports on the cross talk between Vpu and interferon regulatory factor 3 (IRF3) have been conflicting (10, 16–18). Doehle et al. reported that HIV NL4.3 Vpu induces IRF3 degradation by a lysosome-dependent pathway, thus blocking type I interferon production in infected cells (16, 17). Recently, Park et al. reported that Vpu induces a caspase-dependent cleavage of IRF3 (19). In contrast, Hotter et al. did not observe any changes in IRF3 levels upon infection with either wild-type HIV (WT) or Δvpu HIV but confirm that Vpu hampers IFN-β expression (18). The authors show that Vpu has an inhibitory effect on the NF-κB pathway, which is important for IFN expression. These data suggest that Vpu mediated inhibition of IFN expression is due to the presence of NF-κB binding sites within the IFN-β promoter rather than IRF3 degradation (18). Because of these contradictory results, we decided therefore to investigate the extent to which HIV Vpu modulates IRF3 and NF-κB in the context of viral infection of human primary blood lymphocytes (PBLs), purified CD4+ T cells and human primary monocyte derived macrophages, as well as in established reporter model systems (20–22).
MATERIALS AND METHODS
Cell isolation and cell culture.
Human embryonic kidney 293T cells (HEK293T) and TZM-bl cells were cultured in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum (FBS) and penicillin-streptomycin. TZM-bl cells were obtained through the NIH AIDS Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Disease (NIAID), National Institutes of Health (NIH; (J. C. Kappes, Xiaoyun Wu, and Tranzyme, Inc.).
Human peripheral blood mononuclear cells (PBMCs) were obtained by using Ficoll (Ficoll Hystopaque; Sigma) density centrifugation from anonymous healthy blood donors (New York Blood Center). CD4+ T cells were negatively selected using magnetic beads (CD4+ T cell isolation kit II; Miltenyi Biotec) and cultured in Roswell Park Memorial Institute medium 1640 (RPMI) supplemented with 10% FBS, penicillin-streptomycin, 0.1 M HEPES, 2 mM l-glutamine, and interleukin-2 (IL-2) at 20 U/ml (NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH; human rIL-2 was obtained from M. Gately, Hoffmann-La Roche, Inc.) (23). Lymphocytes were activated with 1 μg of phytohemagglutinin-P (P; Sigma)/ml for 48 h.
CD14+ cells were isolated from PBMCs using a MACS CD14 isolation kit (Miltenyi Biotec). CD14+ cells were differentiated into macrophages by culturing them in RPMI supplemented with 2,000 U of human granulocyte-macrophage colony-stimulated factor (GM-CSF)/ml for 10 days. The GM-CSF containing medium was replenished at days 2, 5, and 8 as previously described (24).
Plasmids.
pHR-vpu-IRES-EGFP and the control pHR-IRES-EGFP were kindly provided by P. E. Klotman (25). The AU1-tagged Vpu alleles and the green fluorescent protein (GFP) control were kindly provided by F. Kirchhoff and D. Sauter (26). The Vpu alleles derived from primary isolates (subtype A A1.SE, subtype B WITO, subtype B CH106, subtype C ZM246F, and subtype C ZM247F1) and lab strain NL4.3 were cloned into the cytomegalovirus promoter-based pCG vector that coexpresses GFP via an internal ribosome entry site (IRES) (18).
The pCG-AU1 NL4.3 Vpu S52/S56N mutant was generated using the QuikChange II XL site-directed mutagenesis kit (Agilent Technologies) with the following two primers: forward, 5′-GAGCAGAAGACAATGGCAATGAGAATGAAGGAGAAGTATCAG-3′; and reverse, 5′-CTGATACTTCTCCTTCATTCTCATTGCCATTGTCTTCTGCTC-3′.
The IFN-β-luciferase reporter, the IRF3 responsive p55C1 promoter (p55C1-Luc), the nuclear factor κB (NF-κB) responsive synthetic promoter, and the c-fos gene NF-κB responsive promoter (c-fos-Luc) were previously described (20, 27, 28). The plasmids encoding RIG-I 2CARD, TBK1, and IKKβ constitutively active (IKKβca) were previously described (20, 21).
The replication-competent HIV molecular clone pLAI.2 was obtained from the AIDS Research and Reference Reagent Program (29). The pBR HIV-1 NL4-3 Nef+ IRES Renilla luciferase with or without vpu (HIV WT/R5 and HIV-Δvpu/R5), and the pBR HIV NL4.3 nef-IRES-Renilla Δenv with or without vpu (VSV-G HIV WT or VSV-G HIV-Δvpu) were modified from pBR HIV-1 NL4-3 nef-IRES GFP (30). HIV NL4.3 nef-IRES-GFP construct was kindly provided by B. K. Chen (31). HIV R7/3 nef-IRES-GFP was kindly provided by C. Cheng-Mayer (32). pSIV3+ (VLP Vpx) was previously described (33, 34). The pHCMV-G coding for the VSV-G envelope was used to pseudotype viruses lacking envelope (pBR HIV NL4.3 nef-IRES-Renilla Δenv WT and Δvpu) (35).
Production of viral stocks.
All of the viral stocks were generated by transfection of HEK293T cells using 3 μg of polyethyleneimine (Polysciences)/ml. The cell culture medium was replaced 12 h posttransfection (36), and after 2 days the transfection supernatants were collected, clarified by centrifugation, filtered (0.45-μm pore size), and stored at −80°C. The 50% tissue culture infective doses were determined on TZM-bl reporter cells as previously described (37–42).
Immunoblotting.
Cells were lysed in radioimmunoprecipitation assay buffer supplemented with complete protease inhibitor (Roche). Proteins were separated by SDS-PAGE (Invitrogen) and transferred to polyvinylidene difluoride membranes (Pierce). The following antibodies were used for detection: anti-IRF3 (Cell Signaling, catalog no. 4302), anti-GAPDH (Santa Cruz, catalog no. 32233), anti-p24 (m183 clone; NIH AIDS Research Program), anti-SAMHD1 (Cell Signaling, catalog no. 12361), anti-tubulin (Invitrogen), anti-GFP (Santa Cruz), and anti-AU (Covance). All antibodies were used at a 1:1,000 dilution. Western blot signals were visualized using SuperSignal West Pico or Femto (Pierce) on a Fluorochem E System protein simple machine. Densitometric analysis of Western blots using Alpha View software (Protein Simple).
Flow cytometry.
CD4+ T cells and macrophages were fixed and permeabilized using the Cytofix/Cytoperm kit (BD Biosciences), followed by staining with anti-p24 antibody KC57 RD1 (1:100 dilution; Coulter Clone) and anti-IRF3 antibody (1:100 dilution; Santa Cruz), followed by anti-rabbit Alexa 647 (1:500; Life Technologies). Gates were set by comparison with isotype-unrelated antibody control MslgG1-RD1 (1:100 dilution; Coulter Clone) and rabbit isotype-unrelated antibody control (Invitrogen). In the case of infection with GFP reporter viruses, only IRF3 intracellular staining was performed. HIV-positive cells were gated on the fluorescein isothiocyanate channel. An average of 5 × 104 cells were collected on a BD LSR II flow cytometer and analyzed using FlowJo.
HEK293T were fixed and permeabilized using the Cytofix/Cytoperm kit after a 15-min treatment with tumor necrosis factor alpha (TNF-α) at 10 ng/ml. The cells were stained with anti-IKBα antibody (1:50 dilution; Cell Signaling, catalog no. 9242), followed by anti-rabbit Alexa 647 antibody (1:500). The gates were set by comparison with a rabbit isotype-unrelated antibody control (Invitrogen). An average of 3 × 104 cells were collected on a BD LSR II flow cytometer and analyzed using FlowJo.
Luciferase reporter assay.
HEK293T cells were plated in 24-well plates and transfected with 100 ng of firefly luciferase reporter plasmids (IFN-β-Luc, p55C1-Luc, NF-κB-Luc, and c-fos-Luc) and 50 ng of p-RL-TK Renilla-luciferase plasmid as a control. The stimuli used included the RIG-I two CARD construct (RIG-I 2CARD, 10 to 50 ng) for IFN-β-Luc, the TANK-binding kinase 1 (TBK1, 10 ng) for p55C1-Luc, the constitutively active form of IκB kinase β (IKKβca, 50 ng), and 50 ng of TNF-α/ml for NF-κB-Luc and c-fos-Luc. Cells were also transfected with Vpu-coding plasmids and respective controls coding for GFP alone. The cells were lysed at 24 h posttransfection with passive lysis buffer (Promega). The firefly luciferase values were measured using a luciferase assay system (Promega), and the Renilla luciferase values were measured using a Renilla assay system (Promega). The values of firefly luciferase were normalized against the expression of a TK-Renilla luciferase vector, which is not responsive to IRF3 or NF-κB. The values were expressed as the fold induction over samples lacking stimuli (unstimulated was set as 1) and transfected with the IRES-EGFP coding plasmid. All experiments were performed in duplicate or triplicate.
Statistical analysis.
Statistical analysis was performed using GraphPad Prism 5 software. P values are two sided, and values of <0.05 were considered significant.
RESULTS
NL4.3 Vpu inhibits IFN-β promoter dependent gene expression.
Two different mechanisms have been proposed to explain how Vpu inhibits IFN-β production (16–18). Doehle et al. suggested that Vpu hampers IFN-β signaling by mediating IRF3 degradation (16, 17). Hotter et al. proposed that suppression of IFN-β is due to inhibition of the NF-κB signaling pathway (18).
Both Doehle and Hotter performed IFN-β promoter reporter assays using Sendai virus infection as stimuli. In order to avoid any possible interaction between proteins from Sendai Virus and host proteins or between Sendai Virus and HIV Vpu itself, we decided to perform IFN-β promoter reporter assays in the presence of the two CARD domains of retinoic acid inducible gene I (RIG-I), which is sufficient to induce IFN-β transcription (Fig. 1A) (22, 27, 43). In good agreement with published data using Sendai virus as a stimulus, the induction of the IFN-β promoter by RIG-I 2CARD was reduced by 2-fold when HIV NL4.3 Vpu protein was coexpressed (Fig. 1B shows the results of one representative experiment of three; P < 0.01 for 10 ng of RIG-I2 CARD and P < 0.001 for 50 ng of RIG-I 2CARD) (16, 18). Of note, we used two different amounts of plasmid expressing the RIG-I 2CARD construct to achieve induction of the IFN-β reporter. In both cases, we observed that overexpression of Vpu-IRES-EGFP, but not the IRES-EGFP coding plasmid, has a negative effect on luciferase expression.
FIG 1.
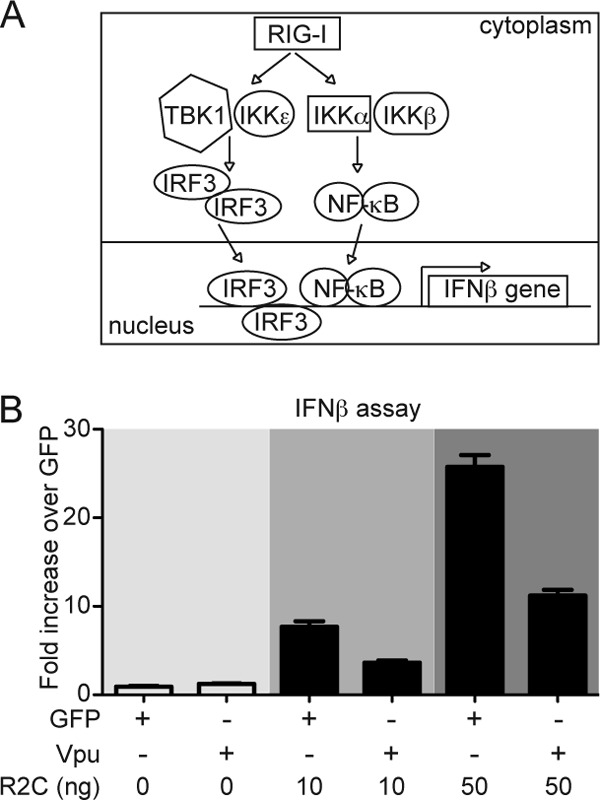
Vpu inhibits IFN-β promoter-dependent gene expression. (A) Schematic representation of the RIG-I pathway. (B) HEK293T cells were cotransfected with vectors expressing Vpu and GFP as a negative control in the presence or absence of the constitutively active RIG-I (2CARD) plasmid, together with IFN-β firefly and Renilla luciferase vectors. The induction of the IFN-β promoter was determined by a luciferase assay. Firefly luciferase values were normalized to Renilla luciferase values. The data were represented as the fold change over unstimulated GFP transfected cells (set as 1). All experiments were performed in duplicate or triplicate. The results of one representative experiment of three are shown.
HIV Vpu fails to degrade IRF3 in primary blood lymphocytes and CD4+ T cells.
We next investigated the possibility that Vpu induces the degradation of the transcription factor IRF3 in primary blood lymphocytes (PBLs) and in primary purified CD4+ T cells. We purified PBLs from five healthy donors and infected them with VSV-G-pseudotyped HIV NL4.3 nef-IRES Renilla luciferase (pBR HIV NL4.3 nef-IRES-Renilla Δenv, HIV WT) or with the Vpu deletion isogenic molecular clone (HIV-Δvpu) (30). The levels of IRF3, HIV p24, and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) were determined by Western blot analysis at 5 days postinfection. In all five donors, the levels of IRF3 were comparable between mock-infected cells and cells infected with HIV WT or HIV-Δvpu (Fig. 2A shows a representative Western blot and Fig. 2B depicts the normalized average IRF3 expression of the five independent experiments). The single cycle infection efficiency for both viruses was comparable in the different donors (Fig. 2C).
FIG 2.

HIV Vpu fails to degrade IRF3 in primary blood lymphocytes. (A) PBLs were infected with VSV-G HIV WT virus or VSV-G HIV-Δvpu virus. At 5 days postinfection, cellular lysates were probed for IRF3, p24, and GAPDH expression (the results shown are representative of five donors). (B) IRF3 levels were determined by densitometric analysis and normalized for GAPDH. The levels of IRF3 in uninfected cells were set as 1. Values are means plus the standard error of the mean (SEM) from five independent experiments. (C) The levels of infection of VSV-G WT HIV or VSV-G HIV-Δvpu virus in the different donors were assessed by a Renilla luciferase assay. Each symbol represents one donor. (D) Primary CD4+ T lymphocytes were activated for 48 h with phytohemagglutinin and infected with WT HIV/R5 virus or HIV-Δvpu/R5 virus. At 5 days postinfection, the cellular lysates were probed for IRF3, p24, and GAPDH expression (the results shown are representative of two donors). (E) IRF3 levels were determined by densitometric analysis and normalized for GAPDH. The levels of IRF3 in uninfected cells were set as 1. Values are means plus the SEM from two independent experiments. (F) Clarified supernatants were collected every 2 days and infectivity was determined by a TZM-bl infectivity assay.
To examine a possible role of Vpu in the context of a spreading HIV infection, primary CD4+ T lymphocytes from two healthy donors were infected with a CCR5-tropic HIV Renilla Luc virus (containing the V3 loop region of the CCR5-tropic HIV-1 92TH014-2 strain) with (HIV WT/R5) or without (HIV-Δvpu/R5) vpu (30, 44). The levels of IRF3, HIV p24 and GAPDH were determined by Western blot analysis at 5 days postinfection. We observed that the levels of IRF3 protein in mock-infected CD4+ T cells was similar to that of cells infected with HIV WT/R5 or with HIV-Δvpu/R5 (Fig. 2D shows the results of one representative experiment of two, and Fig. 2E illustrates the averages of the two independent experiments). Replication of both viruses was measured over 6 days by quantifying infectious particle release in the culture supernatants using a TZM-bl reporter assay (45). Of note, replication of HIV-Δvpu/R5 virus was slightly impaired compared to the WT virus, likely due to the reduced efficacy of the virus to counteract tetherin (Fig. 2F) (8, 9).
Since the Western blot analysis does not discriminate between infected and uninfected cells within a culture, we analyzed the IRF3 levels at the single CD4+ T cell level using flow cytometry analysis. Activated CD4+ T cells, purified from three healthy donors, were infected with replication-competent viruses: HIV NL4.3 nef-IRES-GFP, HIV R7/3 nef-IRES-GFP, and HIV LAI. At 5 days after infection, the cells were permeabilized and stained for IRF3 (18). The expression of the reporter GFP encoded by NL4.3 and R7/3 allowed us to identify productively infected cells. In the case of LAI, we discriminated infected cells from uninfected by intracellular p24 staining. We observed that intracellular IRF3 expression levels were independent of whether or not the cells were productively infected (in Fig. 3A, the left panels show isotype controls, the middle panels show the fluorescence-activated cell sorting profile of a representative experiment of three, and the right panels show the percentage of IRF3+ cells in the uninfected and infected populations in different donors).
FIG 3.
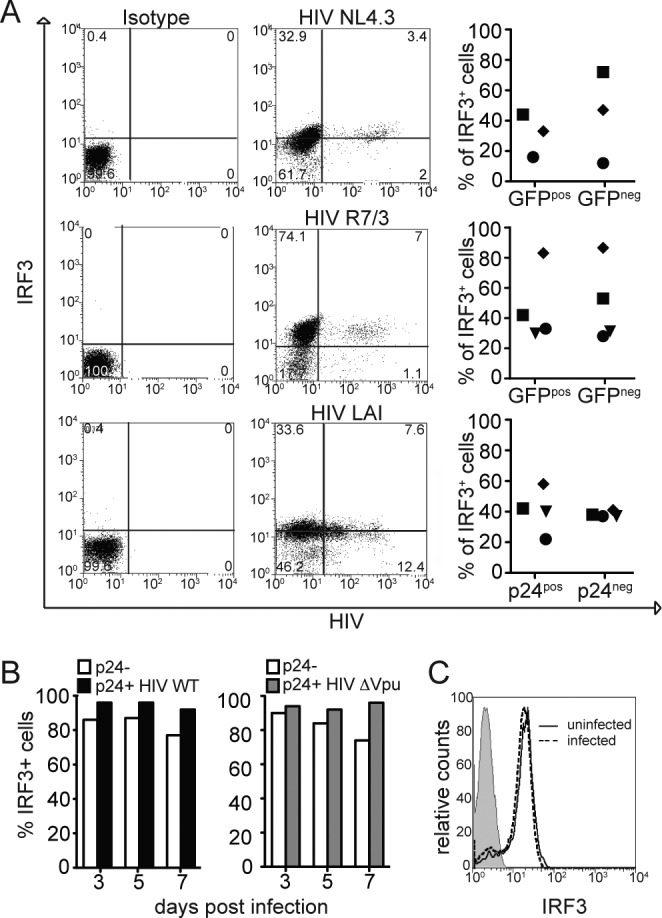
The levels of IRF3 are comparable in infected and uninfected primary CD4+ T lymphocytes. (A) Primary CD4+ T cells were infected with the indicated viruses, and the levels of infection were determined by GFP for NL4.3 and R7/3 expression or p24 intracellular staining for LAI. The levels of IRF3 were determined by intracellular staining (left panels, isotype control; the middle panel is representative of three independent experiments). IRF3 levels were analyzed by flow cytometry in both infected and uninfected populations in three different donors (right panels). (B) CD4+ T cells were infected with HIV WT or HIV-Δvpu viruses and stained for IRF3 and p24 at the indicated time points of infection. Histogram representing the percentage of IRF3+ CD4+ T cells (infected and uninfected) was prepared. (C) Intensity of IRF3 signal in HIV-infected CD4+ T cells and uninfected cells.
We next infected primary CD4+ T cells with HIV WT or with HIV-Δvpu, and followed the variation of IRF3 levels in infected cells for 7 days. We observed that the levels of IRF3 protein remained constant, similar not only between infected and uninfected cells but also between cells infected with HIV WT and HIV-Δvpu (Fig. 3B). Of note, the intensity of IRF3 signal was comparable between the infected population and the uninfected (Fig. 3C).
Taken together, these data suggest that the amount of IRF3 protein in the cells is not affected by an ongoing HIV infection. We used three different HIV strains, which all failed to downregulate IRF3 protein levels.
IRF3 protein levels are comparable in infected and uninfected primary macrophages.
We next tested the hypothesis that the effect of Vpu on IRF3 levels could be cell type specific. We infected primary human macrophages with VSV-G HIV WT-Renilla Luc (VSV-g HIV WT), and VSV-G HIV-Δvpu-Renilla Luc (VSV-g HIV-Δvpu). In agreement with our T cell infection experiments, we observed that the levels of IRF3 in macrophages infected with WT or vpu-deficient virus and uninfected cells remained comparable (Fig. 4A). Of note, we provided Vpx containing viral particles (Vpx-VLPs) in trans at the time of infection in order to increase the efficiency of infection (46–48). The levels of IRF3, HIV p24, SAMHD1, and tubulin were determined by Western blot analysis at 5 days postinfection. The expression of IRF3 remained comparable for all of the conditions even when the levels of infection were increased 8-fold by the addition of Vpx (Fig. 4A and B).
FIG 4.
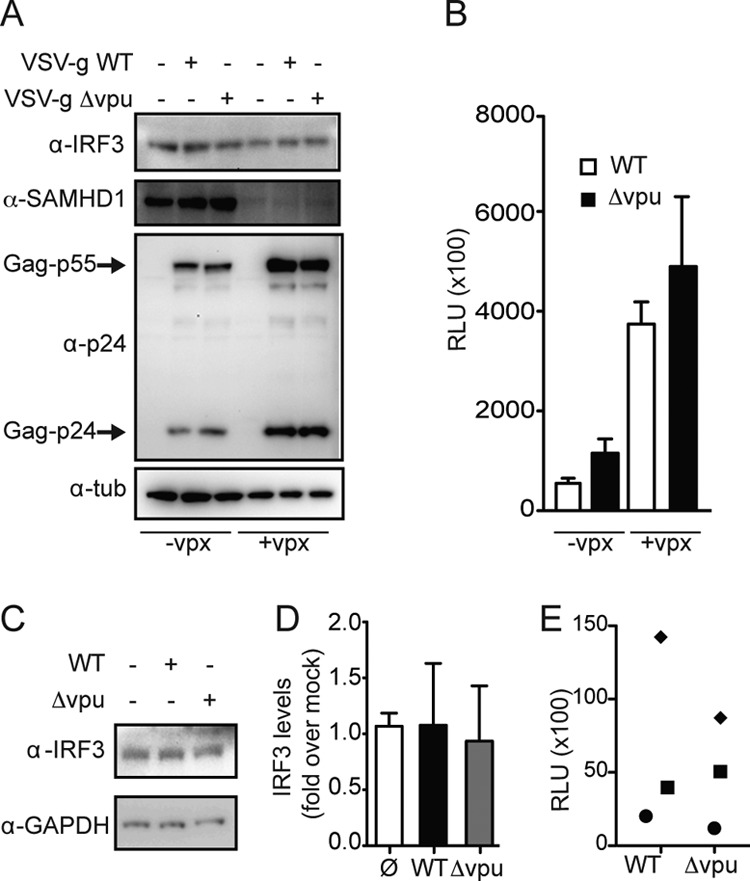
HIV infection has no impact on IRF3 protein levels in primary macrophages. (A) Primary human macrophages were infected with VSV-G WT HIV (HIV WT) virus or VSV-G HIV-Δvpu (HIV-Δvpu) virus in the presence absence of vpx VLPs. At 5 days postinfection, cellular lysates were probed for IRF3, SAMHD1 (Cell Signaling), p24, and tubulin expression. (B) The levels of infection of VSV-G WT HIV or VSV-G HIV-Δvpu virus in the presence or in the absence of vpx VLPs were assessed by Renilla luciferase assay. Values represent the means plus the standard deviations of relative luciferase units (RLU) from three technical replicates. (C) Primary human macrophages were infected with R5-WT HIV or R5-HIV-Δvpu virus. At 5 days postinfection, cellular lysates were probed for IRF3 and GAPDH expression (the results shown are representative of three independent experiments). (D) IRF3 levels were determined by densitometric analysis and normalized for GAPDH. The levels of IRF3 in uninfected cells were set as 1. Values are means plus the SEM from three independent experiments (lower panel). (E) The levels of infection of VSV-G WT HIV or VSV-G HIV-Δvpu in the different donors were determined by a Renilla luciferase assay.
We next determined the effect of Vpu in the context of a spreading HIV infection in human macrophages. Macrophages from three healthy donors were infected with HIV WT-Renilla Luc virus (WT) or with HIV-Δvpu/R5-Renilla Luc virus (Δvpu). Cell lysates were collected 5 days postinfection, and the levels of IRF3, p24, and GAPDH were determined by Western blotting. As shown in Fig. 4C, the levels of IRF3 protein were similar in macrophages infected with HIV WT or HIV-Δvpu or in uninfected macrophages (Fig. 4C shows a representative Western blot of three; Fig. 4D depicts the average of three independent experiments). The infection rates of the two viruses in three independent donors were comparable (Fig. 4E).
To better discriminate variation in IRF3 levels in infected versus uninfected macrophages, we measured intracellular IRF3 expression by flow cytometry. Primary human macrophages from two healthy donors were infected with VSV-G HIV WT in the presence Vpx-VLPs (46–48). At 5 days postinfection, the cells were permeabilized and stained for IRF3 and p24 in order to identify productively infected cells (Fig. 5A shows the results from a representative experiment) (49). We observed that the IRF3 levels were comparable between the p24-positive and the p24-negative cell populations in both donors (Fig. 5B, left panel). Of note, the percentage of tetherin-positive cells is lower in the infected population compared to the uninfected cells (Fig. 5B, right panel) (8). We next infected primary macrophages from different donors with VSV-G HIV WT or with VSV-G HIV-Δvpu and monitored the levels of IRF3 in the infected cells at 5 days postinfection. We found that the amount of IRF3 protein in cells infected with VSV-G HIV WT was comparable to the amount in cells infected with VSV-G HIV-Δvpu in all three donors (Fig. 5C). Similar results were obtained when macrophages were infected with replication-competent viruses (Fig. 5D).
FIG 5.

IRF3 protein levels are comparable in infected and uninfected primary macrophages. (A) Primary human macrophages were infected with VSV-G WT HIV in the presence of Vpx-VLPs. The levels of p24 and IRF3 expression were determined by intracellular staining at 5 days postinfection (representative plot). (B) Graph representing percentage of IRF3+ cells in p24neg and p24pos cells of two different donors (left panel). Graph representing percentage of tetherin+ cells in p24neg and p24pos cells of two different donors (right panel). (C) Primary human macrophages were infected with VSV-G WT HIV or VSV-G Δvpu in the presence of Vpx-VLPs. The levels of p24 and IRF3 expression were monitored 5 days postinfection (the left panel shows a representative plot, and the right panel shows the percentage of IRF3+ cells in VSV-G WT HIV- and VSV-G HIV-Δvpu-infected cells from three different donors). (D) Primary human macrophages were infected with replication-competent WT HIV or HIV-Δvpu in the presence of Vpx-VLPs. The levels of p24 and IRF3 expression were monitored 5 days postinfection (the left panel shows a representative plot, and the right panel shows the percentage of IRF3+ cells in WT HIV- and HIV-Δvpu-infected cells of three different donors). (E) Primary human macrophages were infected with VSV-G WT HIV (left panel) or VSV-G ΔVpu (right panel). The levels of p24 and IRF3 expression were monitored at the indicated days postinfection. (F) Primary human macrophages were infected with WT HIV or HIV-Δvpu. The levels of p24 and IRF3 expression were monitored in the indicated days postinfection.
It is conceivable that the effect of infection on IRF3 levels is only detectable during a particular point of infection. In order to test whether IRF3 degradation in infected macrophages was time dependent, primary macrophages infected with VSV-G HIV WT or with Vpu deleted were monitored for 5 days. We observed that the levels of IRF3 between uninfected and infected cell populations remained comparable over the observed period. This was true for both WT and Vpu deletion viruses (Fig. 5E). We performed a similar analysis in the context of a 7-day spreading HIV infection and observed that there were no significant changes in IRF3 levels in p24pos and p24neg macrophages (Fig. 5F).
In conclusion, endogenous IRF3 protein expression remained stable in primary human macrophages infected with HIV throughout the course of infection. In addition, the amount of IRF3 protein was unchanged in cells infected with HIV WT or Vpu deletion viruses.
NL4.3 Vpu inhibits NF-κB but not IRF3 signaling.
Since the IFN-β promoter contains binding sites for both IRF3 and NF-κB, we next investigated the effect of Vpu in the context of IRF3 or NF-κB specific transcription. We used specific reporter constructs that drive the expression of the firefly luciferase reporter gene under the control of IRF3 or NF-κB. Specifically, we tested to what extent NL4.3 Vpu inhibits activation of the IRF3 responsive p55C1 promoter (p55C1-Luc), the nuclear factor κB (NF-κB) responsive synthetic promoter, and the c-fos gene NF-κB responsive promoter (c-fos-Luc) (50, 51). An empty vector expressing only EGFP (GFP) was used as a control.
We expressed p55 C1-Luc, together with HIV NL4.3 Vpu or GFP alone, and induced specific IRF3-dependent transcription by cotransfecting a plasmid coding for TANK-binding kinase 1 (TBK1) (52). We observed that the levels of luciferase were comparable between cells transfected with Vpu and cells expressing GFP alone, suggesting that NL4-3 Vpu does not exert an inhibitory effect on the IRF3 expression under these experimental conditions (Fig. 6A).
FIG 6.

Vpu inhibits NF-κB but not IRF3 signaling. (A) HEK293T cells were cotransfected with vectors expressing Vpu and GFP as a negative control in the presence or absence of TBK1 plasmid, together with the p55 C1 promoter firefly luciferase and Renilla luciferase vectors. (B) IKKβca expression plasmid was used as a stimulus to activate synthetic NF-κB promoter when cotransfected with Vpu or GFP control plasmid (as described in panel A). (C) HEK293T cells were cotransfected with c-fos promoter firefly luciferase and Renilla luciferase constructs together with Vpu or GFP control vectors and stimulated by TNF-α at 24 h posttransfection. (D) The levels of IκBα were monitored by intracellular staining in cells expressing GFP or NL4.3 Vpu before and after TNF-α treatment. In all of the experiments, firefly luciferase values were normalized to Renilla luciferase values. The data were represented as the fold change over unstimulated GFP-transfected cells (set as 1). All experiments were performed in duplicate or triplicate. The results of one representative experiment of three are shown.
The effect of Vpu on the NF-κB pathway was determined using a synthetic NF-κB-Luc reporter plasmid. Activation of NF-κB was achieved by expressing the constitutively active form of IκB kinase β (IKKβca) (53). In line with previous reports, we detected a 5-fold reduction of the NF-κB promoter activation (P < 0.001) (Fig. 6B) (10, 12, 18). To confirm the reduction of NF-κB activation by Vpu, we cotransfected the c-fos-Luc reporter plasmid, together with Vpu or GFP, and stimulated the transfected cells 24 h later with TNF-α (20). Activation was measured 1 day after treatment and showed that Vpu reduced the NF-κB activation by 4-fold (P < 0.01) (Fig. 6C). These results suggest that HIV NL4.3 Vpu downmodulates NF-κB downstream IKKβ. Previous studies reported that Vpu could stabilize the nuclear factor of kappa light polypeptide gene enhancer in B cells inhibitor alpha (IκBα), thus inhibiting NF-κB activation (12, 54). Since TNF-α stimulation induces IκBα rapid degradation (55), we next compared the levels of IκBα in response to TNF-α in 293T expressing Vpu or GFP. After 15 min of TNF-α treatment, the levels of IκBα are reduced only in cells expressing GFP, whereas in cells expressing Vpu the IκBα remains stable (Fig. 6D). Taken together, these data indicate that Vpu downmodulates NF-κB activation by stabilizing its inhibitor IκBα but has no effect on IRF3 activation.
Serines in position 52 and 56 within NL4.3 Vpu are necessary for its inhibitory effect on NF-κB signaling.
Vpu downregulates the CD4 receptor from the cell surface and counteracts tetherin (8, 9, 13). In both cases, Vpu functions as an adapter bridging the ubiquitin-ligase βTrCP with its targets (56–58). This interaction has been proposed to be responsible for the inhibitory effect of Vpu on NF-κB signaling also, with a slightly different mechanism. In the case of CD4 and tetherin, Vpu links the target proteins to βTrCP inducing their degradation (57, 58). In the case of NF-κB, Vpu binds and sequesters βTrCP at the plasma membrane, hampering its ability to ubiquitinate IKBα upon external stimuli (12, 59). Lack of IKBα ubiquitination blocks its degradation and interferes with NF-κB nuclear translocation (60–62). Interaction between Vpu and βTrCP requires the phosphorylation of serines 52 and 56 in the cytoplasmic domain of Vpu (12).
To test the hypothesis that interaction of Vpu with βTrCP is important for the inhibitory effect of Vpu on NF-κB-dependent transcription, we performed reporter assays in the presence of the NL4.3 Vpu mutant S52N/S56N, which is expressed at the same levels of NL4.3 Vpu WT (Fig. 7A). We transfected the firefly luciferase under the control of the IFN-β promoter together with NL4.3 Vpu WT, NL4.3 Vpu S52N/S56N, or GFP coding plasmid. Activation of the IFN-β reporter construct was achieved by transfecting the RIG-I-2CARD construct. Induction of IFN-β promoter by RIG-I 2CARD was reduced by 2-fold in the presence of Vpu-WT (P < 0.01). On the other hand, expression of Vpu-S52N/S56N had no effect on luciferase expression (Fig. 7B).
FIG 7.
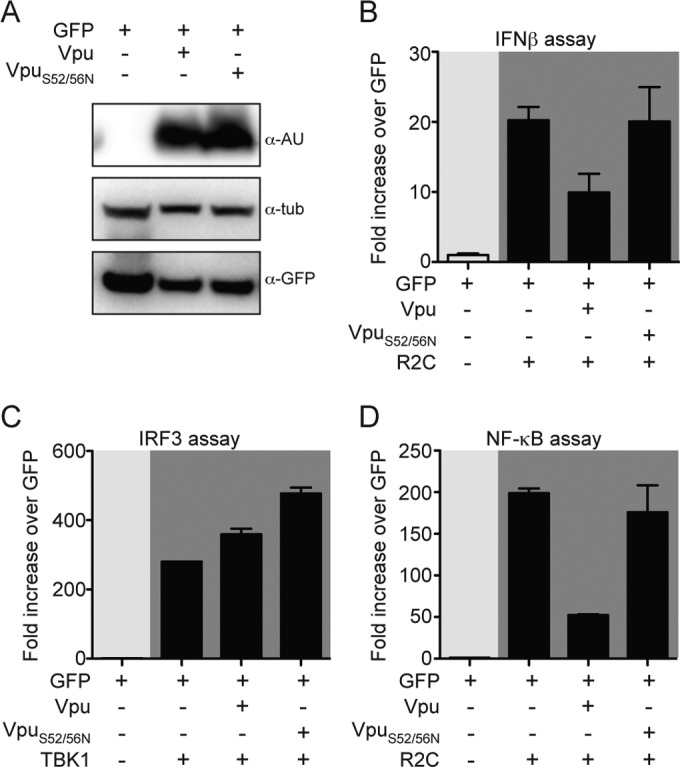
Serines at positions 52 and 56 within NL4.3 Vpu are necessary for its inhibitory effect on NF-κB signaling. (A) HEK293T cells were transfected with WT Vpu, S52N/S56N Vpu, and GFP. The expression levels of the three constructs were monitored by Western blotting. (B) HEK293T cells were cotransfected with vectors expressing WT-Vpu, S52N/S56N Vpu, and GFP as a negative control in the presence or absence of RIG-I 2CARD plasmid, together with IFN-β promoter firefly luciferase and TK-Renilla luciferase vectors. (C) The activity of WT Vpu, S52N/S56N Vpu, and GFP on induction of p55 C1 promoter firefly luciferase in the presence of TBK1 plasmid was assessed in HEK293T cells. The data were analyzed as in Fig. 1B. (D) IKKβca expression plasmid was used as a stimulus to activate synthetic NF-κB promoter when cotransfected with WT Vpu, S52N/S56N Vpu, and GFP.
In order to determine whether IRF3 binding sites or NF-κB binding sites or both were involved, we used reporter plasmids coding for the firefly luciferase under the control of IRF3 alone or NF-κB alone. Neither expression of Vpu-WT nor expression of Vpu-S52N/S56N affected the induction of IRF3-dependent gene expression by TBK1 (Fig. 7C). When we analyzed the effect of Vpu-S52N/S56N on NF-κB-driven luciferase expression, we observed that this mutant lost its ability to inhibit this pathway, whereas Vpu-WT induced a 5-fold reduction of NF-κB-driven transcription (P < 0.001; Fig. 7D). Taken together, these results suggest that the phosphorylation of serines 52 and 56 is required for the ability of NL4.3 Vpu to alter the NF-κB activation pathway, likely via binding to βTrCP.
Different Vpu alleles inhibit NF-κB-dependent gene expression.
To explore the extent to which Vpu-induced NF-κB inhibition is strain specific, we performed reporter assays with six different alleles of Vpu. We included Vpu derived from the lab-adapted strain NL4.3, together with five Vpu alleles derived from primary isolates (Fig. 8A) (18). We cotransfected HEK293T with the different alleles of Vpu and a NF-κB synthetic promoter firefly luciferase reporter construct. Activation of NF-κB was achieved by expressing IKKβca. The reporter assay shows that all of the different Vpu alleles reduce NF-κB activity by 5- to 10-fold (Fig. 8A). The Vpu proteins were all expressed as shown by Western blot analysis (Fig. 8B).
FIG 8.
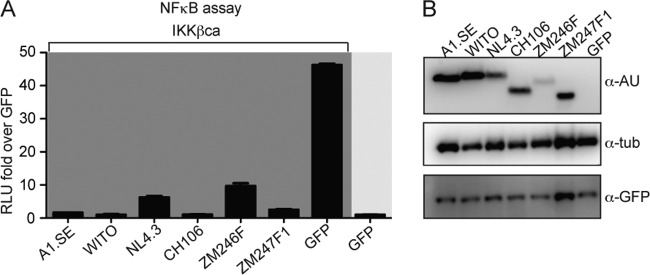
Different vpu alleles inhibit NF-κB-dependent gene expression. (A) HEK293T cells were cotransfected with vectors expressing different variants of Vpu and GFP as a negative control in the presence or absence of the IKKβca expressing vector, together with NF-κB firefly and Renilla luciferase vectors. Induction of the NF-κB promoter was determined by luciferase assay as before. (B) The samples were analyzed by Western blotting using anti-AU, anti-GFP, and anti-GAPDH antibodies.
In conclusion, the Vpu alleles from primary isolates tested exert an inhibitory effect on NF-κB signaling pathway similarly to those observed for NL4.3 Vpu (10, 12, 18).
DISCUSSION
Signaling of innate immune sensing pathways converges to trigger the transcription of proinflammatory and antiviral genes through activation of NF-κB and IRF3. HIV developed different strategies to evade and block such innate immune activation (reviewed in references 63 and 64). We show in the present study that NL4.3 Vpu has the ability to downmodulate IFN-β expression in reporter assays without affecting IRF3 levels. Indeed, the levels of IRF3 protein in primary CD4+ T cells and macrophages were not affected by HIV infection. Since activation of the innate immune response is time dependent, we measured IRF3 expression and HIV infection at different time points in both human primary CD4+ T lymphocytes and macrophages. The proportion of IRF3+ cells in infected and uninfected populations remained comparable at all of the examined time points (Fig. 3B and 5D). We also analyzed different viral strains, with or without Vpu, but failed to detect IRF3 degradation (Fig. 3A).
Although reporter assays using a promoter specific for IRF3 failed to reveal any inhibition of the pathway, we found that Vpu inhibited NF-κB activation downstream of IKKβ activation. HIV Vpu can antagonize NF-κB activation by stabilizing IκBα through its binding with the ubiquitin ligase β-TrCP (12). In agreement with this report, mutating the β-TrCP binding motif within NL4.3 Vpu abolished its ability to downmodulate NF-κB pathway (Fig. 7D). Taken together, our data suggest that the lack of IFN pathway induction during HIV infection in primary cells is caused by the inhibition of NF-κB signaling rather than IRF3 degradation.
The interplay between HIV Vpu and IRF3 has been subject of several studies from different groups. Although our data are in agreement with the findings of Hotter and coworkers, our results disagree with those of Doehle and Park (16–19). A possible explanation is the high level of Vpu expression and the high multiplicity of infection (MOI) in the experiments that can report an effect on IRF3. For example, Park et al. show that the overexpression of Vpu, Vif, and Vpr induces caspase-dependent cleavage of IRF3, suggesting that these proteins can cause activation of the apoptotic pathway that ultimately acts on IRF3 (19). Similarly, infection at a very high MOI can also induce some degree of apoptosis that leads to caspase activation and IRF3 cleavage and/or degradation (65). In the present study we, took great case to use experimental conditions that did not induce any cellular stress despite efficient HIV infection.
We found that the integrity of βTrCP binding sites within NL4.3 Vpu (e.g., the presence of phosphoserines at positions 52 and 56) were required for NF-κB downmodulation. These results are in agreement with recent findings, suggesting that Vpu from NL4.3 strain required the presence of an intact βTrCP consensus motif, whereas Vpu alleles from other strains use additional mechanisms to inhibit NF-κB (54).
It is conceivable that HIV developed cell-type-specific mechanisms to dampen immune responses. For example, TBK1 autophosphorylation, a modification required for an efficient signal transduction, is inhibited during HIV infection in dendritic cells and macrophages (66). In particular, infection of dendritic cells with HIV lacking functional Vif or Vpr alleles induced IFN-β expression (66). Further studies are required to understand whether Vif and Vpr are also required to block IFN-β expression in T cells and macrophages.
The interplay between HIV and NF-κB pathway is essential for efficient viral replication, especially in T cells (reviewed in reference 67). Although HIV needs NF-κB activation for efficient transcription, activation of proinflammatory genes is detrimental for viral replication. The balance needed to achieve optimal viral transcription with a minimum of proinflammatory activation may be obtained through partial inhibition of NF-κB activation by Vpu. In fact, inhibition of NF-κB in the presence of Vpu is not complete, suggesting that some residual activity is preserved. The remaining activity may be sufficient for an efficient HIV transcription but inadequate for an effective immune response. It is conceivable that one of the roles of HIV Vpu is the maintenance of a NF-κB activation level that is favorable for the virus. The different results obtained with different viral strains and cell types suggest a complex relationship between the virus and the infected cell. Further studies are required to dissect the specific mechanisms used by different HIV strains to quench innate immune response in a cell-type-specific manner.
ACKNOWLEDGMENTS
We thank F. Kirchhoff and D. Sauter (Ulm University Medical Center) for sharing the Vpu plasmids. The anti-tetherin antibody and the HIV NL4.3 nef-IRES-GFP construct were kindly provided by B. K. Chen (Icahn School of Medicine at Mount Sinai). We thank R. A. Alvarez (Icahn School of Medicine at Mount Sinai) for technical support.
This study was funded by NIH grants PO1 AI090935, AI089246, and AI72645 (V.S.), NIH/NIAID grants PO1AI90935 and R01AI073450, and DARPA grant HR0011-11-C-0094 (A.F.-S.).
REFERENCES
- 1.Kumar H, Kawai T, Akira S. 2011. Pathogen recognition by the innate immune system. Int Rev Immunol 30:16–34. doi: 10.3109/08830185.2010.529976. [DOI] [PubMed] [Google Scholar]
- 2.Newton K, Dixit VM. 2012. Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol 4:a006049. doi: 10.1101/cshperspect.a006049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ranjan P, Bowzard JB, Schwerzmann JW, Jeisy-Scott V, Fujita T, Sambhara S. 2009. Cytoplasmic nucleic acid sensors in antiviral immunity. Trends Mol Med 15:359–368. doi: 10.1016/j.molmed.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 4.Iwasaki A, Medzhitov R. 2010. Regulation of adaptive immunity by the innate immune system. Science 327:291–295. doi: 10.1126/science.1183021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iannello A, Debbeche O, Martin E, Attalah LH, Samarani S, Ahmad A. 2006. Viral strategies for evading antiviral cellular immune responses of the host. J Leukoc Biol 79:16–35. [DOI] [PubMed] [Google Scholar]
- 6.Landau NR. 2014. The innate immune response to HIV-1: to sense or not to sense. DNA Cell Biol 33:271–274. doi: 10.1089/dna.2014.2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roy N, Pacini G, Berlioz-Torrent C, Janvier K. 2014. Mechanisms underlying HIV-1 Vpu-mediated viral egress. Front Microbiol 5:177. doi: 10.3389/fmicb.2014.00177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neil SJ, Zang T, Bieniasz PD. 2008. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451:425–430. doi: 10.1038/nature06553. [DOI] [PubMed] [Google Scholar]
- 9.Van Damme N, Goff D, Katsura C, Jorgenson RL, Mitchell R, Johnson MC, Stephens EB, Guatelli J. 2008. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 3:245–252. doi: 10.1016/j.chom.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Galao RP, Le Tortorec A, Pickering S, Kueck T, Neil SJ. 2012. Innate sensing of HIV-1 assembly by Tetherin induces NF-κB-dependent proinflammatory responses. Cell Host Microbe 12:633–644. doi: 10.1016/j.chom.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tokarev A, Suarez M, Kwan W, Fitzpatrick K, Singh R, Guatelli J. 2013. Stimulation of NF-κB activity by the HIV restriction factor BST2. J Virol 87:2046–2057. doi: 10.1128/JVI.02272-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bour S, Perrin C, Akari H, Strebel K. 2001. The human immunodeficiency virus type 1 Vpu protein inhibits NF-κB activation by interfering with beta TrCP-mediated degradation of IκB. J Biol Chem 276:15920–15928. doi: 10.1074/jbc.M010533200. [DOI] [PubMed] [Google Scholar]
- 13.Willey RL, Maldarelli F, Martin MA, Strebel K. 1992. Human immunodeficiency virus type 1 Vpu protein induces rapid degradation of CD4. J Virol 66:7193–7200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moll M, Andersson SK, Smed-Sorensen A, Sandberg JK. 2010. Inhibition of lipid antigen presentation in dendritic cells by HIV-1 Vpu interference with CD1d recycling from endosomal compartments. Blood 116:1876–1884. doi: 10.1182/blood-2009-09-243667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shah AH, Sowrirajan B, Davis ZB, Ward JP, Campbell EM, Planelles V, Barker E. 2010. Degranulation of natural killer cells following interaction with HIV-1-infected cells is hindered by downmodulation of NTB-A by Vpu. Cell Host Microbe 8:397–409. doi: 10.1016/j.chom.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doehle BP, Chang K, Fleming L, McNevin J, Hladik F, McElrath MJ, Gale M Jr. 2012. Vpu-deficient HIV strains stimulate innate immune signaling responses in target cells. J Virol 86:8499–8506. doi: 10.1128/JVI.00424-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doehle BP, Chang K, Rustagi A, McNevin J, McElrath MJ, Gale M Jr. 2012. Vpu mediates depletion of interferon regulatory factor 3 during HIV infection by a lysosome-dependent mechanism. J Virol 86:8367–8374. doi: 10.1128/JVI.00423-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hotter D, Kirchhoff F, Sauter D. 2013. HIV-1 Vpu does not degrade interferon regulatory factor 3. J Virol 87:7160–7165. doi: 10.1128/JVI.00526-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park SY, Waheed AA, Zhang ZR, Freed EO, Bonifacino JS. 2014. HIV-1 Vpu accessory protein induces caspase-mediated cleavage of IRF3 transcription factor. J Biol Chem 289:35102–35110. doi: 10.1074/jbc.M114.597062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Versteeg GA, Rajsbaum R, Sanchez-Aparicio MT, Maestre AM, Valdiviezo J, Shi M, Inn KS, Fernandez-Sesma A, Jung J, Garcia-Sastre A. 2013. The E3-ligase TRIM family of proteins regulates signaling pathways triggered by innate immune pattern-recognition receptors. Immunity 38:384–398. doi: 10.1016/j.immuni.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rajsbaum R, Versteeg GA, Schmid S, Maestre AM, Belicha-Villanueva A, Martinez-Romero C, Patel JR, Morrison J, Pisanelli G, Miorin L, Laurent-Rolle M, Moulton HM, Stein DA, Fernandez-Sesma A, ten Oever BR, Garcia-Sastre A. 2014. Unanchored K48-linked polyubiquitin synthesized by the E3-ubiquitin ligase TRIM6 stimulates the interferon-IKKεkinase-mediated antiviral response. Immunity 40:880–895. doi: 10.1016/j.immuni.2014.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nistal-Villan E, Gack MU, Martinez-Delgado G, Maharaj NP, Inn KS, Yang H, Wang R, Aggarwal AK, Jung JU, Garcia-Sastre A. 2010. Negative role of RIG-I serine 8 phosphorylation in the regulation of interferon-beta production. J Biol Chem 285:20252–20261. doi: 10.1074/jbc.M109.089912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lahm HW, Stein S. 1985. Characterization of recombinant human interleukin-2 with micromethods. J chromatography 326:357–361. doi: 10.1016/S0021-9673(01)87461-6. [DOI] [PubMed] [Google Scholar]
- 24.Aguirre S, Maestre AM, Pagni S, Patel JR, Savage T, Gutman D, Maringer K, Bernal-Rubio D, Shabman RS, Simon V, Rodriguez-Madoz JR, Mulder LC, Barber GN, Fernandez-Sesma A. 2012. DENV inhibits type I IFN production in infected cells by cleaving human STING. PLoS Pathog 8:e1002934. doi: 10.1371/journal.ppat.1002934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Husain M, Gusella GL, Klotman ME, Gelman IH, Ross MD, Schwartz EJ, Cara A, Klotman PE. 2002. HIV-1 Nef induces proliferation and anchorage-independent growth in podocytes. J Am Soc Nephrol 13:1806–1815. doi: 10.1097/01.ASN.0000019642.55998.69. [DOI] [PubMed] [Google Scholar]
- 26.Sauter D, Hue S, Petit SJ, Plantier JC, Towers GJ, Kirchhoff F, Gupta RK. 2011. HIV-1 Group P is unable to antagonize human tetherin by Vpu, Env, or Nef. Retrovirology 8:103. doi: 10.1186/1742-4690-8-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, Jung JU. 2007. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446:916–920. doi: 10.1038/nature05732. [DOI] [PubMed] [Google Scholar]
- 28.Yoneyama M, Suhara W, Fukuhara Y, Fukuda M, Nishida E, Fujita T. 1998. Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J 17:1087–1095. doi: 10.1093/emboj/17.4.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peden K, Emerman M, Montagnier L. 1991. Changes in growth properties on passage in tissue culture of viruses derived from infectious molecular clones of HIV-1LAI, HIV-1MAL, and HIV-1ELI. Virology 185:661–672. doi: 10.1016/0042-6822(91)90537-L. [DOI] [PubMed] [Google Scholar]
- 30.Munch J, Rajan D, Schindler M, Specht A, Rucker E, Novembre FJ, Nerrienet E, Muller-Trutwin MC, Peeters M, Hahn BH, Kirchhoff F. 2007. Nef-mediated enhancement of virion infectivity and stimulation of viral replication are fundamental properties of primate lentiviruses. J Virol 81:13852–13864. doi: 10.1128/JVI.00904-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cohen GB, Gandhi RT, Davis DM, Mandelboim O, Chen BK, Strominger JL, Baltimore D. 1999. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity 10:661–671. doi: 10.1016/S1074-7613(00)80065-5. [DOI] [PubMed] [Google Scholar]
- 32.Chakrabarti LA, Ivanovic T, Cheng-Mayer C. 2002. Properties of the surface envelope glycoprotein associated with virulence of simian-human immunodeficiency virus SHIV(SF33A) molecular clones. J Virol 76:1588–1599. doi: 10.1128/JVI.76.4.1588-1599.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goujon C, Jarrosson-Wuilleme L, Bernaud J, Rigal D, Darlix JL, Cimarelli A. 2006. With a little help from a friend: increasing HIV transduction of monocyte-derived dendritic cells with virion-like particles of SIV(MAC). Gene Ther 13:991–994. doi: 10.1038/sj.gt.3302753. [DOI] [PubMed] [Google Scholar]
- 34.Negre D, Mangeot PE, Duisit G, Blanchard S, Vidalain PO, Leissner P, Winter AJ, Rabourdin-Combe C, Mehtali M, Moullier P, Darlix JL, Cosset FL. 2000. Characterization of novel safe lentiviral vectors derived from simian immunodeficiency virus (SIVmac251) that efficiently transduce mature human dendritic cells. Gene Ther 7:1613–1623. doi: 10.1038/sj.gt.3301292. [DOI] [PubMed] [Google Scholar]
- 35.Yee JK, Friedmann T, Burns JC. 1994. Generation of high-titer pseudotyped retroviral vectors with very broad host range. Methods Cell Biol 43(Pt A):99–112. [DOI] [PubMed] [Google Scholar]
- 36.Boussif O, Lezoualc'h F, Zanta MA, Mergny MD, Scherman D, Demeneix B, Behr JP. 1995. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc Natl Acad Sci U S A 92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ooms M, Majdak S, Seibert CW, Harari A, Simon V. 2010. The localization of APOBEC3H variants in HIV-1 virions determines their antiviral activity. J Virol 84:7961–7969. doi: 10.1128/JVI.00754-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Platt EJ, Bilska M, Kozak SL, Kabat D, Montefiori DC. 2009. Evidence that ecotropic murine leukemia virus contamination in TZM-bl cells does not affect the outcome of neutralizing antibody assays with human immunodeficiency virus type 1. J Virol 83:8289–8292. doi: 10.1128/JVI.00709-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Platt EJ, Wehrly K, Kuhmann SE, Chesebro B, Kabat D. 1998. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J Virol 72:2855–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takeuchi Y, McClure MO, Pizzato M. 2008. Identification of gammaretroviruses constitutively released from cell lines used for human immunodeficiency virus research. J Virol 82:12585–12588. doi: 10.1128/JVI.01726-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wei X, Decker JM, Liu H, Zhang Z, Arani RB, Kilby JM, Saag MS, Wu X, Shaw GM, Kappes JC. 2002. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob Agents Chemother 46:1896–1905. doi: 10.1128/AAC.46.6.1896-1905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Derdeyn CA, Decker JM, Sfakianos JN, Wu X, O'Brien WA, Ratner L, Kappes JC, Shaw GM, Hunter E. 2000. Sensitivity of human immunodeficiency virus type 1 to the fusion inhibitor T-20 is modulated by coreceptor specificity defined by the V3 loop of gp120. J Virol 74:8358–8367. doi: 10.1128/JVI.74.18.8358-8367.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol 5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 44.Papkalla A, Munch J, Otto C, Kirchhoff F. 2002. Nef enhances human immunodeficiency virus type 1 infectivity and replication independently of viral coreceptor tropism. J Virol 76:8455–8459. doi: 10.1128/JVI.76.16.8455-8459.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harari A, Ooms M, Mulder LC, Simon V. 2009. Polymorphisms and splice variants influence the antiretroviral activity of human APOBEC3H. J Virol 83:295–303. doi: 10.1128/JVI.01665-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hrecka K, Hao C, Gierszewska M, Swanson SK, Kesik-Brodacka M, Srivastava S, Florens L, Washburn MP, Skowronski J. 2011. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 474:658–661. doi: 10.1038/nature10195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, Segeral E, Yatim A, Emiliani S, Schwartz O, Benkirane M. 2011. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 474:654–657. doi: 10.1038/nature10117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Manel N, Hogstad B, Wang Y, Levy DE, Unutmaz D, Littman DR. 2010. A cryptic sensor for HIV-1 activates antiviral innate immunity in dendritic cells. Nature 467:214–217. doi: 10.1038/nature09337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mascola JR, Louder MK, Winter C, Prabhakara R, De Rosa SC, Douek DC, Hill BJ, Gabuzda D, Roederer M. 2002. Human immunodeficiency virus type 1 neutralization measured by flow cytometric quantitation of single-round infection of primary human T cells. J Virol 76:4810–4821. doi: 10.1128/JVI.76.10.4810-4821.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Snyder A, Alsauskas ZC, Leventhal JS, Rosenstiel PE, Gong P, Chan JJ, Barley K, He JC, Klotman ME, Ross MJ, Klotman PE. 2010. HIV-1 viral protein r induces ERK and caspase-8-dependent apoptosis in renal tubular epithelial cells. AIDS 24:1107–1119. doi: 10.1097/QAD.0b013e328337b0ab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fujita T, Shibuya H, Hotta H, Yamanishi K, Taniguchi T. 1987. Interferon-beta gene regulation: tandemly repeated sequences of a synthetic 6-bp oligomer function as a virus-inducible enhancer. Cell 49:357–367. doi: 10.1016/0092-8674(87)90288-1. [DOI] [PubMed] [Google Scholar]
- 52.Sharma S, ten Oever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. 2003. Triggering the interferon antiviral response through an IKK-related pathway. Science 300:1148–1151. doi: 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- 53.Uchil PD, Hinz A, Siegel S, Coenen-Stass A, Pertel T, Luban J, Mothes W. 2013. TRIM protein-mediated regulation of inflammatory and innate immune signaling and its association with antiretroviral activity. J Virol 87:257–272. doi: 10.1128/JVI.01804-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sauter D, Hotter D, Van Driessche B, Sturzel CM, Kluge SF, Wildum S, Yu H, Baumann B, Wirth T, Plantier JC, Leoz M, Hahn BH, Van Lint C, Kirchhoff F. 2015. Differential regulation of NF-κB-mediated proviral and antiviral host gene expression by primate lentiviral Nef and Vpu proteins. Cell Reports 10:586–599. doi: 10.1016/j.celrep.2014.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Beg AA, Finco TS, Nantermet PV, Baldwin AS Jr. 1993. Tumor necrosis factor and interleukin-1 lead to phosphorylation and loss of IκBα: a mechanism for NF-κB activation. Mol Cell Biol 13:3301–3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Blanchet FP, Mitchell JP, Piguet V. 2012. beta-TrCP dependency of HIV-1 Vpu-induced downregulation of CD4 and BST-2/tetherin. Curr HIV Res 10:307–314. doi: 10.2174/157016212800792441. [DOI] [PubMed] [Google Scholar]
- 57.Margottin F, Bour SP, Durand H, Selig L, Benichou S, Richard V, Thomas D, Strebel K, Benarous R. 1998. A novel human WD protein, h-beta TrCp, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol Cell 1:565–574. doi: 10.1016/S1097-2765(00)80056-8. [DOI] [PubMed] [Google Scholar]
- 58.Mangeat B, Gers-Huber G, Lehmann M, Zufferey M, Luban J, Piguet V. 2009. HIV-1 Vpu neutralizes the antiviral factor Tetherin/BST-2 by binding it and directing its beta-TrCP2-dependent degradation. PLoS Pathog 5:e1000574. doi: 10.1371/journal.ppat.1000574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Besnard-Guerin C, Belaidouni N, Lassot I, Segeral E, Jobart A, Marchal C, Benarous R. 2004. HIV-1 Vpu sequesters β-transducin repeat-containing protein (βTrCP) in the cytoplasm and provokes the accumulation of beta-catenin and other SCFβTrCP substrates. J Biol Chem 279:788–795. doi: 10.1074/jbc.M308068200. [DOI] [PubMed] [Google Scholar]
- 60.Beg AA, Baldwin AS Jr. 1993. The IκB proteins: multifunctional regulators of Rel/NF-κB transcription factors. Genes Dev 7:2064–2070. doi: 10.1101/gad.7.11.2064. [DOI] [PubMed] [Google Scholar]
- 61.Brown K, Park S, Kanno T, Franzoso G, Siebenlist U. 1993. Mutual regulation of the transcriptional activator NF-κB and its inhibitor, IκBα. Proc Natl Acad Sci U S A 90:2532–2536. doi: 10.1073/pnas.90.6.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Henkel T, Machleidt T, Alkalay I, Kronke M, Ben-Neriah Y, Baeuerle PA. 1993. Rapid proteolysis of IκBα is necessary for activation of transcription factor NF-κB. Nature 365:182–185. [DOI] [PubMed] [Google Scholar]
- 63.Altfeld M, Gale M Jr. 2015. Innate immunity against HIV-1 infection. Nat Immunol 16:554–562. doi: 10.1038/ni.3157. [DOI] [PubMed] [Google Scholar]
- 64.Simon V, Bloch N, Landau NR. 2015. Intrinsic host restrictions to HIV-1 and mechanisms of viral escape. Nat Immunol 16:546–553. doi: 10.1038/ni.3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cummins NW, Badley AD. 2010. Mechanisms of HIV-associated lymphocyte apoptosis: 2010. Cell Death Dis 1:e99. doi: 10.1038/cddis.2010.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Harman AN, Nasr N, Feetham A, Galoyan A, Alshehri AA, Rambukwelle D, Botting RA, Hiener BM, Diefenbach E, Diefenbach RJ, Kim M, Mansell A, Cunningham AL. 2015. HIV blocks interferon induction in human dendritic cells and macrophages by dysregulation of TBK1. J Virol 89:6575–6584. doi: 10.1128/JVI.00889-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chan JK, Greene WC. 2012. Dynamic roles for NF-κB in HTLV-1 and HIV-1 retroviral pathogenesis. Immunol Rev 246:286–310. doi: 10.1111/j.1600-065X.2012.01094.x. [DOI] [PubMed] [Google Scholar]