

Abstract
Background
Sustained changes in network activity cause homeostatic synaptic plasticity in part by altering the postsynaptic accumulation of N-methyl-D-aspartate receptors (NMDAR) and α-amino-3-hydroxyle-5-methyl-4-isoxazolepropionic acid receptors (AMPAR), which are primary mediators of excitatory synaptic transmission. A key trafficking modulator of NMDAR and AMPAR is STriatal-Enriched protein tyrosine Phosphatase (STEP61) that opposes synaptic strengthening through dephosphorylation of NMDAR subunit GluN2B and AMPAR subunit GluA2. However, the role of STEP61 in homeostatic synaptic plasticity is unknown.
Findings
We demonstrate here that prolonged activity blockade leads to synaptic scaling, and a concurrent decrease in STEP61 level and activity in rat dissociated hippocampal cultured neurons. Consistent with STEP61 reduction, prolonged activity blockade enhances the tyrosine phosphorylation of GluN2B and GluA2 whereas increasing STEP61 activity blocks this regulation and synaptic scaling. Conversely, prolonged activity enhancement increases STEP61 level and activity, and reduces the tyrosine phosphorylation and level of GluN2B as well as GluA2 expression in a STEP61–dependent manner.
Conclusions
Given that STEP61-mediated dephosphorylation of GluN2B and GluA2 leads to their internalization, our results collectively suggest that activity-dependent regulation of STEP61 and its substrates GluN2B and GluA2 may contribute to homeostatic stabilization of excitatory synapses.
Electronic supplementary material
The online version of this article (doi:10.1186/s13041-015-0148-4) contains supplementary material, which is available to authorized users.
Keywords: STEP, GluN2B, GluA2, Tyrosine phosphorylation, Tetrodotoxin, Bicuculline, Hippocampal neurons, Homeostatic plasticity, Synaptic scaling
Findings
Background
In response to sustained changes in neuronal activity, homeostatic synaptic plasticity maintains synaptic strength and flexibility within physiological limit. This plasticity is expressed in part by dynamic changes in the postsynaptic levels of NMDARs and AMPARs that mediate excitatory synaptic transmission [1]. A key trafficking modulator of both NMDAR and AMPAR is STEP61, a protein tyrosine (Tyr) phosphatase in the central nervous system that has two main alternatively spliced forms, the cytosolic STEP46 and the membrane-associated STEP61 [2]. Tightly associated with the postsynaptic density, STEP61 regulates the Tyr phosphorylation and surface density of NMDARs and AMPARs [3–6]. This regulation contributes to Hebbian long-term potentiation [4, 7] and several neuropsychiatric disorders most notably Alzheimer’s disease [4] and Fragile X-syndrome [2].
We have previously identified mRNA transcripts whose expressions are regulated by prolonged activity perturbation [8] due to a critical role of transcription in homeostatic synaptic plasticity [9, 10]. Of these activity-regulated transcripts, we identified PTPN5 that encodes STEP [8]. The present study investigated whether STEP61 contributes to homeostatic synaptic plasticity.
Results and discussion
Prolonged alterations of hippocampal network activity regulate STEP61 level and activity
Prolonged blockade of network activity for 48 h with the sodium channel blocker tetrodotoxin (TTX) induced synaptic scaling in dissociated hippocampal cultured neurons as demonstrated previously [11, 12] (Fig. 1a–d), and reduced STEP61 mRNA and protein expression compared to CTL treatment (Fig. 1e, f). Conversely, prolonged activity enhancement for 48 h using the GABAA receptor antagonist bicuculline (BC) increased STEP61 protein level (Fig. 1g), but did not alter its mRNA level and the miniature excitatory postsynaptic current (mEPSC) (Fig. 1a–e).
Fig. 1.
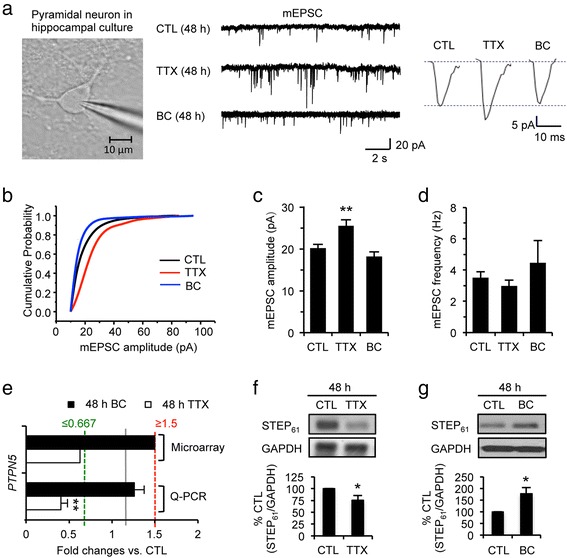
Prolonged alterations of hippocampal network activity induce homeostatic synaptic plasticity and regulate STEP61 level. a–d Whole-cell patch clamp recording of miniature excitatory postsynaptic currents (mEPSCs) from rat dissociated hippocampal neurons cultured in high density that were treated for 48 h with vehicle control (CTL, 0.1 % H2O), TTX (1 μM), or BC (20 μM) at 12–14 days in vitro. a Representative traces of mEPSCs. b Normalized cumulative fraction of the mEPSC amplitudes. c–d Summary plots of average mEPSC amplitudes (c) and frequencies (d) for CTL (n = 19), TTX (n = 20), or BC (n = 16). TTX treatment for 48 h induced synaptic scaling whereas 48 h BC treatment had no effect. e Microarray (n = 4) and QPCR (n = 5) analyses revealed that 48 h application of TTX but not BC reduced the expression of PTPN5, which encodes STEP. f–g Immunoblot analysis of STEP61 following 48 h administration of CTL, TTX (F), BC (g) (n = 9 per treatment). Data shown represent the mean ± SEM (*p < 0.05; **p < 0.01)
To test whether prolonged TTX or BC treatment affects STEP61 activity, we examined the phosphorylation of STEP61 at Ser221 within its kinase-interactive motif domain, which prevents STEP61 interaction with all known substrates (Fig. 2a) [13]. TTX treatment for 36–48 h enhanced Ser221–phosphorylation of STEP61, indicating decreased STEP61 activity (Fig. 2b, d). In contrast, 36–48 h BC treatment reduced Ser221–phosphorylation, indicating increased STEP61 activity (Fig. 2c, d).
Fig. 2.
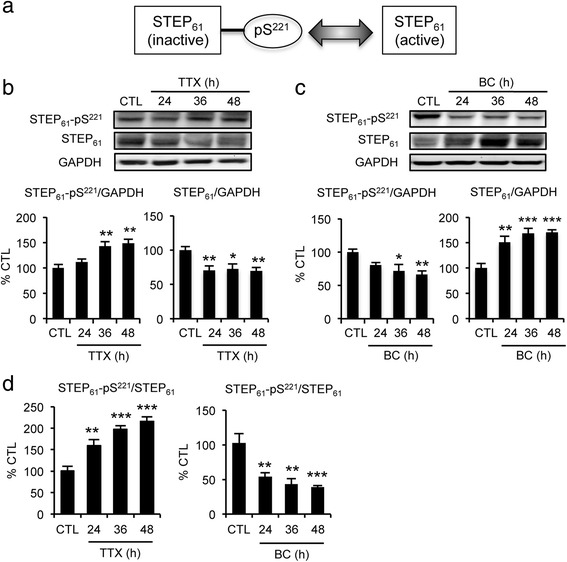
Prolonged alterations of hippocampal network activity regulate STEP61 activity. a A schematic depicting the regulation of STEP61 activity by its phosphorylation at Ser221 within its kinase-interactive motif, a binding site for all STEP substrates. b–d Immunoblot analysis of STEP61 and Ser221–phosphorylated STEP61 (STEP61-pS221) in hippocampal cultured neurons following CTL, TTX, or BC treatment for 24–48 h (n = 3 per treatment). b, d Prolonged TTX treatment reduced STEP61 protein level and activity. c, d Prolonged BC treatment enhanced STEP61 protein level and activity. d The relative phosphorylation state of STEP61 as calculated by the ratio of STEP61-pS221 level over total STEP61 level. Data shown represent the mean ± SEM (*p < 0.05; **p < 0.01; ***p < 0.005)
Prolonged alterations of hippocampal network activity regulate Tyr-phosphorylation of GluN2B and GluA2 in a STEP61-dependent manner
STEP61 dephosphorylates the NMDAR subunit GluN2B at Tyr1472 [5, 6] and reduces Tyr-phosphorylation of the AMPAR subunit GluA2 following group 1 metabotropic glutamate receptor (mGluR) stimulation [3]. Although the specific Tyr residues on GluA2 regulated by STEP61 are unknown, the GluA2 phosphorylation state at Tyr869, Tyr873, and Tyr876 (3Tyr) regulates AMPAR trafficking [14]. To determine if the TTX- or BC-induced changes in STEP61 alter Tyr-phosphorylation of GluN2B and GluA2, we performed immunoblot analyses using specific antibodies to phosphorylated Tyr1472 of GluN2B [6] and phosphorylated 3Tyr of GluA2 [14] (Fig. 3).
Fig. 3.
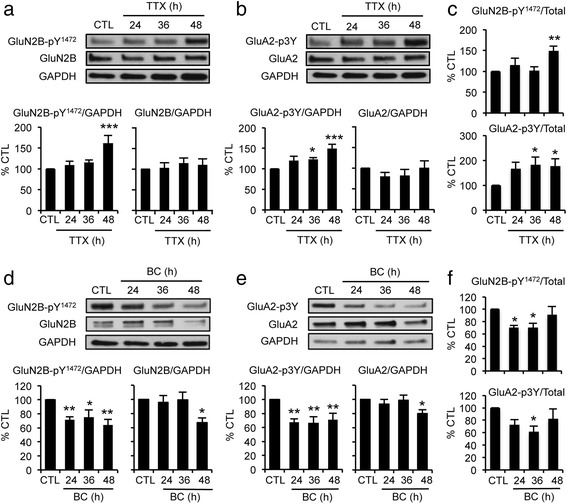
Prolonged alterations of hippocampal network activity regulate Tyr-phosphorylation and levels of GluN2B and GluA2. Immunoblot analysis of hippocampal cultured neurons that were treated for 48 h with CTL, 24–48 h TTX (a–c), or 24–48 h BC treatment (d–f) (n = 6 per treatment). a–c Prolonged TTX treatment increased the level of Tyr1472–phosphorylated GluN2B (GluN2B-pY1472) (a) and the level of GluA2 that were phosphorylated at Tyr 869, Tyr 873, and Tyr 876 (3Tyr) (GluA2-p3Y) (b). d–f Prolonged BC treatment decreased the levels of GluN2B-pY1472 (d) and GluA2-p3Y (e). Total GluN2B and GluA2 levels were reduced at 48 h BC application. c, f The relative phosphorylation state of GluN2B and GluA2 as calculated by the ratio of phosphorylated proteins over total proteins followed by normalization to GAPDH. Data shown represent the mean ± SEM (*p < 0.05; **p < 0.01)
Consistent with the TTX-induced decrease in STEP61 level and activity (Fig. 2b, d), prolonged TTX treatment increased the levels of Tyr1472-phosphorylated GluN2B (GluN2B-pY1472) and 3Tyr-phopshorylated GluA2 (GluA2-p3Y) compared to CTL treatment without affecting their total protein expression (Fig. 3a–c). In contrast, BC treatment for 24–48 h decreased the levels of GluN2B-pY1472 and GluA2-p3Y (Fig. 3d–f), concurrently with an increase in STEP61 level and activity (Fig. 2c, d). Interestingly, total levels of GluN2B and GluA2 were reduced by 48 h BC application (Fig. 3d, e).
We next examined if STEP61 mediates the TTX- or BC-induced changes in Tyr-phosphorylation of GluN2B and GluA2. Transactivator of transcription (TAT) sequence was fused to STEP46 and a myc tag (Fig. 4a), allowing the TAT fusion proteins to be membrane permeable (Additional file 1: Figure S1A, B) [15]. Preincubation for 30 min with active TAT-STEP wild-type (WT) but not control inactive TAT-myc reduced the levels of GluN2B-pY1472 and GluA2-p3Y in CTL-treated neurons (Fig. 4b, c, Additional file 1: Figure S1C) and occluded the TTX-induced increase in GluN2B-pY1472 and GluA2-p3Y levels compared to CTL application (Fig. 4b, c), suggesting that the increase in Tyr-phosphorylation of GluN2B and GluA2 is mediated by the TTX-induced reduction in STEP61.
Fig. 4.
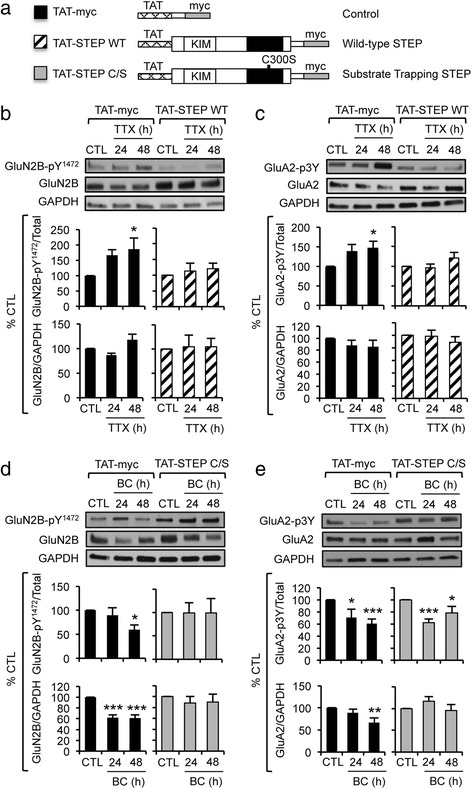
TAT-STEP WT or C/S blocks the activity-induced bidirectional changes in GluN2B and GluA2. a A simplified schematic (not to scale) of cell-permeable TAT-STEP molecular tools. b–e Immunoblot analysis of hippocampal cultured neurons that were treated for CTL, TTX (b–c), or BC (d–e) for 24 h and 48 h (n = 8 per treatment). Prior to neuronal lysate preparation, neurons were preincubated for 30 min with TAT-myc, TAT-STEP WT, or TAT-STEP C/S proteins. b–c TAT-STEP WT blocked the TTX-induced increase in Tyr1472–phosphorylation of GluN2B (GluN2B-pY1472) (b) and 3Tyr-phosphorylation of GluA2 (GluA2-p3Y) (c). d–e TAT-STEP C/S blocked the BC-induced reduction in Tyr1472–phosphorylation and level of GluN2B (d) as well as GluA2 level but not 3Tyr–phosphorylation of GluA2 (e). Data shown represent the mean ± SEM following normalization to CTL values in the presence of TAT-myc (black bars), TAT-STEP WT (striped bars), and TAT-STEP C/S (gray bars) (*p < 0.05; **p < 0.01; ***p < 0.005)
In TAT-STEP C/S, a C300S point mutation inactivates STEP46, allowing it to bind constitutively to substrates but not to dephosphorylate them [13, 15, 16]. Consistently, introduction of TAT-STEP C/S in CTL-treated neurons significantly increased the levels of GluN2B-pY1472 and GluA2-p3Y compared to TAT-myc application (Fig. 4d, e, Additional file 1: Figure S1D). Preincubation with TAT-STEP C/S but not TAT-myc blocked the BC-induced reduction in the levels of GluN2B-pY1472, total GluN2B, and total GluA2 but not GluA2-p3Y (Fig. 4d, e). Since specific Tyr residues regulated by STEP61 remain unknown, our analyses for GluA2-p3Y may not have revealed the effect of TAT-STEP C/S if STEP61 causes dephosphorylation of only one Tyr. Nonetheless, these results suggest that STEP61 mediates the BC-induced changes in Tyr1472-phosphorylation of GluN2B and abundance of GluN2B and GluA2.
Enhancement of STEP activity blocks synaptic scaling
Dephosphorylation of Tyr1472 within a conserved endocytic motif of GluN2B [17] via STEP61 reduces surface NMDAR level [4, 6] by clathrin-mediated internalization [18]. Furthermore, AMPAR internalization can be induced by mGluR stimulation through STEP61 [3] and by dephosphorylation of GluA2 at 3Tyr [14]. We hypothesized that prolonged activity blockade induces synaptic scaling (Fig. 1a–d) by inhibiting endocytosis of synaptic NMDARs and AMPARs upon STEP61 reduction (Fig. 1f, Fig 2b). To test this, we enhanced STEP activity by administering TAT-STEP WT for 30 min prior to recording. In the presence of TAT-myc, 48 h TTX treatment increased the mEPSC amplitude but not frequency compared to CTL application (Fig. 5a–d). However, this TTX-induced synaptic scaling was abolished by TAT-STEP WT preincubation (Fig. 5a–d), indicating that STEP61 reduction contributes to synaptic scaling.
Fig. 5.
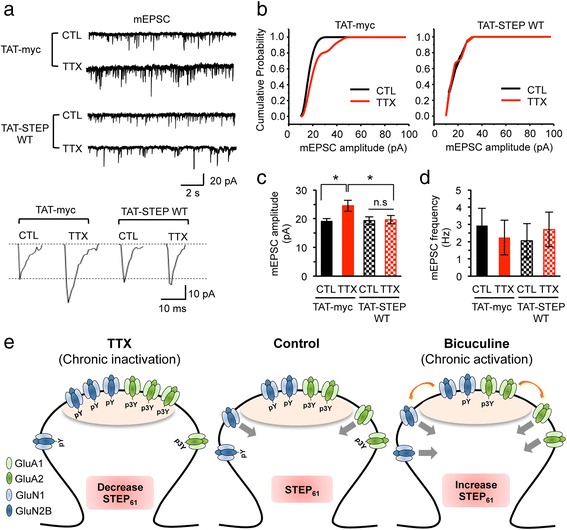
TAT-STEP WT blocks synaptic scaling induced by prolonged inhibition of hippocampal network activity. a–d Whole-cell patch clamp recording of mEPSCs from cultured hippocampal neurons that were treated for 48 h with vehicle control (CTL, 0.1 % H2O) and TTX (1 μM). Prior to mEPSCs recording, neurons were preincubated for 30 min with TAT-myc and TAT-STEP WT protein. a Representative traces of mEPSCs. b Normalized cumulative fraction of the mEPSC amplitudes. c TAT-STEP WT abolished the TTX-induced increase in the mEPSC amplitudes. d TAT-STEP WT did not affect the mEPSC frequencies. Data shown (c, d) represent the mean ± SEM (*p < 0.05). e Model by which activity-dependent changes in STEP61 level and activity regulate Tyr-dephosphorylation of GluA2 and GluN2B, leading to changes in surface AMPAR and NMDAR expression during homeostatic synaptic plasticity. Gray arrows indicate internalization. Orange arrows indicate lateral movement of surface AMPAR and NMDAR out of postsynaptic density (light pink)
Interestingly, prolonged activity enhancement increased STEP61 (Fig. 1g, Fig. 2c) and decreased Tyr-phosphorylation of GluN2B and GluA2 (Fig. 3d–f, Fig. 4d, e) without inducing synaptic down-scaling (Fig. 1a–d), suggesting that this STEP61 upregulation may cause internalization of extrasynaptic GluN2B and GluA2. Indeed, activity-dependent AMPAR endocytosis requires GluA2 [19] and occurs extrasynaptically [20]. Similarly, GluN2B-containing NMDARs enriched in extrasynaptic sites [21] undergo robust endocytosis [17, 18]. The BC-induced STEP61-dependent decrease in GluA2 and GluN2B abundance (Fig. 3d, e, Fig. 4d, e) may provide an additional homeostatic defense to limit membrane depolarization and overstimulation of extrasynaptic GluN2B-containing NMDARs, which is shown to cause excitotoxicity [22].
It remains unknown how prolonged activity perturbation regulates STEP61. Previous studies have reported that Ser221 of STEP61 is dephosphorylated by calcium-dependent calcineurin upon NMDAR activation [13] and phosphorylated by protein kinase A (PKA) upon stimulation of dopamine D1 receptor [23]. Interestingly, synaptic scaling is shown to involve reduced calcium influx to the postsynaptic neuron [9], reduced calcineurin activity [24], and enhanced PKA activity at excitatory synapses [25]. Hence, prolonged activity blockade could increase Ser221-phosphorylation of STEP61 (Fig. 2b, d) by reduced calcineurin activity and/or enhanced PKA activity, in addition to decreasing STEP61 level by transcriptional down-regulation (Fig. 1e, f). Considering that a loss of PKA from synapses was found during synaptic downscaling [25], reduced PKA activity may contribute to the BC-induced decrease in Ser221-phosphorylation of STEP61 (Fig. 2c, d).
Conclusions
In summary, we demonstrate a bidirectional modulation of STEP61 level and activity by prolonged alterations of hippocampal network activity, resulting in correlative changes in Tyr-phosphorylation of STEP61 substrates, GluN2B and GluA2. We also show that the reduction in STEP61 contributes to synaptic scaling. Future studies should test if this regulation alters NMDAR and AMPAR surface density during homeostatic plasticity (Fig. 5e). Investigating how prolonged activity perturbation regulates STEP61 should provide mechanistic insights into the dysregulation of STEP61 expression, which are present in multiple neuropathologies [2].
Materials and methods
Hippocampal neuronal culture
The Institutional Animal Care and Use Committee at the University of Illinois Urbana-Champaign approved all experimental procedures involving animals. Primary dissociated hippocampal cultures were prepared from Sprague–Dawley rat embryos at embryonic day 18 and plated at high density (330 cells/mm2) as described [8]. At 10–13 days in vitro, neurons were treated for 24–48 h with vehicle control (0.1 % dH2O), TTX (1 μM), and BC (20 μM) (all Tocris).
Electrophysiology
Whole-cell patch-clamp recordings of mEPSCs (>150 events per neuron) were performed at 23–25 °C from pyramidal neurons held at −60 mV in external solution containing 1 μM TTX and 20 μM BC as described [12, 25] using a Multiclamp 700B amplifier, Digidata1440A, and the pClamp 10.2 (Molecular Devices). Signals were acquired 3 min after making the whole-cell configuration, filtered at 1 kHz, and sampled at 10 kHz on gap free mode (5 min). The mEPSCs were detected with a 10 pA thresholds and analyzed by Mini Analysis (Synaptosoft).
QPCR
The QPCR was performed with the StepOnePlus real-time PCR system (Applied Biosystems) using total RNA (1–2 μg) as described [8]. The forward and reverse primer sequences for PTPN5 were 5’-GGAGTCAGCCCATGAATACC-3’ and 5’-CAGACGTACCCTGCTGTGAG-3’ respectively. The primer sequences for GAPDH has been previously described [8]. Following normalization to control GAPDH cDNA levels, the fold change of PTPN5 cDNA levels for each treatment compared to control was determined.
Immunoblot analysis
Neuronal lysate samples were prepared in RIPA buffer supplemented with protease inhibitors and Tyr phosphatase inhibitors (1 mM NaVO3, 10 mM Na4O7P2, and 50 mM NaF) as described [8] and were subjected to immunoblot analysis with primary antibodies against STEP61 (Santa Cruz), STEP61-pS221([13]), GluN2B and GluA2 (Millipore), GluN2B-pY1472 (PhosphoSolutions), GluA2-p3Y and GAPDH (Cell Signaling). Densitometric quantification following normalization to GAPDH was performed with ImageJ software (National Institutes of Health).
Immunocytochemistry
Permeabilized immunostaining were performed with anti-myc antibodies (Thermo-Scientific) as described [12, 25]. Fluorescence images of the neurons were acquired using the same exposure time and analyzed with ImageJ to compare their background-subtracted fluorescence intensities.
Statistical analyses
Using Origin 9.1 (Origin Lab), the Student’s t test and one-way ANOVA with Tukey’s and Fisher’s multiple comparison tests were performed to identify the statistically significant difference with a priori value (p) < 0.05 between 2 groups and for >3 groups, respectively.
Acknowledgements
This work is supported by ICR start-up funding from the University of Illinois at Urbana Champaign (HJC), an Epilepsy Foundation Predoctoral Fellowship (SER), and NIH funding MH052711 and MH091037 (PJL).
Abbreviations
- STEP
STriatal-Enriched protein tyrosine Phosphatase
- Ser
Serine
- Tyr
Tyrosine
- AMPAR
α-amino-3-hydroxyle-5-methyl-4-isoxazolepropionic acid receptor
- NMDAR
N-methyl-D-aspartate receptor
- CTL
Control
- TTX
Tetrodotoxin
- BC
Bicuculline
- PKA
Protein kinase A
- mEPSC
miniature excitatory postsynaptic current
- QPCR
real-time quantitative polymerase chain reaction
Additional file
Membrane-permeable TAT-STEP WT or C/S proteins alter STEP61–dependent Tyr-phosphorylation of GluN2B and GluA2. (A) Permeabilized immunostaining of cultured hippocampal neurons at 12 days in vitro that were incubated for 30 min with no fusion proteins (None), TAT-myc, TAT-STEP WT, or TAT-STEP C/S. Scale bars are 20 μm. (B) Background subtracted, mean intensity of myc fluorescence (n = 10–19 images per treatment). AU, arbitrary unit. (C–D) Quantification of the levels of Tyr1472–phosphorylated GluN2B (GluN2B-pY1472) and the level of GluA2 that were phosphorylated at Tyr 869, Tyr 873, and Tyr 876 (GluA2-p3Y) in CTL-treated neurons (from Fig. 4b–e) that were incubated with TAT-fusion proteins for 30 min. (C) TAT-STEP WT decreases basal Tyr-phosphorylation of GluN2B and GluA2, confirming that TAT-STEP WT increases STEP activity. (D) TAT-STEP C/S increases basal Tyr-phosphorylation of GluN2B and GluA2, confirming its ability to block dephosphorylation of STEP substrates. Data shown represent the mean ± SEM (*p < 0.05; **p < 0.01). (PDF 969 kb)
Footnotes
Sung-Soo Jang and Sara E. Royston contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Under HJC’s direction, SSJ and SER designed the study and drafted the manuscript. HJC, SER and SSJ participated in its design and coordination. SSJ, SER, KYL, and JPC prepared and maintained hippocampal culture. SSJ and KYL performed electrophysiological recordings and analysis. SER, SSJ, SBL, and JX performed immunoblot analysis. MOV and JPC isolated total RNA and performed QPCR. HGJ performed immunostaining. HJC directed the study and edited the manuscript. PL and JX provided anti-STEP61-pS221 antibodies and TAT-fusion proteins. All authors read and approved the final manuscript.
Contributor Information
Sung-Soo Jang, Email: sjang16@illinois.edu.
Sara E. Royston, Email: sroysto2@illinois.edu
Jian Xu, Email: jx38@connect.yale.edu.
John P. Cavaretta, Email: jpcav@yahoo.com
Max O. Vest, Email: vest2@illinois.edu
Kwan Young Lee, Email: red3327@gmail.com.
Seungbae Lee, Email: xbee888@gmail.com.
Han Gil Jeong, Email: hjeong10@illinois.edu.
Paul J. Lombroso, Email: paul.lombroso@yale.edu
Hee Jung Chung, Email: chunghj@life.illinois.edu.
References
- 1.Turrigiano G. Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harb Perspect Biol. 2012;4(1):a005736. doi: 10.1101/cshperspect.a005736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goebel-Goody SM, Lombroso PJ. Taking STEPs forward to understand fragile X syndrome. Results Probl Cell Differ. 2012;54:223–41. doi: 10.1007/978-3-642-21649-7_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang Y, Venkitaramani DV, Gladding CM, Zhang Y, Kurup P, Molnar E, Collingridge GL, Lombroso PJ. The tyrosine phosphatase STEP mediates AMPA receptor endocytosis after metabotropic glutamate receptor stimulation. J Neurosci. 2008;28(42):10561–6. doi: 10.1523/JNEUROSCI.2666-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Y, Kurup P, Xu J, Carty N, Fernandez SM, Nygaard HB, Pittenger C, Greengard P, Strittmatter SM, Nairn AC, Lombroso PJ. Genetic reduction of striatal-enriched tyrosine phosphatase (STEP) reverses cognitive and cellular deficits in an Alzheimer’s disease mouse model. Proc Natl Acad Sci U S A. 2010;107(44):19014–9. doi: 10.1073/pnas.1013543107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kurup P, Zhang Y, Xu J, Venkitaramani DV, Haroutunian V, Greengard P, Nairn AC, Lombroso PJ. Abeta-mediated NMDA receptor endocytosis in Alzheimer’s disease involves ubiquitination of the tyrosine phosphatase STEP61. J Neurosci. 2010;30(17):5948–57. doi: 10.1523/JNEUROSCI.0157-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8(8):1051–8. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- 7.Pelkey KA, Askalan R, Paul S, Kalia LV, Nguyen TH, Pitcher GM, Salter MW, Lombroso PJ. Tyrosine phosphatase STEP is a tonic brake on induction of long-term potentiation. Neuron. 2002;34(1):127–38. doi: 10.1016/S0896-6273(02)00633-5. [DOI] [PubMed] [Google Scholar]
- 8.Lee K, Royston SE, Vest MO, Ley DJ, Lee S, Bolton EC, Chung H. N -methyl-D-aspartate receptors mediate activity-dependent down-regulation of potassium channel genes during the expression of homeostatic intrinsic plasticity. Mol Brain. 2015;8(1):4. doi: 10.1186/s13041-015-0094-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ibata K, Sun Q, Turrigiano GG. Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron. 2008;57(6):819–26. doi: 10.1016/j.neuron.2008.02.031. [DOI] [PubMed] [Google Scholar]
- 10.Goold CP, Nicoll RA. Single-cell optogenetic excitation drives homeostatic synaptic depression. Neuron. 2010;68(3):512–28. doi: 10.1016/j.neuron.2010.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shepherd JD, Rumbaugh G, Wu J, Chowdhury S, Plath N, Kuhl D, Huganir RL, Worley PF. Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron. 2006;52(3):475–84. doi: 10.1016/j.neuron.2006.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shin SM, Zhang N, Hansen J, Gerges NZ, Pak DT, Sheng M, Lee SH. GKAP orchestrates activity-dependent postsynaptic protein remodeling and homeostatic scaling. Nat Neurosci. 2012;15(12):1655–66. doi: 10.1038/nn.3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paul S, Nairn AC, Wang P, Lombroso PJ. NMDA-mediated activation of the tyrosine phosphatase STEP regulates the duration of ERK signaling. Nat Neurosci. 2003;6(1):34–42. doi: 10.1038/nn989. [DOI] [PubMed] [Google Scholar]
- 14.Hayashi T, Huganir RL. Tyrosine phosphorylation and regulation of the AMPA receptor by SRC family tyrosine kinases. J Neurosci. 2004;24(27):6152–60. doi: 10.1523/JNEUROSCI.0799-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paul S, Olausson P, Venkitaramani DV, Ruchkina I, Moran TD, Tronson N, Mills E, Hakim S, Salter MW, Taylor JR, Lombroso PJ. The striatal-enriched protein tyrosine phosphatase gates long-term potentiation and fear memory in the lateral amygdala. Biol Psychiatry. 2007;61(9):1049–61. doi: 10.1016/j.biopsych.2006.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu J, Kurup P, Zhang Y, Goebel-Goody SM, Wu PH, Hawasli AH, Baum ML, Bibb JA, Lombroso PJ. Extrasynaptic NMDA receptors couple preferentially to excitotoxicity via calpain-mediated cleavage of STEP. J Neurosci. 2009;29(29):9330–43. doi: 10.1523/JNEUROSCI.2212-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roche KW, Standley S, McCallum J, Dune Ly C, Ehlers MD, Wenthold RJ. Molecular determinants of NMDA receptor internalization. Nat Neurosci. 2001;4(8):794–802. doi: 10.1038/90498. [DOI] [PubMed] [Google Scholar]
- 18.Lavezzari G, McCallum J, Lee R, Roche KW. Differential binding of the AP-2 adaptor complex and PSD-95 to the C-terminus of the NMDA receptor subunit NR2B regulates surface expression. Neuropharmacology. 2003;45(6):729–37. doi: 10.1016/S0028-3908(03)00308-3. [DOI] [PubMed] [Google Scholar]
- 19.Lee SH, Simonetta A, Sheng M. Subunit rules governing the sorting of internalized AMPA receptors in hippocampal neurons. Neuron. 2004;43(2):221–36. doi: 10.1016/j.neuron.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 20.Ashby MC, De La Rue SA, Ralph GS, Uney J, Collingridge GL, Henley JM. Removal of AMPA receptors (AMPARs) from synapses is preceded by transient endocytosis of extrasynaptic AMPARs. J Neurosci. 2004;24(22):5172–6. doi: 10.1523/JNEUROSCI.1042-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tovar KR, Westbrook GL. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci. 1999;19(10):4180–8. doi: 10.1523/JNEUROSCI.19-10-04180.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2010;11(10):682–96. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paul S, Snyder GL, Yokakura H, Picciotto MR, Nairn AC, Lombroso PJ. The Dopamine/D1 receptor mediates the phosphorylation and inactivation of the protein tyrosine phosphatase STEP via a PKA-dependent pathway. J Neurosci. 2000;20(15):5630–8. doi: 10.1523/JNEUROSCI.20-15-05630.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim S, Ziff EB. Calcineurin mediates synaptic scaling via synaptic trafficking of Ca2 + −permeable AMPA receptors. PLoS Biol. 2014;12(7) doi: 10.1371/journal.pbio.1001900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Diering GH, Gustina AS, Huganir RL. PKA-GluA1 coupling via AKAP5 controls AMPA receptor phosphorylation and cell-surface targeting during bidirectional homeostatic plasticity. Neuron. 2014;84(4):790–805. doi: 10.1016/j.neuron.2014.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]