

Abstract
We report a simple temperature-responsive bioconjugate system comprising superfolder green fluorescent protein (sfGFP) decorated with poly[(oligo ethylene glycol) methyl ether methacrylate] (PEGMA) polymers. We used amber suppression to site-specifically incorporate the non-canonical azide-functional amino acid p-azidophenylalanine (pAzF) into sfGFP at different positions. The azide moiety on modified sfGFP was then coupled using copper-catalyzed “click” chemistry with the alkyne terminus of a PEGMA synthesized by reversible addition–fragmentation chain transfer (RAFT) polymerization. The protein in the resulting bioconjugate was found to remain functionally active (i.e., fluorescent) after conjugation. Turbidity measurements revealed that the point of attachment of the polymer onto the protein scaffold has an impact on the thermoresponsive behavior of the resultant bioconjugate. Furthermore, small-angle X-ray scattering analysis showed the wrapping of the polymer around the protein in a temperature-dependent fashion. Our work demonstrates that standard genetic manipulation combined with an expanded genetic code provides an easy way to construct functional hybrid biomaterials where the location of the conjugation site on the protein plays an important role in determining material properties. We anticipate that our approach could be generalized for the synthesis of complex functional materials with precisely defined domain orientation, connectivity, and composition.
Introduction
Ever since the pioneering work of Davis and co-workers,1,2 the conjugation of synthetic macromolecules with proteins to enhance the chemical properties and functions of the latter has been demonstrated for a variety of systems and a range of applications. Some illustrative examples include increased protein activity, proteolytic resistance, and thermal and pH stability,3,4 properties that have been attributed to the careful selection of the molecular characteristics of the polymer and the conjugation site.3 The vast majority of reports involve proteins conjugated with poly(ethylene glycol) (PEG),5−8 as it is a biocompatible polymer with a proven record of applications.3,4,8−11 Noteworthy also is the use of branched PEG analogues that have been shown to further enhance the biocompatibility of their protein bioconjugates.12 Nevertheless, the use of polymers that endow the protein with more intricate properties has been sought, such as polymers that respond to external stimuli.13,14
Stimuli-responsive polymers can be used to expand the properties of protein–polymer systems,15−17 owing to their ability to change their physicochemical properties as a response to small changes in their environment (i.e., temperature, pH, light, etc.) and their corresponding bioconjugates inherit that ability, obtaining a triggered (and commonly reversible) amphiphilic character.18−24 Hoffman and co-workers pioneered the use of stimuli-responsive polymers for conjugation with proteins that allowed their isolation and reuse, or modulation of their activity.25−29 In other examples, permanently amphiphilic bioconjugates,18,19,30−32 whereby the protein is conjugated with a hydrophobic polymer, have shown potential in improving the protein activity (such as inhibition of tumor cell growth),33 although in some cases the opposite effect was observed.34,35 Similarly, stimuli-responsive bioconjugates (frequently referred to as “smart” bioconjugates21) are often studied as potential “on/off” systems,36−38 whereby the solvation of the polymer dictates the protein activity.39 In addition to the effect on protein activity, bioconjugates with an amphiphilic character (often referred to as “giant amphiphiles”) form elaborate nanostructures as a result of their self-assembly in water.30,32,34,40,41
In building protein–polymer macromolecules, several design decisions must be considered. First, the strategy to attach the polymer to the protein must be defined. The most used conjugation method involves the functionalization of all available natural amino acid target moieties on the protein,42−45 commonly lysine or cysteine residues. Other approaches have targeted the N-terminus of the protein,46,47 while in other examples a single available functional amino acid is targeted.32,48−51 These synthetic approaches to decorating proteins have been extensively presented in numerous noteworthy reviews.6,7,52−63 A recurring limitation in a large number of the reports in the literature is that covalent coupling leads to a heterogeneous mixture of products with varying conjugation degrees. This has been found to be related to the polymer molecular weight,64 polymer docking location onto the protein, and also heterogeneities in the protein which affect the availability of the modification sites,65 thus highlighting the need for complete control over the conjugation site.20
Recent studies have shown that the introduction of noncanonical amino acids (ncAAs) into a functional protein expands the available chemistries for conjugation,66 allowing a higher degree of precision and minimization of side-reactions and byproducts.67 In one approach, pioneered by Tirrell and colleagues,68−72 all natural amino acids (typically methionine, isoleucine, or leucine) are globally replaced by a ncAA. While powerful, changing all occurrences of a natural amino acid in a protein may unfavorably affect protein folding and activity. In addition, the chemical diversity introduced via ncAAs in this procedure is limited since the ncAA must be a close analogue of the natural amino acid it replaces. In an alternative approach, ncAAs are quantitatively installed at defined sites in a protein through genetic code expansion. The most widely used strategy for expanding the genetic code is based on the amber suppression technique using orthogonal aminoacyl-tRNA synthetase/tRNA pairs.73 Many seminal works from Schultz and others have established and driven the field forward, and more than 150 different ncAAs have been site-specifically incorporated into proteins to date.66,74−76 These ncAAs normally carry functional moieties (e.g., aryl-azide) that do not exist in the canonical 20 amino acids and that are easy to chemically modify (e.g., using copper-catalyzed alkyne–azide cycloaddition77−81), although the success of the modification also relies on the conjugation site.82 A notable example is the incorporation of a polymerization-initiating ncAA into green fluorescent protein (GFP) and the subsequent growth of a polymer from the surface of the protein.83 Such modifications can thus allow conjugation with polymers which, as previously mentioned, could protect the protein from degradation or prevent the polymer interfering with the protein activity. Another significant advantage of this approach is that it can allow the introduction of ncAAs without altering the net charge or the redox potential of the protein, as is often the result of functionalizing lysine and cysteine residues, respectively.
After deciding how to precisely link proteins to polymers, the second key design consideration is polymer conjugation strategy. In one approach, the presynthesized polymer can be “grafted to” the protein. The major drawback of this approach stems from the difficulty removing the high-molecular-weight byproducts (i.e., excess polymer). An alternative strategy is to “graft from”, where the protein is functionalized with a moiety that participates in the polymer synthesis, such as a polymerization initiator/mediator. Recently, such “grafting from” approaches have become more accessible since the development of reversible deactivation radical polymerizations,57,58 such as atom transfer radical polymerization (ATRP), reversible addition–fragmentation chain transfer (RAFT) polymerization, and nitroxide-mediated polymerization (NMP) which allow the reaction to occur under conditions suitable for retention of the protein stability.84 However, “grafting to” is still a popular conjugation method as it allows the fine-tuning of the molecular characteristics of the polymer before its conjugation.
Recently, several reports have begun to make possible new types of protein–polymer bioconjugates using GFP as a model protein.85 Nolte and co-workers, for example, studied the self-assembly of conjugated biohybrid copolymers comprising GFP and poly[(oligo ethylene glycol) methyl ether acrylate] (PEGMA) and showed that the resulting biohybrid amphiphiles were thermoresponsive.86 However, their study was limited to the use of natural amino acid handles for conjugation (i.e., cysteine) and was insufficient to study the impact of multiple polymers attached to the compact protein core. Similarly, Olsen et al. reported the conjugation of thermoresponsive polymers with a GFP via thiol–maleimide ligation. The resulting bioconjugates formed micelles when the solution temperature was increased.87 In another example, Matyjaszewski and co-workers reported the incorporation of pAzF into a GFP and its subsequent bioconjugation with a PEG containing two alkyne functionalities. This resulted in a “step-growth” formation of micron-sized fibers that were attributed to the dimerization of GFP.88 This was a significant advance in the study of properties of polymer–protein bioconjugates and it demonstrates the infinite potential applications that will emerge once more intricate polymers are explored in such systems. However, our understanding of how the location on the protein surface of conjugation can affect the resultant properties of the protein–polymer bioconjugate material remains incomplete.
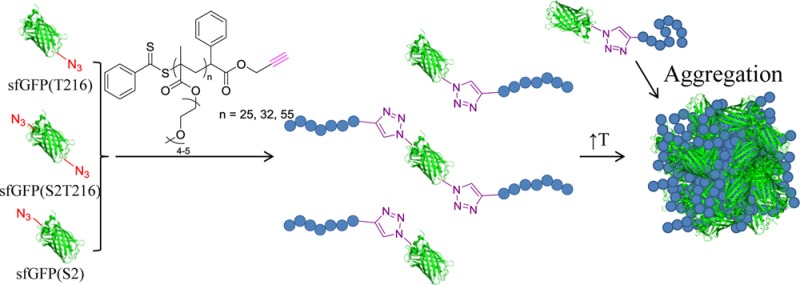
Here, we sought to build on these recent reports to demonstrate a simple bioconjugate protein–polymer system that would allow us to study the impact of site-specific conjugation on self-assembly and responsiveness. Our goal was to produce and study biomacromolecules comprising superfolder GFP (sfGFP) decorated with temperature-responsive poly[(oligo ethylene glycol) methyl ether methacrylate] (PEGMA) chains of different molecular weights on more than one site, by copper-catalyzed azide–alkyne cycloaddition reaction (CuAAc). Similarly to PEG, PEGMA has been shown to be biocompatible33 and, additionally, exhibits a lower critical solution temperature (LCST) in water.89 Our study involved three steps. First, the sfGFP molecules were functionalized with azide groups (at amino acid residues 2, 216, or 2 and 216). Second, the reactive azide moieties were conjugated with an alkyne-containing PEGMA synthesized by RAFT polymerization. Third, we characterized the reversible transition of the protein–polymer structures from a water-soluble to a water-insoluble state upon heating above a critical temperature (namely the cloud point). Our results showed that the resultant structures had properties of both the fluorescent sfGFP and the temperature-responsive PEGMA (Figure 1). Additionally, we explored the effect of different attachment positions on the protein on the cloud point of the bioconjugate using turbidimetry, dynamic light scattering (DLS), and small-angle X-ray scattering (SAXS) analysis.
Figure 1.
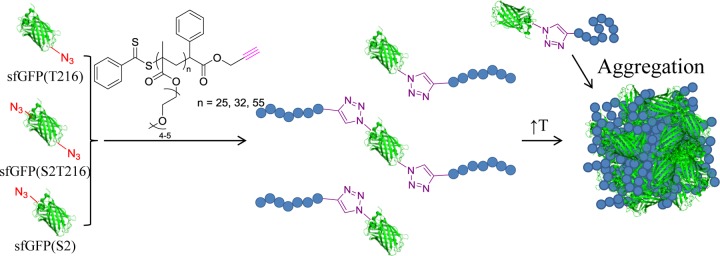
Strategy followed for the synthesis of sfGFP-PEGMA bioconjugates via the engineering of three sfGFP analogues with T216, S2T216, and S2 site modification with pAzF before the CuAAc of alkyne-functional PEGMA (three different molecular weights). Upon an increase of the solution temperature, all bioconjugates were found to aggregate.
Results and Discussion
We began our study by producing the p-azidophenylalanine (pAzF) sfGFP labeled reagents. To incorporate pAzF into sfGFP, Escherichia coli BL21(DE3) cells were first co-transformed with the pEVOL-pAzF plasmid that encodes the aminoacyl-tRNA synthetase/suppressor tRNA pair90 and an appropriate mutant pY71-sfGFP plasmid with amber codon (TAG) at positions of S2, T216, or S2/T216. These locations were chosen as the S2 and T216 residues are located at opposite ends of the protein’s barrel structure on flexible loops that do not affect sfGFP folding. In addition, this design allowed us to introduce two conjugation points on opposite sides of the protein structure (Figure 1; see Figure S1 in the Supporting Information (SI) for more information on the sites of modification).
Then, the desired sfGFP proteins were overexpressed and purified from the BL21 (DE3) cells, noting that T7 RNA polymerase, which drives sfGFP transcription in pY71-sfGFP, was expressed from a DE3 λ prophage under an isopropyl β-d-1-thiogalactopyranoside (IPTG)-inducible lacI promoter in BL21(DE3) (see SI for methods). Protein expression yields were estimated to be ∼20 mg/L by comparison of purified protein to standards of bovine serum albumin at known concentrations. With the purified sfGFP variants in hand, we carried out top-down mass spectrometry (i.e., MS analysis of whole intact proteins) to detect and provide semiquantitative information for the incorporation of pAzF into sfGFP. Figure 2 shows the 32+ charge state of sfGFP and clearly illustrates mass shifts corresponding to the incorporation of each of the specifically incorporated pAzF residues. Site-specific incorporation of pAzF, as detected by MS, was greater than 95% in all samples (Figure 2), noting that the experimental and theoretical protein masses were in good agreement (see SI, Table S2). In summary, we achieved efficient, high yielding, and pure site-specific pAzF incorporation into sfGFP at two different sites at opposite ends of the protein barrel structure.
Figure 2.
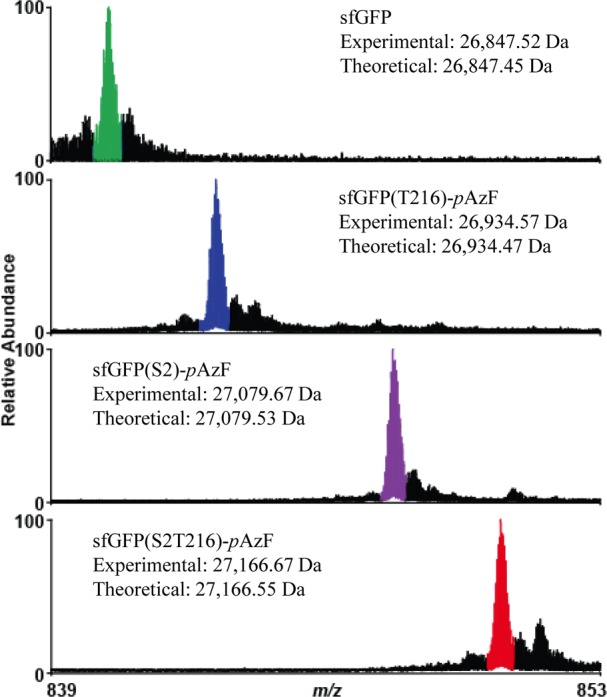
Mass spectrum of the 32+ charge state of sfGFP obtained via top-down mass spectrometry illustrating site-specific incorporation of pAzF at single and multiple sites. Major peaks in each spectrum coincide with the theoretical peaks for each species and have been highlighted. Smaller peaks to the right of the colored peaks are due to oxidation of the protein—a common electrochemical reaction occurring during electrospray ionization.91 Water loss events from the intact sfGFP are detected at minor levels to the left of the major (colored) peaks. Note that the start (N-terminal) methionine of sfGFP is usually cleaved post-translationally by methionine aminopeptidase present in the E. coli proteome. However, the presence of an unnatural amino acid at S2 appears to hinder this enzyme (For more detail, see SI Table S2).
Once the production of pure modified proteins by mass spectrometry was confirmed, the accessibility of the reactive azide moieties was established by exploration of a CuAAC reaction with an alkyne-containing rhodamine B fluorescent dye (1) (see SI). All protein–dye bioconjugates were found to contain the rhodamine B dye by PAGE analysis (see SI, Figure S3), although LC-MS suggested incomplete conjugation (see SI, Figure S4). This highlighted that the two modified positions on the sfGFP were accessible for reaction using CuAAC.
For the conjugation of the protein with a polymer, an alkyne-containing chain transfer agent (CTA, 2) was chosen for the RAFT polymerization of OEGMA300 (Figure 1). Three polymers varying in molecular weight (Table 1) were synthesized by changes in monomer feed and reaction time (see SI for synthetic procedure). Overall, the molecular weight distribution of the polymers was fairly low, while the crucial presence of the alkyne end-group was confirmed by 1H NMR spectroscopy (see SI, Figures S6 and S7). It should be noted that this CTA was chosen as it bears the alkyne functionality on the R-group, thus permitting the bioconjugation regardless of the thiocarbonylthio bond stability.92
Table 1. Number Average Molecular Weights and Molecular Weight Distributions of the Polymers Used for the Synthesis of the Bioconjugates.
| polymer | Mna (g/mol) | ĐM |
|---|---|---|
| PEGMA-1 | 7600 | 1.26 |
| PEGMA-2 | 9600 | 1.32 |
| PEGMA-3 | 16700 | 1.36 |
Determined by SEC in THF (2% triethylamine).
Conjugation of the alkyne-functional polymers with the azide-bearing proteins was carried out in Tris buffer solution using copper sulfate as the catalyst, to make a total of nine protein–polymer structures (three sfGFP constructs plus three different polymer molecular weights). Each bioconjugate was then purified by preparative size exclusion chromatography (SEC), which allowed for assessment of the efficiency of the reaction (Figure 3A). When compared to the unmodified sfGFP, all samples were found to exhibit higher molecular weight peaks, eluting at lower volumes, which were attributed to the polymer–protein bioconjugates. It should also be noted that the bioconjugate retention volume decreased with increasing polymer molecular weight, suggesting that higher molecular weight polymers resulted in higher molecular weight bioconjugates. These data confirmed that decoration of site selective sfGFPs with PEGMA polymers of different molecular weights at both positions 2 and 216 was possible.
Figure 3.

Comparison of the synthesized bioconjugates with their corresponding unfunctionalized protein and polymer: (A) Chromatograms from the crude protein–polymer bioconjugates, and (B) PAGE gels of the proteins upon conjugation with PEGMA-1: lane 1, ladder; lane 2, PEGMA-1; lane 3, sfGFP(S2); lane 4, sfGFP(S2)-PEGMA1; lane 5, sfGFP(T216); lane 6, sfGFP(T216)-PEGMA1; lane 7, sfGFP(S2T216); lane 8, sfGFP(S2T216)-PEGMA1; (C) upon conjugation with PEGMA-2: lane 1, ladder; lane 2, PEGMA-2; lane 3, sfGFP(S2); lane 4, sfGFP(S2)-PEGMA2; lane 5, sfGFP(T216); lane 6, sfGFP(T216)-PEGMA2; lane 7, sfGFP(S2T216); lane 8, sfGFP(S2T216)-PEGMA2; (D) and upon conjugation with PEGMA-3: lane 1, ladder; lane 2, PEGMA-3; lane 3, sfGFP(S2); lane 4, sfGFP(S2)-PEGMA3; lane 5, sfGFP(T216); lane 6, sfGFP(T216)-PEGMA3; lane 7, sfGFP(S2T216); lane 8, sfGFP(S2T216)-PEGMA3.
To confirm the successful formation of the protein–polymer bioconjugates, we next carried out SDS-PAGE analysis on the sfGFP-PEGMA bioconjugates following purification by preparative SEC and sample concentration. Comparison of the unconjugated sfGFP and the product of the CuAAc reactions with the different molecular weight polymers showed that the latter exhibit a significantly broader band at lower mobility, consistent with the presence of the bioconjugate (Figure 3B–D). In the case of PEGMA-1 and PEGMA-2, the broad band with the highest mobility matches that of the neat polymer and is attributed to unreacted polymer chains. This is especially prominent for the PEGMA-1 reactions (Figure 3B), which is due to the fact that removal from the bioconjugate is more challenging for the lowest molecular weight polymer sample. In the case of PEGMA-3, it is hard to determine if there is unconjugated polymer, as the broad polymer band overlaps with the molecular weight assigned to the bioconjugate. However, it was noted that upon heating of these bioconjugate samples, a precipitate was formed which was determined to be unreacted PEGMA (see SI, Figure S8) and hence removal of this by filtration readily allowed for the removal of any unconjugated polymer.
Following production of protein–polymer bioconjugates, we then carried out a series of characterization experiments to assess the impact of conjugation at different sites on the protein surface on the macromolecule properties of the bioconjugates. First, the activity of the protein–polymer bioconjugates was compared with that of the wild type nonconjugated sfGFP, in order to confirm that polymer conjugation does not affect the inherent fluorescence of the protein.93 To assess activity, we determined the quantum yield of the sfGFP fluorescence before and after conjugation (Figure 4).94 Using fluorescein free acid as the standard, sfGFP was found to have a quantum yield of 0.613 (±0.016). Similarly, the quantum yield of the bioconjugated sfGFP with PEGMA-2 at the T216 position (sfGFP(T216)-PEGMA2) was found to be 0.638 (±0.014). The comparable quantum yields for the bioconjugate and the sfGFP protein suggest that the fluorophore of the protein is not affected by polymer conjugation, thus confirming that careful selection of the conjugation site (which is enabled through the site-specific incorporation of a ncAA) allows for the retention of the protein activity. It should be noted that the conjugation did not have an effect on the sfGFP fluorescence even at elevated temperatures, as both the bioconjugate and the wild type sfGFP showed similar fluorescence emissions when cycling the temperature between 25 and 70 °C (see SI, Figure S9). Although we did not test the activity of all the protein–polymer constructs, our data supports an emerging wave of examples showing the ability to maintain protein activity in protein–polymer bioconjugates prepared using site-specifically incorporated ncAAs.88,93
Figure 4.
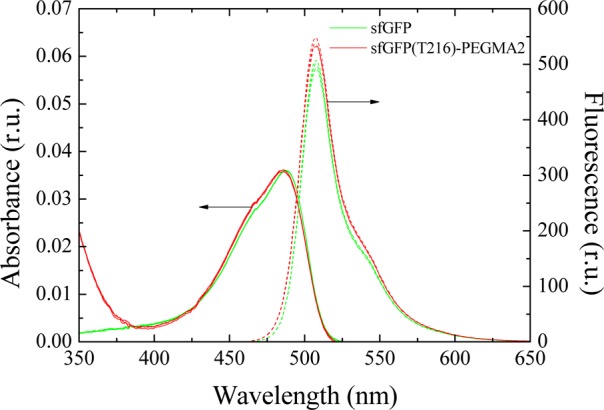
Normalized absorption and fluorescence emission spectra in relative units (r.u.) for the sfGFP and the sfGFP PEGMA-2 bioconjugate with pAzF at position T216 (sfGFP(T216)-PEGMA2), showing retention of the protein fluorescence upon conjugation.
We then set out to explore the properties of this series of bioconjugates. First, we wanted to investigate how the conjugation of a temperature-responsive polymer at different residues in the protein affects the overall bioconjugate thermal properties. PEGMA is a temperature-responsive polymer with its transition temperature depending on the PEG side chain length and the overall polymer molecular weight.89,95 Using turbidimetry, the cloud point of the neat polymers and all nine bioconjugates in Tris buffer was evaluated (Figure 5). As expected due to the hydrophobicity of the polymer end group, the cloud point of the low-molecular-weight PEGMA-1 was at 26.4 °C; however, PEGMA-2 and PEGMA-3 exhibited a hydrophilic–hydrophobic transition at higher temperatures (57.8 and 64.5 °C, respectively).
Figure 5.

Cloud point curves for the three PEGMA solutions (black lines: squares for PEGMA-1, circles for PEGMA-2, and triangles for PEGMA-3) and their corresponding bioconjugates with the grafting position being sfGFP(S2) (red lines), sfGFP(T216) (green lines), and both sfGFP(S2T216) (blue lines), sfGFP (purple line) are also shown for comparison. Note that all measurements are averages of three runs with a standard deviation of ±1 °C.
In the case of the proteins conjugated with PEGMA-1, the cloud point was found to be significantly higher than that of the neat polymer alone, which was attributed to the fact that the protein provides better water solubility than the end group of the polymer itself, thus rendering it more hydrophilic. For the PEGMA-2 and PEGMA-3 bioconjugates, the cloud point was slightly higher than that of the homopolymers. While the transition temperature of the bioconjugates varied from 61 to 67 °C there is a distinct effect on the observed transition temperature through variation of the polymer molecular weight and the conjugation site. As such, the shorter polymer (PEGMA-1) results in bioconjugates that regardless of the conjugation site become insoluble at almost the same temperature (63–65 °C). Increasing the molecular weight of the conjugated polymer (PEGMA-2) results in the hybrid that is conjugated at the S2 position (sfGFP(S2)-PEGMA2) to transition at a lower temperature, compared to that conjugated at the T216 position (sfGFP(T216)-PEGMA2) (ca. 4 °C lower). Although both positions are located in the flexible loops of the sfGFP barrel, we suspect that the local environment of conjugation affects the ability of the PEGMA chains to collapse upon heating above their cloud point. This is again observed when comparing the two conjugation sites for the larger (PEGMA-3) polymers (with a ca. 3 °C difference between sfGFP(S2)-PEGMA3 and sfGFP(T216)-PEGMA3). The consistently higher transition temperature for proteins conjugated at the T216 may be attributed to this site being located in a more highly charged region of the protein compared to the S2 site. Note that as expected the higher molecular weight polymer, PEGMA-3, always afforded bioconjugates with higher transition temperatures compared to the PEGMA-2 conjugates.
Interestingly, the transition temperature for the double-conjugated sfGFPs with the PEGMA-1 and PEGMA-2 polymers (sfGFP(S2T216)-PEGMA1 and sfGFP(S2T216)-PEGMA2) occurs at a temperature intermediate to the observed transition of the single modified protein bioconjugates. In contrast, the transition temperature for the higher molecular weight polymer (PEGMA-3) conjugated in two positions (sfGFP(S2T216)-PEGMA3) is slightly lower than that of the two single-functionalized proteins by ca. 1 °C (for sfGFP(S2)-PEGMA3) and 3 °C (for sfGFP(T216)-PEGMA3). This can be attributed to the two polymer chains reaching a critical molecular weight that allows them to interact and thus decrease the effective transition temperature, as seen in other similar bioconjugate systems.96
The increase in turbidity and the absence of macroscopic precipitation upon heating the bioconjugates above the transition temperature suggests the formation of dispersed aggregates whereby the hydrophobic part consists of the polymer and the hydrophilic is the protein segment of the bioconjugate. The bioconjugates were thus characterized by dynamic light scattering (DLS) over a range of temperatures (Figure 6), whereby upon heating the hydrodynamic size of the bioconjugates dramatically increased but the unmodified sfGFP retained its original size. This supports the hypothesis that due to the now hydrophobic character of the polymer and the amphiphilic character of the overall hybrid, the bioconjugates self-assemble at elevated temperature. It should however be noted that large aggregate populations were also observed by DLS—regardless of the temperature of the measurement (see SI, Figures S10–S11)—attributed to the presence of aggregates which were also observed in neat buffer. In an attempt to further confirm the formation of bioconjugate assemblies, the heated samples were analyzed by transmission electron microscopy (with the sample preparation taking place at 70 °C, see SI, Figure S12). Unfortunately, only large ill-defined aggregates could be identified which were attributed to the difficulty in sample preparation at elevated temperature.
Figure 6.
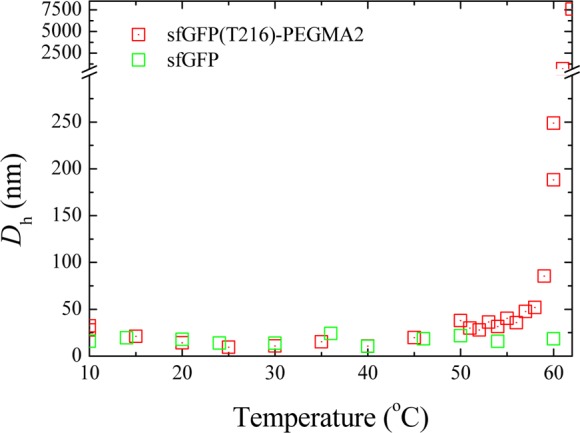
Dependence of the hydrodynamic diameter of sfGFP and the sfGFP(T216)-PEGMA2 bioconjugate on temperature, as determined by DLS analysis.
To gain more information on the solution structure of the protein–polymer bioconjugates, small-angle X-ray scattering (SAXS) experiments were conducted. Data was collected for the sfGFP alone, as well as all the bioconjugates at room and elevated temperature (at 25 and 65 °C). Measurements were performed using dilute solutions (in all cases less than 0.2 mg/mL) to minimize the amount of unwanted aggregation. Fitting analysis (see SI for details) was performed to determine the radius of gyration Rg and shape of the bioconjugate (see Tables S3 and S4). As expected, at 25 °C the bioconjugates all had a larger size than the sfGFP, and furthermore the size of the bioconjugates in solution increased as the molecular weight of the conjugated polymer increased.
A Kratky plot (q2I(q) vs q) for each sample was derived, from the SAXS data, in order to further analyze the bioconjugate morphology. Such plots are often used to emphasize the differences between compact objects such as globular, structured proteins and that of a random chain, such as an unfolded protein.97 A bell-shaped curve is obtained in the first case whereas a plateau is found for the second case, and depending on the local rigidity of the chain, an increase in slope as q increases may also be observed.97 Such a plot however suffers from limitations as it does not allow direct comparison of scattering profiles of objects of different sizes. Moreover, the Kratky plot of partially folded proteins still shows bell-shaped curves owing to the presence of structured regions in the protein. To obviate this problem, a dimensionless Kratky plot was utilized in this work: the intensity I(q) is normalized to the forward scattering intensity I(0), which allows comparison of samples of different molecular weights as I(0) is proportional to the molecular weight; q is normalized to the radius of gyration of the protein, which makes the angular scale independent of the protein size.98 Analysis of the dimensionless Kratky plots at 25 °C indicated that the conjugation of the polymer does not affect the structured domains of the protein for all of the PEGMA bioconjugates, as the plots at low x-axis values are similar before and after conjugation (see SI, Figure S13). The GFP plots show a symmetrical bell-shaped curve as well as a horizontal asymptote at high x-axis values, characteristic of a folded protein. The presence of more unstructured domains after conjugation is proposed as the plots for the bioconjugates appear to have a higher gradient at high qRg values (qRg > 3). By SAXS analysis, no significant difference in solution size or shape for the bioconjugates with different site modifications is observed (see SI, Table S3). However, the length of the polymer which is conjugated to the protein has an effect on the solution structure of the resultant bioconjugate, in that the wrapping of the bioconjugated polymers around the protein is more efficient for the longest polymer, PEGMA-3, as observed by an increase of the Rg and a more spherical morphology after bioconjugation (see SI, Table S3).
The dimensionless Kratky plots at elevated temperature 65 °C (close to or above the cloud point of the bioconjugates) show that the sfGFP is equally or more folded in its native form than when it is conjugated to the polymers (see SI, Figure S14). Moreover, the conjugation of the polymers increases the number of unstructured domains as expected for the conjugation of a polymer with a random coil conformation in a collapsed state. The bioconjugates display a more elongated morphology than the sfGFP at elevated temperature. As the sfGFP by itself does not exhibit a more elongated morphology, the elongation is attributed to the polymer chains. This was also confirmed from analysis of the SAXS curves of the polymers at different temperatures (see SI, Figure S15).
In summary, we have shown the successful incorporation of an azide-functional ncAA into sfGFP at multiple locations, synthesizing three sfGFP analogues which could be readily bioconjugated with one or two alkyne-functional PEGMA polymers. Our work described the combination of chemical and biological approaches to produce synthetic protein–polymer bioconjugates having new structures and reversible self-assembly properties. The resulting bioconjugates exhibited no loss in fluorescence, while an increase in temperature resulted in the reversible increase in turbidity of the bioconjugates solutions, suggesting the formation of aggregates. Additionally, the transition temperature was found to be affected by the molecular weight of the polymer as well as the location of the polymer conjugation. Finally, we demonstrated that using the same responsive polymer and conjugating to different sites of a protein leads to no difference in bioconjugate shape, but it does lead to a discernible difference in thermal properties for the bioconjugates. Our work thus highlights that site-selective polymer conjugation, which is possible using protein engineering alongside common conjugation approaches, can be used to fine-tune functional properties of polymer–protein bioconjugates.
Improvements in modified protein yields will open the way to even broader applications. For example, amber suppression technologies in vivo are still generally limited to expression of proteins containing ncAAs incorporated into a single instance or few instances within a polypeptide chain.99,100 New genomically recoded strains101 lacking release factor 1, cell-free approaches,75,91,93,102,103 and the ability to site-specifically incorporate multiple types of ncAAs per protein with high efficiencies promise to make possible novel synthesis approaches for unique polymeric materials with atomic-scale resolution over composition, architecture, and functionality.
Acknowledgments
We acknowledge the EPSRC, University of Warwick, the National Science Foundation (MCB-0943393), the DARPA YFA Program (N66001-11-1-4137), and the NSF Materials Network Grant (DMR - 1108350) for funding. The research leading to these results has received funding from the European Research Council under the European Union’s Seventh Framework Programme (FP/2007-2013)/ERC Grant Agreement n. 615142. M.C.J. is a Packard Fellow for Science and Engineering. We thank Tom Lawton with assistance in the preparation of some of the figures.
Supporting Information Available
Materials and methods as well as supplementary tables and figures related to the synthesis and characterization of all bioconjugates (PAGE, cloud point, DLS, TEM, SAXS analysis). The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bioconjchem.5b00264.
Author Contributions
# Dafni Moatsou and Jian Li equally contributed to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Abuchowski A.; van Es T.; Palczuk N. C.; Davis F. F. (1977) Alteration of immunological properties of bovine serum albumin by covalent attachment of polyethylene glycol. J. Biol. Chem. 252, 3578–3581. [PubMed] [Google Scholar]
- Abuchowski A.; McCoy J. R.; Palczuk N. C.; van Es T.; Davis F. F. (1977) Effect of covalent attachment of polyethylene glycol on immunogenicity and circulating life of bovine liver catalase. J. Biol. Chem. 252, 3582–3586. [PubMed] [Google Scholar]
- Caliceti P.; Veronese F. M. (2003) Pharmacokinetic and biodistribution properties of poly(ethylene glycol)–protein conjugates. Adv. Drug Delivery Rev. 55, 1261–1277. [DOI] [PubMed] [Google Scholar]
- Alconcel S. N. S.; Baas A. S.; Maynard H. D. (2011) FDA-approved poly(ethylene glycol)-protein conjugate drugs. Polym. Chem. 2, 1442–1448. [Google Scholar]
- Fuertges F.; Abuchowski A. (1990) The clinical efficacy of poly(ethylene glycol)-modified proteins. J. Controlled Release 11, 139–148. [Google Scholar]
- Veronese F. M. (2001) Peptide and protein PEGylation: a review of problems and solutions. Biomaterials 22, 405–417. [DOI] [PubMed] [Google Scholar]
- Roberts M. J.; Bentley M. D.; Harris J. M. (2002) Chemistry for peptide and protein PEGylation. Adv. Drug Delivery Rev. 54, 459–476. [DOI] [PubMed] [Google Scholar]
- Knop K.; Hoogenboom R.; Fischer D.; Schubert U. S. (2010) Poly(ethylene glycol) in Drug Delivery: Pros and Cons as Well as Potential Alternatives. Angew. Chem., Int. Ed. 49, 6288–6308. [DOI] [PubMed] [Google Scholar]
- Harris J. M.; Chess R. B. (2003) Effect of pegylation on pharmaceuticals. Nat. Rev. Drug Discovery 2, 214–221. [DOI] [PubMed] [Google Scholar]
- Veronese F. M.; Pasut G. (2005) PEGylation, successful approach to drug delivery. Drug Discovery Today 10, 1451–1458. [DOI] [PubMed] [Google Scholar]
- Joralemon M. J.; McRae S.; Emrick T. (2010) PEGylated polymers for medicine: from conjugation to self-assembled systems. Chem. Commun. 46, 1377–1393. [DOI] [PubMed] [Google Scholar]
- Veronese F. M.; Caliceti P.; Schiavon O. (1997) Branched and Linear Poly(Ethylene Glycol): Influence of the Polymer Structure on Enzymological, Pharmacokinetic, and Immunological Properties of Protein Conjugates. J. Bioact. Compat. Polym. 12, 196–207. [Google Scholar]
- Pelegri-O’Day E. M.; Lin E.-W.; Maynard H. D. (2014) Therapeutic Protein–Polymer Conjugates: Advancing Beyond PEGylation. J. Am. Chem. Soc. 136, 14323–14332. [DOI] [PubMed] [Google Scholar]
- Cobo I.; Li M.; Sumerlin B. S.; Perrier S. (2015) Smart hybrid materials by conjugation of responsive polymers to biomacromolecules. Nat. Mater. 14, 143–159. [DOI] [PubMed] [Google Scholar]
- Gil E. S.; Hudson S. M. (2004) Stimuli-reponsive polymers and their bioconjugates. Prog. Polym. Sci. 29, 1173–1222. [Google Scholar]
- Stayton P. S.; Shimoboji T.; Long C.; Chilkoti A.; Ghen G.; Harris J. M.; Hoffman A. S. (1995) Control of protein-ligand recognition using a stimuli-responsive polymer. Nature 378, 472–474. [DOI] [PubMed] [Google Scholar]
- Krishna O. D.; Kiick K. L. (2010) Protein- and peptide-modified synthetic polymeric biomaterials. Pept. Sci. 94, 32–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirks A. J.; Nolte R. J. M.; Cornelissen J. J. L. M. (2008) Protein–Polymer Hybrid Amphiphiles. Adv. Mater. 20, 3953–3957. [Google Scholar]
- Velonia K. (2010) Protein-polymer amphiphilic chimeras: recent advances and future challenges. Polym. Chem. 1, 944–952. [Google Scholar]
- Hoffman A. S.; Stayton P. S. (2007) Conjugates of stimuli-responsive polymers and proteins. Prog. Polym. Sci. 32, 922–932. [Google Scholar]
- Hoffman A. S.; Stayton P. S. (2004) Bioconjugates of smart polymers and proteins: synthesis and applications. Macromol. Symp. 207, 139–152. [Google Scholar]
- Borner H. G.; Schlaad H. (2007) Bioinspired functional block copolymers. Soft Matter 3, 394–408. [DOI] [PubMed] [Google Scholar]
- Li H.; Li M.; Yu X.; Bapat A. P.; Sumerlin B. S. (2011) Block copolymer conjugates prepared by sequentially grafting from proteinsvia RAFT. Polym. Chem. 2, 1531–1535. [Google Scholar]
- Boyer C.; Bulmus V.; Liu J.; Davis T. P.; Stenzel M. H.; Barner-Kowollik C. (2007) Well-Defined Protein–Polymer Conjugates via in Situ RAFT Polymerization. J. Am. Chem. Soc. 129, 7145–7154. [DOI] [PubMed] [Google Scholar]
- Chen G.; Hoffman A. S. (1993) Preparation and properties of thermoreversible, phase-separating enzyme-oligo(N-isopropylacrylamide) conjugates. Bioconjugate Chem. 4, 509–514. [DOI] [PubMed] [Google Scholar]
- Ding Z.; Chen G.; Hoffman A. S. (1996) Synthesis and Purification of Thermally Sensitive Oligomer–Enzyme Conjugates of Poly(N-isopropylacrylamide)–Trypsin. Bioconjugate Chem. 7, 121–125. [DOI] [PubMed] [Google Scholar]
- Bulmus V.; Ding Z.; Long C. J.; Stayton P. S.; Hoffman A. S. (1999) Site-Specific Polymer–Streptavidin Bioconjugate for pH-Controlled Binding and Triggered Release of Biotin. Bioconjugate Chem. 11, 78–83. [DOI] [PubMed] [Google Scholar]
- Ding Z.; Long C. J.; Hayashi Y.; Bulmus E. V.; Hoffman A. S.; Stayton P. S. (1999) Temperature Control of Biotin Binding and Release with A Streptavidin-Poly(N-isopropylacrylamide) Site-Specific Conjugate. Bioconjugate Chem. 10, 395–400. [DOI] [PubMed] [Google Scholar]
- Fong R. B.; Ding Z.; Long C. J.; Hoffman A. S.; Stayton P. S. (1999) Thermoprecipitation of Streptavidin via Oligonucleotide-Mediated Self-Assembly with Poly(N-isopropylacrylamide). Bioconjugate Chem. 10, 720–725. [DOI] [PubMed] [Google Scholar]
- Le Droumaguet B.; Mantovani G.; Haddleton D. M.; Velonia K. (2007) Formation of giant amphiphiles by post-functionalization of hydrophilic protein-polymer conjugates. J. Mater. Chem. 17, 1916–1922. [Google Scholar]
- Hannink J. M.; Cornelissen J. J. L. M.; Farrera J. A.; Foubert P.; De Schryver F. C.; Sommerdijk N. A. J. M.; Nolte R. J. M. (2001) Protein–Polymer Hybrid Amphiphiles. Angew. Chem., Int. Ed. 40, 4732–4734. [PubMed] [Google Scholar]
- Dirks A. J.; van Berkel S. S.; Hatzakis N. S.; Opsteen J. A.; van Delft F. L.; Cornelissen J. J. L. M.; Rowan A. E.; van Hest J. C. M.; Rutjes F. P. J. T.; Nolte R. J. M. (2005) Preparation of biohybrid amphiphiles via the copper catalysed Huisgen [3 + 2] dipolar cycloaddition reaction. Chem. Commun. 4172–4174. [DOI] [PubMed] [Google Scholar]
- Maeda H. (2001) SMANCS and polymer-conjugated macromolecular drugs: advantages in cancer chemotherapy. Adv. Drug Delivery Rev. 46, 169–185. [DOI] [PubMed] [Google Scholar]
- Boerakker M. J.; Hannink J. M.; Bomans P. H. H.; Frederik P. M.; Nolte R. J. M.; Meijer E. M.; Sommerdijk N. A. J. M. (2002) Giant Amphiphiles by Cofactor Reconstitution. Angew. Chem., Int. Ed. 41, 4239–4241. [DOI] [PubMed] [Google Scholar]
- Velonia K.; Rowan A. E.; Nolte R. J. M. (2002) Lipase Polystyrene Giant Amphiphiles. J. Am. Chem. Soc. 124, 4224–4225. [DOI] [PubMed] [Google Scholar]
- Ding Z.; Chen G.; Hoffman A. S. (1998) Unusual properties of thermally sensitive oligomer–enzyme conjugates of poly(N-isopropylacrylamide)–trypsin. J. Biomed. Mater. Res. 39, 498–505. [DOI] [PubMed] [Google Scholar]
- Shimoboji T.; Larenas E.; Fowler T.; Kulkarni S.; Hoffman A. S.; Stayton P. S. (2002) Photoresponsive polymer–enzyme switches. Proc. Natl. Acad. Sci. U.S.A. 99, 16592–16596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi M.; Kobayashi M.; Fujii M. (1989) Properties of a reversible soluble–insoluble cellulase and its application to repeated hydrolysis of crystalline cellulose. Biotechnol. Bioeng. 34, 1092–1097. [DOI] [PubMed] [Google Scholar]
- Tae Gwan P.; Hoffman A. S. (1993) Synthesis and characterization of a soluble, temperature-sensitive polymer-conjugated enzyme. J. Biomater. Sci., Polym. Ed. 4, 493–504. [DOI] [PubMed] [Google Scholar]
- Boerakker M. J.; Botterhuis N. E.; Bomans P. H. H.; Frederik P. M.; Meijer E. M.; Nolte R. J. M.; Sommerdijk N. A. J. M. (2006) Aggregation Behavior of Giant Amphiphiles Prepared by Cofactor Reconstitution. Chem.—Eur. J. 12, 6071–6080. [DOI] [PubMed] [Google Scholar]
- Reynhout I. C.; Cornelissen J. J. L. M.; Nolte R. J. M. (2007) Self-Assembled Architectures from Biohybrid Triblock Copolymers. J. Am. Chem. Soc. 129, 2327–2332. [DOI] [PubMed] [Google Scholar]
- Li M.; De P.; Gondi S. R.; Sumerlin B. S. (2008) Responsive Polymer-Protein Bioconjugates Prepared by RAFT Polymerization and Copper-Catalyzed Azide-Alkyne Click Chemistry. Macromol. Rapid Commun. 29, 1172–1176. [Google Scholar]
- Tao L.; Mantovani G.; Lecolley F.; Haddleton D. M. (2004) α-Aldehyde Terminally Functional Methacrylic Polymers from Living Radical Polymerization: Application in Protein Conjugation “Pegylation. J. Am. Chem. Soc. 126, 13220–13221. [DOI] [PubMed] [Google Scholar]
- Lecolley F.; Tao L.; Mantovani G.; Durkin I.; Lautru S.; Haddleton D. M. (2004) A new approach to bioconjugates for proteins and peptides (″pegylation″) utilising living radical polymerisation. Chem. Commun. 2026–2027. [DOI] [PubMed] [Google Scholar]
- Heredia K. L.; Bontempo D.; Ly T.; Byers J. T.; Halstenberg S.; Maynard H. D. (2005) In Situ Preparation of Protein–“Smart” Polymer Conjugates with Retention of Bioactivity. J. Am. Chem. Soc. 127, 16955–16960. [DOI] [PubMed] [Google Scholar]
- Gilmore J. M.; Scheck R. A.; Esser-Kahn A. P.; Joshi N. S.; Francis M. B. (2006) N-Terminal Protein Modification through a Biomimetic Transamination Reaction. Angew. Chem., Int. Ed. 45, 5307–5311. [DOI] [PubMed] [Google Scholar]
- Dixon H. B. F. (1984) N-terminal modification of proteins—a review. J. Protein Chem. 3, 99–108. [Google Scholar]
- Chilkoti A.; Chen G.; Stayton P. S.; Hoffman A. S. (1994) Site-Specific Conjugation of a Temperature-Sensitive Polymer to a Genetically Engineered Protein. Bioconjugate Chem. 5, 504–507. [DOI] [PubMed] [Google Scholar]
- Shimoboji T.; Ding Z.; Stayton P. S.; Hoffman A. S. (2001) Mechanistic Investigation of Smart Polymer–Protein Conjugates. Bioconjugate Chem. 12, 314–319. [DOI] [PubMed] [Google Scholar]
- Ding Z.; Fong R. B.; Long C. J.; Stayton P. S.; Hoffman A. S. (2001) Size-dependent control of the binding of biotinylated proteins to streptavidin using a polymer shield. Nature 411, 59–62. [DOI] [PubMed] [Google Scholar]
- Schoffelen S.; van Eldijk M. B.; Rooijakkers B.; Raijmakers R.; Heck A. J. R.; van Hest J. C. M. (2011) Metal-free and pH-controlled introduction of azides in proteins. Chem. Sci. 2, 701–705. [Google Scholar]
- Lutz J.-F.; Börner H. G. (2008) Modern trends in polymer bioconjugates design. Prog. Polym. Sci. 33, 1–39. [Google Scholar]
- Heredia K. L.; Maynard H. D. (2007) Synthesis of protein-polymer conjugates. Org. Biomol. Chem. 5, 45–53. [DOI] [PubMed] [Google Scholar]
- Johnson R. P.; John J. V.; Kim I. (2013) Recent developments in polymer–block–polypeptide and protein–polymer bioconjugate hybrid materials. Eur. Polym. J. 49, 2925–2948. [Google Scholar]
- Boyer C.; Huang X.; Whittaker M. R.; Bulmus V.; Davis T. P. (2011) An overview of protein-polymer particles. Soft Matter 7, 1599–1614. [Google Scholar]
- Canalle L. A.; Lowik D. W. P. M.; van Hest J. C. M. (2010) Polypeptide-polymer bioconjugates. Chem. Soc. Rev. 39, 329–353. [DOI] [PubMed] [Google Scholar]
- Nicolas J.; Mantovani G.; Haddleton D. M. (2007) Living Radical Polymerization as a Tool for the Synthesis of Polymer-Protein/Peptide Bioconjugates. Macromol. Rapid Commun. 28, 1083–1111. [Google Scholar]
- Le Droumaguet B.; Nicolas J. (2010) Recent advances in the design of bioconjugates from controlled/living radical polymerization. Polym. Chem. 1, 563–598. [Google Scholar]
- Broyer R. M.; Grover G. N.; Maynard H. D. (2011) Emerging synthetic approaches for protein-polymer conjugations. Chem. Commun. 47, 2212–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klok H.-A. (2005) Biological–synthetic hybrid block copolymers: Combining the best from two worlds. J. Polym. Sci., Part A: Polym. Chem. 43, 1–17. [Google Scholar]
- Thordarson P.; Le Droumaguet B.; Velonia K. (2006) Well-defined protein–polymer conjugates—synthesis and potential applications. Appl. Microbiol. Biotechnol. 73, 243–254. [DOI] [PubMed] [Google Scholar]
- Gauthier M. A.; Klok H.-A. (2008) Peptide/protein-polymer conjugates: synthetic strategies and design concepts. Chem. Commun. 2591–2611. [DOI] [PubMed] [Google Scholar]
- Borchmann D. E.; Carberry T. P.; Weck M. (2014) “Bio”-Macromolecules: Polymer-Protein Conjugates as Emerging Scaffolds for Therapeutics. Macromol. Rapid Commun. 35, 27–43. [DOI] [PubMed] [Google Scholar]
- Oshiba Y.; Tamaki T.; Ohashi H.; Hirakawa H.; Yamaguchi S.; Nagamune T.; Yamaguchi T. (2013) Effect of length of molecular recognition moiety on enzymatic activity switching. J. Biosci. Bioeng. 116, 433–437. [DOI] [PubMed] [Google Scholar]
- Kochendoerfer G. G.; Chen S.-Y.; Mao F.; Cressman S.; Traviglia S.; Shao H.; Hunter C. L.; Low D. W.; Cagle E. N.; Carnevali M.; et al. (2003) Design and Chemical Synthesis of a Homogeneous Polymer-Modified Erythropoiesis Protein. Science 299, 884–887. [DOI] [PubMed] [Google Scholar]
- Dumas A.; Lercher L.; Spicer C. D.; Davis B. G. (2015) Designing logical codon reassignment - Expanding the chemistry in biology. Chem. Sci. 6, 50–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Schultz P. G. (2005) Expanding the Genetic Code. Angew. Chem., Int. Ed. 44, 34–66. [DOI] [PubMed] [Google Scholar]
- Wang P.; Tang Y.; Tirrell D. A. (2003) Incorporation of Trifluoroisoleucine into Proteins in Vivo. J. Am. Chem. Soc. 125, 6900–6906. [DOI] [PubMed] [Google Scholar]
- Tang Y.; Wang P.; Van Deventer J. A.; Link A. J.; Tirrell D. A. (2009) Introduction of an Aliphatic Ketone into Recombinant Proteins in a Bacterial Strain that Overexpresses an Editing-Impaired Leucyl-tRNA Synthetase. ChemBioChem 10, 2188–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J. A.; Lu Y. Y.; Van Deventer J. A.; Tirrell D. A. (2010) Residue-specific incorporation of non-canonical amino acids into proteins: recent developments and applications. Curr. Opin. Chem. Biol. 14, 774–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y.; Tirrell D. A. (2002) Attenuation of the Editing Activity of the Escherichia coli Leucyl-tRNA Synthetase Allows Incorporation of Novel Amino Acids into Proteins in Vivo. Biochemistry 41, 10635–10645. [DOI] [PubMed] [Google Scholar]
- Kiick K. L.; Saxon E.; Tirrell D. A.; Bertozzi C. R. (2002) Incorporation of azides into recombinant proteins for chemoselective modification by the Staudinger ligation. Proc. Natl. Acad. Sci. U.S.A. 99, 19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. C.; Schultz P. G. (2010) Adding New Chemistries to the Genetic Code. Annu. Rev. Biochem. 79, 413–444. [DOI] [PubMed] [Google Scholar]
- Chin J. W. (2014) Expanding and Reprogramming the Genetic Code of Cells and Animals. Annu. Rev. Biochem. 83, 379–408. [DOI] [PubMed] [Google Scholar]
- Hong S. H., Kwon Y.-C., and Jewett M. C. (2014) Non-standard amino acid incorporation into proteins using Escherichia coli cell-free protein synthesis. Front. Chem. 2, DOI: 10.3389/fchem.2014.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donoghue P.; Ling J.; Wang Y.-S.; Soll D. (2013) Upgrading protein synthesis for synthetic biology. Nat. Chem. Biol. 9, 594–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb H. C.; Finn M. G.; Sharpless K. B. (2001) Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem., Int. Ed. 40, 2004–2021. [DOI] [PubMed] [Google Scholar]
- Rostovtsev V. V.; Green L. G.; Fokin V. V.; Sharpless K. B. (2002) A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem., Int. Ed. 41, 2596–2599. [DOI] [PubMed] [Google Scholar]
- Huisgen R. (1963) 1,3-Dipolar Cycloadditions. Past and Future. Angew. Chem., Int. Ed. 2, 565–598. [Google Scholar]
- Huisgen R. (1963) Kinetics and Mechanism of 1,3-Dipolar Cycloadditions. Angew. Chem., Int. Ed. 2, 633–645. [Google Scholar]
- Schoffelen S.; Lambermon M. H. L.; Eldijk M. B. v.; Hest J. C. M. v. (2008) Site-Specific Modification of Candida antarctica Lipase B via Residue-Specific Incorporation of a Non-Canonical Amino Acid. Bioconjugate Chem. 19, 1127–1131. [DOI] [PubMed] [Google Scholar]
- Reddington S. C.; Tippmann E. M.; Dafydd Jones D. (2012) Residue choice defines efficiency and influence of bioorthogonal protein modification via genetically encoded strain promoted Click chemistry. Chem. Commun. 48, 8419–8421. [DOI] [PubMed] [Google Scholar]
- Peeler J. C.; Woodman B. F.; Averick S.; Miyake-Stoner S. J.; Stokes A. L.; Hess K. R.; Matyjaszewski K.; Mehl R. A. (2010) Genetically Encoded Initiator for Polymer Growth from Proteins. J. Am. Chem. Soc. 132, 13575–13577. [DOI] [PubMed] [Google Scholar]
- Bulmus V. (2011) RAFT polymerization mediated bioconjugation strategies. Polym. Chem. 2, 1463–1472. [Google Scholar]
- Longo J.; Yao C.; Rios C.; Chau N. T. T.; Boulmedais F.; Hemmerle J.; Lavalle P.; Schiller S. M.; Schaaf P.; Jierry L. (2015) Reversible biomechano-responsive surface based on green fluorescent protein genetically modified with unnatural amino acids. Chem. Commun. 51, 232–235. [DOI] [PubMed] [Google Scholar]
- Lavigueur C.; Garcia J. G.; Hendriks L.; Hoogenboom R.; Cornelissen J. J. L. M.; Nolte R. J. M. (2011) Thermoresponsive giant biohybrid amphiphiles. Polym. Chem. 2, 333–340. [Google Scholar]
- Xia Y.; Tang S.; Olsen B. D. (2013) Site-specific conjugation of RAFT polymers to proteins via expressed protein ligation. Chem. Commun. 49, 2566–2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Averick S.; Karácsony O.; Mohin J.; Yong X.; Moellers N. M.; Woodman B. F.; Zhu W.; Mehl R. A.; Balazs A. C.; Kowalewski T.; et al. (2014) Cooperative, Reversible Self-Assembly of Covalently Pre-Linked Proteins into Giant Fibrous Structures. Angew. Chem., Int. Ed. 53, 8050–8055. [DOI] [PubMed] [Google Scholar]
- Lutz J.-F. (2008) Polymerization of oligo(ethylene glycol) (meth)acrylates: Toward new generations of smart biocompatible materials. J. Polym. Sci., Part A: Polym. Chem. 46, 3459–3470. [Google Scholar]
- Young T. S.; Ahmad I.; Yin J. A.; Schultz P. G. (2010) An Enhanced System for Unnatural Amino Acid Mutagenesis in E. coli. J. Mol. Biol. 395, 361–374. [DOI] [PubMed] [Google Scholar]
- Hong S. H.; Kwon Y.-C.; Martin R. W.; Des Soye B. J.; de Paz A. M.; Swonger K. N.; Ntai I.; Kelleher N. L.; Jewett M. C. (2015) Improving Cell-Free Protein Synthesis through Genome Engineering of Escherichia coli Lacking Release Factor 1. ChemBioChem 16, 844–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De P.; Li M.; Gondi S. R.; Sumerlin B. S. (2008) Temperature-Regulated Activity of Responsive Polymer–Protein Conjugates Prepared by Grafting-from via RAFT Polymerization. J. Am. Chem. Soc. 130, 11288–11289. [DOI] [PubMed] [Google Scholar]
- Albayrak C.; Swartz J. R. (2014) Direct Polymerization of Proteins. ACS Synth. Biol. 3, 353–362. [DOI] [PubMed] [Google Scholar]
- Würth C.; Grabolle M.; Pauli J.; Spieles M.; Resch-Genger U. (2013) Relative and absolute determination of fluorescence quantum yields of transparent samples. Nat. Protoc. 8, 1535–1550. [DOI] [PubMed] [Google Scholar]
- Bebis K.; Jones M. W.; Haddleton D. M.; Gibson M. I. (2011) Thermoresponsive behaviour of poly[(oligo(ethyleneglycol methacrylate)]s and their protein conjugates: importance of concentration and solvent system. Polym. Chem. 2, 975–982. [Google Scholar]
- Liu M.; Tirino P.; Radivojevic M.; Phillips D. J.; Gibson M. I.; Leroux J.-C.; Gauthier M. A. (2013) Molecular Sieving on the Surface of a Protein Provides Protection Without Loss of Activity. Adv. Funct. Mater. 23, 2007–2015. [Google Scholar]
- Receveur-Brechot V.; Durand D. (2012) How Random are Intrinsically Disordered Proteins? A Small Angle Scattering Perspective. Curr. Protein Pept. Sci. 13, 55–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand D.; Vivès C.; Cannella D.; Pérez J.; Pebay-Peyroula E.; Vachette P.; Fieschi F. (2010) NADPH oxidase activator p67phox behaves in solution as a multidomain protein with semi-flexible linkers. J. Struct. Biol. 169, 45–53. [DOI] [PubMed] [Google Scholar]
- O’Donoghue P.; Ling J.; Wang Y. S.; Söll D. (2013) Upgrading protein synthesis for synthetic biology. Nat. Chem. Biol. 9, 594–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Liu C. C. (2014) Biological Applications of Expanded Genetic Codes. ChemBioChem. 15, 2335–2341. [DOI] [PubMed] [Google Scholar]
- Lajoie M. J.; Rovner A. J.; Goodman D. B.; Aerni H.-R.; Haimovich A. D.; Kuznetsov G.; Mercer J. A.; Wang H. H.; Carr P. A.; Mosberg J. A.; et al. (2013) Genomically Recoded Organisms Expand Biological Functions. Science 342, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S. H.; Ntai I.; Haimovich A. D.; Kelleher N. L.; Isaacs F. J.; Jewett M. C. (2014) Cell-free Protein Synthesis from a Release Factor 1 Deficient Escherichia coli Activates Efficient and Multiple Site-specific Nonstandard Amino Acid Incorporation. ACS Synth. Biol. 3, 398–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S. H.; Kwon Y.-C.; Martin R. W.; Des Soye B. J.; de Paz A. M.; Swonger K. N.; Ntai I.; Kelleher N. L.; Jewett M. C. (2015) Improving Cell-Free Protein Synthesis through Genome Engineering of Escherichia coli Lacking Release Factor 1. ChemBioChem 16, 844–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.