

Abstract
Vascular smooth muscle cells (VSMCs) undergo death during atherosclerosis, a widespread cardiovascular disease. Recent studies suggest that oxidative damage occurs in VSMCs and induces atherosclerosis. Here, we analyzed oxidative damage repair in VSMCs and found that VSMCs are hypersensitive to oxidative damage. Further analysis showed that oxidative damage repair in VSMCs is suppressed by a low level of poly (ADP-ribosyl)ation (PARylation), a key post-translational modification in oxidative damage repair. The low level of PARylation is not caused by the lack of PARP-1, the major poly(ADP-ribose) polymerase activated by oxidative damage. Instead, the expression of poly(ADP-ribose) glycohydrolase, PARG, the enzyme hydrolyzing poly(ADP-ribose), is significantly higher in VSMCs than that in the control cells. Using PARG inhibitor to suppress PARG activity facilitates oxidative damage-induced PARylation as well as DNA damage repair. Thus, our study demonstrates a novel molecular mechanism for oxidative damage-induced VSMCs death. This study also identifies the use of PARG inhibitors as a potential treatment for atherosclerosis. [BMB Reports 2015; 48(6): 354-359]
Keywords: DNA damage, Oxidative stress, Poly(ADP-ribosyl)ation, Poly(ADP-ribose) glycohydrolase, Vascular smooth muscle cells
INTRODUCTION
Atherosclerosis, the leading cause of cardiovascular disease, is formerly considered a chronic inflammatory disease. However, increasing evidence suggests that oxidative stress-induced DNA damage induces the apoptosis of VSMCs during the pathogenesis of atherosclerosis (1, 2). For example, the level of 8-oxoG, a DNA adduct from oxidative damage, is significantly higher in VSMCs of the aorta wall (3, 4). However, in response to DNA damage, cells usually activate DNA damage repair systems to repair DNA lesions. Thus, it is unclear why VSMCs are sensitive to oxidative damage.
Oxidative DNA damage is usually induced by reactive oxygen species (ROS) primarily generated from normal intracellular metabolism in mitochondria and peroxisomes. A number of external hazards such as ionizing radiation, chemicals and UVA solar light can also trigger ROS production (5, 6). These active free radicals attack double-stranded DNA, inducing various types of DNA lesions, including DNA single-stand breaks (SSBs) and double-strand breaks (DSBs), which may lead to genomic instability (7, 8). To cope with these threats, cells have evolved DNA damage response systems to detect and repair DNA lesions. As one of the earliest alarm systems and regulators in DNA damage response, poly(ADP-ribose) (PAR) participates in the repair of numerous types of DNA damage including SSBs and DSBs (9, 10). Thus, the cellular metabolism of PAR is critical for DNA damage response and genomic stability. The reaction of poly(ADP-ribosyl)ation (PARylation) is catalyzed by a group of PAR polymerases (PARPs). Using NAD+ as the substrate, PARPs covalently adds ADP-ribose to the side chains of arginine, aspartic acid, and glutamic acid residues in target proteins. After catalyzing the first ADP-ribose onto the proteins, other ADP-riboses can be covalently linked and the continuous reactions produce both linear and branched polymers known as PAR (11, 12). The structure of PAR has been well characterized: the ADP-ribose unit in the polymer is linked by glycosidic ribose-ribose 1’-2’ bonds. The chain length is heterogeneous and can reach around 200 units with 20-50 units in each branch (13). PARylation is regulated not only by PARPs but also by PARG, the major enzyme for hydrolyzing PAR. In response to DNA damage, PARG is recruited to DNA lesions and digest PAR within a few minutes.
Although PARylation has been examined both in vivo and in vitro, the metabolism of PAR in VSMCs remains elusive. In this study, we examined PAR metabolism following oxidative DNA damage in mouse aortic VSMCs (MOVAS), and used mouse embryonic fibroblasts (MEFs) as the control cell line. Similar to MOVAS, MEFs can be used to study DNA damage (14, 15) and originate from mesenchymal stem cells with the ability to differentiate into myocytes (16, 17). With mass spectrometry, we quantitatively measured the level of PAR in MOVAS, and found that that it was relatively low. Our study also suggests that the PARG level in MOVAS is relatively high, which suppresses PARylation following oxidative damage, and thus affect DNA damage repair. Suppression of PARG by the PARG inhibitor facilitates PARylation and DNA damage repair in MOVAS. Thus, PARG inhibitor treatment could be a potential therapeutic approach for arteriosclerosis.
RESULTS AND DISCUSSION
H2O2 induces DNA damage in MOVAS
ROS is one of the most common by-products during metabolism and induces SSBs (18). Under physiological conditions, ROS-induced SSBs can be repaired via the base excision repair pathway (19). However, when two SSBs happen in close proximity, or when the DNA-replication apparatus encounters a SSB, DSBs, the more deleterious genomic lesion, are formed by overwhelming ROS (20, 21). Excessive ROS imposes an oxidative stress condition on vascular cells especially VSMCs, triggering the apoptosis of VSMCs and arteriosclerosis (22, 23). It is well known that ROS can be generated by externally adding H2O2 (24). Thus, to study the oxidative DNA damage in MOVAS, we treated MOVAS with H2O2, and employed alkaline comet assays (25) to detect SSBs and DSBs in the cells. Damaged genomic DNA fragments migrated from nuclei during electrophoresis Fig. 1A). Shorter DNA fragments move faster in electrophoresis, therefore, by measuring the migrated length of DNA fragments, we can quantitatively examine the repair of oxidative damage. To our surprise, we found that the repair in MOVAS was much slower than that in MEFs since much shorter DNA fragments were found in MOVAS especially at 60 minutes (MOVAS: 7.18 ± 0.99, MEFs: 2.68 ± 0.44, P = 0.000) and 120 minutes (MOVAS: 2.87 ± 0.24, MEFs: 0.70 ± 0.16, P = 0.000) following H2O2 treatment (Fig. 1A, B).
Fig. 1. H2O2-induced DSBs activate DNA damage repair in MOVAS. (A) Representative images of alkaline comet assays at 20, 60, and 120 min following H2O2 treatment. Comet tails were found in H2O2 treated cells. (B) Quantification of DNA damage by tail moment of comet assay treated with or without H2O2. Tail moments were measured from three independent experiments with at least 30 cells at single time points per sample. (C) Cells were treated with various concentrations of H2O2, and visualized by immunostaining with anti-γH2AX (red) following 1 h H2O2 treatment. Nuclei were stained with Hoechst 33258 (blue). (D) The number of γH2AX foci in cells was counted (n = 15). The error bars represent the standard deviation, *P < 0.05.
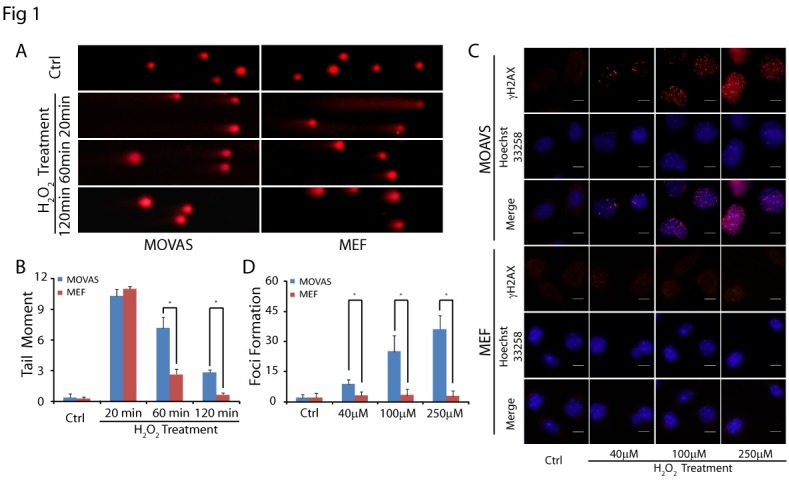
The H2O2-induced DSBs in MOVAS and MEFs were examined by staining the cells with anti-γH2AX antibody. We observed γH2AX nuclear foci in MOVAS, suggesting that DSBs occurred in MOVAS following oxidative damage (the number of foci were 2.00 ± 1.75, 8.86 ± 2.21, 25.03 ± 7.93, 36.13 ± 7.06 per cell from the control group to each H2O2 group). Meanwhile, we found little γH2AX foci in MEFs damage (the number of foci were 2.20 ± 2.10, 3.20 ± 2.05, 3.30 ± 3.06, 2.80 ± 2.77 per cell from the control group to each H2O2 group) (Fig. 1C, D). Thus, these results suggest that oxidative damage-induced DSBs were not repaired in a timely manner in MOVAS unlike in MEFs, which may partly explain the molecular mechanism of VSMC death and arteriosclerosis.
PARylation is suppressed in MOVAS in response to oxidative DNA damage
Next, we asked why MOVAS failed to repair all the oxidative lesions. We examined PARylation, because it is an immediate and dramatic posttranslational modification in response to DNA damage. PAR is a polymer of repeating ADP-ribose units with varying length and branches. Thus, quantitatively measuring PARylation is limited by the heterogeneous format of PAR. To overcome the limitation, we measured the amount of single ADP-ribose using mass spectrometry. We cut phosphate diester bonds within each ADP-ribose and removed the phosphate group by phosphotases to obtain a ribosyladenosine unit from PAR (Fig. 2A). Thus, we were able to accurately measure the amount of ribosyladenosine units from PAR with a molecular weight of 400.146 by Q-TOF mass spectrometry. As seen in Fig. 2B, mass spectrometry peaks from both MOVAS and MEFs showed identical molecular weights of 400.146, corresponding to ribosyladenosine. To compare the relative abundance of ribosyladenosine from MOVAS and MEFs, we used the peak height of ribosyladenosine. We found that H2O2 induced a higher ribosyladenosine peak value in MEFs than in MOVAS [before and after H2O2 treatment ratio of ribosyladenosine peak: MOVAS: 1.61 ± 0.03, MEFs: 3.31 ± 0.05 (Fig. 2B, C)]. This result suggests that the amount of PAR generated in MEFs in response to oxidative DNA damage is greater than in MOVAS. To validate the results, we introduced two other cell lines, mouse aortic endothelial cells (MECs) and skeletal muscle cells (MSCs), and used dot blot assays to determine the amount of PAR in the cells. We found that the amount of PAR in MOVAS was much lower than in the other three cell lines after H2O2 treatment (Fig. 2D). Meanwhile, we investigated PAR synthesis in the wire-injured mice model, which is reported to induce oxidative damage (26, 27). The PAR-positive percentage in MECs was consistently higher than in MOVAS after wire injury (Fig. 2E, F). Thus, a correlation between PAR synthesis and DNA damage repair in VSMCs was observed in both cell and animal experiments, further suggesting that PARylation plays an important role for oxidative DNA damage repair in VSMCs.
Fig. 2. PAR synthesis is suppressed in MOVAS. (A) Structure of PAR. In response to oxidant DNA damage, PARP-1 hydrolyzes NAD+, releases nicotinamide and one proton (H+), and transfers the ADP-ribose moiety (blue) to protein acceptors. Subsequent digestion of PAR with phosphodiesterase (red scissors) and phosphotase (purple scissors) releases ribosyladenosine (green). (B) Ribosyladenosine levels in MOVAS and MEFs were measured by Q-TOF mass spectrometry, and arbitrary units are shown in the histogram (C), the error bars represent the standard deviation, *P<0.05. (D) Following 20 min H2O2 treatment, PAR was purified from MOVAS, MEFs, MECs and MSCs, then examined by dot blot using anti-PAR antibody. β-actin was examined by western blot and used as the loading control. (E) Immunofluorescence staining of PAR in artery tissue (Scale bar = 100 ㎛). (F) The number of PAR positive cells are shown in the histogram. Error bars represent the standard deviation (n = 3), *P<0.05.

High expression of PARG decreases PARylation in MOVAS
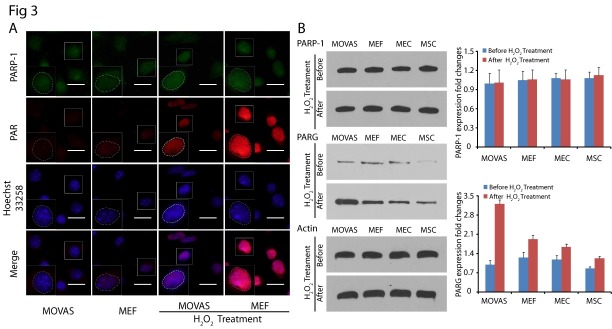
We next examined why the level of PAR in MOVAS was significantly lower than in the other cells. Since DNA damage-induced PAR is mainly synthesized by PARP-1, the founding member in PARPs family (13), we first examined the expression of PARP-1 in MOVAS and MEFs by immunofluorescence staining. We co-stained PAR and PARP-1 in the nuclei of both MOVAS and MEFs. As shown in Fig. 3A, although the level of PAR was significantly higher in MEFs, the expression level of PARP-1 in MOVAS and MEFs did not show significant differences. To further examine the level of PARP-1, we analyzed PARP-1 in the above four cell lines by western blot (Fig. 3B). Again, we did not observe significant differences in PARP-1’s expression in the control and H2O2 treated cells. PARP-1 is the most important enzyme involved in oxidative DNA repair (28), which normally occurs in the cell nucleus. When DNA damage occurs, PARP-1 is activated and recruited to the damage sites, but the amount does not increase (29).Besides PARP-1, PARG, the enzyme that regulates PAR degradation, also plays an important role in PAR metabolism. To characterize PARG, we examined the expression of PARG following H2O2 treatment. Under physiological conditions, the expression level of PARG was relatively low, and there was no significant difference in PARG expression in the MOVAS, MEFs, MECs and MSCs. However, upon treatment with H2O2, we found that the expression of PARG increased in varying degrees in different cells, and oxidative stress induced higher expression of PARG in MOVAS (Fig. 3B). Thus, it is likely that the high level of PARG expression in MOVAS is responsible for the low level of PAR in response to DNA damage.
Fig. 3. High expression of PARG level promotes PAR degradation in MOVAS. (A) Representative immunostaining images of PARP-1 (green) and PAR (red) in H2O2 treated MOVAS and MEFs cells at 20 min. Nuclei were stained with Hoechst 33258 (blue), and outlined by white dashed lines in enlarged boxes to denote the localization of PARP-1 and PAR in nuclei. (B) The expression of PARP-1 and PARG in cells were examined at 1 h following H2O2 treatment, β-actin was used as the protein loading control. The error bars represent the standard deviation.
PARG inhibitor rescues MOVAS from oxidative stress-induced cell death
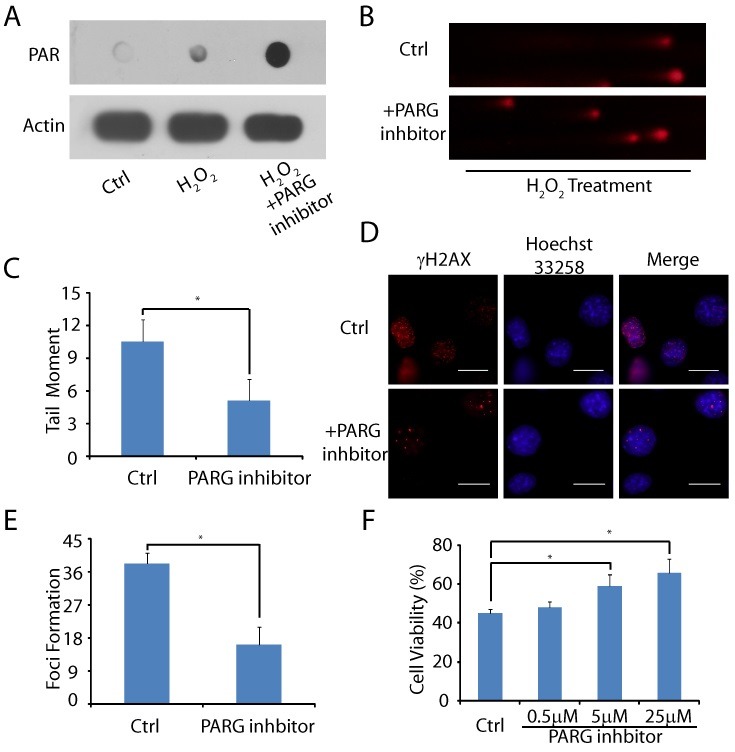
To further examine the role of PARG, we treated the cells with the PARG inhibitor GLTN. In the presence of the PARG inhibitor, the amount of PAR increased significantly in MOVAS following H2O2 treatment (Fig. 4A). Thus, we could use PARG inhibitors to suppress PARG-dependent PAR degradation in MOVAS. Next, we wondered whether the PAR-dependent DNA damage repair is enhanced in the presence of the PARG inhibitor. As seen in Fig. 4B, C, the PARG inhibitor significantly reduced oxidative stress-induced DNA damage in MOVAS as the number of short DNA fragments was significantly reduced during the 30 min recovery following H2O2 treatment (Control group: 11.03 ± 1.98, inhibitor group: 5.11 ± 1.91, P = 0.000). Moreover, the number of oxidative stress-induced DSBs were lower with the PARG inhibitor treatment as the number of γH2AX foci was remarkably reduced (Control group: 38.20 ± 2.77, inhibitor group: 16.20 ± 5.16, P = 0.000) (Fig. 4D, E). In addition, accumulative evidence has proven that oxidative stress induces cell death (30), which appears to be a major molecular pathogenesis of atherosclerosis (31). Thus, we wondered whether inhibition of PARG could suppress MOVAS death from oxidative stress. As shown in Fig. 4F, when MOVAS were incubated with H2O2 for 8 hours, 55.4 ± 2.2% of the cells underwent death as detected by the MTT assay. However, when the cells were pretreated with 0.5, 5, and 25 ㎛ PARG inhibitor (GLTN) for 1 hour, the percentage of cell death decreased to 52.2 ± 3.0% (P = 0.380), 41.6 ± 5.9% (P = 0.041), and 34.0 ± 7.1% (P = 0.000) respectively, suggesting that the inhibition of PARG effectively rescues the cells from oxidative stress-induced death.
Fig. 4. PARG inhibitor reduced oxidative DNA damage level and cell death. (A) Following PARG treatment, PAR synthesis in MOVAS was examined by dot blot. (B, C) MOVAS were pre-treated with or without PARG inhibitor followed by H2O2. DNA breaks were examined at 20 min following H2O2 treatment by alkaline comet assays. Tail moment was measured. (D, E) MOVAS were pre-treated with or without PARG inhibitor. H2O2-induced DSBs were examined and the foci of γH2AX in each cell were counted (n = 15). (F) MOVAS were pre-treated with or without PARG inhibitor and then exposed to 100 ㎛ H2O2. Cell viability was evaluated using an MTT assay. The error bars represent the standard deviation, *P < 0.05.
Normally PARPs and PARG maintain the balance of PARylation. However, during pathological events such as excessive and unrepairable oxidant DNA damage, highly activated PARPs synthesize large amounts of PAR in a few seconds. The PAR synthesis also triggers rapid PAR degradation with the over-activated PARG within a few minutes following DNA damage. This vicious cycle not only impairs DNA damage repair but also leads to intracellular ATP depletion and NAD (32, 33). Our results show that VSMCs death may result from excessive PARG expression to impair PAR-dependence DNA damage repair. In recent years, PARP-1 inhibitors have shown promising therapeutic effects on atherosclerosis, and this effect is mainly associated with the maintenance of cellular NAD and ATP level (33, 34). Similarly, suppression of PARG was likely to protect cells from oxidative DNA damage and facilitated cell survival in our results. In addition, PARG inhibitor protection is supported in the ischemic animal model (35) and in cells (36). Here, we demonstrate that PARG inhibitor treatment contributes to genomic stability and anti-oxidative stress injury, and positively regulates the viability of VSMCs, which reveals a potential therapeutic strategy for arteriosclerosis.
In conclusion, our study suggests that a high level of endogenous PARG in VSMCs impairs oxidative DNA damage repair, inducing the death of VSMCs during atherosclerosis. PARG inhibitor treatment suppresses PARG activity and facilitates DNA damage repair in VSMCs by prolonging PARylation. Thus, PARG acts as a mediator of cardiovascular disease, and PARylation may play a pivotal protective role as an antioxidant defense mechanism by maintaining VSMCs function under pathologic conditions of oxidative stress. Hence, PARG inhibitors may provide a novel clinical treatment for suppressing arteriosclerosis caused by oxidative damage.
MATERIALS AND METHODS
Materials and Methods are described in the online data supplement, available at http://www.bmbreports.org/.
Acknowledgments
This work was supported by the National Natural Science Foundation of PR China Grant (81470587 to T.L.), and the National Institute of Health of the United States (CA132755, CA130899 and CA187209 to X.Y.).
References
- 1.Ragnauth CD, Warren DT, Liu Y, et al. (Prelamin A acts to accelerate smooth muscle cell senescence and is a novel biomarker of human vascular aging. Circulation. (2010);121:2200–2210. doi: 10.1161/CIRCULATIONAHA.109.902056. [DOI] [PubMed] [Google Scholar]
- 2.Gray K, Bennett M. Role of DNA damage in atherosclerosis--bystander or participant? Biochem Pharmacol. (2011);82:693–700. doi: 10.1016/j.bcp.2011.06.025. [DOI] [PubMed] [Google Scholar]
- 3.Martinet W, Knaapen MW, De Meyer GR, Herman AG, Kockx MM. Oxidative DNA damage and repair in experimental atherosclerosis are reversed by dietary lipid lowering. Circ Res. (2001);88:733–739. doi: 10.1161/hh0701.088684. [DOI] [PubMed] [Google Scholar]
- 4.Mahmoudi M, Gorenne I, Mercer J, Figg N, Littlewood T, Bennett M. Statins use a novel Nijmegen breakage syndrome-1-dependent pathway to accelerate DNA repair in vascular smooth muscle cells. Circ Res. (2008);103:717–725. doi: 10.1161/CIRCRESAHA.108.182899. [DOI] [PubMed] [Google Scholar]
- 5.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. (2000);408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 6.Cadet J, Douki T, Ravanat J-L. Oxidatively generated base damage to cellular DNA. Free Radical Biol Med. (2010);49:9–21. doi: 10.1016/j.freeradbiomed.2010.03.025. [DOI] [PubMed] [Google Scholar]
- 7.Olinski R, Gackowski D, Foksinski M, Rozalski R, Roszkowski K, Jaruga P. Oxidative DNA damage: assessment of the role in carcinogenesis, atherosclerosis, and acquired immunodeficiency syndrome. Free Radical Biol Med. (2002);33:192–200. doi: 10.1016/S0891-5849(02)00878-X. [DOI] [PubMed] [Google Scholar]
- 8.Sedelnikova OA, Redon CE, Dickey JS, Nakamura AJ, Georgakilas AG, Bonner WM. Role of oxidatively induced DNA lesions in human pathogenesis. Mutat Res-Rev Mutat. (2010);704:152–159. doi: 10.1016/j.mrrev.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li M, Yu X. Function of BRCA1 in the DNA Damage Response Is Mediated by ADP-Ribosylation. Cancer Cell. (2013);23:693–704. doi: 10.1016/j.ccr.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li M, Lu LY, Yang CY, Wang S, Yu X. The FHA and BRCT domains recognize ADP-ribosylation during DNA damage response. Genes Dev. (2013);27:1752–1768. doi: 10.1101/gad.226357.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol. (2006);7:517–528. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- 12.Gibson BA, Kraus WL. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat Rev Mol Cell Bio. (2012);13:411–424. doi: 10.1038/nrm3376. [DOI] [PubMed] [Google Scholar]
- 13.Kim MY, Zhang T, Kraus WL. Poly(ADP-ribosyl)ation by PARP-1: 'PAR-laying' NAD+ into a nuclear signal. Genes Dev. (2005);19:1951–1967. doi: 10.1101/gad.1331805. [DOI] [PubMed] [Google Scholar]
- 14.de Murcia JM, Niedergang C, Trucco C, et al. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc Natl Acad Sci U S A. (1997);94:7303–7307. doi: 10.1073/pnas.94.14.7303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Celeste A, Fernandez-Capetillo O, Kruhlak MJ, et al. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat Cell Biol. (2003);5:675–679. doi: 10.1038/ncb1004. [DOI] [PubMed] [Google Scholar]
- 16.Beyer Nardi N, da Silva M eirelles L. Mesenchymal stem cells: isolation, in vitro expansion and characterization. Handb Exp Pharmacol. (2006):249–282. [PubMed] [Google Scholar]
- 17.Shen H, McElhinny AS, Cao Y, et al. The Notch coactivator, MAML1, functions as a novel coactivator for MEF2C-mediated transcription and is required for normal myogenesis. Genes Dev. (2006);20:675–688. doi: 10.1101/gad.1383706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Asad NR, Asad LMBO, Almeida CEBd, Felzenszwalb I, Cabral-Neto JB, Leitão AC. Several pathways of hydrogen peroxide action that damage the E. coli genome. Genet Mol Biol. (2004);27:291–303. doi: 10.1590/S1415-47572004000200026. [DOI] [Google Scholar]
- 19.Hegde ML, Mantha AK, Hazra TK, Bhakat KK, Mitra S, Szczesny B. Oxidative genome damage and its repair: implications in aging and neurodegenerative diseases. Mech Ageing Dev. (2012);133:157–168. doi: 10.1016/j.mad.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hegde ML, Hazra TK, Mitra S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. (2008);18:27–47. doi: 10.1038/cr.2008.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.D'Errico M, Parlanti E, Dogliotti E. Mechanism of oxidative DNA damage repair and relevance to human pathology. Mutat Res. (2008);659:4–14. doi: 10.1016/j.mrrev.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 22.Herbert KE, Mistry Y, Hastings R, Poolman T, Niklason L, Williams B. Angiotensin II-mediated oxidative DNA damage accelerates cellular senescence in cultured human vascular smooth muscle cells via telomere-dependent and independent pathways. Circ Res. (2008);102:201–208. doi: 10.1161/CIRCRESAHA.107.158626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang JC, Bennett M. Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ Res. (2012);111:245–259. doi: 10.1161/CIRCRESAHA.111.261388. [DOI] [PubMed] [Google Scholar]
- 24.Matsura T, Kai M, Fujii Y, Ito H, Yamada K. Hydrogen peroxide-induced apoptosis in HL-60 cells requires caspase-3 activation. Free Radic Res. (1999);30:73–83. doi: 10.1080/10715769900300081. [DOI] [PubMed] [Google Scholar]
- 25.Olive PL, Banath JP. The comet assay: a method to measure DNA damage in individual cells. Nat Protoc. (2006);1:23–29. doi: 10.1038/nprot.2006.5. [DOI] [PubMed] [Google Scholar]
- 26.Zhang C, Yang J, Jennings LK. Attenuation of neointima formation through the inhibition of DNA repair enzyme PARP-1 in balloon-injured rat carotid artery. Am J Physiol Heart Circ Physiol. (2004);287:H659–666. doi: 10.1152/ajpheart.00162.2004. [DOI] [PubMed] [Google Scholar]
- 27.Forte A, Finicelli M, Grossi M, et al. DNA damage and repair in a model of rat vascular injury. Clin Sci (Lond) 2009 doi: 10.1042/CS20090416. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 28.Burkle A. Physiology and pathophysiology of poly(ADP-ribosyl)ation. Bioessays. (2001);23:795–806. doi: 10.1002/bies.1115. [DOI] [PubMed] [Google Scholar]
- 29.El-Khamisy SF, Masutani M, Suzuki H, Caldecott KW. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. (2003);31:5526–5533. doi: 10.1093/nar/gkg761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jacobson MD. Reactive oxygen species and programmed cell death. Trends Biochem Sci. (1996);21:83–86. doi: 10.1016/S0968-0004(96)20008-8. [DOI] [PubMed] [Google Scholar]
- 31.Martinet W, Knaapen MW, De Meyer GR, Herman AG, Kockx MM. Elevated levels of oxidative DNA damage and DNA repair enzymes in human atherosclerotic plaques. Circulation. (2002);106:927–932. doi: 10.1161/01.CIR.0000026393.47805.21. [DOI] [PubMed] [Google Scholar]
- 32.Hassa PO, Hottiger MO. The diverse biological roles of mammalian PARPS, a small but powerful family of poly-ADP-ribose polymerases. Front Biosci. (2008);13:3046–3082. doi: 10.2741/2909. [DOI] [PubMed] [Google Scholar]
- 33.Zhang H, Xiong ZM, Cao K. Mechanisms controlling the smooth muscle cell death in progeria via down-regulation of poly(ADP-ribose) polymerase 1. Proc Natl Acad Sci U S A. (2014);111:E2261–2270. doi: 10.1073/pnas.1320843111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sodhi RK, Singh N, Jaggi AS. Poly(ADP-ribose) polymerase-1 (PARP-1) and its therapeutic implications. Vascul Pharmacol. (2010);53:77–87. doi: 10.1016/j.vph.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 35.Lu XC, Massuda E, Lin Q, Li W, Li JH, Zhang J. Post-treatment with a novel PARG inhibitor reduces infarct in cerebral ischemia in the rat. Brain Res. (2003);978:99–103. doi: 10.1016/S0006-8993(03)02774-4. [DOI] [PubMed] [Google Scholar]
- 36.Blenn C, Althaus FR, Malanga M. glycohydrolase silencing protects against H2O2- induced cell death. Biochem J. (2006);396:419–429. doi: 10.1042/BJ20051696. [DOI] [PMC free article] [PubMed] [Google Scholar]