

Abstract
Background
Cardiac oxidative stress, bioenergetics and catecholamine play major roles in heart failure progression. However, the relationships between these three dominant heart failure factors are not fully elucidated. Caffeic acid ethanolamide (CAEA), a synthesized derivative from caffeic acid that exerted antioxidative properties, was thus applied in this study to explore its effects on the pathogenesis of heart failure.
Results
In vitro studies in HL-1 cells exposed to isoproterenol showed an increase in cellular and mitochondria oxidative stress. Two-week isoproterenol injections into mice resulted in ventricular hypertrophy, myocardial fibrosis, elevated lipid peroxidation, cardiac adenosine triphosphate and left ventricular ejection fraction decline, suggesting oxidative stress and bioenergetics changes in catecholamine-induced heart failure. CAEA restored oxygen consumption rates and adenosine triphosphate contents. In addition, CAEA alleviated isoproterenol-induced cardiac remodeling, cardiac oxidative stress, cardiac bioenergetics and function insufficiency in mice. CAEA treatment recovered sirtuin 1 and sirtuin 3 activity, and attenuated the changes of proteins, including manganese superoxide dismutase and hypoxia-inducible factor 1-α, which are the most likely mechanisms responsible for the alleviation of isoproterenol-caused cardiac injury
Conclusion
CAEA prevents catecholamine-induced cardiac damage and is therefore a possible new therapeutic approach for preventing heart failure progression.
Keywords: Bioenergetics, Caffeic acid, Heart failure, Sirtuin
Background
Heart failure (HF) remains a major cause of death in developed nations [1]. It is a complex and multi-causal syndrome characterized by cardiac dysfunction [2–6]. Evidence has shown that catecholamine, oxidative stress and bioenergetic insufficiency contribute to the pathogenesis of HF [7–13]. The increase in sympathetic tone in HF is supposed to compensate for cardiac dysfunction; however, a previous study found that the patients with higher plasma catecholamine concentrations had poorer outcomes [14]. A synthetic catecholamine, isoproterenol (ISO), has also been widely used to induce oxidative stress HF, displaying cardiac remodeling, dysfunction, and bioenergetics insufficiency [15–17]. These observations imply that catecholamine released to counterbalance the cardiac dysfunction could further result in myocardial oxidative injury and bioenergetics impairment in HF.
Mitochondria are responsible for oxidative phosphorylation. Adenosine triphosphate (ATP) is produced from the electron transport chain (ETC) which supplies energy for well-perfused hearts [12, 18, 19]. On the other hand, reactive oxygen species (ROS) leaking from impaired ETC in failing myocardium contributes to mitochondrial and cellular oxidative stress, further deteriorating cardiac bioenergetics [9, 10, 13, 18, 20–29]. Accordingly, amelioration of mitochondrial oxidative stress has been considered as a possible resolution to heart failure [23, 26]. Agents that correct impaired ETC can reduce ROS leakage from mitochondria [30, 31]. Modulation of the cellular oxidative alternation is another possible therapeutic modality [31, 32] and attenuating mitochondrial oxidative stress is yet another [33].
Sirtuins (SIRTs) are family of class III histone deacetylases, which require NAD+ to deacetylate histone and nonhistone lysines [34]. Mammals contain seven sirtuins, SIRT1–7 [35]. SIRT1 and SIRT3 are highly expressed in the nucleus/cytoplasm and mitochondria of the heart, respectively [34–37]. It has been shown that sirtuin 1 (SIRT1) is downregulated in patients with heart failure, and that there is an increase in sirtuin 1 reducing oxidative stress-mediated cardiac reperfusion injury [38, 39]. Meanwhile, sirtuin 3 (SIRT3) has been demonstrated to regulate cardiac energy status and mitochondria tolerance to ischemia-reperfusion injury by deacetylating specific mitochondrial proteins [40, 41]. Therefore, SIRT 1 and SIRT 3 are potential targets for managing catecholamine inducing oxidative stress and bioenergetic insufficiency, thus preventing the progression of HF.
Caffeic acid, a natural phenolic constituent, has antioxidative properties [42, 43]. Its cardiovascular protection has been demonstrated through its free radical scavenging effect [44–50]. However, the exact mechanisms underlying caffeic acid-induced cardio-protection and its therapeutic potential on HF remain unknown. In addition, our preliminary data represented that a new caffeic acid derivate, caffeic acid ethanolamide (CAEA), exerted cardioprotective effects, which was superior to caffeic acid (data will show later). We aimed in our present study to evaluate the effects of CAEA on catecholamine-induced HF, and the involved mechanisms.
Methods
Experimental animals and ethics statement
Eight-week-old male C57BL/6 mice were purchased from the National Laboratory Animal Center of Taiwan. The research was performed according to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication no. 85–23, revised 1996), and was approved by the Institutional Animal Care and Use Committee of the National Taiwan University, Taiwan. ISO (Sigma-Aldrich, St. Louis, MO) 16 mg/kg once daily was subcutaneously injected for 14 days. The control groups received the same volume of isotonic saline. CAEA suspended in isotonic saline was administered subcutaneously as a dose of 1 mg/kg/day after ISO injection. In some experiments, nicotinamide (20 mg/kg/day), a sirtuin inhibitor, was also injected subcutaneously to investigate the mechanism of CAEA.
Caffeic acid ethanolamide preparation
CAEA is synthesized in the laboratory of YH Kuo (Fig. 1). CAEA was produced from caffeic acid (100 mg, 0.56 mmole) dissolved in 1 mL N,N-dimethylformamide and 80 μL triethylamine in a two-necked bottle. The solution was then added into 5 mL dichloromethane containing 41 μL (1.2 eq) ethanolamine, and 298 mg (1.2 eq) (Benzotriazol-1-yloxy)tris- (dimethylamino)phosphonium hexafluorophosphate to react for 30 min in an ice bath, followed by reacting at room temperature for 2 h. After the reaction, dichloromethane was removed with low negative pressure. The residue was then added into water, and then extracted by ethyl acetate. The organic phase was then collected, washed with 3 N HCl, 10 % NaHCO3 and water, and then dried with anhydrous sodium sulfate. After filtration, condensation, and column chromatography, the final product- caffeic acid ethanolamide was obtained.
Fig. 1.
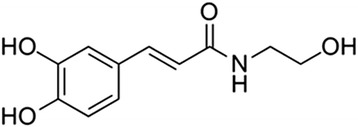
Structure of caffeic acid ethanolamide
Cardiac function assessment
After 14 days of drug administration, small animal ultrasound imaging system (S-Sharp Corporation, Taipei, Taiwan) was used for echocardiography measurements. Transthoracic echocardiography was performed 12 h after the last drug injection. Mice were anesthetized by 2 % isoflurane mixed with 1 L/min O2 in the induction chamber, while the continuous application of anesthesia was dropped to 1 % isoflurane. Cardiac function was calculated, in duplicate, in M-mode images from the parasternal long axis by using the leading-edge technique defined by the American Society of Echocardiography. Left ventricle ejection fraction (EF) is an indicator for the determination of cardiac function.
Cardiac histology
After the echocardiogram was recorded, the heart was excised and perfused with PBS. The weight of heart was measured, and the heart to body weight (HW/BW) ratio was calculated. The hearts were fixed in 4 % paraformaldehyde, embedded in paraffin, and sectioned horizontally in 4 μm slices. Masson’s trichrome stain and Sirius red stain were performed for fibrosis analysis.
Cardiac protein extraction
Left ventricles were homogenized as described previously [51]. Briefly, left ventricles were homogenized in tissues protein extraction buffer (Thermo Fisher Scientific Inc., IL, USA) containing cocktail proteases and phosphatase inhibitors (Sigma, St. Louis, MO, USA). The supernatant of the tissue homogenate was collected after centrifugation (800 × g, 10 min at 4 °C) and was defined as total cardiac protein. Protein concentrations were determined by a BCA protein assay kit (Thermo Fisher Scientific Inc., IL, USA).
ATP content determination
Mouse ventricular tissue lysate was prepared for measuring cardiac ATP content, which was measured by an ELISA kit (Biovision, CA, USA). To detect lactate content, the ATP reaction mix was mixed well with tissue lysate in each well at room temperature protected from light for 30 min, and the fluorescence signals were detected by excitation wavelength of 535 nm and an emission wavelength of 587 nm with a microplate spectrophotometer.
Western blotting
Cardiac protein samples were analyzed for manganese superoxide dismutase (MnSOD), c-Jun N-terminal kinase (JNK), phospho-JNK (p-JNK) and hypoxia-inducible factor 1-α (HIF-1α) expression (Cell Signaling, MA, USA), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Santa Cruz Biotechnology, CA, USA). The methods were described in our previous study [51].
Sirtuin activity detection
Mouse ventricular tissue lysates were prepared for the measurement of SIRT1 and SIRT3 activities, which were measured by kits (Cayman Chemicals, MI, USA). p53 sequence, as the substrate for sirtuin deacetylation, was mixed with tissue lysate in a 96 well microplate, and was then shaken at 25 °C for 45 min. Fluorescence signals were detected by an excitation wavelength of 360 nm and an emission wavelength of 450 nm with a microplate spectrophotometer.
Lactate content and the ratio of oxidized and reduced forms of nicotinamide adenine dinucleotides (NAD+/NADH ratio) detection
Mouse ventricular tissue lysate was prepared for the measurement of lactate content and NAD+/NADH ratio, which were both measured by ELISA kits (Biovision, CA, USA). To detect lactate content, the lactate reaction mix was added to each well along with tissue lysate at room temperature away from light for 30 min, and then read at an optical density of 570 nm. In addition, after centrifuging the samples at 14,000 rpm for 5 min, the supernatant of the heart tissue was transferred into a new tube. To detect total NAD, the supernatant was mixed with an NADH developer in each well of a 96 well microplate at room temperature for 5 min, and then the color was read at an OD of 450 nm. To detect NADH, we heated the supernatant to 60 °C for 30 min to decompose NAD+, and then we followed the steps of the reaction mentioned above. After the standard curve was prepared, the NAD+/NADH ratio was obtained from the total NAD and NADH detected, which is equal to the (total NAD - NADH)/NADH ratio.
Lipid peroxidation determination
Cardiac oxidative stress was represented by lipid peroxidation of mouse ventricular tissue, and determined by a kit (Cayman Chemicals, MI, USA). Briefly, after centrifuging the samples at 1,600 g at 4 °C for 10 min, the supernatant was mixed with sodium dodecyl sulfate solution along with the color reagent in tubes, and then put them into boiling water for 1 h, followed by incubating them on ice for 10 min. After centrifuging the samples at 1,600 g at 4 °C for 10 min, we read the fluorescence signals at the excitation wavelength of 350–360 nm and an emission wavelength of 450–465 nm by using a microplate spectrophotometer.
Cell culture
HL-1 cells, a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte, were obtained from Dr. William C. Claycomb (Louisiana State University Health Sciences Center, New Orleans, LA). Cells were cultured in Claycomb medium supplemented with 10 % FBS (Gibco, Scotland, UK), 2 mM L-glutamine (Gibco, Scotland, UK), 0.1 mM norepinephrine, and antibiotics (100 μg/ml penicillin and 100 μg/ml streptomycin) at 37 °C under a 5 % CO2 − 95 % air atmosphere. The HL-1 cells were used for experimentation after reaching 80 % confluency. ISO was added to induce stress for 24 h. CAEA (1 μM) was pre-incubated 1 h before ISO treatment.
Intracellular free radical determination
Intracellular ROS and mitochondria superoxide generation was detected in cardiomyocytes by labeling with fluorescence dye 5-(and-6)-chloromethyl-2′,7′- dichlorodihydrofluorescein diacetate and MitoSOX™, respectively. By using fluorescence microscopy, intracellular ROS level was monitored at 488 nm excitation and 515 nm emission, and mitochondria superoxide generation was monitored at 510 nm excitation and 580 nm emission, respectively. Fluorescence intensity was calculated by averaging fluorescence intensity of numerous outlined cells using ImageQuant (Molecular Dynamics, Inc., Sunnyvale, CA, USA).
Rate of oxygen consumption detection
To assess the function of the cellular electron transport chain, oxygen consumption rate (OCR) was estimated by a kit (Luxcel Biosciences Ltd., Cork, Ireland). MitoXpressR Xtra was added to each well containing cells after treatment. The dual-read signal was recorded continuously right after mineral oil sealing. Since the detection dye is quenched by O2 through molecular collision, the fluorescence signal is inversely proportional to the amount of extracellular O2 in the sample. Rates of oxygen consumption were determined from the changes in the fluorescence signals over time. The slope between linear regression lifetime of fluorescence and detection period was calculated as OCR. The values of OCR were normalized to protein content.
pH level determination
pH values were measured in cell culture, by adding a pH-sensitive fluorescence dye (Invitrogen, NY, USA). The fluorescence signal is proportionally increased during the lowering of the pH value, and detected by excitation wavelength of 560 nm and an emission wavelength of 585 nm.
Glycolysis detection
Cellular glycolysis was measured by a kit (Luxcel Biosciences Ltd., Cork, Ireland). After several washes, pH-Xtra™ was added to the cells, and the fluorescence signal was recorded in a continuous dual-read manner. The values of the glycolysis rate were normalized to protein content.
Statistical analysis
All values were represented as means ± SE. The results were analyzed using ANOVA followed by Bonferroni's post hoc tests. P <0.05 was considered as a significant difference.
Results
CAEA prevents isoproterenol caused myocardial remodeling
CAEA (1 mg/kg) alone had no significant impact on cardiac morphology and histology (Fig. 2). Conversely, the two-week ISO induced cardiac remodeling- ventricular hypertrophy and myocardial fibrosis was alleviated by CAEA (1 mg/kg). The cardio-protective effects of CAEA were superior to caffeic acid (1 mg/kg) in terms of isoproterenol-induced cardiac remodeling (Fig. 2). The ratio of heart weight to body weight was considerably reduced from 5.95 mg/g in ISO-treated mice (ISO group) to 5.49 mg/g in mice subjected to CAEA and ISO (ISO + CAEA group), while that in the vehicle-treated mice (control group) was 4.95 mg/g (Fig. 2ab). Meanwhile, cardiac fibrosis was significantly attenuated from 11.02 % in the ISO group to 3.67 % in the ISO + CAEA group, when compared with 0.77 % in the control group (Fig. 2cd).
Fig. 2.
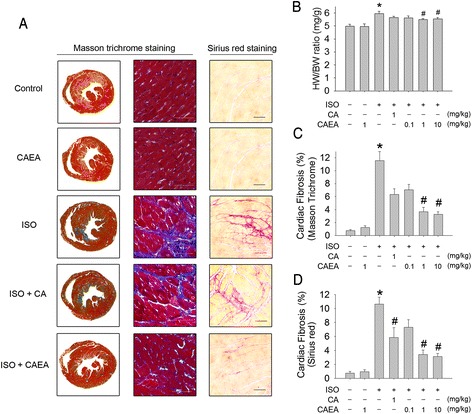
Effects of caffeic acid ethanolamide (CAEA) on cardiac remodeling in mice subjected to two weeks subcutaneous isoproterenol (ISO) injections. a Representative histological sections. b Heart weight to body weight (HW/BW) ratio. c Cardiac fibrosis in blue assessed by Masson’s trichrome staining d Quantification of cardiac fibrosis in pink assessed by Sirius red staining (bar = 100 μm). n = 9, *P <0.05 versus control, # P <0.05 versus ISO
CAEA alleviates isoproterenol induced cardiac dysfunction and bioenergetic insufficiency
CAEA (1 mg/kg) alone did not change left ventricle ejection fraction (LVEF) and cardiac ATP (Fig. 3abc). LVEF declined from 65.8 % in the control group to 48.2 % in the ISO group (Fig. 3ab). The decline of LVEF in the ISO group was significantly attenuated to 66.4 % in the ISO + CAEA group (Fig. 3ab). In the meantime, the drop of cardiac ATP in the ISO group (50.4 %) was preserved in the ISO + CAEA group (86.2 %), when compared with the control group (Fig. 3c). LVEF correlated well with cardiac ATP content in mice (Fig. 3d).
Fig. 3.
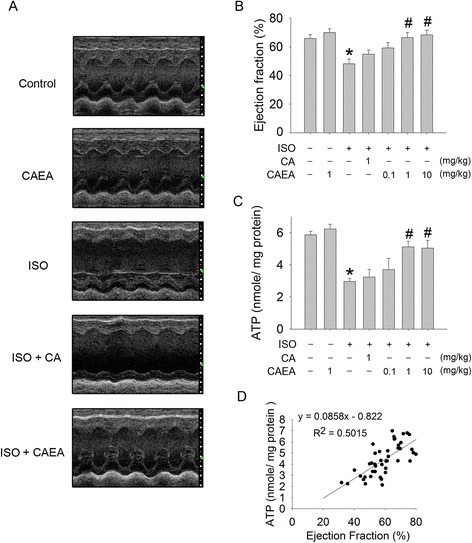
Effects of caffeic acid ethanolamide (CAEA) on cardiac function and cardiac energy in mice subjected to two weeks subcutaneous isoproterenol (ISO) injections. a Representative M-mode echocardiogram. b Quantification of left ventricular ejection fraction. c Quantification of ATP contents. d The correlation between left ventricular ejection fraction and ATP contents (R2: coefficient of determination). n = 9, *P <0.05 versus control, # P <0.05 versus ISO
Again, CAEA showed its superiority over caffeic acid when considering isoproterenol-induced cardiac bioenergetic impairment and dysfunction. Therefore, we chose CAEA for further evaluation (Fig. 3abc).
CAEA recovers cardiac manganese superoxide dismutase and reduces oxidative stress in isoproterenol induced heart failure
ISO increased cardiac oxidative stress, which was measured as lipid peroxidation. CAEA alleviated ISO induced cardiac oxidative stress from 1.65- to 1.23-fold, when compared with control group (Fig. 4a). Cardiac protein expression was analyzed by Western blotting. CAEA ameliorated ISO induced JNK phosphorylation from 1.76- to 1.52-fold higher, compared to the control group (Fig. 4bc), while cardiac MnSOD, which was 67.9 % in ISO group, recovered to 88.9 % in the ISO + CAEA group, compared to the control group (Fig. 4bd).
Fig. 4.
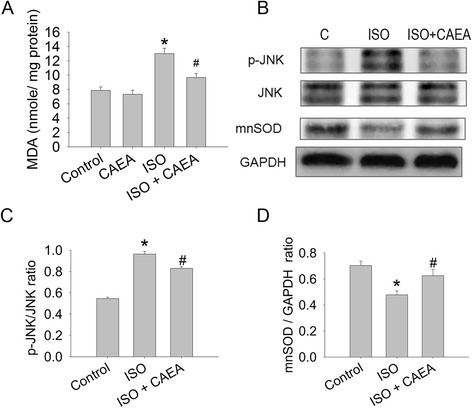
Effects of caffeic acid ethanolamide (CAEA) on cardiac oxidative stress in mice subjected to two weeks subcutaneous isoproterenol (ISO) injections. a Cardiac tissue lipid peroxidation determined by malondialdehyde (MDA). b Representative Western blot image of cardiac tissue phosphorylation of JNK, and manganese superoxide dismutase (mnSOD) expression. c Densitometry of cardiac tissue phosphorylation of JNK and d mnSOD expression. n = 6 ~ 9, *P <0.05 versus control, # P <0.05 versus ISO
Isoproterenol increases cellular and mitochondrial oxidative stress in HL-1 cardiomyocytes, while CAEA reduces them both
ISO induced cellular oxidative stress, which was measured by fluorescence staining in HL-1 cardiomyocytes (Fig. 5). Intracellular ROS (green fluorescence) was 8.93-fold greater in the ISO group than in the control group, and was reduced to 4.81-fold elevation in the ISO + CAEA group (Fig. 5ab). In addition, mitochondrial superoxide (red fluorescence) was 1.23-fold higher in the ISO group than in the control group, and was alleviated to 1.06-fold elevation in the ISO + CAEA group (Fig. 5cd). CAEA reduced ISO caused cellular oxidative stress and mitochondrial superoxide activity.
Fig. 5.
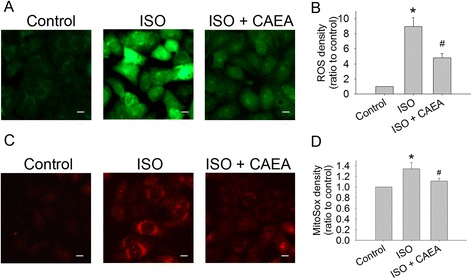
Effects of caffeic acid ethanolamide (CAEA) on oxidative stress in HL-1 cells exposed to isoproterenol (ISO). a Cellular reactive oxygen species (ROS) in green fluorescence (bar = 10 μm). b Quantification of cellular ROS. c Mitochondrial superoxide in red fluorescence (bar = 10 μm). d Quantification of mitochondrial superoxide. n = 4 in triplicate for each group, *P <0.05 versus control, # P <0.05 versus ISO
CAEA preserves oxidative phosphorylation, cellular bioenergetics and cellular redox state in isoproterenol-treated HL-1 cardiomyocytes
Cellular oxidative phosphorylation in HL-1 cardiomyocytes was represented by oxygen consumption rate (OCR). OCR declined from 5.53 μs/h in the control group to 1.09 μs/h in the ISO group, and only declined to 2.67 μs/h in the ISO + CAEA group (Fig. 6ab). The glycolysis rate was 1.9-fold higher in ISO group than control group (Fig. 6d). The ISO group showed the greatest decrease in pH values among the groups (pH 7.20) and recovered to pH 7.33 in the ISO + CAEA group (Fig. 6c). Cellular ATP in the ISO group was 58.5 % of the control group. NAD+ in the ISO group was 66.0 % of the control group, compared to NADH which was 1.42 fold higher than in the control group. The NAD+/NADH ratio (representing cellular redox state [18]) in the ISO group was 45.7 % of the control group (Fig. 6de). Taken together, ISO decreased cellular OCR, elevated the glycolysis rate and NADH, reduced cellular pH, ATP production, NAD+ and NAD+/NADH ratio (Fig. 6). Conversely, CAEA significantly reversed the effects of ISO on cellular glycolysis rate, pH, ATP production, NAD+, NADH and NAD+/NADH ratio, which were 1.23-fold, 83.3 %, 87.3 %, 92.9 %, 1.17-fold and 79.2 % of control group, respectively (Fig. 6). The preservation of cellular oxidative phosphorylation and the alleviation of glycolysis by CAEA in HL-1 cells exposed to ISO could lead to cellular ATP and redox state restoration.
Fig. 6.
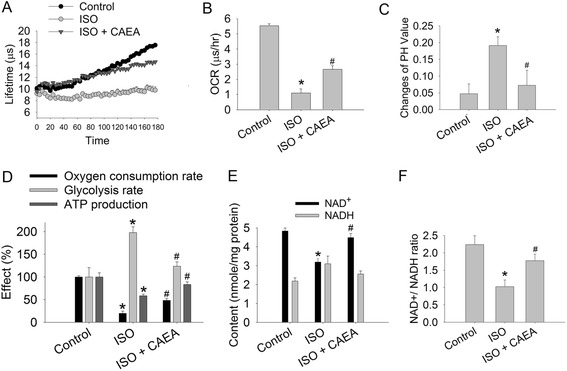
Effects of caffeic acid ethanolamide (CAEA) on cellular bioenergetics in HL-1 cells exposed to isoproterenol (ISO). a Lifetimes of oxygen consumption in fluorescence changes. b Quantification of oxygen consumption rate (OCR). c Quantification of intracellular pH value changes. d Quantification of cellular bioenergetics. e Quantification of cellular NAD+ and NADH. f Quantification of cellular NAD+/NADH. n = 4 in triplicate for each group, *P <0.05 versus control, # P <0.05 versus ISO
CAEA preserved cardiac bioenergetics in isoproterenol induced cardiac dysfunction is sirtuin dependent
CAEA did not change the SIRT1 and SIRT3 expression levels compared to the control group. However, the decline of SIRT1 and SIRT3 activity in the ISO mouse group was preserved in the ISO + CAEA group, which were elevated from 67.7 % to 82.5 % and from 68.5 % to 83.6 % of the control group, respectively (Fig. 7a). The increase in lipid peroxidation and HIF-1α expression in the ISO group (1.65-fold, 2.1-fold of control group) was significantly reduced in the ISO + CAEA group (1.23-fold, 1.4-fold of control group) (Fig. 7bcd). When sirtuin was inhibited by nicotinamide, the CAEA protective effects, including lipid peroxidation, HIF-1α expression, lactate contents, LVEF and ATP production, were all abolished (Fig. 7b ~ h). In summary, the CAEA alleviating effects of ISO induced cardiac injury were sirtuin dependent.
Fig. 7.

Mechanism of caffeic acid ethanolamide (CAEA) affecting cardiac oxidative stress and cardiac bioenergetics in mice subjected to two weeks subcutaneous isoproterenol (ISO) injections. a Quantification of cardiac sirtuin 1 (SIRT1) and sirtuin 3 (SIRT3) activity. b Quantification of cardiac lipid peroxidation. c Representative Western blot image of cardiac HIF-1α expression. d Densitometry of cardiac HIF-1α expression. e Quantification of cardiac lactate content. f Representative M-mode echocardiogram. g Quantification of left ventricular ejection fraction. h Quantification of cardiac ATP content. n = 6 ~ 9, **P <0.01 versus control, *P <0.05 versus control, # P <0.05 versus ISO, @ P <0.05 versus ISO + CAEA. (Nic = nicotinamide)
Discussion
We demonstrated that CAEA alleviates cardiac remodeling and improves cardiac functions in murine ISO induced HF. CAEA recovered the SIRT1, SIRT3 activity, and MnSOD expression and downregulated HIF-1α expression, leading to a reduction in oxidative stress, preserving oxidative phosphorylation, cardiac bioenergetics, and cardiac function.
Cardiac ATP status is linked to cardiac ventricular performance [52]. Normally, two thirds of the ATP hydrolysis in cardiomyocytes is utilized for the contractile apparatus while the rest is used for the ion pumps to maintain the cellular ion concentrations [19]. Cardiac energy is impaired in HF [10, 13, 18, 19, 53, 54]. Elevated catecholamines given to compensate for cardiac dysfunction in HF may do more harm than good and lead to a deterioration in cardiac bioenergetics [13, 55, 56]. Mitochondria are responsible for ATP production through oxidative phosphorylation. Several studies have shown that cardiac ETC is impaired and is accompanied with an increase in mitochondrial ROS generation in HF [20, 23, 24, 32, 57, 58]. Collectively, catecholamine has been shown to cause bioenergetics impairment in HF [10]. Our present study shows that continuous ISO stimulation results in cellular oxidative stress, cardiac remodeling, ETC impairment, mitochondrial superoxide elevation, cardiac bioenergetics alteration, and finally cardiac function deterioration.
Our present study shows that CAEA has anti-oxidative properties. CAEA recovered MnSOD expression and activity in mice subjected to ISO, subsequently alleviated cardiac oxidative stress, preserved cellular oxidative phosphorylation, cardiac energy, and cardiac function. The cardioprotective effect of CAEA was blocked by nicotinamide, inferring a sirtuin-dependent MnSOD restoration. This is in line with a previous study reporting a sirtuin-dependent MnSOD enhancement in AC5 knockout mice [59]. In view of the fact that some general antioxidants fail to treat HF, subcellular compartment signaling is believed to be the target for future drug development [26]. SIRT1 is found mostly in the nucleus and cytoplasm while SIRT3 is predominantly in the mitochondria [35, 36]. CAEA restored both SIRT1 and SIRT3 activity, and reduced cellular and mitochondrial oxidative stress. Hence, CAEA is a potential therapeutic candidate for preventing catecholamine-induced cardiac dysfunction during HF progression.
HIF-1α is a protein that regulates hypoxia-regulated gene expression to mediate cell adaption to low oxygen circumstances [60, 61]. ISO injections in rats have been shown to increase HIF-1α expression [62]. In the present study, ISO impaired cardiac ATP production while increasing the cardiac working load. This may have augmented HIF-1α expression due to relative hypoxia. HIF-1α is further stabilized by ROS or mitochondrial dysfunction [63, 64]. In addition, HIF-1α reprograms glucose metabolism from mitochondrial oxidative phosphorylation to glycolysis [65]. It has shown that metabolic remodeling in advanced HF includes elevated glycolysis, and a reduced respiratory chain activity [18]. This is consistent with the findings of the present study where ISO elevated cardiac HIF-1α expression, and the glycolysis rate. Hence, HIF-1α may be the missing link between oxidative stress and the metabolic shift seen in HF, resulting from chronic catecholamine stimulation.
CAEA reversed the HIF-1α elevation caused by ISO, which may result in the preservation of the cellular redox state. NAD+/NADH ratio represents the cellular redox state [18]. Through mitochondrial oxidative phosphorylation (OXPHOS), NADH produced by glycolysis is normally shuttled into the mitochondrial to generate ATP, H2O, CO2 and NAD+ that are shuttled back into the cytoplasm, maintaining the cellular and mitochondrial NAD+/NADH ratio [18]. Hence, mitochondrial OXPHOS is essential to maintain the cellular redox state. Studies have shown that HIF-1α increases anaerobic glycolysis accompanied with lactate accumulation. A prolonged lactate accumulation inhibits NAD+ regenerated from NADH, which leads to a decline in the NAD+/NADH ratio [18, 65]. In addition, a low NAD+/NADH ratio has been shown to enhance HIF-1α mediated mitochondrial OXPHOS inhibition that could cause a failure in the preservation of the cellular redox state [18, 66, 67]. These are compatible to the findings in the present study where CAEA restored mitochondrial OXPHOS, reduced HIF-1α expression and lactate content, and maintained cellular redox state in mice during chronic ISO treatment. Being NAD+-dependent deacetylases, the maintenance of sirtuins activity in the present study may be cooperatively by the preservation of intracellular NAD+.
Study limitations
Our study does not provided a reason as to why CAEA was superior to caffeic acid in its cardioprotective effects on ISO-induced cardiac dysfunction. The mechanisms of the cardioprotective differences between CAEA and caffeic acid are planned for future studies.
Conclusion
Our study shows that CAEA triggers intrinsic anti-oxidants in the cardiomyocyte, thus preventing oxidative stress-induced heart failure. CAEA also preserved the cardiac bioenergetic functions by oxidative phosphorylation restoration, HIF-1α expression reversal and cellular redox state maintenance. These findings suggest that the regulation of cardiac bioenergetics by SIRT1 and SIRT3 could increase heart tolerance to chronic stress and prevent catecholamine-induced cardiac dysfunction during HF progression.
Acknowledgments
This work was supported by a grant from Ministry of Science and Technology (MOST-102-2325-B-002-095-B4, MOST 103-2325-B-002-020), and CMU under the Aim for Top University Plan of the Ministry of Education, Taiwan, and the Department of Health Clinical Trial and Research Center of Excellent (DOH102-TD-C-111-004), Taiwan. The funding agencies had no role in study design, data collection, decision to publish, or preparation of the manuscript.
Abbreviations
- CAEA
Caffeic acid ethanolamide
- EF
Ejection fraction
- ETC
Electric transport chain
- HF
Heart failure
- ISO
Isoproterenol
- MnSOD
Manganese superoxide dismutase
- OCR
Oxygen consumption rate
- OXPHOS
Oxidative phosphorylation
- ROS
Reactive oxygen species. ATP, Adenosine triphosphate
- JNK
c-Jun N-terminal kinase
- HIF-1α
Hypoxia-inducible factor 1-α
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SYL, MJS, and KCY conceived and designed the experiments. SYL and HCK performed the experiments and analyzed the data. SYL and HCK wrote the manuscript. YHK, PCT, and HLC performed the drug design. All authors read and approved the final manuscript.
References
- 1.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, et al. Heart disease and stroke statistics--2014 update: a report from the American Heart Association. Circulation. 2004;129:e28–292. doi: 10.1161/01.cir.0000441139.02102.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nativi-Nicolau J, Ryan JJ, Fang JC. Current Therapeutic Approach in Heart Failure with Preserved Ejection Fraction. Heart Fail Clin. 2014;10:525–38. doi: 10.1016/j.hfc.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 3.Lupon J, de Antonio M, Vila J, Penafiel J, Galan A, Zamora E, et al. Development of a novel heart failure risk tool: the barcelona bio-heart failure risk calculator (BCN bio-HF calculator) PLoS One. 2014;9 doi: 10.1371/journal.pone.0085466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang S, Wu J, Wang Z, Zou Y. Crosstalk between Src and β-arrestin2 orchestrates cardiac hypertrophic responses under mechanical stresses. Cardiovasc Regen Med. 2014;1 [Google Scholar]
- 5.Lucas E, Jurado-Pueyo M, Vila-Bedmar R, Diez J, Mayor F, Murga C. Linking cardaic insulin resistance and heart failure: GRK2 as an integrative node. Cardiovasc Regen Med. 2015;2 [Google Scholar]
- 6.Pandey R, Ahmed RP. MicroRNAs inducing proliferation of quiescent adult cardiomyocytes. Cardiovasc Regen Med. 2015;2 doi: 10.14800/crm.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Belch JJ, Bridges AB, Scott N, Chopra M. Oxygen free radicals and congestive heart failure. Br Heart J. 1991;65:245–8. doi: 10.1136/hrt.65.5.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol. 2011;301:H2181–90. doi: 10.1152/ajpheart.00554.2011. [DOI] [PubMed] [Google Scholar]
- 9.Ingwall JS. Energy metabolism in heart failure and remodelling. Cardiovasc Res. 2009;81:412–9. doi: 10.1093/cvr/cvn301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosca MG, Hoppel CL. Mitochondrial dysfunction in heart failure. Heart Fail Rev. 2013;18:607–22. doi: 10.1007/s10741-012-9340-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sorescu D, Griendling KK. Reactive oxygen species, mitochondria, and NAD(P)H oxidases in the development and progression of heart failure. Congest Heart Fail. 2002;8:132–40. doi: 10.1111/j.1527-5299.2002.00717.x. [DOI] [PubMed] [Google Scholar]
- 12.Nickel A, Loffler J, Maack C. Myocardial energetics in heart failure. Basic Res Cardiol. 2013;108:358. doi: 10.1007/s00395-013-0358-9. [DOI] [PubMed] [Google Scholar]
- 13.Neubauer S. The failing heart--an engine out of fuel. N Engl J Med. 2007;356:1140–51. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- 14.Jay N, Cohn TBL, Olivari MT, Garberg V, Lura D, Francis GS, Simon AB, Rector T. Plasma Norepinephrine as a Guide to Prognosis in Patients with Chronic Congestive Heart Failure. N Engl J Med. 1984;311:819–23. doi: 10.1056/NEJM198409273111303. [DOI] [PubMed] [Google Scholar]
- 15.Brooks WW, Conrad CH. Isoproterenol-induced myocardial injury and diastolic dysfunction in mice: structural and functional correlates. Comp Med. 2009;59:339–43. [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang GX, Kimura S, Nishiyama A, Shokoji T, Rahman M, Yao L, et al. Cardiac oxidative stress in acute and chronic isoproterenol-infused rats. Cardiovasc Res. 2005;65:230–8. doi: 10.1016/j.cardiores.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 17.Carll AP, Willis MS, Lust RM, Costa DL, Farraj AK. Merits of non-invasive rat models of left ventricular heart failure. Cardiovasc Toxicol. 2011;11:91–112. doi: 10.1007/s12012-011-9103-5. [DOI] [PubMed] [Google Scholar]
- 18.Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005;85:1093–129. doi: 10.1152/physrev.00006.2004. [DOI] [PubMed] [Google Scholar]
- 19.Solaini G, Harris DA. Biochemical dysfunction in heart mitochondria exposed to ischaemia and reperfusion. Biochem J. 2005;390:377–94. doi: 10.1042/BJ20042006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ide T, Tsutsui H, Kinugawa S, Utsumi H, Kang D, Hattori N. Mitochondrial electron transport complex I is a potential source of oxygen free radicals in the failing myocardium. Circ Res. 1999;85:357–63. doi: 10.1161/01.RES.85.4.357. [DOI] [PubMed] [Google Scholar]
- 21.Sawyer DB, Colucci WS. Mitochondrial oxidative stress in heart failure: "oxygen wastage" revisited. Circ Res. 2000;86:119–20. doi: 10.1161/01.RES.86.2.119. [DOI] [PubMed] [Google Scholar]
- 22.Tsutsui H, Ide T, Kinugawa S. Mitochondrial oxidative stress, DNA damage, and heart failure. Antioxid Redox Signal. 2006;8:1737–44. doi: 10.1089/ars.2006.8.1737. [DOI] [PubMed] [Google Scholar]
- 23.Tsutsui H. Mitochondrial oxidative stress and heart failure. Intern Med. 2006;45:809–13. doi: 10.2169/internalmedicine.45.1765. [DOI] [PubMed] [Google Scholar]
- 24.Khechaduri A, Bayeva M, Chang HC, Ardehali H. Heme levels are increased in human failing hearts. J Am Coll Cardiol. 2013;61:1884–93. doi: 10.1016/j.jacc.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kindo M, Gerelli S, Bouitbir J, Charles AL, Zoll J, Hoang Minh T, et al. Pressure overload-induced mild cardiac hypertrophy reduces left ventricular transmural differences in mitochondrial respiratory chain activity and increases oxidative stress. Front Physiol. 2012;3:332. doi: 10.3389/fphys.2012.00332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schwarz K, Siddiqi N, Singh S, Neil CJ, Dawson DK, Frenneaux MP. The breathing heart - mitochondrial respiratory chain dysfunction in cardiac disease. Int J Cardiol. 2014;171:134–43. doi: 10.1016/j.ijcard.2013.12.014. [DOI] [PubMed] [Google Scholar]
- 27.Ingwall JS, Weiss RG. Is the failing heart energy starved? On using chemical energy to support cardiac function. Circ Res. 2004;95:135–45. doi: 10.1161/01.RES.0000137170.41939.d9. [DOI] [PubMed] [Google Scholar]
- 28.Yoshikawa Y, Takaki M. Energy utility of failing heart. Nihon Yakurigaku Zasshi. 2004;123:77–86. doi: 10.1254/fpj.123.77. [DOI] [PubMed] [Google Scholar]
- 29.Majzunova M, Dovinova I, Barancik M, Chan JY. Redox signaling in pathophysiology of hypertension. J Biomed Sci. 2013;20:69. doi: 10.1186/1423-0127-20-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alehagen U, Johansson P, Bjornstedt M, Rosen A, Dahlstrom U. Cardiovascular mortality and N-terminal-proBNP reduced after combined selenium and coenzyme Q10 supplementation: a 5-year prospective randomized double-blind placebo-controlled trial among elderly Swedish citizens. Int J Cardiol. 2013;167:1860–6. doi: 10.1016/j.ijcard.2012.04.156. [DOI] [PubMed] [Google Scholar]
- 31.Gomes KM, Campos JC, Bechara LR, Queliconi B, Lima VM, Disatnik MH, et al. Aldehyde dehydrogenase 2 activation in heart failure restores mitochondrial function and improves ventricular function and remodelling. Cardiovasc Res. 2014;498–508. [DOI] [PMC free article] [PubMed]
- 32.Dedkova EN, Seidlmayer LK, Blatter LA. Mitochondria-mediated cardioprotection by trimetazidine in rabbit heart failure. J Mol Cell Cardiol. 2013;59:41–54. doi: 10.1016/j.yjmcc.2013.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dai DF, Chen T, Szeto H, Nieves-Cintron M, Kutyavin V, Santana LF, et al. Mitochondrial targeted antioxidant Peptide ameliorates hypertensive cardiomyopathy. J Am Coll Cardiol. 2011;58:73–82. doi: 10.1016/j.jacc.2010.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Webster KA. A sirtuin link between metabolism and heart disease. Nat Med. 2012;18:1617–9. doi: 10.1038/nm.2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol. 2010;5:253–95. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tanno M, Kuno A, Horio Y, Miura T. Emerging beneficial roles of sirtuins in heart failure. Basic Res Cardiol. 2012;107:273. doi: 10.1007/s00395-012-0273-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hall JA, Dominy JE, Lee Y, Puigserver P. The sirtuin family's role in aging and age-associated pathologies. J Clin Invest. 2013;123:973–9. doi: 10.1172/JCI64094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu T-M, Tsai J-Y, Chen Y-C, Huang C-Y, Hsu H-L, Weng CF, et al. Downregulation of Sirt1 as aging change in advanced heart failure. J Biomed Sci. 2014;21:57. doi: 10.1186/1423-0127-21-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hsu CP, Zhai P, Yamamoto T, Maejima Y, Matsushima S, Hariharan N, et al. Silent information regulator 1 protects the heart from ischemia/reperfusion. Circulation. 2010;122:2170–82. doi: 10.1161/CIRCULATIONAHA.110.958033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, et al. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci U S A. 2008;105:14447–52. doi: 10.1073/pnas.0803790105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shinmura K, Tamaki K, Sano M, Nakashima-Kamimura N, Wolf AM, Amo T, et al. Caloric restriction primes mitochondria for ischemic stress by deacetylating specific mitochondrial proteins of the electron transport chain. Circ Res. 2011;109:396–406. doi: 10.1161/CIRCRESAHA.111.243097. [DOI] [PubMed] [Google Scholar]
- 42.Xiang L, Xiao L, Wang Y, Li H, Huang Z, He X. Health benefits of wine: don't expect resveratrol too much. Food Chem. 2014;156:258–63. doi: 10.1016/j.foodchem.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 43.Jung UJ, Lee MK, Park YB, Jeon SM, Choi MS. Antihyperglycemic and antioxidant properties of caffeic acid in db/db mice. J Pharmacol Exp Ther. 2006;318:476–83. doi: 10.1124/jpet.106.105163. [DOI] [PubMed] [Google Scholar]
- 44.Spilioti E, Jaakkola M, Tolonen T, Lipponen M, Virtanen V, Chinou I, et al. Phenolic Acid composition, antiatherogenic and anticancer potential of honeys derived from various regions in Greece. PLoS One. 2014;9 doi: 10.1371/journal.pone.0094860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang X, Stavchansky S, Zhao B, Bynum JA, Kerwin SM, Bowman PD. Cytoprotection of human endothelial cells from menadione cytotoxicity by caffeic acid phenethyl ester: the role of heme oxygenase-1. Eur J Pharmacol. 2008;591:28–35. doi: 10.1016/j.ejphar.2008.06.017. [DOI] [PubMed] [Google Scholar]
- 46.Koriem KM, Abdelhamid AZ, Younes HF. Caffeic acid protects tissue antioxidants and DNA content in methamphetamine induced tissue toxicity in Sprague Dawley rats. Toxicol Mech Methods. 2013;23:134–43. doi: 10.3109/15376516.2012.730561. [DOI] [PubMed] [Google Scholar]
- 47.Mansour HH, Tawfik SS. Early treatment of radiation-induced heart damage in rats by caffeic acid phenethyl ester. Eur J Pharmacol. 2012;692:46–51. doi: 10.1016/j.ejphar.2012.06.037. [DOI] [PubMed] [Google Scholar]
- 48.Kumaran KS, Prince PS. Caffeic acid protects rat heart mitochondria against isoproterenol-induced oxidative damage. Cell Stress Chaperones. 2010;15:791–806. doi: 10.1007/s12192-010-0187-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee SY, Ku HC, Kuo YH, Chiu HL. Su MJ Pyrrolidinyl caffeamide against ischemia/reperfusion injury in cardiomyocytes through AMPK/AKT pathways. J Biomed Sci. 2015;22:18. doi: 10.1186/s12929-015-0125-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ku HC, Lee SY, Chen CH, Wang YH, Lin CT, Lee SS, et al. TM-1-1DP exerts protective effect against myocardial ischemia reperfusion injury via AKT-eNOS pathway. Naunyn Schmiedebergs Arch Pharmacol. 2015;388:539–48. doi: 10.1007/s00210-015-1098-1. [DOI] [PubMed] [Google Scholar]
- 51.Ku HC, Chen WP, Su MJ. DPP4 deficiency exerts protective effect against H2O2 induced oxidative stress in isolated cardiomyocytes. PLoS One. 2013;8 doi: 10.1371/journal.pone.0054518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reibel DK, Rovetto MJ. Myocardial ATP synthesis and mechanical function following oxygen deficiency. Am J Physiol. 1978;234:H620–4. doi: 10.1152/ajpheart.1978.234.5.H620. [DOI] [PubMed] [Google Scholar]
- 53.Anderson M, Moore D, Larson D. Comparison of isoproterenol and dobutamine in the induction of cardiac hypertrophy and fibrosis. Perfusion. 2008;23:231–5. doi: 10.1177/0267659108100708. [DOI] [PubMed] [Google Scholar]
- 54.Herrmann JE, Heale J, Bieraugel M, Ramos M, Fisher RL, Vickers AE. Isoproterenol effects evaluated in heart slices of human and rat in comparison to rat heart in vivo. Toxicol Appl Pharmacol. 2014;274:302–12. doi: 10.1016/j.taap.2013.11.011. [DOI] [PubMed] [Google Scholar]
- 55.Murakami Y, Zhang Y, Cho YK, Mansoor AM, Chung JK, Chu C, et al. Myocardial oxygenation during high work states in hearts with postinfarction remodeling. Circulation. 1999;99:942–8. doi: 10.1161/01.CIR.99.7.942. [DOI] [PubMed] [Google Scholar]
- 56.Gong G, Liu J, Liang P, Guo T, Hu Q, Ochiai K, et al. Oxidative capacity in failing hearts. Am J Physiol Heart Circ Physiol. 2003;285:H541–8. doi: 10.1152/ajpheart.01142.2002. [DOI] [PubMed] [Google Scholar]
- 57.Bayeva M, Gheorghiade M, Ardehali H. Mitochondria as a therapeutic target in heart failure. J Am Coll Cardiol. 2013;61:599–610. doi: 10.1016/j.jacc.2012.08.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen YR, Zweier JL. Cardiac mitochondria and reactive oxygen species generation. Circ Res. 2014;114:524–37. doi: 10.1161/CIRCRESAHA.114.300559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lai L, Yan L, Gao S, Hu CL, Ge H, Davidow A, et al. Type 5 adenylyl cyclase increases oxidative stress by transcriptional regulation of manganese superoxide dismutase via the SIRT1/FoxO3a pathway. Circulation. 2013;127:1692–701. doi: 10.1161/CIRCULATIONAHA.112.001212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jain K, Suryakumar G, Prasad R, Ganju L. Upregulation of cytoprotective defense mechanisms and hypoxia-responsive proteins imparts tolerance to acute hypobaric hypoxia. High Alt Med Biol. 2013;14:65–77. doi: 10.1089/ham.2012.1064. [DOI] [PubMed] [Google Scholar]
- 61.Chiu CZ, Wang BW, Shyu KG. Molecular regulation of the expression of leptin by hypoxia in human coronary artery smooth muscle cells. J Biomed Sci. 2015;22:5. doi: 10.1186/s12929-014-0109-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tanaka T, Yamaguchi J, Shoji K, Nangaku M. Anthracycline inhibits recruitment of hypoxia-inducible transcription factors and suppresses tumor cell migration and cardiac angiogenic response in the host. J Biol Chem. 2012;287:34866–82. doi: 10.1074/jbc.M112.374587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest. 2005;115:500–8. doi: 10.1172/JCI200524408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Archer SL, Gomberg-Maitland M, Maitland ML, Rich S, Garcia JG, Weir EK. Mitochondrial metabolism, redox signaling, and fusion: a mitochondria-ROS-HIF-1alpha-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am J Physiol Heart Circ Physiol. 2008;294:H570–8. doi: 10.1152/ajpheart.01324.2007. [DOI] [PubMed] [Google Scholar]
- 65.Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–85. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 66.Kerbey AL, Randle PJ, Cooper RH, Whitehouse S, Pask HT, Denton RM. Regulation of pyruvate dehydrogenase in rat heart. Mechanism of regulation of proportions of dephosphorylated and phosphorylated enzyme by oxidation of fatty acids and ketone bodies and of effects of diabetes: role of coenzyme A, acetyl-coenzyme A and reduced and oxidized nicotinamide-adenine dinucleotide. Biochem J. 1976;154:327–48. doi: 10.1042/bj1540327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Whitehouse S, Cooper RH, Randle PJ. Mechanism of activation of pyruvate dehydrogenase by dichloroacetate and other halogenated carboxylic acids. Biochem J. 1974;141:761–74. doi: 10.1042/bj1410761. [DOI] [PMC free article] [PubMed] [Google Scholar]