

Abstract
Models of reabsorptive barriers require both a means to provide realistic physiologic cues to and quantify transport across a layer of cells forming the barrier. Here we have topographically-patterned porous membranes with several user-defined pattern types. To demonstrate the utility of the patterned membranes, we selected one type of pattern and applied it to a membrane to serve as a cell culture support in a microfluidic model of a renal reabsorptive barrier. The topographic cues in the model resemble physiological cues found in vivo while the porous structure allows quantification of transport across the cell layer. Sub-micron surface topography generated via hot-embossing onto a track-etched polycarbonate membrane, fully replicated topographical features and preserved porous architecture. Pore size and shape were analyzed with SEM and image analysis to determine the effect of hot embossing on pore morphology. The membrane was assembled into a bilayer microfluidic device and a human kidney proximal tubule epithelial cell line (HK-2) and primary renal proximal tubule epithelial cells (RPTEC) were cultured to confluency on the membrane. Immunofluorescent staining of both cell types revealed protein expression indicative of the formation of a reabsorptive barrier responsive to mechanical stimulation: ZO-1 (tight junction), paxillin (focal adhesions) and acetylated α-tubulin (primary cilia). HK-2 and RPTEC aligned in the direction of ridge/groove topography of the membrane in the device, evidence that the device has mechanical control over cell response. This topographically-patterned porous membrane provides an in vitro platform on which to model reabsorptive barriers with meaningful applications for understanding biological transport phenomenon, underlying disease mechanisms, and drug toxicity.
Introduction
Reabsorptive barriers in vivo govern many physiological processes, and are formed by a single layer of polarized epithelial cells supported by a basement membrane (BM). Solutes and molecules cross the epithelial barrier by transcellular or paracellular pathways to the interstitial space and surrounding blood vessels, resulting in reabsorption of essential water and solutes.1,2 Common examples of reabsorptive or absorptive barriers in the body include those of the respiratory, gastrointestinal, and urinary tracts.3–5 Fluid and solute transport across these barriers make them particularly susceptible to injury by circulating toxins, pathogenic antibodies, or certain drugs.6,7 In vitro models of such barriers offer platforms to better understand the biology and function of reabsorptive barriers, interrogate underlying disease mechanisms affecting those barriers, and provide rapid screening of drugs for toxic effects to and excretion by organs containing those barriers. In particular, since the kidney is susceptible to drug toxicity and governs excretion of drugs, its renal epithelial structures provide valuable test cases for in vitro models of reabsorptive barriers.
Creating an in vitro reabsorptive barrier model requires replicating physiologically-relevant cues and architectures found in vivo. In addition to its chemical components of proteins and matrix-bound growth factors,8–10 the in vivo BM structural network of proteins and sulphated proteoglycans yields a surface topography as well as a porous architecture.11–13 The surface topography of the BM consists of a complex organization of pores, ridges and fibers with sizes in the nanometer to submicron range.12,14–16 In vitro, synthetic nano- and micro-scale topography influences cell morphology, migration, adhesion and differentiation,14,16–21 providing a means to mechanically cue cells in a fashion similar to the in vivo case. Experimental evidence suggests that submicron ridge/groove features control renal epithelial tissue organization by inducing cellular alignment and enhancing tight junction formation in the presence of flow-induced shear stress (FSS).22 Clearly, topography is critical for influence of barrier-forming cells and will likely provide a cue to control barrier function. It would be advantageous for a model of a reabsorptive barrier to couple topographic cues with a porous cell support structure to allow direct interrogation of the transport occurring across the adherent epithelial barrier.
Various experimental studies of renal epithelial barriers use permeable membranes as cell culture platforms to study transport characteristics of renal tubule epithelium.23–26 Porous membranes commonly used as cell support structures can be fabricated using various methods such as foaming,27,28 particulate leaching,29,30 immersion precipitation,31 and freeze drying32 which generate porosity and a consequent topography dependent on the porous features. Although these methods generate membranes with topography and porosity, the two parameters cannot be independently controlled or tailored. Generating a topography independent of the pore formation would allow creation of a porous membrane with both user-defined topography and pores, to tailor each to facilitate a BM-like architecture or enable control of experimental variables. Recent work has demonstrated the feasibility of creating pores in a topographically-patterned sheet by leaching micro-particles,33 which lacks control over pore size and shape, and by phase separation micromolding (PSμM),34 which depends heavily upon polymer type, solvent, and non-solvent component, and temperature. Additionally, the high porosity and interconnectivity of these porous scaffolds do not accurately mimic BM in vivo, as the latter is more likely to contain single, isolated trans-membrane pores as is seen in the renal tubular BM.35 Creating a membrane with single, isolated trans-membrane pores of well-controlled size with an independent set of topographical patterns would allow a close approximation of BM with the ability to regulate mechanical signalling to adherent cells and to evaluate barrier formation.
In this work, we have used a hot-embossing method to mold sub-micron topographic features onto a track-etched porous membrane, thereby creating a membrane with isolated trans-membrane pores and a controlled topography. The topographically-patterned membrane presents two areas of novelty: (1) The topographic feature geometry can be selected independently of the pore shape and size and the membrane porosity and (2) The membrane can be used to demonstrate influence of the topography on adherent cells while generating a barrier in an architecture which permits evaluation of transport. The topographic membrane can be integrated into microfluidic devices which would provide simulated renal filtrate flow, thereby establishing a platform for an in vitro reabsorptive barrier model of renal proximal tubule tissue.
Experimental
Topographic membrane fabrication
Hot-embossing, shown in Fig. 1, patterned thermoplastic porous membranes with topographic features in the sub-micron to microscale range. Polycarbonate track-etched (PCTE) membranes (GE Power & Water, Tervose, PA) with nominal pore sizes between 3 μm and 12 μm were embossed with topographic patterns from a master mold. The master mold was fabricated using a two step molding process, as described previously.22 First, a silicon mold was fabricated using standard photolithography and reactive ion etching (RIE) to produce four distinct topographic geometries of linear ridge/groove and post patterns, described in Table 1. Second, a nickel mastermold was electroformed from the silicon mold (NiCoForm, Inc., Rochester, NY). The original silicon wafer was attached to a negative pole of a DC power supply and submerged in a plating solution of nickel and cobalt ions. A current of 10–20 mA cm−2 was used to produce a 0.5 mm-thick electroform of approximately 98% nickel and 2% cobalt in 24 h. Upon completion, the silicon wafer was chemically dissolved in a solution of KOH.36 The resulting nickel mold was used to emboss the thermoplastic membranes. Prior to embossing, the patterned face of the nickel mold was soaked in a 1 mM solution of hexadecanethiol (HDT), which formed a self-assembled monolayer (SAM) to decrease surface energy to aid in subsequent polymer release.
Fig. 1.
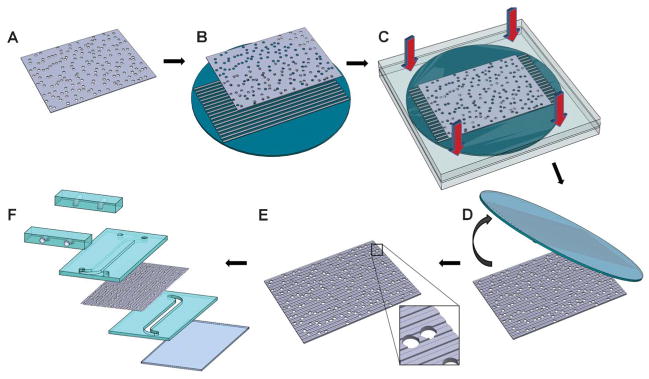
Hot embossing was used to topographically pattern the porous membranes. An un-patterned polycarbonate membrane (A) was placed on a nickel mold (B). The nickel mold contained relief structures of the topographical features of interest and was fabricated using photolithography, etching and electroforming processes. The membrane and nickel mold were brought in contact between two platens and placed under elevated temperatures and pressure (C) and the membrane was de-molded (D) from the nickel mold. The result was a topographically-patterned membrane (E) with intact pore structure. The membrane was then assembled into a multichannel microfluidic device (F) for the study of cell culture and transport.
Table 1.
Four distinct topographic geometries were patterned via hot embossing to PCTE membranes. The geometries ranged from 0.5–1.0 μm ridge/groove features to 1.0 μm posts. All features were designed to be evenly spaced with a 1 : 1 ratio of feature width to spacing and to have a height of 0.75 μm. Non-embossed PCTE membranes served as control samples
| Name | Feature description | Pattern type | Feature width | Depth |
|---|---|---|---|---|
| 05Grat | ridge/groove | linear | 0.5 μm | 0.75 μm |
| 075Grat | ridge/groove | linear | 0.75 μm | 0.75 μm |
| 10Grat | ridge/groove | linear | 1.0 μm | 0.75 μm |
| 10Post | post | radial | 1.0 μm | 0.75 μm |
| Control | flat surface | none | — | — |
The PCTE membranes were placed with one surface of the membrane in contact with the topographically patterned face of the nickel mold. The mold and membrane were then sandwiched between two Kapton polyimide films and silicone rubber sheets to decrease sticking and add compliance. The stack was placed in a uniformly heated, temperature- and pressure-controlled automatic hydraulic press (Carver Inc., Wabash, IN). A light load was applied to the stack- while the temperature was ramped to 150 °C. The specified pressure was then applied for the specified dwell time before being cooled to 60 °C under constant pressure. Upon cooling, the newly patterned membrane was carefully released from the nickel mold and analyzed for changes in pore size and geometry. Membranes were then assembled into a microfluidic device to serve as a cell culture platform. The nickel mold was sonicated in acetone and an HDT SAM was reapplied approximately every 20 embossing runs to clear residual PC membrane debris.
Device assembly
Embossed membranes were assembled into a multilayer microfluidic device developed previously,37 with the patterned side of the membrane serving as the floor to one of the cell chambers. The membrane was bonded to each polymer layer with an RTV silicone adhesive (Dow Corning, Midland, MI). The bottom microfluidic channel layer was oxygen plasma bonded to a glass coverslip. Completed devices were sterilized with ethylene oxide (EtO), allowed to de-gas, rinsed with 70% ethanol, and purged with phosphate buffered saline (PBS) before use. The patterned side of the membrane was coated with an extracellular matrix coating by infusing a solution of 60 μg mL−1 collagen IV (human, C5533, Sigma-Aldrich, St. Louis, MO) through the channel and letting it soak for 3 h at room temperature. Both channels were rinsed with PBS and primed with media pre-conditioned in incubator conditions prior to cell seeding.
Cell culture and device seeding
Cells from a human renal proximal tubule epithelial cell line (HK-2) (ATCC, Manassas, VA) and primary renal proximal tubule epithelial cells (RPTEC) (Lonza Walkersville Inc., Walkersville, MD) were cultured at 37 °C and 5% CO2 in renal tubular epithelial medium (DMEM/F12 supplemented with 0.5% FBS, 10 ng mL−1 hEGF, 5 μg mL−1 insulin, 0.5 μg mL−1 hydrocortisone, 0.5 μg mL−1 epinephrine, 6.5 ng mL−1 triiodothyronine, 10 μg mL−1 transferrin, 100 U ml−1 penicillin and 100 μg mL−1 streptomycin). Media was renewed every other day until the cells reached 80% confluency. HK-2 cells were passaged no more than 15 times and RPTECs were passaged only once before being seeded into the device. To seed cells in the device, ports to the channel containing the unpatterned face of the membrane were blocked, while a concentrated cell suspension solution of approximately 2.5 × 106 cells mL−1 was injected into the open channel via a syringe. Cells were allowed to settle and adhere under static conditions. Media was renewed twice a day until 24 h after cells reached confluency.
Cell characterization
Immunofluorescent labeling
Confluent cell populations were rinsed, fixed, and fluorescently labelled within the channel using a syringe pump to deliver reagents to the channel. The following reagents were used in this order: PBS to rinse away media, 3.7% paraformaldehyde solution (w/v in PBS), PBS to rinse, 0.1% Triton-X to permeabilize the cell membranes, PBS to thoroughly rinse the channel, and 2% FBS in PBS, which was allowed to sit in the channel for 30 min. Using the same method, primary antibodies were perfused through the channel: anti-ZO1 mouse IgG (610967, BD Biosciences, San Jose, CA), diluted 1 : 200 in 2% FBS to label tight junctions, anti-paxillin rabbit monoclonal IgG1 (04-581, Millipore, Billerica, MA) diluted 1 : 250 to label focal adhesions, and anti-acetylated tubulin mouse monoclonal IgG2b, (T6793, Sigma, St. Louis, MO) diluted 1 : 500 to label primary cilia and cytoplasmic microtubules. The cells were incubated in the primary antibody for 2 h at room temperature. The channel was then rinsed with PBS and perfused with a secondary antibody solution in 2% FBS, which consisted of a 1 : 200 dilution of an anti-mouse IgG conjugated to an Alexa fluor 488 dye (A11029, Invitrogen, Carlsbad, CA) to label the mouse primary antibodies, and an anti-rabbit Alexa Fluor 568 (A10042, Invitrogen, Carlsbad, CA) to label paxillin. Lastly, a 1 : 1000 dilution of Hoechst dye was used to label nuclei. The cells were rinsed with a final PBS wash followed by deionized water, all channel inlets and outlets were sealed, and the cells were imaged within the device. Images were collected with a confocal laser microscope (LSM 510, Carl Zeiss, Jena, Germany). Due to the thin nature of the device, a 40× oil immersion lens had a suitable working distance and was used to capture all cell images.
Cellular alignment
Nuclear alignment was used as a metric for overall cell alignment to groove topography as used by other studies.22,38 Alignment was quantified in cell populations cultured on stand-alone membranes with and without topography and on topographical membranes in a microfluidic device. Cells were fixed, labelled with the nuclei stain as described above, and imaged. The image processing software, CellProfiler,39 developed by the Broad Institute, Cambridge, MA, was used to analyze nuclear alignment with respect to topographical groove direction. Groups were compared for statistical significance using a one-way ANOVA test with a paired Tukey analysis.
Image collection and pore geometry analysis
Embossing pressure, temperature and dwell time were characterized to achieve full topographical feature replication while preserving pore structure. Membranes were sputter coated with a layer of gold and imaged using SEM. Three trials were conducted for each pore size and embossing parameter, and three samples were taken from each trial, resulting in hundreds of pores being analyzed per trial. Membrane pore size and shape were analyzed with CellProfiler. The program was used to accurately identify each pore, calculate the area, the major and minor axes, and the derived diameter, which was calculated from the area. To observe the effect of dwell time on pore diameter, membranes were embossed over four time points at a constant temperature and pressure over all topographical feature geometries. The elongation fraction was determined by subtracting the minor axis from the major axis and normalizing with respect to the major axis. Pore derived diameter and pore elongation were measured with respect to the nominal pore diameter among all topographical feature geometries. The control pore dimensions were measured over several areas from three non-embossed membranes. For each data point at least 6 membranes were embossed and evaluated. Groups were compared for statistical significance using a one-way ANOVA test with a paired Tukey analysis.
Results and discussion
Topographic membrane fabrication
Four distinct topographical feature geometries were replicated in PCTE membranes, with resulting feature dimensions within 5% of the original feature dimensions present in the nickel mold. An embossing dwell time of 15 min, 820 kPa of pressure and a temperature of 150 °C, resulted in preservation of pore architecture for all pore sizes tested. The four topographical geometries with preserved porous architecture are shown in Fig. 2. A range of user-defined topographies can be embossed onto the membrane and chosen based on cell type and application. The membranes released easily from the nickel mold due to the SAM of HDT molecules applied to the nickel surface prior to embossing.
Fig. 2.

Hot-embossing created well-defined topographical features while maintaining pore architecture in polycarbonate membranes with 8 μm diameter pores. Representative illustrations (top row) and corresponding SEM images of the membranes (bottom row) show the five distinct topographic patterns created using the hot-embossing process. The blank, or control, sample contains a flat surface. The topographically patterned membranes contain a variety of evenly spaced ridges with increasing ridge widths of 0.5 μm, 0.75 μm, and 1.0 μm, respectively, and evenly spaced 1.0 μm pits. All topographical features have a depth of 0.75 μm
A variety of porous membranes are suitable for topographical patterning via hot embossing. Polycarbonate and polyester membranes are typical because they are thermoplastic and lend themselves well to embossing and they provide an adequate cell culture surface for epithelial cells. We chose to use GE™ polycarbonate membranes for this study because they are commonly used for cell culture applications, such as in transwell dishes, and microfluidic cell analysis applications.22,23,25,37 PCTE membranes are available in a choice of diameters and pore sizes with a narrow pore size distribution and low non-specific binding. They are semi-translucent, offer superior strength and thermal stability, and are biologically inert. The mechanical and physical properties of these membranes make them ideal for our embossing procedure, allowing selective permeability to be integrated with surface topography on a cell culture platform without destroying the membrane’s innate characteristics.
Analysis of embossing parameters on pore size and shape
Embossing control of pore diameter
Membrane pore diameter depended on embossing dwell time in a controlled and repeatable manner. For a constant load of 820 kPa and temperature of 150 °C, pore diameter decreased as dwell time increased. For dwell times of 15 min or less, pore diameters were not significantly affected by embossing, as shown in Fig. 3A. PC membranes embossed with a dwell time of 20 min or greater had significantly reduced pore diameters compared to unembossed samples. A clear window of embossing dwell time from 10 to 15 min afforded a robust process which replicated topography, yet did not significantly alter the pore diameter. Alternatively, pore diameters could be reduced in a controlled fashion by extending dwell times beyond 20 min, creating a method to reduce pore size as needed. As shown in Fig. 3B, the unembossed control membranes exhibited pore sizes at or below their specified nominal diameters, supporting the suppliers’ claims. For a dwell time of 20 min under 820 kPa at 150 °C, embossing reduced pore diameter over all feature geometries for membranes with large pores, such as 8 and 12 μm nominal diameter, but did not reduce pore diameter for pores with small nominal pore diameters, such as 3 and 5 μm.
Fig. 3.
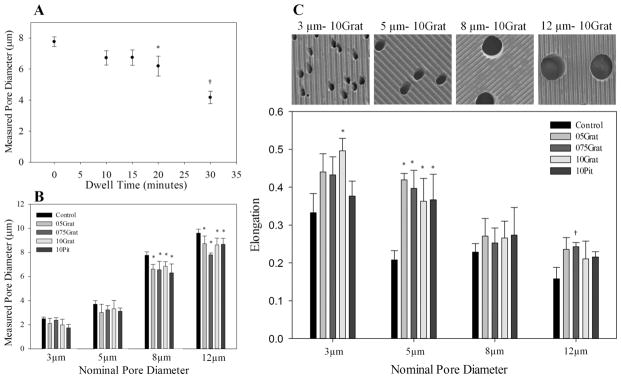
Pore diameter and geometry can be controlled with embossing parameters. (A) Pore diameter decreased as embossing dwell time increased. Dwell times between 10–15 min did not change pore diameters significantly from nominal pore size. *, p < 0.01 compared to 0 min dwell time, †p < 0.001, compared to 20 min dwell time. (B) For larger nominal pore diameters and a 20 min dwell time, embossing decreased pore size slightly over all topographical feature geometries, although the fractional change between the control and embossed pore diameters decreased as nominal pore diameter increased. *, p < 0.05 compared to control. (C) SEM images above illustrate the differences in elongation of 3–12 μm pores embossed with 10Grat topography. Smaller nominal pore sizes were more elongated than larger nominal pore sizes. Control membranes, regardless of pore size, have elongated pore morphology, with 3 μm pores having an elongation of almost 35%. Hot embossing elongated the pores significantly, although this was not consistent for all patterns and nominal pore sizes. In general, smaller pore sizes elongated due to embossing more often than larger pore sizes. *p < 0.02 compared to control pore size; †, p < 0.05 compared to control pore size. All samples represented in (B) and (C) were embossed for 20 min under 820 kPa of pressure at 150 °C. Measured pore diameters were derived from the calculated pore area using image analysis software. Data is presented as mean ± standard deviation.
Some decrease in pore diameter was expected after embossing due to the flow of the polymer under high temperature and pressure. For our patterns, significant pore diameter decrease occurred at a 20 min dwell time and the amount of diameter reduction increased with increasing dwell time. Pore diameter decrease was independent of pattern type, although patterns used in the experiments were of a uniform and shallow depth. In addition, the fractional area of the protruding features on the mold, which determines the amount of polymer displaced during embossing, did not vary significantly for our patterns. Increasing depth and fraction area of protruding features will likely increase pore diameter sensitivity to embossing parameters. The amount of pore diameter change can be limited based on the embossing recipe, with a dwell time of 10–15 min providing a good balance of replication of topographical features without significant changes to pore diameter. Since the pores in the membrane are mainly columnar in cross section, the permeability and transport properties will be largely dependent on pore diameter and the number of pores, both of which can be changed predictably or unchanged by the topographic patterning process.
Embossing control of pore elongation
Pores in control PCTE membranes were elongated circles, a condition enhanced in some cases by the embossing procedure. The average pore in non-embossed control membranes exhibited a slightly elongated shape with a fraction of elongation, found by subtracting the minor axis from the major axis and normalizing with respect to the major axis, ranging from 0.16 for 12 μm pores to 0.34 for 3 μm pores. Fig. 3C shows how the fraction of elongation for pores after embossing was dependent on nominal pore diameter and in some cases, topographical feature geometry. Membranes with smaller pore diameters, i.e. 3 and 5 μm, yielded significantly higher fractions of pore elongation compared to larger pore sizes. Membranes with a pore size of 3 μm exhibited pores with an average elongation fraction of almost 0.5 when embossed with a 1 μm ridge/groove pattern, a 49% increase from the control. However, elongation of the 3 μm pores did not increase significantly for other topographical feature geometries. For larger pore sizes, 8 μm and 12 μm, the change in pore elongation became insignificant across most topographical feature geometries when compared to the control pore geometry. The one exception was membranes with 12 μm pores, which had significantly increased elongation of pores when embossed with the 75 μm ridge/groove pattern. The 5 μm pores increased elongation fraction for all embossed geometries. For the embossed geometries here, elongation of pores did not trend clearly with pattern geometry and was most significant for pore sizes of 5 μm. Although hot embossing can change pore geometry, this effect can be greatly reduced when working with pore sizes other than 5 μm.
Microfluidic device assembly and operation
A bilayer microfluidic device was fabricated such that the topographically-patterned membrane formed a permeable barrier separating two channels. The finished device offered a cell culture platform with well controlled topographical geometry, porous architecture, and two channels that provided microscopic and fluidic access to both apical and basal sides of a cell monolayer. The thin nature of the device resulted in a short working distance to the cell layer for high-resolution imaging of cells on either side of the membrane. A full device summary can be seen in Fig. 4, which shows the bilayer channel architecture with the patterned porous membrane integrated between the channels. For a model of kidney tissue such as the proximal tubule, this architecture allowed one chamber to model the tubular lumen, a membrane support to grow an epithelial reabsorptive barrier, and a second chamber to serve as the interstitial space and/or surrounding blood vessels. The result was a platform which allowed physiologically-realistic arrangement of cells and stimuli with the capacity to study transport phenomenon across epithelial tissue.
Fig. 4.

The topographically-patterned membrane assembled into a microfluidic device. The overall cross-sectional architecture of a device (A) was formed by a glass coverslip and two PDMS cell culture chambers separated by the patterned porous membrane. The thin nature of the device (B) allowed high-resolution imaging of cell populations on either side of the porous membrane. An SEM cross section (C) of the device shows the porous nature of the membrane separating the top and bottom chambers, while insets (D) and (E) show close-ups of well-defined groove topography coexisting with the porous architecture.
Expression of reabsorptive epithelial barrier markers on porous membrane topography
To establish the device as a platform for a reabsorptive barrier, the basic elements of a cellular barrier were characterized for renal epithelial cells cultured on the patterned porous membrane in the microfluidic device. For formation of a cellular barrier, cells must exhibit appropriate anchoring to the substrate, generate cell–cell junctions such as tight junctions, express mechanosensory machinery appropriate for the given tissue type, and physically occlude pores of the cell substrate. For renal proximal tubule tissue, appropriate mechanosensory machinery are primary cilia which interact with filtrate flow in the tubule in vivo. Immunofluorescent techniques labeled markers indicative of an epithelial layer for cells cultured in the device. HK-2 cells and RPTECs proliferated from initial seeding to confluency within the device over approximately 4 days. A uniform initial seeding density and appropriate culture time yielded complete confluency of both HK-2 and RPTECs over the microfluidic device channel area of 1.25 mm2, seen respectively in the bright field composites in Fig. 5A and 5B. HK-2 and RPTEC monolayers expressed paxillin, a typical epithelial marker of focal adhesions, ZO-1 tight junction complexes, and acetylated tubulin, an indicator of primary cilia and cytoplasmic microtubules, also shown in Fig. 5. Paxillin expression in HK-2 samples signified focal adhesions that were less discrete with a weaker signal than RPTEC samples. Both HK-2s and RPTECs expressed ZO-1 in fairly distinct borders outlining the perimeter of cells as observed in 2D, indicating initial formation of a tight-junction-based sealed epithelial barrier. Acetylated tubulin morphology differed between the HK-2s and RPTECs. The HK-2s showed somewhat distributed cytoplasmic microtubules, but distinct primary cilia were not expressed on the apical surface. The RPTECs expressed acetylated tubulin in cytoplasmic micro-tubules and also as a single punctuate spot on the apical surface of each cell, indicating formation of a primary cilia as seen in vitro.25,40,41 For both cell types, SEM images showed the pores of the membrane fully covered and occluded by the confluent monolayer.
Fig. 5.

Cells proliferated, maintained a monolayer, and expressed markers of a reabsorptive epithelial barrier on a topographically-patterned membrane within a microfluidic device. (A) and (B) HK-2 cells and RPTECs, respectively, were seeded into the top channel of a bilayer microfluidic device and grew to a confluent monolayer on the topographically-patterned membrane 1.25 mm2; 10× magnification, scale bar: 200 μm. Insets (C) and (D) show a closer view of the cobble-stone appearance of a confluent monolayer within the channel. Bright spots are pores. Ridge/groove topography runs parallel to the channel. Each cell type expressed paxillin as a marker of focal adhesions (A, B) (i), ZO-1 tight junctions (A, B) (ii), and acetylated tubulin indicating cytoplasmic microtubules and primary cilia (A, B) (iii), Nuclei are labelled blue. (E) Isolated transmembrane pores were fully occluded by an HK-2 monolayer, shown by SEM, as compared to (F) an acellular membrane
Formation of the complete, confluent monolayer within the channel layer allows interrogation of the layer for permeability, a requisite for a reabsorptive barrier. As the layer is confluent, fluidic or electrical access to any point on the microchannel will enable future experiments, which directly measure transport, allowing quantification of the reabsorptive properties. Formation of ZO-1 junctions indicated progress of the cells towards an epithelial barrier capable of active transport. Some inconsistency of the ZO-1 delineated borders still existed, as did some diffuse presence of ZO-1 within the cell body, indicating the cell layer has further potential for enhanced tight junctions. Conditioning of the cells through mechanical and potentially other stimuli, will likely improve the junctions.22,42 The HK-2 cells formed a more mature monolayer than the RPTECs due to a longer culture time, causing paxillin expression to be less distinct in HK-2 monolayers. The lack of primary cilia in HK-2 cells was not abnormal. Primary cilia may not be fully expressed in HK-2 populations if their formation was not enhanced by serum starvation or FSS.43 Cytoplasmic tubulin was more diffuse for HK-2 cells compared with RPTEC, with signs of a microtubule-organizing center that may nucleate cilia development.43 The presence of primary cilia in the RPTECs indicated the cells will likely be responsive to mechanical stimuli, such as FSS, as the cilia can serve to transduce mechanical signals to chemical activity.44–46 Continuous flow in the device will mimic the filtrate flow seen by PTCs in vivo. Finally, as shown by the SEMs, the cells block the pores of the membrane, which indicates transport across the membrane-cell layer construct can be limited to transcellular transport if paracellular transport is limited through tight junction and other cell–cell junction formation. With the ability to stimulate cells mechanically, interrogate them via microscopy and electrical and fluidic means, and support growth of an epithelial layer expressing indications of a mechanically-responsive reabsorptive barrier, the patterned porous membrane integrated with the microfluidic device will allow future experiments quantifying reabsorptive barrier function.
Mechanical control of cell response
As an example of the device stimulating cells via a mechanical stimulus, both renal cell types responded to the membrane topography by aligning in the direction of the topographical grooves as shown in Fig. 6. Outside the device, non-patterned (blank) membranes elicited a random alignment from the cells, indicated by an average of 11% of cells being aligned within 10° of the groove direction. Cellular alignment increased to 32% on membranes patterned with 1.0 μm groove topography. To further validate this response, HK-2 and RPTEC alignment was also measured on topographical membranes within the device. Fig. 6 shows that alignment of both HK-2 and RPTEC increased significantly from that observed in the control membranes, although the RPTEC alignment was significantly higher than the alignment seen in HK-2 cell populations within the device. Verification of cell alignment and expression of epithelial cell markers observed in the RPTEC population indicated response of cells to a mechanical stimulus and the potential to create a kidney-like reabsorptive barrier tissue in vitro.
Fig. 6.

Renal epithelial cell nuclear alignment was quantified on topographically-patterned membranes outside the device and on membranes integrated in the device. HK-2 cells showed random alignment when cultured on non-patterned (1) PC membranes outside of the device. Alignment increased to 32% within 10° of the gratings on topographically-patterned membranes (2). Membranes assembled into the device also elicited alignment of cells to the gratings, with 25% of HK-2s (3) and 34% of RPTECs (4) aligning to the gratings. *, p < 0.05 compared to control samples, †, p < 0.05 compared to RPTECs. The images (top row) correspond to each bar of the graph; cell nuclei are labelled blue and tight junctions green. Scale bar: 50 μm. Arrows represent the direction of groove topography.
Organized monolayers of renal epithelial tissue have been generated in a microfluidic device, evidenced by focal adhesions, TJ and cilia expression, and by increased cellular alignment. The microfluidic nature of the device and its topographically-patterned culture surfaces provided at least two ways to mechanically stimulate cells within the device. Surface topography stimulates a range of mechanical responses from cells, the most visible being changes in cell morphology and alignment. Ridge/groove topography, in particular, induces alignment of the cell body to the ridge/groove axis for many cell types.18,47–49 It is important to note, even general morphological response to substrate topography is not universally observed across all cell types.14,18,50,51 A different barrier model may require a topographically-patterned membrane with pits,20 for example, or deep grooves with a small pitch.51 These features are user-controlled and depend upon the user’s model cell type. In our example model, well-controlled ridge/groove topography signalled cell alignment in both HK-2 and RPTECs, forming a monolayer of aligned renal proximal tubule cells, demonstrating that the device not only sustained cell growth but also controlled cell alignment in a monolayer with mechanical cues. The highly organized cell monolayer lends itself well to receiving additional mechanical cues, such as FSS, to further encourage in vivo function in an in vitro microenvironment.22
Conclusions
We have developed a membrane with defined topographical patterns and distinct trans-membrane pores to model transport across a renal epithelial monolayer in vitro. A permeable membrane with independently-controlled sub-micron topography and isolated pores was fabricated using a hot-embossing process and integrated into a bilayer device. The hot-embossing process dwell time was controlled to limit its impact on pore geometry. HK-2 cells and primary RPTECs were grown to confluency within the device, and expressed markers for differentiated epithelia indicative of a reabsorptive renal epithelial barrier responsive to mechanical stimuli. Cells aligned to the topographic ridge and groove patterns, indicating the ability of the patterned membrane to mechanically stimulate cell function. The resulting platform provides a means to characterize transport across a renal cell monolayer while independently controlling mechanical signals to the cells. Both surface topography and FSS can be used to mimic the mechanical stimuli renal epithelia receive in vivo, while fluidic access allows application of chemical stimuli and perfusion of nutrients. Independent access to both sides of the membrane will allow direct interrogation of active and passive trans-barrier transport, consequently enabling quantification of the reabsorptive function of the barrier. Adjustment of the parameters will drive the renal epithelia towards a more physiologically-accurate function, providing a means to create realistic tissue with organ-specific reabsorptive properties. For example, in kidney tissue models, sodium and water reabsorption may be monitored using fluorescent sodium indicators, water transport can be measured by quantifying the monolayer’s hydraulic permeability, and general transport can be quantified by examining permeability to labelled macromolecules such as fluorescently-labelled dextrans. Extension of the experimental system can mimic a wide range of reabsorptive barriers in the body, thus providing a model to study disease states that affect transportation barriers, screen drugs for toxicity and excretion, and provide a pathway towards a realistic construct of therapeutic tissue with organ-specific function.
Acknowledgments
This work was funded by the Draper Laboratory Internal Research and Development program for Draper and a Draper University Research and Development program for MGH.
References
- 1.Garcia NH, Ramsey CR, Knox FG. News Physiol Sci. 1998;13:38–43. doi: 10.1152/physiologyonline.1998.13.1.38. [DOI] [PubMed] [Google Scholar]
- 2.Mostov KE, Verges M, Altschuler Y. Curr Opin Cell Biol. 2000;12:483–490. doi: 10.1016/s0955-0674(00)00120-4. [DOI] [PubMed] [Google Scholar]
- 3.Gehr P, Bachofen M, Weibel ER. Respir Physiol. 1978;32:121–140. doi: 10.1016/0034-5687(78)90104-4. [DOI] [PubMed] [Google Scholar]
- 4.Reed KK, Wickham R. Semin Oncol Nurs. 2009;25:3–14. doi: 10.1016/j.soncn.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 5.Kriz W, Kaissling B. In: The Kidney. Geibisch Sa., editor. Elsevier Inc; 2008. pp. 479–563. [Google Scholar]
- 6.Pfaller W, Gstraunthaler G. Environ Health Perspect. 1998;106:559–569. doi: 10.1289/ehp.98106559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choudhury D, Ahmed Z. Nat Clin Pract Nephrol. 2006;2:80–91. doi: 10.1038/ncpneph0076. [DOI] [PubMed] [Google Scholar]
- 8.LeBleu VS, Macdonald B, Kalluri R. Exp Biol Med. 2007;232:1121–1129. doi: 10.3181/0703-MR-72. [DOI] [PubMed] [Google Scholar]
- 9.Yurchenco PD. Cold Spring Harbor Perspect Biol. 2011;3:a004911. doi: 10.1101/cshperspect.a004911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alberts B. Molecular Biology of the Cell. Garland; New York: 2000. [Google Scholar]
- 11.Rowe RG, Weiss SJ. Trends Cell Biol. 2008;18:560–574. doi: 10.1016/j.tcb.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 12.Hironaka K, Makino H, Yamasaki Y, Ota Z. Kidney Int. 1993;43:334–345. doi: 10.1038/ki.1993.51. [DOI] [PubMed] [Google Scholar]
- 13.Ferrell N, Groszek J, Li L, Smith R, Butler RS, Zorman CA, Roy S, Fissell WH. Am J Physiol: Renal Physiol. 2011;300:F86–90. doi: 10.1152/ajprenal.00484.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Flemming RG, Murphy CJ, Abrams GA, Goodman SL, Nealey PF. Biomaterials. 1999;20:573–588. doi: 10.1016/s0142-9612(98)00209-9. [DOI] [PubMed] [Google Scholar]
- 15.Yamasaki Y, Makino H, Ota Z. Nephron. 1994;66:189–199. doi: 10.1159/000187800. [DOI] [PubMed] [Google Scholar]
- 16.Teixeira AI, Abrams GA, Bertics PJ, Murphy CJ, Nealey PF. J Cell Sci. 2003;116:1881–1892. doi: 10.1242/jcs.00383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Curtis ASG, Casey B, Gallagher JO, Pasqui D, Wood MA, Wilkinson CDW. Biophys Chem. 2001;94:275–283. doi: 10.1016/s0301-4622(01)00247-2. [DOI] [PubMed] [Google Scholar]
- 18.Bettinger CJ, Langer R, Borenstein JT. Angew Chem, Int Ed. 2009;48:5406–5415. doi: 10.1002/anie.200805179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yim EK, Leong KW. Nanomedicine. 2005;1:10–21. doi: 10.1016/j.nano.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 20.Dalby MJ, Gadegaard N, Tare R, Andar A, Riehle MO, Herzyk P, Wilkinson CD, Oreffo RO. Nat Mater. 2007;6:997–1003. doi: 10.1038/nmat2013. [DOI] [PubMed] [Google Scholar]
- 21.Bettinger CJ, Zhang Z, Gerecht S, Borenstein JT, Langer R. Adv Mater. 2008;20:99–103. doi: 10.1002/adma.200702487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frohlich EM, Zhang X, Charest JL. Integr Biol. 2012;4:75–83. doi: 10.1039/c1ib00096a. [DOI] [PubMed] [Google Scholar]
- 23.Jang KJ, Suh KY. Lab Chip. 2010;10:36–42. doi: 10.1039/b907515a. [DOI] [PubMed] [Google Scholar]
- 24.Jang KJ, Cho HS, Kang DH, Bae WG, Kwon TH, Suh KY. Integr Biol. 2011;3:134–141. doi: 10.1039/c0ib00018c. [DOI] [PubMed] [Google Scholar]
- 25.Ferrell N, Desai RR, Fleischman AJ, Roy S, Humes HD, Fissell WH. Biotechnol Bioeng. 2010;107:707–716. doi: 10.1002/bit.22835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dankers PY, Boomker JM, der Vlag AH, Smedts FM, Harmsen MC, van Luyn MJ. Macromol Biosci. 2010;10:1345–1354. doi: 10.1002/mabi.201000146. [DOI] [PubMed] [Google Scholar]
- 27.Kim TK, Yoon JJ, Lee DS, Park TG. Biomaterials. 2006;27:152–159. doi: 10.1016/j.biomaterials.2005.05.081. [DOI] [PubMed] [Google Scholar]
- 28.Wang X, Li W, Kumar V. Biomaterials. 2006;27:1924–1929. doi: 10.1016/j.biomaterials.2005.09.029. [DOI] [PubMed] [Google Scholar]
- 29.Katoh K, Tanabe T, Yamauchi K. Biomaterials. 2004;25:4255–4262. doi: 10.1016/j.biomaterials.2003.11.018. [DOI] [PubMed] [Google Scholar]
- 30.Ma PX, Choi JW. Tissue Eng. 2001;7:23–33. doi: 10.1089/107632701300003269. [DOI] [PubMed] [Google Scholar]
- 31.Wan AC, Mao HQ, Wang S, Leong KW, Ong LK, Yu H. Biomaterials. 2001;22:1147–1156. doi: 10.1016/s0142-9612(00)00355-0. [DOI] [PubMed] [Google Scholar]
- 32.Ho MH, Kuo PY, Hsieh HJ, Hsien TY, Hou LT, Lai JY, Wang DM. Biomaterials. 2004;25:129–138. doi: 10.1016/s0142-9612(03)00483-6. [DOI] [PubMed] [Google Scholar]
- 33.Sarkar S, Lee GY, Wong JY, Desai TA. Biomaterials. 2006;27:4775–4782. doi: 10.1016/j.biomaterials.2006.04.038. [DOI] [PubMed] [Google Scholar]
- 34.Papenburg BJ, Vogelaar L, Bolhuis-Versteeg LA, Lammertink RG, Stamatialis D, Wessling M. Biomaterials. 2007;28:1998–2009. doi: 10.1016/j.biomaterials.2006.12.023. [DOI] [PubMed] [Google Scholar]
- 35.Blattmann A, Denk L, Strehl R, Castrop H, Minuth WW. Biomaterials. 2008;29:2749–2756. doi: 10.1016/j.biomaterials.2008.03.019. [DOI] [PubMed] [Google Scholar]
- 36.Stein B, Kunnavakkam M, Stelick S, Batt C. IEEE Sensors. IEEE; Toronto, Canada: 2003. pp. 679–683. [Google Scholar]
- 37.Epshteyn AA, Maher S, Taylor AJ, Holton AB, Borenstein JT, Cuiffi JD. Biomicrofluidics. 2011;5:1–6. doi: 10.1063/1.3647824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Charest JL, Bryant LE, Garcia AJ, King WP. Biomaterials. 2004;25:4767–4775. doi: 10.1016/j.biomaterials.2003.12.011. [DOI] [PubMed] [Google Scholar]
- 39.Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, Golland P, Sabatini DM. Genome Biology. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Radford R, Slattery C, Jennings P, Blaque O, Pfaller W, Gmuender H, Van Delft J, Ryan MP, McMorrow T. Am J Physiol: Renal Physiol. 2012;302:F905–F916. doi: 10.1152/ajprenal.00427.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Babbey CM, Bacallao RL, Dunn KW. Am J Physiol: Renal Physiol. 2010;299:F495–F506. doi: 10.1152/ajprenal.00198.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Duan Y, Gotoh N, Yan Q, Du Z, Weinstein AM, Wang T, Weinbaum S. Proc Natl Acad Sci U S A. 2008;105:11418–11423. doi: 10.1073/pnas.0804954105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hartman TR, Liu D, Zilfou JT, Robb V, Morrison T, Watnick T, Henske EP. Hum Mol Genet. 2009;18:151–163. doi: 10.1093/hmg/ddn325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Praetorius HA, Spring KR. J Membr Biol. 2001;184:71–79. doi: 10.1007/s00232-001-0075-4. [DOI] [PubMed] [Google Scholar]
- 45.Veland IR, Awan A, Pedersen LB, Yoder BK, Christensen ST. Nephron Physiol. 2009;111:39–53. doi: 10.1159/000208212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bisgrove BW, Yost HJ. Development. 2006;133:4131–4143. doi: 10.1242/dev.02595. [DOI] [PubMed] [Google Scholar]
- 47.Charest JL, Eliason MT, Garcia AJ, King WP. Biomaterials. 2006;27:2487–2494. doi: 10.1016/j.biomaterials.2005.11.022. [DOI] [PubMed] [Google Scholar]
- 48.Charest JL, Garcia AJ, King WP. Biomaterials. 2007;28:2202–2210. doi: 10.1016/j.biomaterials.2007.01.020. [DOI] [PubMed] [Google Scholar]
- 49.Wan CR, Frohlich EM, Charest J, Kamm RD. Cell Mol Bioeng. 2011;4:56–66. [Google Scholar]
- 50.Charest JL, King WP. In: BioNanoFluidic MEMS. Hesketh P, editor. Springer Science; New York: 2008. [Google Scholar]
- 51.Biela SA, Su Y, Spatz JP, Kemkemer R. Acta Biomater. 2009;5:2460–2466. doi: 10.1016/j.actbio.2009.04.003. [DOI] [PubMed] [Google Scholar]