

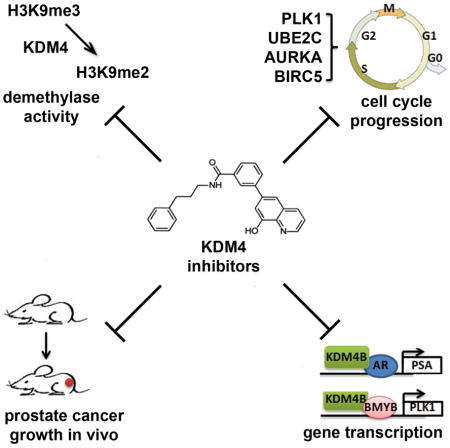
Summary
Histone lysine demethylase KDM4/JMJD2s are overexpressed in many human tumors including prostate cancer (PCa). KDM4s are co-activators of androgen receptor (AR) and thus potential therapeutic targets. Yet to date few KDM4 inhibitors that have anti-prostate tumor activity in vivo have been developed. Here we report the anti-tumor growth effect and molecular mechanisms of three novel KDM4 inhibitors (A1, I9, and B3). These inhibitors repressed the transcription of both AR and BMYB-regulated genes. Compound B3 is highly selective for a variety of cancer cell lines including PC3 cells that lack AR. B3 inhibited the in vivo growth of tumors derived from PC3 cells and ex vivo human PCa explants. We identified a novel mechanism by which KDM4B activates the transcription of polo-like kinase 1 (PLK1). B3 blocked the binding of KDM4B to the PLK1 promoter. Our studies suggested a potential mechanism-based therapeutic strategy for PCa and tumors with elevated KDM4B/PLK1 expression.
Graphical Abstract
Introduction
Histone methylation is an emerging epigenetic mechanism involved in tumorigenesis (1). KDM4/JMJD2s are histone demethylases that act on di- and tri-methylated histone H3 lysine 9 (H3K9me2/me3). Some KDM4s can also demethylate methylated H3K36 in vitro (2). H3K9me3 is normally associated with constitutive and facultative heterochromatins that are condensed and transcriptionally silent. Facultative heterochromatin can de-condense upon loss of H3K9me3 and become transcriptionally permissive in response to specific developmental and environmental cues such as growth factor and/or stress signalings (3). Thus, overexpression of KDM4 is usually associated with downregulation of H3K9me3 and gene activation. The human KDM4 family consists of four members, KDM4A, 4B, 4C and 4D, and two pseudo-genes KDM4E and KDM4F (4). KDM4A, 4B, and 4C contain a catalytic histone demethylase domain (jmj domain), and double PHD and Tudor domains, whereas KDM4D contains only a catalytic domain and lacks PHD and Tudor domains. KDM4s are ferrous iron- and 2-oxoglutarate-dependent oxygenases whose enzymatic activities are amenable to inhibition by small molecules (5). KDM4 proteins are overexpressed in a variety of human pathologies including cancer, mental retardation, and cardiovascular diseases, and are emerging drug targets for cancers (6). KDM4 proteins are co-activators of androgen receptor (AR) (7–11). Overexpression of KDM4 proteins in PCa cells was hypothesized to heighten the sensitivity of AR signaling in castration-resistant prostate cancer (CRPC) under castrated levels of androgen (7). Many small molecule inhibitors of KDM4 have been identified (5, 12–16). However, few compounds have data available on their anti-cancer properties and mechanisms of action. Major challenges to develop KDM4 inhibitors are their specificity and selectivity. Such challenge underscores the need to better understand the mechanism of action of KDM4 in tumor cells and to test any KDM4 inhibitors in the context of such knowledge.
A high-throughput screen for inhibitors of KDM4E was performed as part of the lead optimization projects in NIH for epigenetic targets (12). One of the inhibitors identified, 8-hydroxyquinoline (8HQ), has a 10-fold higher selectivity for the KDM4 family of histone demethylases over other 2-OG oxygenases such as prolyl hydroxylase domain 2 (PHD2). A series of chemical compounds were generated based on the 8HQ chemotype (17). However, the anti-tumor activity and specificity of these compounds remain to be determined. Here, we identified three KDM4 inhibitors (B3, A1, and I9) derived from 8HQ and characterized their anti-tumor growth activities. Among them, compound B3 is the most potent one; it is highly selective for PC3 cells that are androgen-independent. More importantly, B3 is also an active agent in an ex vivo human PCa explant model containing heterogeneous tumors and an in vivo xenograft tumor model. Using these compounds, we uncovered a previously unrecognized mechanism by which KDM4B promotes PCa tumorigenesis. Uniquely, we show that KDM4B appears to be a potent factor in prostate tumorigenesis compared to other KDM4 isoforms. In addition to regulating transcription of AR-dependent genes, KDM4B also regulates gene transcription in an AR-independent manner. KDM4B binds the transcription factor BMYB to activate BMYB-targeted cell cycle genes including polo-like kinase 1 (PLK1). We show that these compounds had similar effects on tumor cell cycle progression and gene transcription as KDM4B knockdown, demonstrating a mechanistic action of novel small molecule inhibitors of KDM4 in targeting prostate tumor growth.
Results
Identification and characterization of novel KDM4 inhibitors that blocked prostate tumor cell growth
Based on Structure-Activity relationship studies, a series of chemical compounds were derived from 8HQ and shown to be active inhibitors of KDM4E and KDM4A (17). We tested the effect of these compounds on the growth of LNCaP cells (Fig. S1A) and selected NCGC00247751 (A1), NCGC00244536 (B3), and NCGC00247743 (I9), which inhibited LNCaP cell growth with IC50s in the μM range (Fig. 1A). These inhibitors suppressed the catalytic activity of KDM4B effectively and among them B3 was the most potent, with an IC50 of ca. 10 nM (Fig. 1B). These compounds also inhibited the enzymatic activity of other KDM4 isoforms although, interestingly, the potency and efficacy of B3 and A1 for KDM4A, 4C, and 4D are lower compared to that of I9, suggesting a potential selectivity of the inhibitors for different isoforms of KDM4. These inhibitors had no or weak activity for KDM5A/JARID1A. KDM5A, like KDM4B, is a trimethyllysine dioxygenase but demethylates H3K4me3 specifically. The three inhibitors had little effect on lysine-specific demethylase 1 (LSD1) (Fig. S1B). LSD1, unlike jmj-C containing lysine demethylases, is a flavin-dependent monoamine oxidase that demethylates mono- and di-methylated lysines on H3K4 and H3K9. To verify that the B3 and A1 compounds act by binding specifically to KDM4s, we used transverse relaxation enhanced spectroscopy (TROSY)-based 1H-15N heteronuclear single quantum correlation (HSQC) spectra. These NMR spectra are like protein fingerprints with one cross-peak for each amide group in a protein and provide a powerful tool to analyze protein-ligand interactions (18). Indeed, addition of B3 and A1 caused shifts in specific cross-peaks of the 1H-15N TROSY-HSQC spectrum of the catalytic domain of KDM4B, and the shift patterns were markedly different for the two compounds (Fig. 1C), showing that both compounds bind specifically to the domain and suggesting that the binding modes are distinct. Similar binding of B3 and A1 to KDM4A was also observed (Fig. S1C).
Figure 1. Novel KDM4 inhibitors selectively inhibit prostate tumor growth.

(A) Cell viability response of LNCaP cells to compound B3, A1, and I9. The chemical structures of B3, A1, and I9 are shown. (B) Dose-response curves of KDM4B, 4A, 4C, 4D, and KDM5A demethylase activities to various concentrations of B3, A1, and I9. (C) Expansions of 1H-15N TROSY-HSQC spectra of KDM4B in the presence (red contours) and absence (black contours) of compounds B3 (left panel) or A1 (right panel). (D) IC50 values reflecting the effects of B3 on the viability of various prostate immortalized epithelial and cancer cell lines, as well as cervical and breast cancer cell lines. Viable cells were quantified by MTT assays. The IC50 values of the compounds were calculated by curve-fitting using Graphpad (N=6, mean ± SD). (E) LNCaP cells were cultured in the presence or absence of the androgen agonist R1881 and/or compound B3 as indicated. (F) PC3 xenograft tumor volume in SCID mice after treatment with vehicle or compound B3. N=9 (vehicle treated) and 10 (B3 treated), mean ± SD. *, p<0.05, compared over vehicle-treated tumors. (G) H&E of representative tumors treated with vehicle or B3.
We tested the inhibitory effect of B3 on a panel of prostate cell lines. B3 displayed high selectivity for the fast-growing AR-negative PC3 cells (IC50 40 nM) and over 100-fold selectivity against the immortalized prostate epithelial cell lines PrEC1 and PrEC4 (Fig. 1D, Fig. S1D). Also, B3 effectively inhibited AR-positive cell lines, including LNCaP and VCaP, with IC50s in the sub-micromolar range (Fig. 1D), and abolished androgen-stimulated LNCaP cell growth (Fig. 1E). In addition, B3 is also potent in inhibiting the growth of other cancer cell lines, including the breast cancer cell lines MDA-MB2 and MCF-7, with micromolar IC50s (Fig. 1D). We further tested whether B3 can inhibit tumor growth in vivo by using a xenograft model with tumors derived from injection of PC3 cells. PC3 cells were injected to the planks of SCID mice. After the tumor volume reached 50 mm3, we began daily treatment with 20 mg/kg of B3 or vehicle control. Treatment with B3 resulted in significant inhibition of tumor growth (Fig. 1F) and animals did not exhibit any major toxicity and appeared to be normal. Histological data clearly indicated that B3-treated tumors had significant amount of cell apoptosis, necrosis, and fibrosis (Fig. 1G).
KDM4 inhibitors had similar inhibitory effects on prostate tumor cell cycle progression as KDM4B knockdown
We were intrigued by the sensitivity of PC3 cells to compound B3 since PC3 cells have no AR. We reasoned that KDM4B may have AR-independent mechanism or may not target KDM4 proteins in PC3 cells. To explore these two possibilities, we first tested whether KDM4 inhibitor-treated PCa cells and KDM4 knockdown cells have similar cellular phenotypes, and then tested whether KDM4 inhibitors and KDM4-knockdown have common transcriptional targets.
We tested the role of individual KDM4 isoform in prostate cell growth as they are all expressed in PCa cells (Fig. S2A). We surveyed the expression levels of KDM4 isoforms in previously published gene expression datasets that contained normal prostate tissue, primary PCa, and metastatic CRPC (GEO accession GDS2545 and GEO accession GSE32269; 19–21). KDM4B is the major isoform expressed in prostate tissues and its expression is upregulated in primary PCa and further upregulated in CRPCs significantly whereas the expression of KDM4A, 4C, and 4D remain similar among normal and cancerous tissues (Fig. 2A). To test the effect of different KDM4 isoforms on the growth of LNCaP and PC3 cell lines, we knocked down 4A, 4B, and 4C with two specific siRNAs for each isoform (Fig. 2B, Fig. S2, A & B). Knockdown of KDM4A, 4B or 4C inhibited the cell growth of both LNCaP and PC3 cells significantly, and the strongest effects on growth were caused by the KDM4B siRNAs (Fig. 2C). We next investigated the effect of KDM4A or KDM4B knockdown on cell cycle progression using flow cytometry (FACS) analysis. Since two independent siRNA duplexes resulted in similar effect on PCa cell growth, we focused on one of the siRNA duplexes in the following experiments. LNCaP and PC3 cells were transfected with control, KDM4A, or KDM4B siRNA, pulse labeled with Bromodeoxyuridine (BrdU), stained with Propidium Iodide (PI) and anti-BrdU antibody, and then subjected to FACS analysis (Fig. S2D). The percentage of cells in S phase was significantly decreased in LNCaP cells transfected with KDM4B siRNA compared to those transfected with control siRNA (Fig. 2D, left panel). Downregulation of S-phase cells by KDM4B siRNA is not due to the off-target effect of siRNA as adding back exogenously expressed siRNA-resistant KDM4B in KDM4B siRNA-treated cells rescued the cell cycle phenotype (Fig. S2E).
Figure 2. KDM4 siRNA and inhibitors inhibited DNA replication of PCa cells.

(A) Relative expression levels of KDM4 isoforms in normal and cancerous prostate tissue samples. Error bars are SEM. *, p<0.05, **, p<0.01. (B) Western blots of LNCaP cells transfected with control (ctl), KDM4A (4A), KDM4B (4B), or KDM4C (4C) specific siRNA and probed with antibodies against KDM4A, KDM4B, and GAPDH. (C) Growth curves of LNCaP (left panel) and PC3 cells (right panel) transfected with the indicated siRNA duplexes. (D) FACS analysis of LNCaP cells (left panel) and PC3 cells (right panel) transfected with control (ctl), 4A, or 4B specific siRNA. Two different siRNA duplexes were used. Representative ones are shown here. N=3, mean ± SD. *, p<0.05, compared to control-siRNA transfected cells. (E) FACS analysis of LNCaP and PC3 cells treated with vehicle DMSO, compound I9 (10 μM), A1 (5 μM), or B3 (1 μM). N=3, mean ± SD. *, p<0.05, compared to DMSO-treated cells. (E) Percentage of apoptotic cells treated with vehicle, A1, or B3 as assayed by Annexin V staining.
Downregulation of S phase cells is even more dramatic in fast cycling PC3 cells that were transfected with either KDM4B or KDM4A siRNA. Knockdown of KDM4B resulted in cell cycle arrest at G1/S (Fig. 2D). Knockdown of KDM4B also led to cell cycle arrest at G2/M in PC3 cells (Fig. 2D, right panel). Taken together, these data suggest that among different KDM4 isoforms, KDM4B plays a major role in the growth of LNCaP and PC3 cells.
To test whether KDM4 inhibitors have similar effects on cell cycle progression as KDM4B siRNA, LNCaP and PC3 cells were treated with B3, A1, and I9 (Fig. 2E). Indeed, we observed similar decreases in the number of S phase cells when cells were treated with compounds B3 and A1. Cell cycle arrest at G2/M by compound A1 in fast cycling PC3 cells is also observed (Fig. 2E, right panel). Moreover, we observed increased cell apoptosis of both LNCaP and PC3 cells after treatment with inhibitors (Fig. 2F). The more sever effect of the inhibitors on PCa cells in comparison to KDM4B-knockdown alone may be due to the combined action of the compound on various KDM4B isoforms. Taken together, our data indicate that either KDM4B knockdown or KDM4 inhibitors had similar inhibitory effect on cell cycle progression.
Inhibition of KDM4B either by siRNA-depletion or inhibitors down-regulates the expression of critical cell cycle gene PLK1
To investigate whether KDM4B knockdown and KDM4 inhibitors have common transcriptional target, we first performed microarray gene expression profiling with cells treated with compound B3, A1, or I9. We reasoned that the common targets of these three compounds may represent targets of KDM4B. All three compounds had similar effects on alteration of gene expression pattern (Fig. S3A). We focused on downregulated genes as KDM4B is a transcriptional co-activator. There were 186 common downregulated genes (Fig. S3B). Consistent with their effect on cell cycle progression, gene ontology analysis indicated that a majority of the genes were nuclear proteins involved in the cell cycle (Fig. S3C). We noticed that some of the most-downregulated genes including PLK1 are not AR-targeted genes and has been shown to be regulated by transcription factor BMYB (22) (Fig. 3A). We compared the 186 genes with published AR- and BMYB-targeted genes identified by AR- and BMYB-ChIP-seq experiments (21, 22). We found that 15% and 18% of 186 downregulated-genes are potential targets of the transcription factors AR and BMYB, respectively (Fig. S3D). We focused on cell cycle regulator PLK1, AURKA, BIRC5, and UBE2C (highlighted in red in Figure 3A) as they have not been shown to be regulated by KDM4B previously. We confirmed their downregulation in both inhibitor-treated and KDM4B siRNA-transfected cells using qRT-PCR (Fig. 3B, 3C). Downregulation of PLK1 in KDM4A, 4B siRNA, or B3-treated cells was further confirmed by western blot analysis (Fig. 3D).
Figure 3. Inhibition of KDM4B by either siRNA or inhibitors down-regulated the expressions of cell cycle genes including PLK1.

(A) Heat map of selected downregulated genes from LNCaP cells treated with compound I9, A1, or B3. Cell cycle genes that are also downregulated in KDM4B knockdown cells are highlighted in red. (B) qRT-PCR analysis of selected differentially expressed transcripts in compound-treated LNCaP and PC3 cells. mRNAs are expressed relative to vehicle (ctl) treated cells. (C) qRT-PCR analysis of selected genes downregulated in KDM4B siRNA-transfected LNCaP and PC3 cells. mRNAs were normalized against GAPDH and expressed relative to that in control siRNA-transfected cells. (D) Western blots of indicated proteins in LNCaP and PC3 cells treated without or with compound B3 (left panel), control, KDM4A, or KDM4B siRNA (right panel).
Downregulation of cell cycle gene PLK1 in KDM4B siRNA-treated cells suggests that PLK1 may be activated by KDM4B. To test this possibility, we measured the expression of KDM4B at different cell cycle phases in LNCaP cells (Fig. 4A). Unlike KDM4A, which is mainly expressed in the G1/S and early S phases but little in late S-phase and G2/M, KDM4B is expressed throughout the cell cycle. We next assayed the KDM4 demethylase activities and the expression of PLK1 and other cell cycle genes at different time points of the cell cycle after synchronized cells were released from G1/S. The activities of KDM4 peaked at 4 and 9 h after entering the cell cycle (Fig. 4B). Coinciding with the maximal expression of PLK1 at 9 h post-G1/S, we observed maximal binding of KDM4B to the PLK1 promoter around the same time point (Fig. 4C). As expected, upregulation of KDM4B binding at the PLK1 promoter correlated with downregulation of H3K9me3 occupancy at the binding site. The peak activities of KDM4 also coincided with mRNA peaks of UBE2C and BIRC5 at 4 h, and PLK1, AURKA, and CDK1 at 9 h (Fig. 4D). Compound B3 effectively suppressed the expression of PLK1, AURKA, and BIRC5 at the right time point. Taken together, these correlative data suggest that KDM4B could regulate transcription of PLK1 and B3 could inhibit PLK1 transcription via KDM4B.
Figure 4. Cell cycle-dependent PLK1 expression requires KDM4 activity.

(A) Western blot of indicated proteins from thymidine-released LNCaP cells. (B) Relative KDM4 demethylase activity at various time points of double thymidine released LNCaP cells. The activities were expressed relative to asynchronized (asy) cells. (C) Relative occupancy of H3K9me3, KDM4B, and BMYB on the PLK1 promoter at various time points of double thymidine-released LNCaP cells. The amounts of immunoprecipitated complex were normalized against DNA input and expressed relative to that from asynchronized cells. (D) Relative fold change of mRNA of the indicated genes from LNCaP cells described in (B) in the presence or absence of compound B3. mRNAs were expressed relative to the 0 h time point.
KDM4B activates PLK1 transcription via BMYB and its recruitment to PLK1 promoter is blocked by compound B3
BMYB is part of the MuvB complex during early S phase and of the MuvB/FoxM1 complex in late S phase to promote expression of early G1/S phase and G2/M phase-expressed cell cycle genes, respectively (23). PLK1 is a master regulator of mitosis (24) and is expressed maximally in late S phase (Fig. 4A). To test whether PLK1 is a direct transcriptional target of KDM4B and understand the molecular mechanism underlying KDM4B-activated PLK1 transcription, we examined the genomic sequence of the human PLK1 promoter (25) and found two highly conserved BMYB binding sites at −490bp (II) and −1181bp (I) 5′-upstream of the transcriptional start site (Fig. 5A, circles). Reporter assays with PLK1 promoter-driven luciferase (PLK1-luc) showed that KDM4B could activate PLK1 transcription in a dose-dependent manner (Fig. 5A). Activation of PLK1 transcription by KDM4B requires its demethylase activity as the demethylase-inactive mutant KDM4BH189G failed to activate PLK1 transcription. Interestingly, KDM4BH189G acted as a dominant-negative form in LNCaP cell growth assay (Fig. S4A). Further reporter assays with deletion and point-mutation variants of the PLK1 promoter indicated that KDM4B activated PLK1 transcription through BMYB binding site II at −490bp (Fig. 5A). Maximal activation of PLK1 transcription by BMYB requires endogenous KDM4B as depletion of KDM4B by siRNA resulted in a significant loss of BMYB-activated PLK1 transcription (Fig. 5B). We next tested whether BMYB can interact with KDM4B, therefore recruiting it to the PLK1 promoter. Indeed, we observed endogenous interaction between BMYB and KDM4B in a co-immunoprecipitation assay (Fig. 5C). Further co-immunoprecipitation assays suggested that the region between the N-terminal jmjC domain and C-terminal PHD domain of KDM4B (residues 430–710) was sufficient to mediate the interaction between KDM4B and BMYB (Fig. 5D). We further tested the effect of B3 on the binding of KDM4B to the promoter of PLK1 in LNCaP cells. B3 disrupted the loading of KDM4B to the PLK1 promoter around the BMYB binding site without affecting BMYB binding (Fig. 5E). Downregulation of KDM4B binding to the PLK1 promoter is not due to a loss of KDM4B protein as B3 treatment at the concentration used (1μM) had little effect on KDM4B protein level (Fig. 3D). Loss of KDM4B on the PLK1 promoter is associated with upregulation of H3K9me3 and downregulation of H3K4me3 on the PLK1 promoter (Fig. 5E). Upregulation of H3K9me3 is likely due to decreased binding of KDM4B on the PLK1 promoter since H3K9me3 is a substrate of KDM4B whereas downregulation of H3K4me3 may be an indirect effect and consequence of transcriptionally less-active gene. B3 also significantly diminished the binding of KDM4B to the promoter/enhancer regions of several other genes that are important for cell cycle progression (Fig. S4B).
Figure 5. KDM4B interacts with BMYB and activates BMYB-targeted PLK1 transcription.

(A) PLK1-luc reporter activities in cells transfected with full-length (1.5 kb), deletion (1.0, 0.5 kb) or point-mutant (x) of a PLK1-promoter construct and a vector that expresses KDM4B, KDM4BH189G, or BMYB as indicated. (B) PLK1-luc activity in control, KDM4A (2A), or KDM4B (2B) knocked down cells that were transfected with various amount of BMYB. (C) LNCaP cell lysates were immunoprecipitated with control IgG or anti-KDM4B antibody, respectively. Immunoprecipitates were Western blotted with antibodies against BMYB and KDM4B. (D) 293T cells were transfected with Flag-BMYB and HA-KDM4B (left lower panel) or various deletion mutants of KDM4B (right panel). Whole cell lysates were immunoprecipitated with anti-Flag antibody and immunoprecipitates were Western blotted with anti-HA antibody. (E) ChIP-qPCR assays showing the relative occupancy of H3K9me3, KDM4B, BMYB, H3K36me3, and H3K4me3 at the PLK1 promoter in LNCaP cells treated with compound B3 (1 μM) or vehicle DMSO. N=3, mean ± SD. *, p<0.05.
B3 is active in human ex vivo prostate tumor explants
To test whether B3 has the same effect on human prostate tumor tissue in terms of target gene expression, an ex vivo explant model that contains both human tumor tissue and its surrounding “native” microenvironment (26) was employed. B3 effectively inhibited the expression of AR (Fig. 6A, upper panel) and other KDM4B-targeted genes in the prostate tumors (Fig. 6B). No global change of H3K9me3 was observed in B3-treated tumors compared to control vehicle treated tumors (Fig. 6A, middle panel). It is noted that B3 had no effect on the morphology of stromal cells surrounding the tumors (Fig. 6A, lower panel, arrows), suggesting that the inhibitory effect of B3 is specific to tumor cells.
Figure 6. Compound B3 inhibits AR and critical cell cycle genes in ex vivo solid human prostate tumors and growth of PC3 xenograft tumor in vivo.

(A) Immunohistochemical staining of AR (upper panel) and H3K9me3 (middle panel), and H & E staining (lower panel) in sections of human prostate tumor in ex vivo cultures in the presence of vehicle DMSO or B3. (B) Relative fold change of mRNA of genes as indicated from tissues described in (A). mRNA levels were normalized against GAPDH and expressed relative to those in cultures in the presence of DMSO. N=5, mean ± SD.
Discussion
Epigenetic control via chromatin modification is recognized as a fundamental mechanism for regulating gene expression throughout development and in human diseases, yet we have only just begun to take advantage of it for therapeutic uses. KDM4 proteins are overexpressed in variety of human pathologies, which made them desirable targets for interventional therapy. Moreover KDM4 proteins are idea targets for small molecule inhibitors compared to transcription factors for their enzymatic activities are amenable for inhibition. In this study, we identified and characterized three novel KDM4 inhibitors (A1, I9, and B3). Our data suggest that these compounds hold potential promise for therapeutic uses. First, these compounds have nanomolar IC50s for inhibiting KDM4B demethylase activity, which is significantly lower than those of previously identified inhibitors. Furthermore they have over 100-fold selectivity between cancerous and non-cancerous cells, although whether this selectivity can be translated into clinical advantage remains to be seen. Importantly, compound B3 has expected inhibitory effects on cell cycle genes in solid ex vivo human prostate tumors that are highly heterogeneous, implicating that the cell cycle genes identified here may be used as biomarkers for evaluating the efficacy of compounds in future cancer therapy. Lastly, while the B3 compound has not yet been optimized for systemic administration, localized administration of B3 near the tumor site blocked tumor growth in an in vivo xenograft model using human prostate cancer cells. This should encourage further medicinal chemistry efforts to explore the full potential of this compound class as drug in future pre-clinical application.
Although KDM4A, 4B, and 4C are shown to be overexpressed in many human cancers and promote tumorigenesis through variety of mechanisms (1, 6, 27, 28), the mechanism by which KDM4B activates PLK1 transcription via BMYB in the late S-phase of PCa cells has not been reported before, thus we focused on this mechanism in this study. We found that KDM4B is the major isoform in PCa tumorigenesis compared to KDm4A and 4C. KDM4B is significantly upregulated in metastatic PCa and has the strongest effect on PCa growth (Fig. 2). This may be due to its unique role in regulating the expression of PLK1 at late S-phase since KDM4B is expressed throughout the cell cycle including the late S-phase where PLK1 is maximally expressed whereas KDM4A is mainly expressed in G1 and early S phase (Fig. 4A). Although it is possible that the effects of KDM4 inhibition on cell cycle may be stress responses and there is a possibility that the core of the cellular effects of the compounds may result from non-KDM4 mediated metallo-enzyme inhibition, several lines of evidence in our study suggest that the inhibitors we identified here can target KDM4B specifically in late S-phase; (1) PCa cells treated with either KDM4 inhibitors or KDM4 siRNA had similar cell cycle defects (Fig. 2); (2) both inhibitors and KDM4B siRNA inhibited the expressions of similar sets of genes that include critical cell cycle regulator PLK1 (Fig. 3); (3) the peak level of KDM4 demethylase activity coincides with a maximal binding of KDM4B to the PLK1 promoter (Fig. 5) and is associated with the expression of PLK1 (Fig. 4); (4) KDM4B interacts with BMYB and activates PLK1 transcription (Fig. 5); finally (5) compound B3 blocked the recruitment of KDM4B to the PLK1 promoter, which is associated with upregulation of repressive mark H3K9me3.
The AR-independent mechanism of KDM4B in PCa cells implicates that KDM4B may play an important role in PCa progression towards castration resistance, albeit it remains to be tested in the future. KDM4B may utilize this AR-independent mechanism to promote PCa cell growth when androgen or AR activity is low, thus select for castration-resistant phenotype. 8HQ-derived chemical compounds identified here thus may be useful for CRPC that are lethal at present. Indeed, we show that compounds B3 is effective in inhibiting growth of CRPC cells such as PC3 and VCaP cells (Fig. 1D).
Activation of PLK1 by KDM4B has also other implications in cancer therapy, for it may provide alternatives over those targeting PLK1 or BMYB. Both PLK1 and BMYB are implicated in human tumorigenesis (22, 23, 27–29). PLK1 is a critical regulator for both DNA replication and chromosome segregation in mitosis (24, 29). Overexpression of PLK1 has been observed in a variety of solid tumors as well as in acute myeloid leukemia, and has been correlated with poor prognosis and metastatic potential (30). BMYB is part of the MuvB complex during S phase and of the MuvB/FoxM1 complex in late S phase to promote expression of early G1/S phase and G2/M phase-expressed cell cycle genes, respectively (23). BMYB is frequently overexpressed in tumors and forms part of the proliferation signature that is characteristic of high-grade tumors with poor prognosis. Upregulation of BMYB can drive cells out of quiescence and towards proliferation. Because of these tumorigenic roles of PLK1 and BYMB, they are attractive molecular targets for cancer therapy. Various PLK1 inhibitors have been developed and are being tested in clinical trials (31). BMYB is a component of the breast cancer gene expression signature in the Oncotype Dx (Genomic Health) clinical biomarker test. However, PLK1 and BMYB are double-edged swords as they are both required for normal cell cycle progression. Inhibition of PLK1 and BMYB could have undesirable side effects in organs with high cell proliferation such as bone marrow. In contrast, no individual KDM4 isoform is required for normal development as mice lacking it are viable (32–35), which could provide a strategy of targeting KDM4B without interfering with its normal function. Indeed, we noted that the B3 inhibited PLK1 expression by suppressing the recruitment of KDM4B to the PLK1 promoter without affecting binding of BMYB to the PLK1 promoter (Fig. 5). Thus, it will be worth investigating in the future how KDM4B differentially regulates BMYB-activated gene transcription between normal and cancer cells.
H3K9me3 is a heterochromatin mark and its level varies between and within each cell cycle phase (36, Fig. 4A). Although it is possible that inhibition of KDM4 may alter the global H3K9me3 level, we did not observe significant changes of global H3K9me3 by KDM4B siRNA knockdown. In synchronized LNCaP cells we observed two peaks of KDM4 activity that occurred at 4 and 9 h after release from double thymidine block (Fig. 4B) and had no correlation with changes of global H3K9me3 during the cell cycle (Fig. 4A). On the other hand, changes of KDM4 demethylase activity are tightly correlated with the peaks of mRNA and protein expression of multiple cell cycle genes including PLK1 and UBE2C in S phase (Fig. 4D). These results suggest that KDM4B regulates H3K9me3 at specific loci rather than the global H3K9me3 level during S phase. It is worth noting that majority of the genes downregulated by KDM4B siRNA or KDM4 inhibitors such as PLK1, AURKA/B, UBE2C, CENPA, and TPX2 are related to DNA replication and centrosome amplification and maturation. These genes regulate cell cycle progression epistatically and/or in coordination. The effect of inhibition of KDM4 activity on the expression of cell cycle genes therefore appears to be highly specific.
In summary, the studies presented here strongly suggest that targeting KDM4 with small molecule inhibitors may provide a therapeutic strategy for prostate cancer, especially CRPCs that are refractory to anti-androgen therapies. Our discovery that KDM4B can function as a BMYB co-activator also suggests that inhibition of KDM4 may be an effective therapeutic strategy for other cancers.
Significance
The progression of prostate cancer from an androgen-dependent to an androgen-independent/castration resistant state marks its lethal progression. Histone demethylase KDM4s are known co-activators of the AR and is believed to promote CPRC through an AR-dependent mechanism. Here, we identified a set of novel KDM4 inhibitors that inhibited the growth of a variety of prostate cancer cell lines including AR-negative PC3 cells and PC3 xenografts in vivo. Using these inhibitors, we uncovered a novel AR-independent mechanism by which KDM4B promotes prostate tumorigenesis. KDM4B activates the transcription of BMYB-targeted genes such as PLK1 that are critical for cell cycle progression and may be one of the mechanisms underlying castration-resistance. Our studies suggest that targeting KDM4B may provide an effective therapeutic strategy for prostate cancers that are refractive to anti-androgen therapies.
Experimental Procedures
General Chemistry
Preparative purification was run on a Waters semi-preparative HPLC system using a Phenomenex Luna C18 (5 micron, 30 × 75 mm) at a flow rate of 45 mL/min. A gradient of 10% to 50% acetonitrile in water over 8 minutes (each containing 0.1% trifluoroacetic acid) was used as a mobile phase during the purification. Fraction collection was triggered by UV detection (220 nm). Analytical analysis was performed on an Agilent LC/MS (Agilent Technologies, Santa Clara, CA). Method t1: A 7 minute gradient of 4% to 100% Acetonitrile (containing 0.025% trifluoroacetic acid) in water (containing 0.05% trifluoroacetic acid) was used with an 8 minute run time at a flow rate of 1 mL/min. A Phenomenex Luna C18 column (3 micron, 3 × 75 mm) was used at a temperature of 50° C. Method t2: A 3 minute gradient of 4% to 100% Acetonitrile (containing 0.025% trifluoroacetic acid) in water (containing 0.05% trifluoroacetic acid) was used with a 4.5 minute run time at a flow rate of 1 mL/min. A Phenomenex Gemini Phenyl column (3 micron, 3 × 100 mm) was used at a temperature of 50° C. Purity was determined using an Agilent Diode Array Detector for both Method 1 and Method 2. Mass determination was performed using an Agilent 6130 mass spectrometer with electrospray ionization in the positive mode. 1H NMR spectra were recorded on Varian 400 MHz spectrometer. Chemical shifts are reported in ppm with undeuterated DMSO-d6 at 2.49 ppm as internal standard. High resolution mass spectrometry was recorded on Agilent 6210 Time-of-Flight LC/MS system. Confirmation of molecular formula was accomplished using electrospray ionization in the positive mode with the Agilent Masshunter software (version B.02).
General experimental procedures
The detailed experimental procedures for the intermediates were described previously (17).
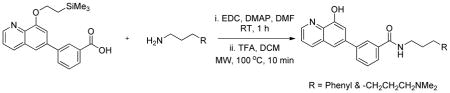
A mixture of 3-(8-(2-(trimethylsilyl)ethoxy)quinolin-6-yl)benzoic acid (0.075 g, 0.205 mmol, 1 eq), EDC (0.079 g, 0.410 mmol, 2 eq) and DMAP (0.013 g, 0.103 mmol, 0.5 eq) in DMF (0.5 mL) was added the amine (0.308 mmol, 1.5 eq) and stirred at room temperature for 2 h. The reaction mixture was filtered through a syringe filter and purified on a preparative HPLC. The product obtained after liophilization of the fractions was dissolved in dichloromethane/trifluroacetic acid (2 mL, 1/2 by volume) and heated in a microwave for 15 minutes at 100°C. The crude product obtained after removing the solvent was purified on a preparative HPLC to obtain the products as TFA salts.
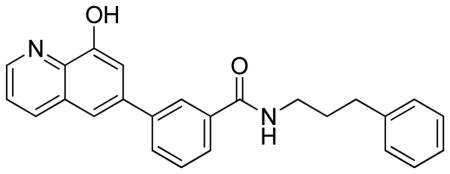
3-(8-Hydroxyquinolin-6-yl)-N-(3-phenylpropyl)benzamide.TFA
LC-MS Retention Time: t1 = 4.401 min and t2 = 2.717 min; 1H NMR (400 MHz, DMSO-d6) δ 10.59 (brs, 1H), 8.92 (dd, J = 4.5, 1.6 Hz, 1H), 8.70 – 8.57 (m, 2H), 8.24 (d, J = 1.9 Hz, 1H), 7.98 – 7.85 (m, 3H), 7.73 (dd, J = 8.5, 4.4 Hz, 1H), 7.66 – 7.54 (m, 2H), 7.34 – 7.21 (m, 4H), 7.23 – 7.13 (m, 1H), 3.38 – 3.28 (m, 2H), 2.66 (t, J = 7.7 Hz, 2H), 1.93 – 1.81 (m, 2H); HRMS (ESI) m/z (M+H)+ calcd. for C25H23N2O2, 383.1754; found 383.1762.
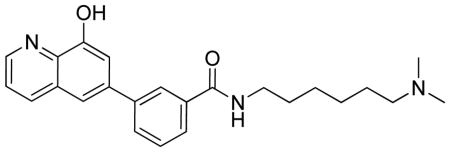
N-(6-(dimethylamino)hexyl)-3-(8-hydroxyquinolin-6-yl)benzamide.TFA
LC-MS Retention Time: t1 = 3.162 min and t2 = 2.529 min; 1H NMR (400 MHz, DMSO-d6) δ 10.34 (s, 1H), 9.36 (s, 1H), 8.90 (dq, J = 5.3, 2.8, 2.3 Hz, 1H), 8.64 (d, J = 5.4 Hz, 1H), 8.52 (dt, J = 10.1, 2.9 Hz, 1H), 8.24 (p, J = 2.0 Hz, 1H), 7.98 – 7.79 (m, 3H), 7.77 – 7.50 (m, 4H), 3.31 (dd, J = 8.1, 4.5 Hz, 2H), 3.02 (dt, J = 9.2, 4.3 Hz, 2H), 2.79 – 2.71 (m, 6H), 1.61 (dd, J = 15.5, 9.0 Hz, 4H), 1.40 – 1.31 (m, 4H); HRMS (ESI) m/z (M+H)+ calcd. for C24H30N3O2, 392.2333; found 392.2325.

Synthesis of 8-hydroxy-3-(4-(2-(naphthalen-1-yl)ethyl)-1,4-diazepan-1-yl)quinoline-5-carboxylic acid. TFA
A mixture of methyl 3-iodo-8-(2-(trimethylsilyl)ethoxy)quinoline-5-carboxylate (0.2 g, 0.466 mmol, 1 eq), 1-(2-(naphthalen-1-yl)ethyl)-1,4-diazepane, 2HCl (0.229 g, 0.699 mmol, 1.5 eq), xantphos (0.013 g, 0.023 mmol, 5 mol %), Pd2(dba)3 (10.66 mg, 0.012 mmol, 2.5 mol %) and t-BuONa (0.157 g, 1.630 mmol, 3.5 eq) in toluene (3 mL) was bubbled with argon for 5 minutes. The vial was sealed and stirred at 110°C in a preheated heating block for 4 h. After completion of the reaction, the solvent was removed by forced air. The crude mixture was taken up in DMSO and stirred with palladium scavenger for 30 minutes. The solution was then filtered through a syringe filter and purified on a preparative HPLC to get pure 3-(4-(2-(naphthalen-1-yl)ethyl)-1,4-diazepan-1-yl)-8-(2-(trimethylsilyl)ethoxy)quinoline-5-carboxylic acid (0.1 g, 0.185 mmol). The above product was dissolved in DCM/TFA (3 mL, 2/1 by volume) and stirred at 75 °C in a sealed tube for 1 h. The crude product was taken up in DMSO (2mL) and purified on a preparative HPLC to obtain the product as TFA salt. Retention Time: t1 = 3.781 min and t2 = 2.726 min; 1H NMR (400 MHz, DMSO-d6) δ 12.48 (brs, 1H), 9.83 (brs, 1H), 8.71 (dt, J = 31.1, 2.8 Hz, 2H), 8.35 – 8.06 (m, 2H), 7.92 (ddd, J = 38.6, 7.7, 2.0 Hz, 2H), 7.73 – 7.34 (m, 4H), 6.82 (dd, J = 8.1, 2.5 Hz, 1H), 4.23 – 3.55 (m, 4H), 3.44 – 3.25 (m, 6H), 2.40 – 2.23 (m, 4H); HRMS (ESI) m/z (M+H)+ calcd. for C27H28N3O3
Cell lines and antibodies used were described in detail in supplemental material. Plasmids, site-directed mutagenesis, lentiviruses, transfection, and luciferase-reporter assays
KDM4A plasmids were described previously (32). KDM4B was obtained from Addgene. PLK1-luc was constructed by subcloning PCR-amplified human PLK1 promoter fragments (25) into pGL2-basic (Promega, USA) using KpnI and XhoI sites. Site-specific mutations were made using quick-change kit (Stratagene). Mutations were confirmed by DNA sequencing. siRNA duplexes were purchased from Sigma and sequences were listed in Table S1. Plasmids and siRNA transfection was performed using lipofectamine 2000 and RNAimax, respectively. Transfections with LNCaP cells were done using EZPlex (Ascension Bio). Luciferase activities were measured with total cell lysates after 48 h transfection using luciferase assay kit (Promega). Relative promoter activities were expressed as relative luminescence units normalized for cotransfected β-galactosidase activities in the cell.
Cell proliferation assay
MTT cell proliferation assay kit (ATCC) and trypan blue staining of live cells were used to measure viable cells.
KDM4 demethylase activities were measured using Epigenase JMJD2/KDM4 demethylase assay kit (Epigentek, P-3081). Human KDM4B(1–500) purified from SF9 cells was purchased from BPS bioscience. KDM4A(1–350), KDM4C(1–350), and KDM4D(1–350) were purified as recombinant proteins from E. coli. These proteins were used to obtain the dose-response curves of KDM4B enzymatic activity upon exposure to different concentrations of KDM4 inhibitors. To measure total KDM4 activity in cells, LNCaP cells were synchronized with the double thymidine block and harvested after they were released for indicated time points. Nuclear proteins were isolated using standard protocol (Abcam) and quantified using Bradford assays. A total of 10 μg nuclear extracts were used to measure the KDM4 demethylase activity.
RNA and cDNA preparation, real-time qRT-PCR, and microarray analysis
Total RNA was isolated using an RNeasy Mini Kit (Qiagen). First-strand cDNA was made using Superscript III Reverse Transcriptase (Invitrogen). SYBR-based qRT-PCR was used to examine relative levels of selected mRNAs. All data were normalized to an internal standard (GAPDH; CT method). Sequences for gene-specific primer pairs are listed in Table S1. Microarray analyses were performed using Illumina bead array platform.
Cell synchronization, BrdU labeling, and FACS
Cells were synchronized at the G1/S boundary by double-thymidine treatment. In brief, cells were incubated for 19 h in 2 mM thymidine (Sigma), released for 9 h and then incubated for another 16 h in 2 mM thymidine. For BrdU labeling, asynchronous cells in culture were incubated with 20 μM BrdU for 20 min before harvested for FACS analysis. Flow cytometry was carried out using a FACSCalibur (BD Biosciences) and data were analyzed using Flowjo software.
ChIP-qPCR
ChIP was carried out using the antibodies indicated in the figures and a protocol described previously (32). Immunoprecipitated chromatin fragments were quantified by SYBR-based qPCR, normalized using the percent input method (Invitrogen). qPCR primers were listed in Table S1.
Western blot, immunoprecipitation, protein proximity ligation assay, immunofluorescence, and immunohistochemistry
Western blot and immunoprecipitation were performed following standard protocols. Immunohistochemical staining of AR and H3K9me3 on paraffin-embedded human prostate tumors was performed following a previously described procedure (37).
In vivo tumor model, ex vivo culture of human prostate tumors, and drug administration
For xenograft animal model, PC3 cell suspension was injected into 4–6 weeks old severe combined immunodeficient (SCID) mice subcutaneously at the concentration of 1×106 per site. When tumors become palpable, animals were randomly grouped for drug treatment. Alzet osmotic minipump containing B3 (20 mg/kg) was subcutaneously inserted into each animal, which allowed continuous drug delivery to the tumor site for up to 5 days. Tumor volume was recorded every other days and calculated by using the ellipsoid formula. All experimental procedures were approved by the Institutional Animal Care and Use Committee. For ex vivo culture, fresh prostate cancer tissues were obtained with informed consent from men undergoing radical prostatectomy at the Hospitals of the UT southwestern see Table S2 for clinicopathological characteristics). The procedure for establishment of explant followed the previous description (38). Tissues were cultured at 373°C with vehicle (DMSO) alone or B3 (2.5 μM) for 483h and then formalin-fixed and paraffin embedded for immunohistochemistry analyses or preserved in RNAlater (Invitrogen).
NMR
All NMR spectra were acquired at 25 °C on Varian INOVA 600 MHz spectrometer. Samples for 1H-15N TROSY-HSQC measurements contained 65 μM protein and 100 μM of compounds B3 and A1 dissolved in 25 mM Tris (pH 7.4), 125 mM NaCl and 1 mM TCEP with 5% D2O. Total acquisition times were 2–10 h. NMR data were processed with NMRPipe (39) and analyzed with NMRView (40).
Statistics
All data are shown as mean ± SD or mean ± SEM as indicated in the figure. Student’s t test (2-tailed) was used to compare the difference between 2 groups. p < 0.05 was considered statistically significant. Gaphpad was used to calculate IC50.
Supplementary Material
Highlights.
We identified several novel inhibitors of histone trimethyllysine demethylase KDM4
Inhibitors blocked the growth of prostate cancer cells and PC3 xenograft in vivo
Inhibitors suppressed cell cycle progression via inhibition of cell cycle gene PLK1
KDM4B activates the transcription of both AR and BMYB-targeted cell cycle genes
Acknowledgments
We thank Yilun Sun for excellent technical assistance.
Funding: Supported by NIHRO1 (Z.P.L. and E.D.M.), Welch foundation (E.D.M), CPRIT-MIRA RP120717-P1 (Z.P.L.), CPRIT-MIRA RP120717-P3 (J.R.), and CPRIT-MIRA RP120717-C1 (J.T.H) G.R., A.J., A.S., and D.J.M. were supported by the intramural research program of the National Center for Advancing Translational Sciences and the Molecular Libraries Program of the National Institutes of Health Common Fund (U54MH084681).
Footnotes
Author contributions: L.L.D. performed MTT, luciferase-reporter, coimmunoprecipitation, Western blot, immunofluorescence, FACS, qPCR, ChIP, demethylase activity, and inhibitor studies. C.R. and J.R. designed and performed NMR studies. Q.J.Z. performed initial MTT assays to screen the bioactive inhibitors. Q.W. performed proximity ligation and IHC assays. S.H.M. and G.V.R. designed and performed ex vivo prostate explants assay. G.R., A.J., A.S., and D.J.M provided JMJD2 inhibitors and controls. E.D.M. designed demethylase assay and provided reagents and technical assistance. J.S. and J.T.H. designed and performed PC3 xenograft tumor studies. Y.Y.Z. and G.H.X. performed microarray data analysis. Z.P.L. designed the study and wrote paper. All authors discussed and commented on the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Berry WL, Janknecht R. KDM4/JMJD2 histone demethylases: epigenetic regulators in cancer cells. Cancer Res. 2013;73:2936–2942. doi: 10.1158/0008-5472.CAN-12-4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, Spooner E, Li E, Zhang G, Colaiacovo M, Shi Y. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell. 2006;125:467–481. doi: 10.1016/j.cell.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 3.Zhang QJ, Liu ZP. Histone methylations in heart development, congenital and adult heart diseases. Epigenetics. 2015;7:321–30. doi: 10.2217/epi.14.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Katoh M, Katoh M. Identification and characterization of JMJD2 family genes in silico. Int J Oncol. 2004;24:1623–1628. [PubMed] [Google Scholar]
- 5.Rose NR, McDonough MA, King ON, Kawmura A, Schofield CJ. Inhibition of 2-oxoglutarate dependent oxygenases. Chem Soc Rev. 2011;40:4364–4397. doi: 10.1039/c0cs00203h. [DOI] [PubMed] [Google Scholar]
- 6.Young LC, Hendzel MJ. The oncogenic potential of Jumonji D2 (JMJD2/KDM4) histone demethylase overexpression. Biochem Cell Biol. 2013;91:369–377. doi: 10.1139/bcb-2012-0054. [DOI] [PubMed] [Google Scholar]
- 7.Gao L, Alumkal J. Epigenetic regulation of androgen receptor signaling in prostate cancer. Epigenetics. 2010;5:100–104. doi: 10.4161/epi.5.2.10778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Metzger, Wissmann M, Yin N, Müller JM, Schneider R, Peters AH, Günther T, Buettner R, Schüle R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437:436–439. doi: 10.1038/nature04020. [DOI] [PubMed] [Google Scholar]
- 9.Shin S, Janknecht R. Activation of androgen receptor by histone demethylases JMJD2A and JMJD2D. Biochem Biophys Res Commun. 2007;359:742–746. doi: 10.1016/j.bbrc.2007.05.179. [DOI] [PubMed] [Google Scholar]
- 10.Wissmann M, Yin N, Müller JJ, Greschik H, Fodor BD, Jenuwein T, Vogler C, Schneider R, Günther T, Buettner R, Metzger E, Schüle R. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat Cell Biol. 2007;9:347–353. doi: 10.1038/ncb1546. [DOI] [PubMed] [Google Scholar]
- 11.Coffey K, Rogerson L, Ryan-Munden C, Alkharaif D, Stockley J, Heer R, Sahadevan K, O’Neill D, Jones D, Darby S, Staller P, Mantilla A, Gaughan L, Robson CN. The lysine demethylase, KDM4B, is a key molecule in androgen receptor signalling and turnover. Nucleic Acids Res. 2013;41:4433–4446. doi: 10.1093/nar/gkt106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.King ON, Li XS, Sakurai M, Kawamura A, Rose NR, Ng SS, Quinn AM, Rai G, Mott BT, Beswick P, Klose RJ, Oppermann U, Jadhav A, Heightman TD, Maloney DJ, Schofield CJ, Simeonov A. Quantitative high-throughput screening identifies 8-hydroxyquinolines as cell-active histone demethylase inhibitors. PLoS One. 2010;23:e15535. doi: 10.1371/journal.pone.0015535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thalhammer A, Mecinović J, Loenarz C, Tumber A, Rose NR, Heightman TD, Schofield CJ. Inhibition of the histone demethylase JMJD2E by 3-substituted pyridine 2,4-dicarboxylates. Org Biomol Chem. 2011;9:127–135. doi: 10.1039/c0ob00592d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hamada S, Suzuki T, Mino K, Koseki K, Oehme F, Flamme I, Ozasa H, Itoh Y, Ogasawara D, Komaarashi H, Kato A, Tsumoto H, Nakagawa H, Hasegawa M, Sasaki R, Mizukami T, Miyata N. Design, synthesis, enzyme-inhibitory activity, and effect on human cancer cells of a novel series of jumonji domain-containing protein 2 histone demethylase inhibitors. J Med Chem. 2010;53:5629–5638. doi: 10.1021/jm1003655. [DOI] [PubMed] [Google Scholar]
- 15.Wang L, Chang J, Varghese D, Dellinger M, Kumar S, Best AM, Ruiz J, Bruick R, Peña-Llopis S, Xu J, Babinski DJ, Frantz DE, Brekken RA, Quinn AM, Simeonov A, Easmon J, Martinez ED. A small molecule modulates Jumonji histone demethylase activity and selectively inhibits cancer growth. Nat Commun. 2013;4:2035. doi: 10.1038/ncomms3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chu H, Wang LY, Hsu KC, Chen CC, Cheng HH, Wang SW, Wu CM, Chen TJ, Li LT, Liu R, Hung CL, Yang JM, Kung HJ, Wang WC. KDM4B as a Target for Prostate Cancer: Structural Analysis and Selective Inhibition by a Novel Inhibitor. J Med Chem. 2014;57:5975–5985. doi: 10.1021/jm500249n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rai G, Kawamura A, Tumber A, Liang Y, Vogel JL, Arbuckle JH, Rose NR, Dexheimer TS, Foley TL, King ON, Quinn A, Mott BT, Schofield CJ, Oppermann U, Jadhav A, Simeonov A, Kristie TM, Maloney DJ. Probe Reports from the NIH Molecular Libraries Program [Internet] Bethesda (MD): National Center for Biotechnology Information (US); 2010–2012. Discovery of ML324, a JMJD2 demethylase inhibitor with demonstrated antiviral activity. [PubMed] [Google Scholar]
- 18.Rizo J, Rosen MK, Gardner KH. Enlightening molecular mechanisms through study of protein interactions. J Mol Cell Biol. 2012;4:270–283. doi: 10.1093/jmcb/mjs036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chandran UR, Ma C, Dhir R, Bisceglia M, Lyons-Weiler L, Liang W, Michalopoulos G, Becich M, Monzon FA. Gene expression profiles of prostate cancer reveal involvement of multiple molecular pathways in the metastatic process. BMC Cancer. 2007;7:64. doi: 10.1186/1471-2407-7-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu YP, Landsittel D, Jing L, Nelson J, et al. Gene expression alterations in prostate cancer predicting tumor aggression and preceding development of malignancy. J Clin Oncol. 2004;22:2790–9. doi: 10.1200/JCO.2004.05.158. [DOI] [PubMed] [Google Scholar]
- 21.Cai C, Wang H, He HH, Chen S, He L, Ma F, Mucci L, Wang Q, Fiore C, Sowalsky AG, Loda M, Liu XS, Brown M, Balk SP, Yuan X. ERG induces androgen receptor-mediated regulation of SOX9 in prostate cancer. J Clin Invest. 2013;123:1109–22. doi: 10.1172/JCI66666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sadasivam S, Duan S, DeCaprio JA. The MuvB complex sequentially recruits B-Myb and FoxM1 to promote mitotic gene expression. Genes Dev. 2012;26:474–89. doi: 10.1101/gad.181933.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sadasivam S, DeCaprio JA. The DREAM complex: master coordinator of cell cycle-dependent gene expression. Nat Rev Cancer. 2013;13:585–95. doi: 10.1038/nrc3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yuan K, Huang Y, Yao X. Illumination of mitotic orchestra during cell division: a Polo view. Cell Signal. 2011;23:1–5. doi: 10.1016/j.cellsig.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bräuninger A, Strebhardt K, Rübsamen-Waigmann H. Identification and functional characterization of the human and murine polo-like kinase (Plk) promoter. Oncogene. 1995;11:1793–1800. [PubMed] [Google Scholar]
- 26.Centenera MM, Gillis JL, Hanson AR, Jindal S, Taylor RA, Risbridger GP, Sutherland PD, Scher HI, Raj GV, Knudsen KE, Yeadon T. Evidence for Efficacy of New Hsp90 Inhibitors Revealed by Ex Vivo Culture of Human Prostate Tumors. Clin Cancer Res. 2012;18:3562–3570. doi: 10.1158/1078-0432.CCR-12-0782. [DOI] [PubMed] [Google Scholar]
- 27.Van Rechem C, Black JC, Greninger P, Zhao Y, Donado C, Burrowes PD, Ladd B, Christiani DC, Benes CH, Whetstine JR. A Coding Single-Nucleotide Polymorphism in Lysine Demethylase KDM4A Associates with Increased Sensitivity to mTOR Inhibitors. Cancer Discov. 2015;5:245–54. doi: 10.1158/2159-8290.CD-14-1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guerra-Calderas L, González-Barrios R, Herrera LA, Cantú de León D, Soto-Reyes E. The role of the histone demethylase KDM4A in cancer. Cancer Genet. 2014 doi: 10.1016/j.cancergen.2014.11.001. S2210-7762(14)00245-2. [DOI] [PubMed] [Google Scholar]
- 29.Song B, Liu XS, Liu X. Polo-like kinase 1 (Plk1): an Unexpected Player in DNA Replication. Cell Div. 2012;7(3) doi: 10.1186/1747-1028-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cholewa D, Liu X, Ahmad N. The role of polo-like kinase 1 in carcinogenesis: cause or consequence? Cancer Res. 2013;73:6848–6855. doi: 10.1158/0008-5472.CAN-13-2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Craig SN, Wyatt MD, McInnes C. Current assessment of polo-like kinases as anti-tumor drug targets. Expert Opin Drug Discov. 2014;9:773–789. doi: 10.1517/17460441.2014.918100. [DOI] [PubMed] [Google Scholar]
- 32.Zhang QJ, Chen HZ, Wang L, Liu DP, Hill JA, Liu ZP. The histone trimethyllysine demethylase JMJD2A promotes cardiac hypertrophy in response to hypertrophic stimuli in mice. J Clin Invest. 2011;121:2447–56. doi: 10.1172/JCI46277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pedersen MT, Agger K, Laugesen A, Johansen JV, Cloos PA, Christensen J, Helin K. The demethylase JMJD2C localizes to H3K4me3-positive transcription start sites and is dispensable for embryonic development. Mol Cell Biol. 2014;34:1031–45. doi: 10.1128/MCB.00864-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kawazu M, Saso K, Tong KI, McQuire T, Goto K, Son DO, et al. Histone demethylase JMJD2B functions as a co-factor of estrogen receptor in breast cancer proliferation and mammary gland development. PLoS One. 2011;6:e17830. doi: 10.1371/journal.pone.0017830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iwamori N, Zhao M, Meistrich ML, Matzuk MM. The testis-enriched histone demethylase, KDM4D, regulates methylation of histone H3 lysine 9 during spermatogenesis in the mouse but is dispensable for fertility. Biol Reprod. 2011;84:1225–34. doi: 10.1095/biolreprod.110.088955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Black JC, Van Rechem C, Whetstine JR. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell. 2012;48:491–507. doi: 10.1016/j.molcel.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wei Q, Zhou W, Wang W, Gao B, Wang L, Cao J, Liu ZP. Tumor-suppressive functions of leucine zipper transcription factor-like 1. Cancer Res. 2010;70:2942–2950. doi: 10.1158/0008-5472.CAN-09-3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ravindranathan P, Lee TK, Yang L, Centenera MM, Butler L, Tilley MD, Hsieh JT, Ahn JM, Raj GV. Peptidomimetic targeting of critical androgen receptor-coregulator interactions in prostate cancer. Nat Commun. 2013;4:1923. doi: 10.1038/ncomms2912. [DOI] [PubMed] [Google Scholar]
- 39.Delaglio F, et al. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 40.Johnson BA, Blevins RA. NMR View: A computer program for the visualization and analysis of NMR data. J Biomol NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.