

Abstract
Post-translational modification through protein acetylation is emerging as an important mode of cellular regulation. We have previously demonstrated the role that glucose-regulated protein 78 kDa (GRP78) acetylation and subsequent activation of the unfolded protein response (UPR) play in the antitumor activity of class I histone deacetylase (HDAC) inhibitors, which primarily target class I HDACs. In this study, we explored the contributory role these class I HDACs may play in UPR regulation. Binding studies were performed using immunoprecipitation/immunoblotting following dual-transfection with HA-tagged GRP78 and FLAG-tagged HDACs. Subcellular localization was performed using Western blot of fractionated cell lysates and confocal microscopy. Individual HDACs were inhibited using RNA interference. We identified the potential of HDACs 1, 2, and 3 to bind to GRP78. These HDACs colocalized with GRP78 in the endoplasmic reticulum (ER). Inhibition of individual HDACs resulted in GRP78 acetylation and selective activation of the UPR. Although traditionally viewed as nuclear enzymes, we demonstrate that Class I HDACs localize to the ER, bind to GRP78, and selectively activate the UPR, representing a novel mode of UPR regulation and mechanism of action of HDAC inhibitors.—Kahali, S., Sarcar, B., Prabhu, A., Seto, E., Chinnaiyan, P. Class I histone deacetylases localize to the endoplasmic reticulum and modulate the unfolded protein response.
Keywords: HDAC inhibitor, GRP78, acetylation
Although oncogenesis has traditionally been considered a disease of genetic mutations and chromosomal abnormalities, a growing body of evidence suggests transcriptional control through aberrant epigenetic states also plays an important role in tumor formation (1, 2). On the basis of their central role in influencing transcriptional programs by modulating chromatin structure, histone deacetylase (HDAC) enzymes have been recognized as promising molecular targets to reverse these epigenetic alterations associated with cancer (3). A total of 18 HDAC enzymes have been identified and classified on the basis of homology to yeast HDACs. Class I HDACs, which are primarily nuclear in localization, include HDACs 1, 2, 3, and 8. They are related to the yeast RPD3 deacetylase and have close homology in their catalytic sites. Class II HDACs are related to yeast Hda 1 (histone deacetylase 1) and include HDACs 4, 5, 6, 7, 9, and 10. Unlike Class I HDACs, Class II HDACs are primarily cytoplasmic and/or migrate between the cytoplasm and nucleus. Class III HDACs comprise the sirtuins, which unlike Class I and II HDACs, which are zinc-dependent enzymes, require NAD+ for their enzymatic activity. Lastly, HDAC11 represents the single member of the class IV HDACs, containing conserved residues in the catalytic core region shared by both class I and II enzymes (4).
Interestingly, phylogenetic analyses of bacterial HDACs suggest that all four HDAC classes preceded the evolution of histone proteins, suggesting that the primary activity of HDACs may be directed against nonhistone substrates (5, 6). In support of this, in addition to transcriptional control through core histones, post-translational modification through protein acetylation has emerged as an important mode of cellular regulation; thereby, extending the mechanistic relevance and research interest in HDACs beyond the field of chromatin biology (1, 7). The chaperone protein glucose-regulated protein 78 kDa (GRP78), which plays a central regulatory role in the unfolded protein response (UPR), represents a protein recently identified to be acetylated following HDAC inhibition, leading to UPR activation, and more importantly, contributing to the antitumor activity of this agent (8–10). However, the specific HDACs that may be regulating GRP78 and the UPR remain undefined. Further, although HDAC6 is known to primarily function in the cytoplasm and modulate chaperone protein activity (10, 11), class I HDACs, which recent chemical phylogenetics (12) suggest as the other primary targets of the specific HDAC inhibitor used in these studies, have traditionally been thought of as nuclear (3, 4); therefore, the potential localization and function of this class of HDACs in other subcellular fractions have yet to be studied in detail. In the current study, we identify the novel function of class I HDACs in GRP78 acetylation and selective UPR activation, providing further insight into the complex signaling associated with UPR regulation.
MATERIALS AND METHODS
Cell lines and reagents
U251 and DU145 were obtained from American Type Culture Collection (Manassas, VA, USA). Cells were maintained in RPMI 1640 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 5% heat-inactivated FBS (Gibco, Carlsbad, CA, USA) and 5 mM l-glutamine (Cellgro, Manassas, VA, USA). Cell cultures were maintained at 37°C under a humidified atmosphere with 5% CO2. Vorinostat was provided by Merck Research Laboratories (Whitehouse Station, NJ, USA) through the National Cancer Institute–Cancer Therapy Evaluation Program and was dissolved in DMSO at 1 mM and stored in aliquots at −80°C. Vorinostat was added to the cell treatment at 1 μM final concentration. Thapsigargin was purchased from Sigma-Aldrich (St. Louis, MO, USA) and dissolved in DMSO at 1 mM, portioned into aliquots, and stored at −20°C.
Plasmid construction and transfection
pcDNA3.1(+)-GRP78/binding immunoglobulin protein (BiP) plasmid was a gift from Dr. Richard Austin (McMaster University and the Hamilton Civic Hospitals Research Centre, Hamilton, ON, Canada). The human GRP78/BiP cDNA was inserted as a 1.95-kb fragment into the HindIII and XhoI sites of the mammalian expression vector pcDNA3.1(+) (Invitrogen).
In PCR reaction, the DNA oligomer CGGGTACCTTAATGAAGCTCTCC, with a KpnI restriction site (underscored), was introduced as the 5′-end forward primer, and the DNA oligomer TAGAAGGCACAGTCGAGG was introduced as the 3′-end reverse primer [bovine growth hormone (BGH) primer]. Full-length GRP78 was amplified using the pcDNA3.1(+)-GRP78/BiP plasmid as a template. The PCR reaction was carried out using the Phusion high-fidelity DNA polymerase kit (New England Biolabs, Ipswich, MA, USA). The reaction was cycled at 98°C for 1 min, then 98°C for 5 s, 52°C for 20 s, and 72°C for 90 s, followed by a final extension of 72°C for 10 min. The expected amplicon size for human GRP78/BiP is 2023 bp. The PCR-amplified product was electrophoresed onto 1% agarose gel for confirmation, gel-purified using Promega Wizard SV PCR and Gel Clean up System (Promega, Madison, WI, USA), and sequenced to confirm that it contained the correct full-length coding sequence. The hemagglutinin (HA) epitope sequence was introduced by PCR into the multiple cloning site of the pcDNA3.1(+) vector. To drive the expression of HA, the modified pcDNA3.1(+) vector had an HA epitope downstream of the T7 sequence. HA was then fused on the N terminus of the modified pcDNA3.1(+) vector. After sequence verification, GRP78/BiP cDNA was subcloned into a derivative of pcDNA3.1(+) vector ligated via the KpnI and XbaI site for expression of HA-tagged GRP78 fusion protein. Finally, sequence confirmation of the pcDNA3.1(+) HA-GRP78 construct was done using primer pairs for both upstream and downstream directions (T7 and BGH primers). FLAG epitope-tagged HDAC expression plasmids were generated as described previously (13).
Generation of pcDNA3.1(+) HA-GRP78 stable cell lines
U251 cells were seeded onto 100-mm dishes and transfected with 5 μg of expression plasmid pcDNA3.1(+) HA-GRP78 into 80% confluent U251 cells using Fugene HD transfection reagent (Roche, Indianapolis, IN, USA), according to manufacturer's instructions. G418 (Cellgro, Manassas, VA) was used for the selection. After 24 h post-transfection, and every 48 h thereafter for 2 wk, culture medium was replaced with fresh medium containing 400 μg/ml of G418. A total of 25 clones were isolated as stable transfectants with HA-tagged GRP78 expression vector in U251 glioma cells for further studies. G418-resistant clones were screened by Western blotting against HA monoclonal antibody purchased from Sigma-Aldrich. HA-tagged GRP78 stably transfected U251 cell lines were generated and used during this study.
GRP78 binding study using HA-GRP78 stable cell lines
HA-GRP78 stable U251 cell lines were maintained under G418 selection, plated onto 100-mm dishes until 80% confluence. Cells were harvested, and lysates were prepared in CellLytic M Cell lysis reagent from Sigma-Aldrich with Complete Mini Protease Inhibitor cocktail (Roche Diagnostics). The lysate was precleared for 30 min with 30 μl of protein-A/G-agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA, USA). GRP78 was immunoprecipitated from cell lysates containing 300 μg of protein with 30 μl of monoclonal anti-HA agarose conjugate beads (clone HA-7, Sigma-Aldrich) on a rotation shaker with gentle shaking at 4°C for 1 h. Agarose beads with bound proteins were washed 4 times with lysis buffer. The washed beads were resuspended in 2× Laemmli sample buffer, boiled for 5 min at 100°C for 3 min, and loaded onto SDS-PAGE gel, followed by immunoblotting. Immunoblotting was carried out with anti-HDAC1 (Millipore, Billerica, MA, USA), anti-HDAC2, anti-HDAC3 (Cell Signaling, Danvers, MA, USA), and anti-HDAC8 (Santa Cruz Biotechnology) antibodies.
Transient plasmid transfection and coimmunoprecipitation
Interactions of class1 HDACs were assessed by cotransfecting individual FLAG-tagged HDAC 1, 2, 3, and 8 construct in combination with pcDNA3.1(+) HA-GRP78 construct into U251 cells. U251 cells were seeded onto 100-mm dishes and transfected with 4 μg of each expression plasmid using Fugene HD transfection reagent (Roche) following the manufacturer's protocol. An equal amount of pcDNA3.1(+) HA-GRP78 vector was transfected into control cells. About 48-h post-transfection, cells were washed twice with 1× PBS, harvested, and lysed in CelLytic M cell lysis reagent (Sigma-Aldrich) containing a protease inhibitor cocktail, 2 mM PMSF, 2 mM Na3VO4, and protease inhibitors (Sigma-Aldrich). The lysate was precleared for 30 min with 30 μl of protein-A/G-agarose beads (Santa Cruz Biotechnology). Lysates were incubated overnight on an end-over-end shaker with 40 μl HA-tagged agarose beads (Sigma-Aldrich) at 4°C for 60 min with gentle shaking for 2 h at 4°C and then washed 4 times with an excess of lysis buffer. Samples were then boiled at 100°C for 5 min in 2× sodium dodecyl sulfate sample buffer and analyzed by Western blotting, as described below.
Immunoblot analysis
Total proteins were prepared from whole-cell lysates. Exponentially growing cells were dissolved in ice-cold cell lysis buffer, as described previously (9). Lysates were centrifuged at 13,000 rpm for 20 min to remove the nuclear and cellular debris. Total cell lysates (30–50 μg) were resolved on a 10% Tris-glycine SDS-PAGE gel, electrotransferred to PVDF membranes (Millipore), and electrophoresed. The blot was analyzed with rabbit monoclonal antibodies against anti-HDAC1 (1:1000, Millipore), anti-HDAC2, anti-HDAC3, anti-protein kinase-like ER kinase (PERK), rabbit polyclonal antibodies against human anti-acetyl lysine (Cell Signaling), anti-HDAC6, anti-HDAC8, anti-Bip/GRP78, anti-phosphospecific PERK (Santa Cruz Biotechnology), anti activating transcription factor 6 (ATF6) Mab (Imgenex, San Diego, CA, USA), and mouse anti-β-actin (Sigma-Aldrich). Secondary antibodies conjugated to horseradish peroxidase (GE Healthcare, Piscataway, NJ, USA) were used, and chemiluminescence (Thermo Fisher Scientific, Waltham, MA, USA) was used for detection. Densitometric scanning and acquisition of images was done using Adobe Photoshop (Adobe Systems Inc., San Jose, CA, USA). Image J (U.S. National Institutes of Health, Bethesda, MD, USA) analysis was used for quantification of all the immunoblots.
Protein subcellular fractionation
Protein subcellular fractions from the cytosol, membrane, organelle, nucleus, and cytoskeleton were prepared using the ProteoExtract Subcellular Proteome Extraction kit (S-PEK) from EMD Biosciences and Calbiochem (San Diego, CA, USA) following manufacturer's recommendations. About 3 × 106 U251 cells were harvested, and the pellet was fractionated to sequential subcellular extraction steps that yield protein fractions from the cytosol, membrane, organelle, soluble nuclear proteins, and finally cytoskeletal proteins. Protease inhibitors and benzonase, a nonspecific nuclease, were added freshly to the appropriate fractions. The fractionated extracts were kept at −80°C until use. Protein concentrations were determined, and then equal amounts of cytosolic fractions were mixed with membrane and organelle fractions, and nuclear fractions with cytoskeleton fractions, and subjected to Western blot analysis, as described earlier. Digitonin release experiments to differentiate subcellular localization of GRP78 and HA-tagged GRP78 were performed as described by Ni et al. (14).
Small-interfering RNA (siRNA)
U251 cells were seeded at 1 × 105 cells/well in 6-well plates and allowed to reach 70% confluency at the day of transfection. The siRNA construct used was purchased from Dharmacon (ThermoFisher Scientific) and was obtained as siGenome SMARTpool reagents human HDAC1 (M-003493-02-0010), human HDAC2 (M-003495-02-0010), human HDAC3 (M-003496-02-0010), and human HDAC6 (M-003499-02-0010). The nontargeting siRNA control was SiConTRolNon-targeting siRNA pool (D-001206-13-20; Dharmacon). The siRNA duplexes were reconstituted in 1× siRNA buffer, diluted from 5× siRNA buffer (Dharmacon) to 20 μM. Cells were transfected with 100 nM siRNA in Opti-Mem medium with 5% FCS using DharmaFECT transfection reagent (Dharmacon), according to the manufacturer's protocol. At 48 h after transfection, the cells were trypsinized, counted, and plated as designed. Efficiency of siRNA knockdown was measured by Western blot analysis.
Immunoprecipitation of GRP78 for acetylation
U251 cells were plated onto 100-mm dishes at 2 × 106 and grown until 80% confluence at the day of the experiment. Exponentially grown cells were transiently transfected with human HDAC1, HDAC2, HDAC3, HDAC6 using X-tremeGENE Transfection reagent (Roche), as per the manufacturer's instructions. The nontargeting siRNA control, siConTRolNon-targeting siRNA was used as negative control. Cells were transfected with 100 nM siRNA in Opti-Mem medium with 5% FCS. Cells were harvested at 48 h post-transfection, and cellular extracts were prepared. Immunoprecipitation from total cell protein was done using GRP78 antibody, essentially as described previously (9), and immunoblots were prepared using anti-acetyl lysine (Cell Signaling) and anti-GRP78 antibody (Santa Cruz Biotechnology).
Confocal microscopy
U251 or DU145 cells were cultured in a Lab-Tek II standard tissue culture slides (Thermo Fisher) in RPMI with 5% FBS. U251 or DU145 cells were seeded onto chamber slides (75,000 cells/well) at least 24 h before use in an experiment. Cells were fixed with 4% paraformaldehyde for 10 min, permeabilized with 0.2% Triton X-100, and blocked with 2% BSA in 1× PBS for 30 min. The slides were then stained overnight at 4°C with anti-HDAC1 (Millipore), anti HDAC2, anti-HDAC3 (Cell Signaling), anti-HDAC6, anti-GRP78/BiP, and anti-calnexin antibodies (Santa Cruz Biotechnology). Cells were washed 3 times with PBS, and were incubated with secondary antibodies (Alexa Fluor 488 donkey anti-mouse IgG, Alexa Fluor 488 donkey anti-goat IgG, or Alexa Fluor 594 anti-rabbit IgG; Molecular Probes, Invitrogen) at 1:500 for 2 h. The cells were counterstained and mounted with antifade containing 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen). Similar methods were used in cells transiently transfected with the plasmid pDsRed2-ER (Clontech, Mountain View, CA) to evaluate for endoplasmic reticulum (ER) luminal localization of individual HDACs. Micrograph images were acquired in the Moffitt Analytical Microscopy Core (H. Lee Moffitt Cancer Center) using a Leica DMI6000 inverted microscope and TCS SP5 tandem confocal scanner, through a ×63/1.40 NA Plan Apochromat oil-immersion objective lens (Leica Microsystems, Wetzlar, Germany) with dual photomultiplier tube detectors.
RT-PCR
Activation of inositol-requiring transmembrane kinase and endonuclease 1 (Ire1) was determined by quantitatively measuring the splicing of its substrate, the mRNA encoding the X-box binding protein 1 (XBP1) transcription factor. A 456-bp PCR product was expected if the Xbp1 cDNA fragment was derived from the unspliced form, spanning a 26-bp nucleotide intron, while a 430-bp PCR product was expected if the Xbp1 amplicon was derived from the spliced form. The housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the internal loading control (amplicon size 450 bp). Total cellular RNA was extracted by Qiagen RNeasy (Qiagen, Valencia, CA, USA) extraction kit. Transcript levels of mRNA were analyzed using 500 ng of total RNA from U251 cells using the TaKaRa RNA PCR kit (AMV) 3.0 (Takara Bio USA, Madison WI, USA), according to the manufacturer's instruction. Briefly, RT-PCR products of XBP1 mRNA were obtained from total RNA extracted using the primers Xbp1-F, 5′-GTTGAGAACCAGGAGTTAAGACAG-3′ (sense), and Xbp1-R, 5′-CAGAGGGTATCTCAAGACTAGG-3′ (antisense). The primers used to measure relative steady-state mRNA levels for GAPDH were GAPDH-F, 5′-CTCAGACACCATGGGGAAGGTGA-3′ (sense), and GAPDH-R, 5′-ATGATCTTGAGGCTGTTGTCATA-3′ (antisense). PCR cycling conditions included denaturing at 94°C for 1 min, annealing at 55°C for 1 min, elongation at 72°C for 1 min, for a total of 30 cycles, with a final extension step at 72°C for 7 min for all amplifiable products. The numbers of PCR amplification cycles for XBP1 and GAPDH were 35 and 25, respectively. The PCR reaction products were analyzed by 2.5% agarose gel electrophoresis and visualized under UV and digitized in gel documentation system. The experiment was performed in triplicate, and similar results were obtained.
RESULTS
We have recently demonstrated GRP78 acetylation and chaperone-client protein dissociation following exposure to the HDAC inhibitor vorinostat, leading to cytotoxicity mediated through UPR pathway activation (8, 9). Although cytoplasmic HDAC6, which has been previously attributed to modulating chaperone protein function (10, 11), has been implicated in this response, the potential role of class I HDACs, the key targets of vorinostat and other hydroxamic acid HDAC inhibitors (12), in regulating this process has not been explored. Therefore, as an initial investigation, we sought to determine whether class I HDACs had the capacity of binding to GRP78. Using the human glioblastoma line U251, we dual-transfected cells with FLAG-tagged class I HDACs (HDACs 1, 2, 3, and 8) and HA-tagged GRP78. Cell lysates were immunoprecipitated for HA, followed by immunoblot analysis using antibodies specific for FLAG and HA to determine which HDACs could bind to GRP78. Of the class I HDACs examined, HDACs 1, 2, and 3 demonstrated the ability of binding to GRP78, with lysates immunostaining for their respective FLAG constructs (Fig. 1A). Interestingly, these represent the 3 class I HDACs that are most sensitive to inhibition by vorinostat (12). Next, we examined the potential of GRP78 to bind to endogenous HDACs. Cell lysates from stable U251 cells expressing HA-GRP78 (which is expressed in both the cytosolic and organelle fractions; Supplemental Fig. S1A) were immunoprecipitated for HA, and the immunoblot was performed using antibodies specific for HDACs 1, 2, and 3. These studies confirmed the potential of GRP78 to bind to endogenous HDACs 1, 2, and 3 (Fig. 1B). HDAC8 did not demonstrate the capacity of binding with GRP78, and HDAC1 appeared to have the strongest interaction in these experiments. To further validate ER localization of the HA-tagged GRP78 construct used in these studies, digitonin release experiments were performed to differentiate subcellular localizations, as described by Ni et al. (14). As demonstrated in Supplemental Fig. S1B, although cytoplasmic localization of endogenous and HA-tagged GRP78 was present in both parental U251 and stable U251 cells expressing HA-GRP78, respectively, ER localization was confirmed in both systems.
Figure 1.

Class 1 HDACs interact with GRP78. A) U251 cells were transiently transfected with an expression plasmid of HA-tagged GRP78 alone (HA-GRP78) or in combination with FLAG-tagged HDAC1, HDAC2, HDAC3, or HDAC8 (F-H1, H2, H3, H8) expression plasmids. Untransfected cells served as controls (B). Cells were lysed 48 h post-transfection in CelLytic M cell lysis reagent for immunoprecipitation with HA-tagged agarose beads followed by immunoblotting with anti-FLAG or with anti-HA antibody. The precision plus dual-color protein standard was used as protein molecular size marker (in kilodaltons). B) GRP78 binding study using HA-GRP78 stable cell lines for class 1 HDACs. U251 (B) and HA-GRP78 stable U251 cell extracts were prepared in CelLytic M cell lysis reagent. The lysates were immunoprecipitated with HA-tagged agarose beads and then immunoblotted and probed for anti-HDAC1, anti-HDAC2, or anti-HDAC3 and anti-HA.
Because class I HDACs have been traditionally viewed as functioning within the nucleus (3), we next evaluated the potential of these HDACs to reside within the ER, the primary cellular compartment where GRP78 regulates UPR activity (15). As an initial investigation, cell lysates were fractionated, separating nuclear and cytoskeletal components (fraction A) from cytoplasm and membrane-bound organelles (including mitochondria and ER; fraction B). Western blot analysis was then performed on respective cell fractions. After confirming fractionation using antibodies specific for nuclear (lamin A/C), cytoskeletal (vimentin), mitochondrial [cytochrome c oxidase (Cox IV)], and endoplasmic reticular (calreticulin) fractions, lysates were probed for individual HDACs (Fig. 2). As described previously, these studies confirmed that HDAC6 was primarily localized outside the nucleus (fraction B). Although HDACs 1, 2, and 3 were expressed in the nucleus (fraction A), a considerable level of expression was also identified in fraction B, further supporting the potential of these class I HDACs to function extranuclearly. Next, we used similar fractionation techniques to determine whether HDACs found in this fraction could bind to GRP78. Cells were cotransfected with HA-tagged GRP78 and individual FLAG-tagged HDACs (HDAC1, HDAC2, and HDAC3). Following fractionation, cell lysates were immunoprecipitated for HA-GRP78 and immunoblotted for individual FLAG-HDACs, confirming the potential for HDACs to bind to GRP78 in this subcellular fraction (Supplemental Fig. S1C). As HDAC expression has been previously shown to be modulated by cellular density (16), we evaluated for HDAC expression in our models under conditions used for transfection, which included 80% confluent and 100% confluent cells harvested 48 h later. These studies confirmed stable HDAC expression in the cell transfection conditions used in our studies (Supplemental Fig. S1D).
Figure 2.
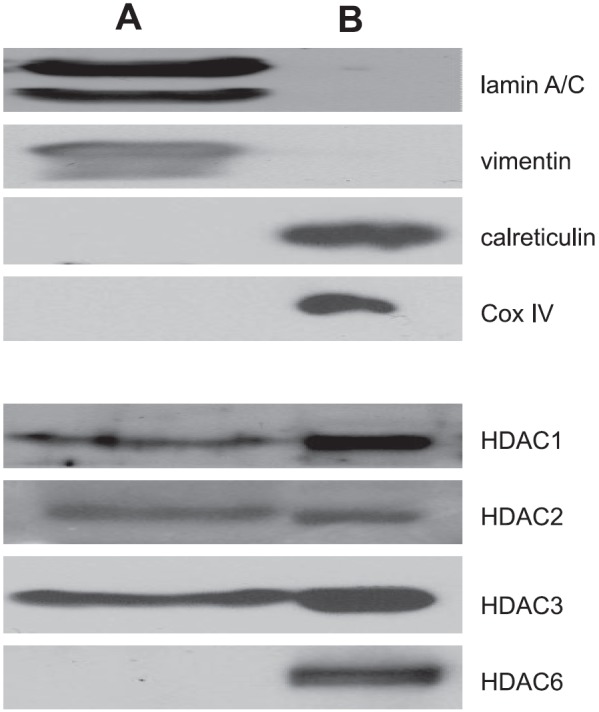
Subcellular fractionation of class I HDACs. Cell pellets were fractionated using sequential subcellular extraction steps, yielding protein fractions from the nucleus and cytoskeleton (fraction A) and cytosol and organelles (fraction B). Western blot analysis was performed using indicated antibodies. The typical nuclear marker lamin A/C, cytoskeletal marker vimentin, ER marker calreticulin, and mitochondrial marker Cox IV were used to verify the purity of each fraction.
We then used confocal microscopy to visualize the potential of these specific HDACs to colocalize with GRP78 in the ER. As demonstrated in Fig. 3, both intranuclear and extranuclear localization were identified for all 3 HDACs, supporting the above fractionation studies. There appeared to be consistent perinuclear localization of the individual HDACs (red) that colocalized with GRP78 (yellow). Similar findings were observed with calnexin (Supplemental Fig. S2A), confirming ER localization of HDACs 1, 2, and 3. In addition, these findings were recapitulated in the prostate cancer cell line DU145 (Supplemental Fig. S2B, C), suggesting these results are not specific to a single cell line. Further studies were performed, confirming colocalization of HDACs to the ER lumen using U251 cells transiently transfected with the pDsRed2 plasmid (Supplemental Fig. S2D). With the above-described experiments confirming the binding of class I HDACs to GRP78 in the ER, we next evaluated their potential role in modulating GRP78 acetylation. Expression of individual HDACs were attenuated using RNA interference (Fig. 4A) and cell lysates immunoprecipitated for GRP78. Immunoblot analysis was then performed using an antibody that recognizes anti-acetylated lysine residues. As shown in Fig. 4B, GRP78 displayed baseline levels of acetylation, further suggesting the potential role post-translational modification may play in regulating its chaperone function. Attenuating expression of individual HDACs using RNA interference led to increased GRP78 acetylation, with HDAC1 appearing to play the most significant role, increasing GRP78 acetylation 3.7-fold, which is consistent with the GRP78/HDAC binding studies (Fig. 1B). In accord with our previously described findings, treatment of U251 cells with the HDAC inhibitor vorinostat leads to a dose-dependent increase in GRP78 acetylation (Fig. 4C and ref. 9). Interestingly, the level of GRP78 acetylation following exposure to vorinostat (7.7-fold increase at 2.5 μM), which inhibits HDACs 1, 2, 3, 6, and 8 was approximately equivalent to the summation of levels of GRP78 acetylation following attenuation of individual HDACs using RNA interference (Fig. 4C), suggesting redundancy in the regulation of GRP78 by HDACs.
Figure 3.
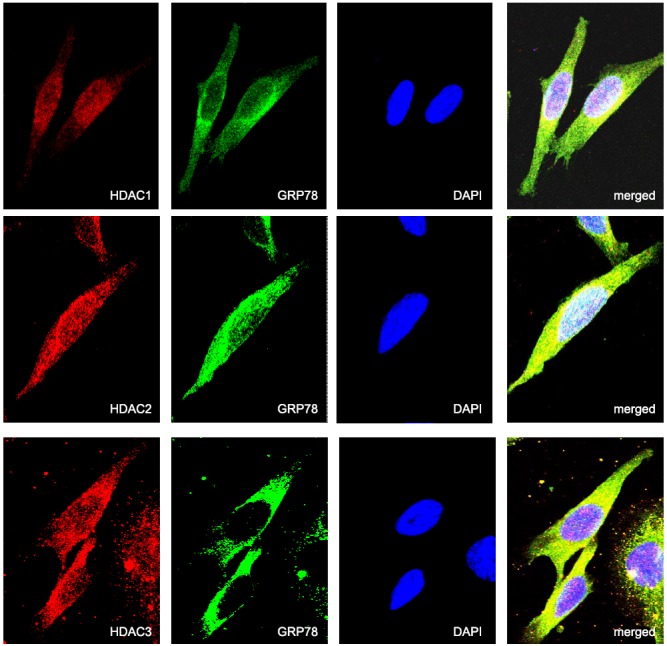
Confocal microscopy demonstrating colocalization of HDAC1, HDAC2, and HDAC3 with GRP78. U251 cells were fixed in paraformaldehyde and immunostained for GRP78 (green) and HDAC1, HDAC2, and HDAC3 (red). Cell nuclei were counterstained and mounted with antifade containing DAPI (blue). Colocalization of HDAC1, HDAC2, HDAC3, and GRP78 were visualized by confocal microscopy (×63 view). Merged images show the colocalization of HDAC1, HDAC2, or HDAC3 with GRP78 (yellow).
Figure 4.
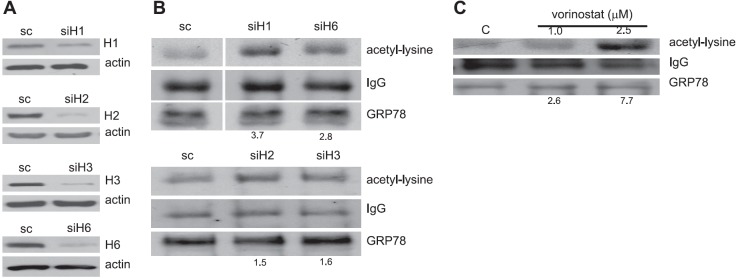
Knockdown of HDACs using RNA interference leads to GRP78 acetylation. A) U251 cells were knocked down for HDAC1 (siH1), HDAC2 (siH2), HDAC3 (siH3), HDAC6 (siH6), or scrambled nontargeting siRNA (sc) for 48 h. Immunoblotting against HDAC1 (H1), HDAC2 (H2), HDAC3 (H3), and HDAC6 (H6) antibodies validated significant knockdown of the respective HDACs. Actin served as loading control. B) These lysates were then immunoprecipitated with anti-GRP78 and immunoblotted with either anti-acetyl lysine or anti-GRP78. Images were quantified using densitometric scanning, normalizing samples to GRP78 expression. C) Similar studies were performed on cells exposed to the HDAC inhibitor vorinostat at described concentrations for 24 h.
Lastly, we explored the potential for individual HDACs to modulate UPR activation. Under nonstressed conditions, GRP78 binds to its client proteins PERK, ATF6, and Ire1, preventing their signaling. However, when the ER is overloaded by newly synthesized proteins or is “stressed” by agents that cause accumulation of unfolded proteins, GRP78 binds to the unfolded proteins in the ER, freeing its client proteins, which then serve as the primary mediators of UPR signaling (17, 18). PERK is released from its chaperone protein GRP78 to permit homodimerization and autophosphorylation, leading to activation. Ire1 similarly oligomerizes in ER membranes when released by GRP78. A key downstream target of Ire1 is XBP1 mRNA. Its endoribonuclease domain splices its mRNA, rendering it competent for translation (19–21). Instead of oligomerizing in the ER as PERK and Ire1, release of ATF6 permits transport to the Golgi compartment, where it is cleaved to generate the cytosolic-activated form of ATF6 that translocates to the nucleus, leading to selective gene activation (19–21). Therefore, these 3 pathways of UPR activation were evaluated following inhibition of individual HDACs. Activation of 2 arms of the UPR, including phosphorylation of PERK and ATF6 cleavage, was demonstrated following inhibition of HDACs 1, 2, 3, and 6 using RNA interference (Fig. 5A, B). Interestingly, of the HDACs tested, HDAC6 appeared to play the least significant role in UPR activation, demonstrating only minimal increases in PERK phosphorylation and ATF6 cleavage when compared to HDACs 1, 2, and 3. However, none of the HDACs appeared to activate Ire1 signaling, as measured by XBP-1 splicing (Fig. 5C), which is in accord with results obtained following vorinostat exposure, including expression of the downstream mediator of PERK, C/EBP homologous protein (CHOP; Supplemental Fig. S3). These findings reiterate the potential redundancy in HDAC-mediated UPR regulation and provide a potential mechanism to allow cells to differentially activate individual arms of UPR signaling relative to the environmental stress.
Figure 5.

Knockdown of HDACs using RNA interference activates the UPR. U251 cells were knocked down for HDAC1 (siH1), HDAC2 (siH2), HDAC3 (siH3), HDAC6 (siH6) or scrambled nontargeting siRNA (sc) for 48 h. Immunoblotting was then performed on cell lysates using antibodies specific for phospho-PERK (pPERK) and PERK (A), cleaved ATF6 and actin (B), and XBP-1 mRNA splicing and GAPDH was determined by RT-PCR (C). Cells treated with thapsigargin (1 μM) served as a positive control (+).
DISCUSSION
Post-translational acetylation, or the reversible process by which acetyl groups are enzymatically placed onto the ε-amino group of lysine residues of target proteins, is a well-studied event that occurs in histone tails during transcription. However, recent investigations have identified this mode of regulation to extend beyond chromatin dynamics (22, 23). The first nonhistone substrate shown to be acetylated was p53, a process that was determined to be central to protein activation (24). Recent scientific advances have since provided further insight into the increasingly important role lysine acetylation plays in protein regulation. Using high-resolution mass spectrometry, 3600 modifiable lysine acetylation sites on 1750 proteins were identified. These proteins had diverse cellular functions beyond chromatin remodeling, including cell cycle regulation, slicing, nuclear transport, and actin nucleation. Of the identified proteins in this high-throughput analysis, the chaperone protein GRP78 was determined to be reversibly acetylated following HDAC inhibition (25). These findings involving GRP78 acetylation have been validated, along with determining its functional consequence, including dissociation with client proteins and pathway activation (9, 10). This potential of chaperone protein acetylation leading to client dissociation has also been described with the chaperone protein heat-shock protein 90 (Hsp90). Investigations identified the potential for HDAC inhibitors to acetylate Hsp90, leading to dissociation and degradation of common oncoproteins, including ErbB1/2, Akt, c-Raf, BCR-ABL, and Fms-like tyrosine kinase 3 (FLT3) (11, 26–28). Studies went on to demonstrate that the specific target was HDAC6, which was shown to bind to Hsp90, and that inhibition using siRNA attenuated binding of Hsp90 to ATP and reduced chaperone association with client oncoproteins (11, 26).
In addition to modulating Hsp90, HDAC6, which primarily resides in the cytoplasm, has also been implicated as the primary enzyme contributing to the acetylation of the ER-localized chaperone protein GRP78 (10). In this report, in addition to HDAC6, we identify class I HDACs 1, 2, and 3, which have been previously considered to be exclusively nuclear, localizing in the ER, binding to GRP78, and contributing to the selective activation of the UPR. These findings represent a novel function of these class I HDACs, further supporting the growing, multifaceted regulatory role that these enzymes play in cell physiology. Although previous studies suggest that these class I HDACs are exclusively nuclear (29, 30), the localization studies used different cell types and did not focus on the ER. In addition, more recent data suggest a potential role for these HDACs in the cytoplasm (31, 32).
These findings also represent a novel mode of UPR regulation. Considerable information is known about the specific proteins involved in UPR signaling; however, steps leading toward activation remain unclear (33). Specifically, activation of the UPR is currently thought to be a passive process, with GRP78, one of the most abundant ER luminal chaperones, binding to the accumulating unfolded proteins following ER stress, thereby dissociating from, and allowing activation of, the 3 membrane-bound proximal ER stress sensors: PERK, ATF6, and Ire1 (15, 18, 33). Further, events leading to the respective chaperone protein dissociating from its client protein remain undefined. Our findings involving the post-translational modification of GRP78 may allow a cell to use HDACs as a mechanism to actively modulate the UPR, thereby fine-tuning a signaling response based on the specific stress, providing another layer of complexity underlying UPR regulation. This is further supported by our findings demonstrating the potential of HDACs to selectively activate two of the three arms of the UPR rather than just a generalized stress response, a process that has not been previously described.
Although the acetylation of GRP78, along with subsequent client protein dissociation and UPR activation following HDAC inhibition, is well supported by our work, a limitation is that a causal link between protein acetylation and the modulation of chaperone has yet to be definitively established. In addition, the importance of the acetylation state of individual residues of GRP78 has yet to be queried. Therefore, current studies are focused on comprehensively defining acetylation sites within the GRP78 sequence and generating point mutations to mimic acetylated/unacetylated states to establish a direct role for a specific modification with chaperone function. In addition, although these studies support the hypothesis that HDACs regulate the UPR directly through GRP78 modulation, the potential of these enzymes to interface with other components of this pathway has been previously described. Baumeister et al. (34) demonstrated the potential of HDAC 1 to regulate the UPR at the level of transcription. In these studies, they showed that HDAC1 represses expression of GRP78 by binding to its promoter before, but not after, ER stress. Inhibition with HDAC inhibitors induced GRP78 expression, leading to therapeutic resistance, which was attenuated when GRP78 was knocked down using RNA interference. In addition to directly modulating the UPR, investigators have also demonstrated the potential of HDACs to indirectly influence UPR activation. Kawaguchi et al. (35) discovered that HDAC6, a recognized microtubule-associated deacetylase, was a crucial player in the cellular management of misfolded protein-induced stress. Specifically, they identified HDAC6 to bind both polyubiquitinated misfolded proteins and dynein motors, thereby acting to recruit misfolded protein cargo to dynein motors for transport to aggresomes. Cells deficient in HDAC6 failed to clear misfolded protein aggregates from the cytoplasm, could not form aggresomes properly, and were hypersensitive to the accumulation of misfolded proteins.
In summary, our results demonstrate that class I HDACs 1, 2, and 3 localize to the ER and modulate the UPR. These findings, along with other recent investigations linking HDACs with the UPR, further strengthen the multifaceted role HDACs play in regulating UPR signaling. Further studies are required to more definitively define the role acetylation plays in modulating chaperone function within the ER and the specific role HDAC signaling plays in the integrated stress response.
Supplementary Material
Acknowledgments
The authors acknowledge Joseph Johnson (Moffitt Imaging Core Facility, H. Lee Moffitt Cancer Center) for assistance with confocal microscopy.
This work was funded by the American Cancer Society (RSG-11-029-01-CSM).
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- ATF6
- activating transcription factor 6
- BGH
- bovine growth hormone
- BiP
- binding immunoglobulin protein
- Cox IV
- cytochrome c oxidase
- DAPI
- 4′,6-diamidino-2-phenylindole
- ER
- endoplasmic reticulum
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- GRP78
- glucose-regulated protein 78 kDa
- HA
- hemagglutinin
- HDAC
- histone deacetylase
- Hsp90
- heat-shock protein 90
- Ire1
- inositol-requiring transmembrane kinase and endonuclease 1
- PERK
- protein kinase-like ER kinase
- siRNA
- small interference RNA
- UPR
- unfolded protein response
- XBP1
- X-box binding protein 1
REFERENCES
- 1. Bolden J. E., Peart M. J., Johnstone R. W. (2006) Anticancer activities of histone deacetylase inhibitors. Nat. Rev. 5, 769–784 [DOI] [PubMed] [Google Scholar]
- 2. Glozak M. A., Seto E. (2007) Histone deacetylases and cancer. Oncogene 26, 5420–5432 [DOI] [PubMed] [Google Scholar]
- 3. Lane A. A., Chabner B. A. (2009) Histone deacetylase inhibitors in cancer therapy. J. Clin. Oncol. 27, 5459–5468 [DOI] [PubMed] [Google Scholar]
- 4. Yang X. J., Seto E. (2007) HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene 26, 5310–5318 [DOI] [PubMed] [Google Scholar]
- 5. Gregoretti I. V., Lee Y. M., Goodson H. V. (2004) Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J. Mol. Biol. 338, 17–31 [DOI] [PubMed] [Google Scholar]
- 6. Xu W. S., Parmigiani R. B., Marks P. A. (2007) Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene 26, 5541–5552 [DOI] [PubMed] [Google Scholar]
- 7. Minucci S., Pelicci P. G. (2006) Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 6, 38–51 [DOI] [PubMed] [Google Scholar]
- 8. Kahali S., Sarcar B., Chinnaiyan P. (2011) The emerging role of histone deacetylases (HDACs) in UPR regulation. Methods Enzymol. 490, 159–174 [DOI] [PubMed] [Google Scholar]
- 9. Kahali S., Sarcar B., Fang B., Williams E. S., Koomen J. M., Tofilon P. J., Chinnaiyan P. (2010) Activation of the unfolded protein response contributes toward the antitumor activity of vorinostat. Neoplasia 12, 80–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rao R., Nalluri S., Kolhe R., Yang Y., Fiskus W., Chen J., Ha K., Buckley K. M., Balusu R., Coothankandaswamy V., Joshi A., Atadja P., Bhalla K. N. (2010) Treatment with panobinostat induces glucose-regulated protein 78 acetylation and endoplasmic reticulum stress in breast cancer cells. Mol. Cancer Ther. 9, 942–952 [DOI] [PubMed] [Google Scholar]
- 11. Bali P., Pranpat M., Bradner J., Balasis M., Fiskus W., Guo F., Rocha K., Kumaraswamy S., Boyapalle S., Atadja P., Seto E., Bhalla K. (2005) Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J. Biol. Chem. 280, 26729–26734 [DOI] [PubMed] [Google Scholar]
- 12. Bradner J. E., West N., Grachan M. L., Greenberg E. F., Haggarty S. J., Warnow T., Mazitschek R. (2010) Chemical phylogenetics of histone deacetylases. Nat. Chem. Biol. 6, 238–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang X., Wharton W., Yuan Z., Tsai S. C., Olashaw N., Seto E. (2004) Activation of the growth-differentiation factor 11 gene by the histone deacetylase (HDAC) inhibitor trichostatin A and repression by HDAC3. Mol. Cell. Biol. 24, 5106–5118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ni M., Zhou H., Wey S., Baumeister P., Lee A. S. (2009) Regulation of PERK signaling and leukemic cell survival by a novel cytosolic isoform of the UPR regulator GRP78/BiP. PLoS ONE 4, e6868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ma Y., Hendershot L. M. (2004) The role of the unfolded protein response in tumour development: friend or foe? Nat. Rev. Cancer 4, 966–977 [DOI] [PubMed] [Google Scholar]
- 16. Dangond F., Henriksson M., Zardo G., Caiafa P., Ekstrom T. J., Gray S. G. (2001) Differential expression of class I HDACs: roles of cell density and cell cycle. Int. J. Oncol. 19, 773–777 [PubMed] [Google Scholar]
- 17. Lee H. K., Xiang C., Cazacu S., Finniss S., Kazimirsky G., Lemke N., Lehman N. L., Rempel S. A., Mikkelsen T., Brodie C. (2008) GRP78 is overexpressed in glioblastomas and regulates glioma cell growth and apoptosis. Neuro-oncology 10, 236–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schroder M., Kaufman R. J. (2005) The mammalian unfolded protein response. Annu. Rev. Biochem. 74, 739–789 [DOI] [PubMed] [Google Scholar]
- 19. Lin J. H., Li H., Yasumura D., Cohen H. R., Zhang C., Panning B., Shokat K. M., Lavail M. M., Walter P. (2007) IRE1 signaling affects cell fate during the unfolded protein response. Science 318, 944–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ye J., Rawson R. B., Komuro R., Chen X., Dave U. P., Prywes R., Brown M. S., Goldstein J. L. (2000) ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 6, 1355–1364 [DOI] [PubMed] [Google Scholar]
- 21. Yoshida H., Matsui T., Yamamoto A., Okada T., Mori K. (2001) XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891 [DOI] [PubMed] [Google Scholar]
- 22. Brooks C. L., Gu W. (2011) The impact of acetylation and deacetylation on the p53 pathway. Protein Cell 2, 456–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. MacDonald V. E., Howe L. J. (2009) Histone acetylation: where to go and how to get there. Epigenetics 4, 139–143 [DOI] [PubMed] [Google Scholar]
- 24. Gu W., Roeder R. G. (1997) Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 90, 595–606 [DOI] [PubMed] [Google Scholar]
- 25. Choudhary C., Kumar C., Gnad F., Nielsen M. L., Rehman M., Walther T. C., Olsen J. V., Mann M. (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840 [DOI] [PubMed] [Google Scholar]
- 26. Kovacs J. J., Murphy P. J., Gaillard S., Zhao X., Wu J. T., Nicchitta C. V., Yoshida M., Toft D. O., Pratt W. B., Yao T. P. (2005) HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol. Cell 18, 601–607 [DOI] [PubMed] [Google Scholar]
- 27. Scroggins B. T., Robzyk K., Wang D., Marcu M. G., Tsutsumi S., Beebe K., Cotter R. J., Felts S., Toft D., Karnitz L., Rosen N., Neckers L. (2007) An acetylation site in the middle domain of Hsp90 regulates chaperone function. Mol. Cell 25, 151–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yu X., Guo Z. S., Marcu M. G., Neckers L., Nguyen D. M., Chen G. A., Schrump D. S. (2002) Modulation of p53, ErbB1, ErbB2, and Raf-1 expression in lung cancer cells by depsipeptide FR901228. J. Natl. Cancer Inst. 94, 504–513 [DOI] [PubMed] [Google Scholar]
- 29. Rountree M. R., Bachman K. E., Baylin S. B. (2000) DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat. Genet. 25, 269–277 [DOI] [PubMed] [Google Scholar]
- 30. Taplick J., Kurtev V., Kroboth K., Posch M., Lechner T., Seiser C. (2001) Homo-oligomerisation and nuclear localisation of mouse histone deacetylase 1. J. Mol. Biol. 308, 27–38 [DOI] [PubMed] [Google Scholar]
- 31. Yang W. M., Tsai S. C., Wen Y. D., Fejer G., Seto E. (2002) Functional domains of histone deacetylase-3. J. Biol. Chem. 277, 9447–9454 [DOI] [PubMed] [Google Scholar]
- 32. Park J. J., Kim Y. E., Pham H. T., Kim E. T., Chung Y. H., Ahn J. H. (2007) Functional interaction of the human cytomegalovirus IE2 protein with histone deacetylase 2 in infected human fibroblasts. J. Gen. Virol. 88, 3214–3223 [DOI] [PubMed] [Google Scholar]
- 33. Zhang K., Kaufman R. J. (2006) The unfolded protein response: a stress signaling pathway critical for health and disease. Neurology 66, S102–S109 [DOI] [PubMed] [Google Scholar]
- 34. Baumeister P., Luo S., Skarnes W. C., Sui G., Seto E., Shi Y., Lee A. S. (2005) Endoplasmic reticulum stress induction of the Grp78/BiP promoter: activating mechanisms mediated by YY1 and its interactive chromatin modifiers. Mol. Cell. Biol. 25, 4529–4540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kawaguchi Y., Kovacs J. J., McLaurin A., Vance J. M., Ito A., Yao T. P. (2003) The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 115, 727–738 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.