

Abstract
Mixed phenotype acute leukemia (MPAL) is considered as a rare type of leukemia with an incidence of less than 4% of all acute leukemia based on the most recent 2008 WHO classification. Common subtypes are the B/myeloid and T/myeloid; B/T and trilineage MPAL being extremely rare. We present a case of a male in his 20s, whose peripheral blood smears showed 34% blast cells and bone marrow with 70% blasts. Immunophenotyping by multiparametric flow cytometry showed two populations of blasts, the major one with B-lineage and the minor one with T-lineage. Conventional karyotyping revealed complex karyotype with the presence of double Philadelphia chromosome (Ph+). BCR/ABL1 rearrangement was confirmed by fluorescent in situ hybridization (FISH) analysis. The BCR/ABL1 ES probe on interphase cells indicated p190 minor m-BCR/ABL fusion in 46% and a second abnormal clone with double Ph+ in 16% of the cells analyzed confirmed by reverse transcription-PCR (RT-PCR). The case was diagnosed as MPAL with double Philadelphia chromosome Ph+. The patient was treated with dasatinib, four cycle hyper CVAD/methotrexate cytarabin protocol, and allogeneic transplant. He is still alive in complete hematological, cytogenetic, and molecular remission. Mixed phenotype B/T acute leukemia is an extremely rare disease, particularly those with double Philadelphia chromosomes and clinically presents challenges in diagnosis and treatment.
Keywords: mixed phenotype acute leukemia B/T, double Philadelphia
Introduction
The majority of acute leukemia cases are classified as acute lymphoblastic leukemia (ALL) or acute myeloid leukemia (AML), based on morphologic, immunophenotypic, cytochemical, and genetic profiles of the blast cells. However, a minority of acute leukemias show no clear evidence of differentiation along a single lineage. These are now classified under acute leukemias of ambiguous lineage by the most recent WHO classification (2008) and account for <4% of all cases of acute leukemia. They include leukemias with no lineage-specific antigens (acute undifferentiated leukemias) and those with blasts that express antigens of more than one lineage (mixed phenotype acute leukemias [MPALs]). The latter can either be leukemias with two distinct populations of blasts, each expressing antigens of a different lineage (historically referred to as “bilineal” leukemias) or a single blast population coexpressing antigens of multiple lineages (historically referred to as “biphenotypic” acute leukemias).1 Coexpression of myeloid and B-lymphoid antigens is most common in MPAL, followed by coexpression of myeloid and T-lymphoid antigens, accounting for 66%–70% and 23%–24% of MPALs, respectively. Coexpression of B- and T-lineage-associated antigens or antigens of all three lineages is exceedingly rare.2,3 The requirement for assigning more than one lineage to a single blast population has been established by current WHO classification.
The BCR-ABL1 rearrangement is the most frequent recurrent genetic abnormality occurring in MPAL and considered a distinctive entity. It accounts for 20% of all MPAL.1,4 MPAL with t(9:22)(q34:q11.2) meets the diagnostic criteria for MPAL with blasts bearing the t(9:22)(q34:q11.2) translocation or BCR-ABL1 rearrangements in patients with no history of chronic myelogenous leukemia (CML). The majority of cases occurring in adults have B/myeloid phenotype, while some show T/myeloid, B- and T-lineage, or trilineage leukemia. Additional cytogenetic abnormalities are shown in many cases, including complex karyotype. Ph+ is a poor prognostic factor in MPAL, with a reported median survival of eight months, which is significantly worse than that of patients of all types of MPAL.4
We present an unusual case of adult MPAL with immunologically two distinct blast cell subsets expressing B-ALL markers and cortical T markers. Cytogenetic analysis revealed double Philadelphia chromosome, with complex karyotypes.
Case Presentation
A male patient in his 20s with unremarkable previous clinical history presented for preemployment check up in November 2012. Peripheral smear showed acute leukemia. The patient was referred to National Centre for Cancer Care and Research (NCCCR) in Qatar for further management. On admission, the patient looked sick, complaining of fever with chills, fatigue, cough, and dyspnea. His physical examination revealed pallor, fever, generalized mild lymph node enlargement, mild hepatosplenomegaly, and bilateral lung basal crackles.
Chest X-ray showed bilateral lung infiltrates and chest high-resolution computerized tomography (CT) scan showed ground glass opacity with patchy nodular infiltrate that is scattered in both lungs more pronounced in the mid and lower lung zones.
Ultrasound of the abdomen showed an enlarged liver and mild splenomegaly (14 cm).
Viral serology for hepatitis B and C and HIV was negative. Respiratory virus panel and septic workup were negative.
A complete blood count (CBC) revealed anemia (hemoglobin of 9.3 g/dL), thrombocytopenia (platelet count of 28 × 103/μL), and leukocytosis (white blood cell count (WBC) of 11.2 × 103/μL). The differential count showed 13% segmented neutrophils, 48% lymphocytes, 2% monocytes, 1% metamyelocytes, 2% myelocytes, and 34% blasts (Fig. 1A).
Figure 1.
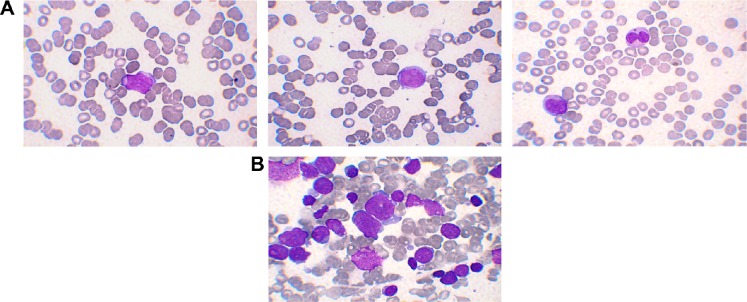
(A) Peripheral blood smear showing example of circulating blasts (100×) (B) Bone marrow aspirate (100×) with many circulating heterogeneous blasts. The majority were medium to large sized with intermediate to fine chromatin with one or more prominent nucleoli and moderate amount of light blue cytoplasm. Some showed azurophilic granules, cytoplasmic blebs and or cytoplasmic vacuolation. Others were smaller with scant cytoplasm, more condensed chromatin and less prominent to inconspicuous nucleoli.
The bone marrow aspirate showed (70%) blasts with heterogeneous morphology. The majority were medium-to-large sized with intermediate-to-fine chromatin with one or more prominent nucleoli and moderate amount of light blue cytoplasm. Some showed azurophilic granules, cytoplasmic blebs, and/or cytoplasmic vacuolation. Others were smaller with scant cytoplasm, more condensed chromatin, and less prominent to inconspicuous nucleoli (Fig. 1B).
The cytochemical staining with myeloperoxidase and nonspecific esterase was negative. Flow cytometry (FCM) was performed on bone marrow aspirate using standard stain/lyse and wash technique. The acute leukemia panel of 22 antibodies in a four color combination (FITC/PE/ECD/PC5 fluorescent conjugates) was used. Eleven combinations were as follows: (1) CD34/CD117/CD45/CD19, (2) CD14/CD13/CD45/CD64, (3) HLADR/CD7/CD45/CD5, (4) CD34/CD33/CD45/CD56/, (5) CD19/CD10/CD45/CD3, (6) CD15/CD33/CD45/CD2, (7) CD9/-/CD45/CD19, (8) CD20/CD10/CD45/CD19, (9) cMPO/cCD79a/cCD3/sCD45, (10) Tdt/sCD19/sCD3/sCD45/, and (11) CD45/CD1a/CD3/CD2.
CD45 vs side scatter (SSC) strategy was used to gate the blast population. Two distinct populations of blasts were identified on CD45 vs SSC plot (Fig. 2).
Figure 2.
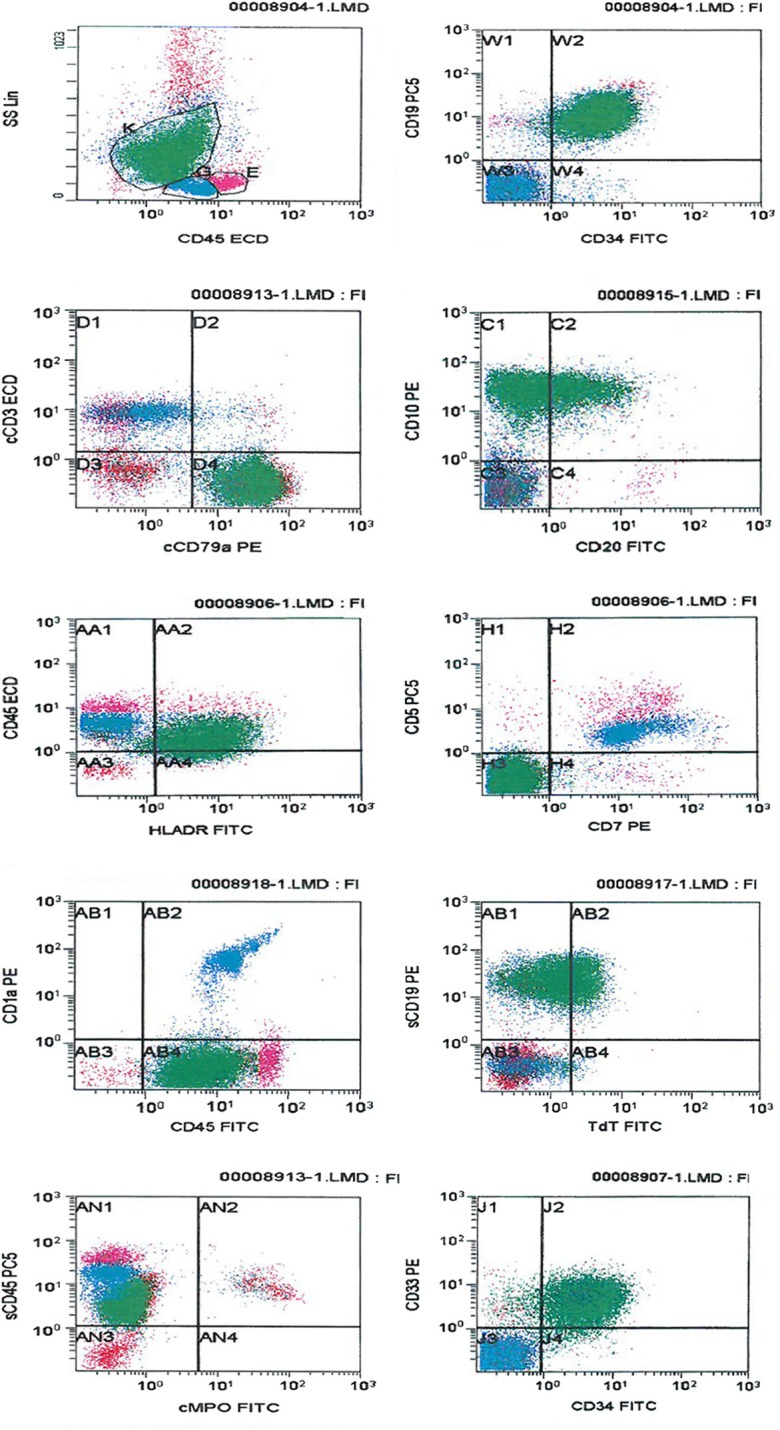
Flow cytometric dot plots: B-lymphoblasts cluster in green color gated (K) on CD45 comprises 60% of total expressing CD19, CD34, CD10, cCD79a, HLADR, and partial Tdt with aberrant expression of CD33 and CD13. T-lymphoblast cluster in blue color gated (G) comprise 19% of the total expressing cCD3, CD7, CD5, CD2, and CD1a; lymphocytes gated (E).
One population comprising 60% of the total cells expressing CD45 (dim), CD34, cCD79a, CD19, CD10, HLADR, CD9 with partial expression of Tdt, and aberrant expression of CD33 and CD13 (partial). This population was negative for T and other myeloid markers. The immunophenotype of these blasts was consistent with B-lymphoblast. The other immature population comprising approximately 19% of total cells expressed CD45 (moderate) and T-cell markers, including cCD3, CD7, CD5, CD2, and CD1a. This population was negative for surface CD3, TdT, CD34, B, and myeloid markers (Fig. 2). Immunohistochemical studies on the core biopsy showed that the blasts were positive for CD34, CD20, Pax 5, CD79a, CD99, CD10, and Tdt. CD1a and CD3 were positive in a subpopulation of the blasts. Based on the morphologic and immunophenotypic profiles, this case was diagnosed as MPAL with two separate populations of blasts belonging to B- and T-lineages.
Conventional cytogenetic analysis was performed on cultured bone marrow cells at the time of diagnosis. The analysis revealed an abnormal karyotype with two clones. The main clone with 46, XY with reciprocal t(9;22)(q34;q11.2) in 22/42 cells (Fig. 3A). The subclone was a near tetraploid (80–88 chromosomes) with multiple structural and numeric abnormalities, including double Ph+ chromosome (Fig. 3B).
Figure 3.

(A) G-banding karyotype showing 46, XY, t(9;22)(q34;q11.2) [22]. (B) Showing a representative cell from minor clone with 80–88, XXYY, −9[5], t(9;22)(q34;q11.2)[20], −11[6], −13[3], −14[3], −15[2], −18[2], −20[6], −21[3], −22, +der(22) t(9;22)[20][cp20].
Fluorescent in situ hybridization (FISH) analysis was performed on unstimulated interphase cells from bone marrow sample using probe LSI BCR-ABL ES Dual color translocation probe (Vysis), which allows the distinction between major M-BCR/ABL and minor m-BCR/ABL gene rearrangements. The FISH study revealed abnormal hybridization pattern with m-BCR/ABL1 rearrangement in 46% (46/100) of examined cells. A second abnormal clone was also detected with a triple fusion pattern (double fusion signal of the minor (m)-BCR/ABL1 rearrangement plus a third extra fusion signal corresponding to the presence of supernumerary Ph chromosome) in 16% of the cells analyzed (Fig. 4).
Figure 4.
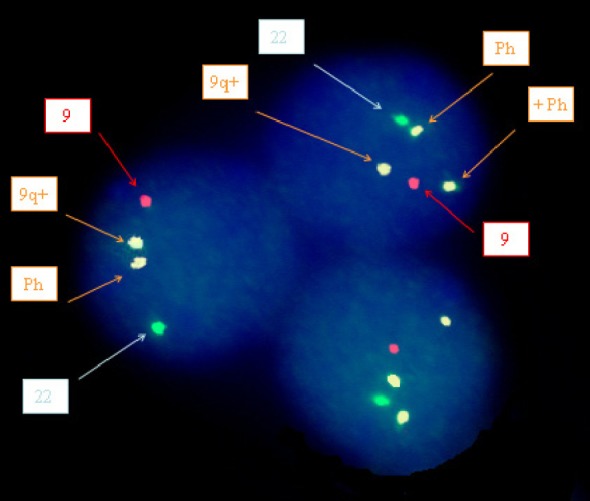
Patterns of BCR/ABL gene rearrangements involving the m-bcr breakpoint (p190 BCR/ABL) as revealed by FISH analysis of interphase-cultured BM nuclei cells showing two different patterns: nuclei with two fusions (typical m-BCR/ABL gene rearrangements) and nuclei with three fusions, including an extra signal corresponding to supernumerary Ph chromosome, in addition to the two fusion signals typical of m-BCR gene rearrangements.
The patient was initially started on supportive measures, including IV fluids 1/2 NS 100 mL/hour, allopurinol 300 mg po daily, tazocin 4.5 g IV every eight hours, ciprofloxacin 400 mg IV every 12 hours, and voriconazole 6 mg/kg IV stat, and then 4 mg/kg. Three days later, he was started on dasatinib 140 mg po daily. After three weeks, he improved from his lung infection and his CBC was normalized. Molecular studies for BCR/ABL p190 mRNA by quantitative, reverse transcription PCR (RT-PCR) was positive and estimated to represent 11.0% of total abl (% bcr/abl (p190:abl)).
Hickman line was inserted and the course A of hyper CVAD/methotrexate, cytrabin regimen (cyclophosphamide 300 mg/m2 IV/12 hour at days 1–3), doxorubicin 50 mg/m2 IV at days 4–5, vincristine 1.4 mg/m2 IV infusion. Maximum 2 mg at days 4 and 11, dexamethasone 40 mg oral, daily at 1 to 4 and day 11–14) was initiated, and then, complicated by febrile neutropenia that was treated empirically with tazocin, amikacin, and voriconazole. He also received valacyclovir 50 mg po three times per day and septrin 960 mg po tree times per week as prophylaxis and pegylated filgrastim 6 mg subcutaneous. The course B (methotrexate 1 g/m2 IV for 24 hours continuously 200 mg/m2 for two hours then 800 mg/m2 for 22 hours at days 1–2, cytarabine 3 g/m2 IV over two hours, 12 hourly total of four doses at days 2–3) of the first cycle was complicated by enterovirus infection and treated by ribavirin and intravenous immunoglobulin (IVIG). He also received broad spectrum antibiotics and antifungals.
The course A of the second cycle was not complicated, whereas the course B was complicated by reactivation of the enterovirus infection with dysphagia grade III and odynophagia and tonsillitis, again treated by ribavirin and IVIG. He also received broad spectrum antibiotics and anti-fungal medication. Upper endoscopy was performed and showed esophagitis and gastritis.
Bone marrow examination and cytogenetic (karyotype/FISH) after two cycles of chemotherapy and dasatinib confirmed complete hematologic and cytogenetic remission; however, he was not yet in molecular remission. The p190 mRNA was detected at 0.1% of total abl (% bcr/abl (p190:abl)).
During the third cycle of hyper CVAD (course A and course B), the patient had no complications. The course A of the fourth cycle was complicated by adenovirus hemorrhagic cystitis and subconjunctival hemorrhage, which was treated by ribavirin, IVIG, and irrigation of the bladder, while course B of the fourth cycle was put on hold. Patient evaluation after four cycles plus dasatinib, including bone marrow aspirate, FCM, and cytogenetic, confirmed the complete remission. BCR/ABL p190.mRNA was not detected (complete molecular remission).
He was started on maintenance dasatinib. Then he had allogeneic bone marrow transplantation (HSCT) from his brother in July 2013, and he is still alive and in complete remission up to the present time.
Discussion
Most acute leukemias can easily be classified as myeloid or lymphoid (including B- or T-lineages). However, a small percentage shows more than one lineage or no specific lineage, which is designated as “acute leukemia of ambiguous lineage,” which can either be an MPAL combining phenotype of two or more lineages or “undifferentiated leukemia” without clear evidence of any lineage commitment. In addition, MPAL may comprise a single population of blast expressing both specific myeloid and lymphoid antigens (biphenotypic) or be made up of two distinct populations, each expressing different lineage specific antigens. Historically, a diagnosis of biphenotypic acute leukemia was made using the few available lineage-associated markers, which had spuriously shown an incidence of acute mixed leukemia to be as high as 20%).5 As more antibodies became available, it became clear that a significant proportion of AML and ALL cases expressed aberrant antigens.6–9 It became necessary to distinguish aberrant expression of an antigen(s) from true “mixed phenotype acute leukemia.” Specific criteria were first proposed by Arnoulet et al10 in the form of a scoring system based on weighted expression of antigens by blasts.
In 2001, the WHO expert group11 proposed a scoring system that is modified from the European Group for the Immunological Characterization of Leukemias (EGIL) scoring12 (where a score of over 2 was required to assign a lineage). The current WHO (2008) classification, however, has replaced these scoring systems and relies upon lineage assignment based on specific criteria. Currently, cytoplasmic CD3 is the most T-lineage specific antigen. To assign B-lineage, multiple antigenic expressions are required, which includes a strong CD19 along with strong expression of any one of CD79, CD10, and cytoplasmic CD22. However, if CD19 is weakly expressed, then, strong expression of at least two of the aforementioned B antigens is necessary.1 Using this recent WHO criterion, prevalence of MPAL has been found to be 1.6% of acute leukemia.3 MPAL usually are either T/myeloid or B/myeloid and less frequently B/T MPAL. Most of the cases of B/T MPAL found in literature describe biphenotypic blasts rather than bilineal blasts4 as seen with our case. Weir et al13 described 19 cases of acute bilineal leukemia over a 10-year period, but they did not report any case of B/T bilineal leukemia. In the present case, two separate populations of blasts were identified. The first population strongly expressed CD19, CD79a, and CD10 without any evidence of cytoplasmic CD3 or other “T” antigens, thus fulfilling the criteria for B-lineage, while the second population expressed cytoplasmic CD3, CD7, CD5, CD2, and CD1a without any evidence CD19 or other “B” antigens, fulfilling the WHO criteria for “T” lineage.
The study conducted by Weir et al13 included only T/myeloid or B/myeloid blasts, where definite morphological differences existed between the two populations. The light scatter properties of these populations were sufficiently different to allow clear-cut recognition among them. The larger myeloid blasts had higher scatter as compared to smaller lymphoid blasts with a lower scatter. In contrast, our case had lymphoid blasts only, which made it difficult to differentiate the two different populations from the FSC vs SSC plot, while CD45 vs SSC discriminated the two populations with T blasts expressing CD45 stronger than B blasts.
Although the putative cell of origin for MPAL is unknown, it is suggested that these arise from very early hematopoietic progenitors with a potential to grow either as myeloid or lymphoid. Accordingly, the cases with B/myeloid and T/myeloid immunophenotypes would be in agreement with findings documented in fetal mice hemopoiesis that suggest the persistence of a myeloid potential in early B- and T-lymphoid precursors14 and the existence of a multipotent progenitor with B-cell, T-cell, and granulocyte/macrophage potential.15
The recognition of MPAL with B- and T-cell lineages would support the proposed model of adult bone marrow hematopoiesis, where a common lymphoid progenitor is present.16
Cytogenetic alterations reported in MPAL are t(9;22) (q34;q11) and 11q23 (MLL) abnormalities. These two abnormalities have been observed so frequently that the newer WHO classification has acknowledged these as separate entities.1
In our case, chromosomal analysis revealed reciprocal t(9;22)(q34;q11.2) in 22/42 (52%) cells analyzed (Fig. 3A). Other additional cytogenetic abnormalities with near tetraploid cells (4n), double Ph+ were identified in another 20 cells (47.6%). The FISH analysis for BCR/ABL rearrangement showed abnormal hybridization pattern in 46% of cells analyzed. In addition, a second abnormal clone was detected with a triple fusion (double Ph+) in 16% of the cells analyzed.
The BCR/ABL1 fusion transcript can be detected by qRT-PCR. The location of the breakpoint within the BCR gene results in either the p190BCR/ABL protein observed in Ph+ ALL (66.3% of the cases) or the p210BCR/ABL protein common in patients with CML and in 31.2% of Ph+ ALL. The remaining cases are associated either with both transcript type or with atypical transcripts.17 In our patient, BCR/ABL p190 mRNA transcript was detected supporting de novo ALL.
Hyperdiploid karyotypes with double Ph+ are common in CML, with over 500 reported cases. In contrast, they are rare in ALL, and a total of 66 cases with double Ph+ are reported in Mitelman’s Catalog of Chromosome Aberrations in Cancer (http://cgap.nci.nih.gov/Chromosomes/CytSearchForm). Of those, only 23 were associated with hyperdiploidy and, interestingly, only three patients showed more than two copies of the Ph+ on conventional karyotyping. Thomas et al18 found seven cases of double Ph+ in 41 ALL (17%), and the presence of an extra Ph+ was considered an initial parameter associated with a statistically significant worse prognosis.
Other additional chromosome aberrations were present in up to 79% of the cases in a large study of 209 patients.19 Yanada et al20 study involving 77 ALL patients with Ph+, additional aberrations with a frequency of greater than 10% included a second Ph chromosome (+der (22) t(9,22) abnormalities involving the short arm of chromosome 9, monosomy 7, and trisomy 8). The presence of additional aberrations was associated with significantly shorter relapse-free survival (RFS) and higher relapse rate. This was particularly pronounced for the +der (22) t(9,22) and abnormalities involving the short arm chromosome 9.19,20 In our case, other additional cytogenetic abnormalities were near tetraploid cells (4n) and double Philadelphia chromosomes were identified in another 20 cells (47.6%).
Since Ph+ cases represent approximately 25% of ALL, cases with double Ph should represent a smaller percentage of adult ALL.5 One case of mixed phenotyping acute leukemia (two distinct population of blasts with B and T blast lineages), which showed t(9;22)(q34;q11) (Ph+) was reported. The present case describes mixed phenotype B/T acute leukemia with double Philadelphia chromosomes and complex karyotype that represent an extremely rare disease, BCR-ABL.
Controversies regarding the treatment of MPAL include which induction protocol is to be used (myeloid or lymphoid) and whether induction is to be followed by stem cell transplantation or not. ALL-based treatments have been found to be more effective with higher response rate as compared to AML or AML ALL schedule.4 Cases of MPAL in pediatric patients showed a better response rate with ALL-based induction protocol compared with AML-based protocol (CR of 83% vs CR of 52%).
Cases of MPAL have shown poor outcomes probably because of adverse cytogenetic abnormalities associated with it, especially the presence of BCR-ABL translocation.21 In addition, high incidence of CD34 positivity, high incidence of extramedullary infiltrate, lack of optimized guidelines for induction, and high incidence of relapse after remission has led to a lower overall survival and disease-free survival in patients of MPAL.22
Philadelphia chromosome-positive ALL have long been recognized as a high-risk subset of ALL. Published data show a disappointing long-term survival of only 20% or less with chemotherapy alone23 with a median survival of eight months. The combination of BCR-ABL1 targeted tyrosine kinase inhibitors (TKIs) (imatinib or dasatinib) chemotherapy has improved outcome with CR of 80%–100% and five years survival in 35%–50% range.24 Nevertheless, Ph+ diseases remain a poor prognosis subgroup with unacceptable high relapse rate.
Because of the poor outcome with chemotherapy, the allogeneic stem cell transplantation hematopoitic stem cell transplant (HSCT) in first complete remission (CR1) remains the standard of care in most centers and is considered to be the treatment of choice in adult Ph+ ALL.4 Cases of MPAL B/T-lymphoid are exceedingly rare and with the exception of the present case not with double Ph have been described.
Our case has responded well with ALL-based treatment plus dasatinib and subsequent allogeneic hematopoietic stem cell transplant and remains in complete remission 18 months following initial diagnosis. But a longer follow-up is required to draw the inference on the treatment effect.
Conclusion
Mixed phenotype B/T acute leukemia is an extremely rare disease, particularly those with double ph chromosomes and clinically presents challenges in diagnosis and treatment.
Chemotherapy (hyper CVAD plus TKIs) followed by allogeneic bone marrow transplant in such patients may give good results in patients with this rare type of acute leukemia.
Acknowledgments
The authors acknowledge the support and guidance of Dr Zafar Nwaz, and are also thankful for use of the Flowcytometry Laboratory at NCCCR.
Footnotes
ACADEMIC EDITOR: Robert E. Richard, Editor in Chief
PEER REVIEW: Four peer reviewers contributed to the peer review report. Reviewers’ reports totaled 489 words, excluding any confidential comments to the academic editor.
FUNDING: Authors disclose no funding sources.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE).
Author Contributions
Analyzed the data: SK. Wrote the first draft of the manuscript: SK, AS, FI, IO, HA. Contributed to the writing of the manuscript: SK, HO, HA, IO, MAY. Agree with manuscript results and conclusions: SK, AS, FI, HO, HA, IO, MAY. Jointly developed the structure and arguments for the paper: SK, HO, MAY. Made critical revisions and approved final version: SK, AS, FI, HA, HO, IO, MAY. All authors reviewed and approved of the final manuscript.
REFERENCES
- 1.Swerdlow SH, Campo E, Harris NL, et al., editors. WHO Classification of Hematopoietic and Lymphoid Tissue. 4th ed. Lyon, France: IARC; 2008. Acute leukemia of ambiguous lineage; pp. 150–155. [Google Scholar]
- 2.Naghashpour M, Lancet J, Moscinski L, Zhang L. Mixed phenotype acute leukemia with t(11;19)(q23;p13.3)/MLL-MLLT1(ENL), B/T-lymphoid type: a first case report. Am J Hematol. 2010;85(6):451–454. doi: 10.1002/ajh.21703. [DOI] [PubMed] [Google Scholar]
- 3.Weinberger OK, Arber DA. Mixed phenotypic acute leukaemia—historical review and a new definition. Leukemia. 2010;24:1844–1851. doi: 10.1038/leu.2010.202. [DOI] [PubMed] [Google Scholar]
- 4.Matutes E, Pickl WF, Van’t Veer M, et al. Mixed phenotype acute leukemia (MPAL): clinal and laboratory features and outcome in 100 patients defined according to the WHO 2008 classification. Blood. 2011;117(11):3163–3171. doi: 10.1182/blood-2010-10-314682. [DOI] [PubMed] [Google Scholar]
- 5.Rahman K, George S, Tewari A, Mehta A. Mixed phenotype acute leukemia with two distinct blast populations. Cytometry B Clin Cytom. 2013;84B:198–201. doi: 10.1002/cyto.b.21086. [DOI] [PubMed] [Google Scholar]
- 6.Della Porta M, Lanza F, Del Vecchio L, Italian Society of Cytometry (GIC) Flow cytometry immunophenotyping for the evaluation of bone marrow dysplasia. Cytometry B Clin Cytom. 2011;80B:201–211. doi: 10.1002/cyto.b.20607. [DOI] [PubMed] [Google Scholar]
- 7.Haycocks N, Lawrence L, Cain J, Zhao F. Optimizing antibody panels for efficient and cost-effective flow cytometric diagnosis of acute leukemia (pages 221–229) article first published online. Cytometry B Clin Cytom. 2011;80B:221–229. doi: 10.1002/cyto.b.20586. [DOI] [PubMed] [Google Scholar]
- 8.Song S. A case report: concurrent chronic myelomonocytic leukemia and T-cell large granular lymphocytic leukemia-type clonal proliferation as detected by multiparametric flow cytometry. Cytometry B Clin Cytom. 2011;80B:126–129. doi: 10.1002/cyto.b.20565. [DOI] [PubMed] [Google Scholar]
- 9.Arnoulet C, Béné MC, Durrieu F, et al. Four- and five-color flow cytometry analysis of leukocyte differentiation pathways in normal bone. Cytometry B Clin Cytom. 2010;78(1):4–10. doi: 10.1002/cyto.b.20484. [DOI] [PubMed] [Google Scholar]
- 10.Mirro J, Kitchingman GR. Leukaemia Diagnosis 3rd. Barbara J. Bain—2010—medical; 1989. The morphology, cytochemistry, molecular characteristics and clinical significance of acute mixed-lineage leukaemia. [Google Scholar]
- 11.Brunning RD, Matutes E, Borowitz M, et al. Acute leukaemias of ambiguous lineage. In: Jaffe ES, Harris NL, Stein H, Vardiman JW, editors. WHO Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC Press; 2001. pp. 106–107. [Google Scholar]
- 12.Bene MC, Castoldi G, Knapp W, et al. European Group for the Immunological Characterization of Leukemias (EGIL) Proposals for the immunological classification of acute leukemias. Leukemia. 1995;9:1783–1786. [PubMed] [Google Scholar]
- 13.Weir EG, Ansari Lari MA, Batista DAS, et al. Acute bilineal leukaemia. Leukemia. 2007;21:2264–2270. doi: 10.1038/sj.leu.2404848. [DOI] [PubMed] [Google Scholar]
- 14.Wada H, Masuda K, Satch R, et al. Adult T-cell progenitors retain myeloid potential. Nature. 2008;452(7188):768–772. doi: 10.1038/nature06839. [DOI] [PubMed] [Google Scholar]
- 15.Luc S, Buza-Vidas N, Jacobsen SE. Delineating the cellular pathways of haemopoietic lineage commitment. Semin Immunol. 2008;20(4):213–230. doi: 10.1016/j.smim.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 16.Kawamoto H. A close developmental relationship between the lymphoid and myeloid lineages. Trends Immunol. 2006;27:169–175. doi: 10.1016/j.it.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 17.Secker-Walker LM, Craig JM, Hawkins JM. Philadelphia positive acute lymphoblastic leukemia in adults: age distributions, BCR breakpoint and prognostic significance. Leukemia. 1991;5:196–199. [PubMed] [Google Scholar]
- 18.Thomas X, Thiebaut A, Olteanu N, et al. Philadelphia chromosome positive adult acute lymphoblastic leukemia: characteristics, prognostic factors and treatment outcome. Hematol Cell Ther. 1998;40:119–128. [PubMed] [Google Scholar]
- 19.Moorman AV, Harrison CJ, Buck GA, et al. Adult Leukaemia Working Party, Medical Research Council/National Cancer Research Institute Karyotype is an independent prognosis factor in adult acute lymphoblastic leukemia (ALL) analysis of cytogenetic data from patients treated on the medical research council (MRC) UKALLXII/Eastern Cooperative Oncology Group (ECOG) 2993 trial. Blood. 2007;109:3189–3197. doi: 10.1182/blood-2006-10-051912. [DOI] [PubMed] [Google Scholar]
- 20.Yanada M, Takeuchi J, Sugiura I, et al. Japan Adult Leukemia Study Group Karyotype at diagnosis is the major prognostic factor predicting relapse-free survival for patients with Philadelphia chromosome-positive acute lymphoblastic leukemia treated with imatinib-combined chemotherapy. Hematologica. 2008;93:287–290. doi: 10.3324/haematol.11891. [DOI] [PubMed] [Google Scholar]
- 21.Killick S, Matutes E, Powles RL, et al. Outcome of biphenotypic acute leukemia. Haematologica. 1999;84:699–706. [PubMed] [Google Scholar]
- 22.Xu XQ, Wang JM, Lu SQ, et al. Clinical and biological characteristic of adult biphenotypic acute leukaemia in comparison with that of acute myeloid leukaemia and acute lymphoblastic leukaemia—a case series of Chinese population. Hematologica. 2009;94:919–927. doi: 10.3324/haematol.2008.003202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Federl S, Brien O, Pui S, et al. Adult acute lymphoblastic leukemia; concepts and stategies. Cancer. 2010;116(5):1165–1176. doi: 10.1002/cncr.24862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thyagu S, Minden MD, Gupta V, et al. Treatment of phiadelphia chromosmepostive acute lymphoblastic leukemia with imatinib combined with a pediarric based prolocol. Br J Haematol. 2012;158:506–514. doi: 10.1111/j.1365-2141.2012.09182.x. [DOI] [PubMed] [Google Scholar]