

SUMMARY
Retinoic acid- (RA-) dependent homeostatic plasticity and NMDA-receptor-dependent LTP, a form of Hebbian plasticity, both enhance synaptic strength by increasing the abundance of postsynaptic AMPA receptors (AMPARs). However, it is unclear whether the molecular mechanisms mediating AMPAR-trafficking during homeostatic and Hebbian plasticity differ, and unknown how RA-signaling impacts Hebbian plasticity. Here, we show that RA increases postsynaptic AMPAR-abundance by an activity-dependent mechanism that requires a unique SNARE-dependent fusion machinery different from that mediating LTP. Specifically, RA-induced AMPAR-trafficking did not involve complexin, which activates SNARE complexes containing syntaxin-1 or -3 but not complexes containing syntaxin-4, whereas LTP required complexin. Moreover, RA-induced AMPAR trafficking utilized the Q-SNARE syntaxin-4 whereas LTP utilized syntaxin-3; both additionally required the Q-SNARE SNAP-47 and the R-SNARE synatobrevin-2. Finally, acute RA treatment blocked subsequent LTP expression, probably by increasing AMPAR-trafficking. Thus, RA-induced homeostatic plasticity involves a novel, activity-dependent postsynaptic AMPAR-trafficking pathway mediated by a unique SNARE-dependent fusion machinery.
INTRODUCTION
The ability of a neuron to change its responsiveness to synaptic inputs based on prior activity and experience is an essential feature of the nervous system. These changes can occur in the properties intrinsic to a neuron (i.e. membrane excitability) or at synapses where communication between two neurons is achieved. Long-term plasticity at synapses is thought to underlie modification of neural circuits that drive behaviors, enable learning, and encode memory. The known forms of long-term synaptic plasticity include at least two main categories: Hebbian and homeostatic. Compared to Hebbian plasticity, the mechanisms and functional significance of homeostatic synaptic plasticity are less understood. One of the main open questions is whether and how homeostatic synaptic plasticity intersects with Hebbian synaptic plasticity, at the functional and molecular levels. Although operating under different computational rules and likely involving distinct molecular mechanisms, homeostatic synaptic plasticity directly impacts the basal state of synapses, and thus is likely to indirectly affect Hebbian plasticity.
We have previously described a critical involvement of retinoic acid (RA) in a form of homeostatic synaptic plasticity that is induced by prolonged reduction in synaptic excitation (Aoto et al., 2008; Chen et al., 2014; Sarti et al., 2013). Acting through a distinct molecular mechanism, RA is capable of rapidly changing excitatory as well as inhibitory synaptic strength (Aoto et al., 2008; Sarti et al., 2013). Thus, through its effects on both excitation and inhibition, the synaptic action of RA may impact Hebbian plasticity due to an altered synaptic excitation/inhibition balance. Indeed, vitamin A deficiency (which depletes RA) impairs hippocampal Hebbian plasticity and learning (Chiang et al., 1998; Cocco et al., 2002; Misner et al., 2001). Moreover, a study using a dominant negative form of RARα expressed in adult forebrain demonstrated impairments in AMPAR-mediated synaptic transmission, hippocampal LTP, hippocampal-dependent social recognition, and spatial memory (Nomoto et al., 2012). However, how RA-induced increases in excitatory synaptic transmission affect Hebbian plasticity has not been investigated.
At the molecular level, changes in synaptic AMPAR abundance have been described for both RA-induced and Hebbian plasticity (e.g. postsynaptic NMDAR-dependent LTP). The molecular machinery that governs AMPAR exocytosis is just beginning to be uncovered. Similar to presynaptic activity-dependent vesicle exocytosis (i.e. calcium-triggered neurotransmitter release)(Sudhof and Rothman, 2009), postsynaptic AMPAR exocytosis during LTP requires a SNARE-dependent membrane-fusion machinery (Ahmad et al., 2012; Jurado et al., 2013). Specifically, both processes require the R-SNARE synaptobrevin-2 (Syb-2) and the SNARE-activating molecule complexin, but differ in the Q-SNAREs. Whereas calcium-triggered synaptic vesicle exocytosis requires the Q-SNAREs SNAP-25 and Syntaxin-1, LTP-induced AMPAR exocytosis requires SNAP-47 and Syntaxin-3 (Stx-3)(Jurado et al., 2013). The question thus arises whether the complex formed by Syb-2, Stx-3, SNAP-47 and complexin represents a SNARE machinery universally used for all forms of postsynaptic activity-dependent AMPAR exocytosis, and whether the similarity and/or differences in the molecular composition of the AMPAR exocytosis machinery between Hebbian and RA-induced plasticity account for a possible functional impact of synaptic RA signaling on Hebbian plasticity.
In the present study, we examined the impact of RA on Hebbian plasticity in the hippocampus. We found that acute RA treatment impairs subsequent expression of LTP, and that this impairment can be reversed by acute genetic deletion of RARα or by inhibiting protein synthesis during RA treatment. Unexpectedly, we found that similar to what occurs during LTP, synaptic incorporation of AMPARs by RA requires synaptic activity and NMDAR activation. We thus examined the potential convergence of the molecular mechanisms underlying the activity-dependent AMPAR insertion between LTP and RA-induced AMPAR exocytosis. Using an shRNA-based approach, we found that RA- and LTP-induced AMPAR insertion are both regulated by activity and both require some of the same SNARE proteins SNAP-47 and Syb-2, but differ in two important components of the SNARE membrane-fusion machinery. First, although complexin is required for LTP, it does not play a role in RA-induced AMPAR exocytosis. Second, whereas Stx-3 (which interacts with complexin) is required for LTP, Stx-4 (which does not bind complexin) is required for RA-induced AMPAR exocytosis. Thus, our findings reveal a previously unidentified, activity-dependent AMPAR exocytosis pathway, which utilizes a unique vesicle fusion machinery that is distinct from that used during LTP or during constitutive AMPAR exocytosis at synapses.
RESULTS
Synaptic RA signaling blocks LTP expression
To directly investigate the impact of synaptic RA signaling on hippocampal LTP, we examined LTP induction at Schaffer collateral-CA1 synapses after acute RA treatment (2–4 hours) in organotypic cultured hippocampal slices. Using a classical pairing protocol, we reliably induced LTP in DMSO-treated slices, but not in RA-treated slices (Figures 1A and S1A: DMSO, 269.91% ± 31.57%; RA, 113.37% ± 7.68%; measured 55–60 min after induction). Given the previously established role of RARα in synaptic RA signaling (Aoto et al., 2008; Poon and Chen, 2008), we asked whether this RA blockade of LTP is also mediated by RARα. We injected lentiviruses expressing either wild type (active) or mutant (inactive) Cre recombinase (Cre or mCre, respectively) into the CA1 pyramidal cell layer of cultured slices obtained from conditional RARα KO mice (Chapellier et al., 2002; Sarti et al., 2012), thus selectively deleting RARα in postsynaptic neurons. Deletion of RARα did not impair LTP (Figures 1B and S1B: mCre/DMSO, 308.77% ± 27.22%; Cre/DMSO, 261.34 ± 24.2%), consistent with previous observations that RA/RARα signaling is specifically involved in homeostatic synaptic plasticity but not LTP (Aoto et al., 2008; Sarti et al., 2012). RA treatment in mCre-infected neurons, similar to uninfected neurons, greatly diminished LTP (Figures 1B and S1B: mCre/RA, 152.22% ± 27.22%), whereas deletion of RARα rescued the ability of neurons to undergo LTP following RA treatment (Figures 1B and S1B: Cre/RA, 322.67% ± 73.21%). Thus, although RA/RARα signaling is not required for LTP, RA’s action on excitatory synapses prevents subsequent induction of LTP, and this effect of RA operates by an RARα-mediated signaling pathway.
Figure 1. Acute RA treatment impairs hippocampal LTP.

(A) Example traces (left) and summary graph (right) of LTP in DMSO- or RA-treated hippocampal CA1 pyramidal neurons from cultured slices (p < 0.0001). (B) Example traces and summary graph of LTP in CA1 pyramidal neurons with RARα deletion. Lentivirus expressing wild-type or mutant inactive Cre recombinase (Cre or mCre) were injected into the CA1 regions of the hippocampal slices obtained from RARα conditional KO mice. LTP was examined 7–10 days after viral injection (mCre/DMSO vs. mCre/RA: p < 0.005; Cre/DMSO vs. Cre/RA: p > 0.5; mCre/RA vs. Cre/RA: p < 0.0001). (C) Example traces and summary graph of LTP in CA1 pyramidal neurons treated with anisomycin, RA or both (RA vs. aniso: p < 0.005; aniso vs. RA+aniso: p > 0.9). Scale bars in(A)–(C): 20 pA, 20 ms. Black bars in all summary graphs indicate the time window for LTP magnitude quantification. All graphs represent average values ± s.e.m.
RA is traditionally considered a key transcription factor during development (Maden, 2007), but also controls synaptic strength by regulating local protein synthesis in neuronal dendrites (Maghsoodi et al., 2008; Poon and Chen, 2008). To test whether RA blocks LTP induction through an action that also requires protein synthesis, we examined the effect of a protein synthesis blocker, anisomycin, on LTP. Pretreating slices with anisomycin for two hours (which blocks the effect of RA on synaptic strength (Aoto et al., 2008)) rescued LTP in RA-treated slices (Figures 1C and S1C: RA, 129.52% ± 12.62%; aniso+RA, 217.22% ± 21.42%), confirming the hypothesis that RA blocks LTP via a protein synthesis-dependent process. Anisomycin treatment, however, did not affect LTP under control conditions, demonstrating that its effect is specific to the RA condition (Figures 1C and S1C: aniso, 225.30% ± 21.99%, measured 55–60 mins after induction). To further control for potential non-specific effects of anisomycin, we repeated the same series of experiments with another protein synthesis inhibitor, cycloheximide. Similar to anisomycin, cycloheximde co-treated with RA reversed the effect of RA on LTP (Figures S1D and S1E: RA, 110.70% ± 7.21%; cyclo, 242.53% ± 23.12%; cyclo+RA, 198.75% ± 17.50%).
RA-induced AMPAR insertion is activity-dependent
One interpretation for the impaired LTP after RA treatment is that RA increases excitatory synaptic strength and thereby prevents the subsequent potentiation of synapses evoked by LTP-induction protocols. We thus wanted to first confirm that RA indeed increased synaptic excitation under our experimental conditions. Consistent with previous results (Aoto et al., 2008), RA increased the amplitude, but not the frequency, of miniature excitatory postsynaptic currents (mEPSCs) from hippocampal CA1 neurons (Figure 2A: DMSO, 12.85 ± 0.33 pA, 0.78 ± 0.12 Hz; RA, 15.10 ± 0.48 pA, 0.70 ±0.09 Hz). We previously reported that RA potentiates the mEPSC amplitude of primary cultured neurons in a multiplicative fashion (Aoto et al., 2008), similar to synaptic scaling in response to activity blockade (Turrigiano et al., 1998). We wondered whether RA-induced potentiation of mEPSC in organotypic slices follows the same pattern. When ranked mEPSC amplitudes of RA-treated responses were plotted against those of DMSO-treated, we noticed that for more than 95% of mEPSCs that exhibit mEPSC amplitudes of < 30pA, the increase in mEPSC amplitude appears to be strictly multiplicative (i.e. scaled, Figure S2A1). However, at large mEPSC amplitudes (30–60 pA, less than 5% of total population), both supralinear and sublinear potentiation was observed (Figure S2A2). Thus, RA-mediated potentiation follows primarily a multiplicative pattern for the majority of mEPSCs. The complexity of the potentiation pattern associated with a small subset of responses at higher amplitudes may reflect RA’s heterogeneous action at a subset of synapses. We speculate that the differences in pathway/input type and the states of the synapses, which are better preserved in organotypic slices, may underlie such complexity.
Figure 2. RA increases AMPAR-mediated excitatory synaptic transmission through an activity-dependent mechanism.

(A) Trace examples (left), amplitude (middle) and frequency (right) quantification of mEPSCs recordings from DMSO- or RA-treated CA1 pyramidal neurons (***, p < 0.0005). Scale bars: 10 pA, 1s. (B) Dual component mEPSC recordings in CA1 pyramidal neurons treated with DMSO or RA. Left: traces examples of AMPA only and dual component mEPSCs. Right: quantification of AMPA and NMDA mEPSC amplitude (*, p < 0.05). NMDA mEPSC component was calculated by subtracting the average AMPA mEPSC component from the average dual mEPSC component. Scale bars: 4 pA, 10 ms. (C) Ratio of AMPAR- to NMDAR-mediated EPSCs in CA1 pyramidal neurons treated with DMSO or RA (*, p < 0.05). Representative EPSCs recorded at −60 mV and +40 mV are shown to the left. Scale bars: 50 pA, 20 ms. (D) Trace examples (left) and quantification of mEPSC amplitude and frequency obtained from CA1 pyramidal neurons treated with four hours of DMSO, RA, RA+TTX, or RA+APV (***, p < 0.0001). Scale bars: 10 pA, 1 s. All graphs represent average values ± s.e.m.
To test whether RA enhances both AMPAR-mediated and NMDAR-mediated components of excitatory synaptic transmission, we recorded dual component mEPSCs in the absence of external Mg2+ (Arendt et al., 2013). RA treatment did not result in any significant increase in the NMDAR-mediated component of mEPSCs (DMSO: 2.03 ± 0.22 pA; RA: 2.38 ± 0.20 pA), despite the persistent increase in the AMPAR-mediated component as measured in this analysis (DMSO: 10.56 ± 0.39 pA; RA: 12.33 ± 0.59pA) (Figure 2B). Additionally, we examined the AMPAR/NMDAR response ratio of evoked EPSCs (eEPSCs), and found that RA significantly increased the AMPAR/NMDAR ratio (Figure 2C: DMSO, 1.12 ± 0.12; RA: 1.63 ± 0.17). We did not observe any effect of RA on the paired-pulse ratio or passive membrane properties (Figures S2B and S2C). Thus RA selectively enhances AMPAR-mediated synaptic responses due to specific promotion of AMPAR insertion (Aoto et al., 2008).
AMPAR insertion during LTP requires activity and NMDAR activation (Collingridge et al., 1983; Huganir and Nicoll, 2013). We next asked whether the RA-mediated increase in the synaptic content of AMPARs also required activity and NMDAR activation. During acute RA treatment, we co-applied hippocampal slices with the voltage-gated sodium channel blocker tetrodotoxin (TTX) to block neuronal activity. Surprisingly, TTX co-applied with RA (< 4 hr) blocked the enhancement of the mEPSC amplitude by RA (Figure 2D: DMSO, 10.58 ± 0.30 pA; RA, 12.65 ± 0.24 pA; RA+TTX, 11.24 ± 0.31 pA). Note that this protocol differs from the TTX-treatment used to induce homeostatic plasticity which involves a chronic pretreatment of neurons, not an acute addition of TTX. To address whether NMDAR activity was also required for AMPAR insertion, in a manner similar to LTP, we co-applied APV acutely with RA (< 4 hr) to specifically block NMDAR activation, and found that this treatment also blocked the increase in mEPSC amplitude by RA (Figure 2D: DMSO, 11.17 ± 0.28 pA; RA, 13.81 ± 0.51 pA; RA+APV, 11.31 ± 0.35 pA). Thus, similar to LTP-induced postsynaptic AMPAR insertion, the RA-mediated increase in post-synaptic AMPARs also requires NMDAR activation. In other words, RA only enhances the strength of actively used synapses, possibly as a mechanism to ensure that the RA-mediated AMPAR insertion does not occur at synapses which are not being used.
RA activates silent synapses by promoting AMPAR insertion
LTP induction is known to not only increase the abundance of AMPARs at synapses that already contain AMPARs, but also to activate “silent” synapse lacking AMPARs (but containing NMDARs) by inserting AMPARs into these synapses (Isaac et al., 1995; Liao et al., 1995). Given the similar requirement of NMDAR activation for both LTP-induced and RA-induced AMPAR insertion at synapses, we were curious whether RA is capable of activating silent synapses by promoting insertion of AMPARs into NMDAR-only synapses. To induce formation of silent synapses, we applied a protocol used previously in cultured hippocampal slices - prolonged silencing of neural network activity with TTX for 60 hrs - that reliably induces formation of silent synapses containing only NMDARs (Arendt et al., 2013) (Figure 3A). To detect silent synapses, cells were clamped at their resting membrane potential (~−60mV), and excitatory synaptic transmission was elicited with a weak stimulus that produced failures in about 50% of trials. Epochs of 50 trials of transmission were recorded at −60 mV and +40 mV for each cell, and the failure rate at each of these two holding potentials was computed.
Figure 3. RA treatment activates postsynaptic silent synapses by promoting AMPAR insertion into the synaptic membrane.

(A) Schematic diagrams of various treatment protocols used in the experiments for this figure. (B–D) Trace examples and scatter plots of eEPSCs recorded from CA1 pyramidal neurons treated with DMSO (B), TTX (60 hours) (C), and RA (4 hr) (D) at −60 mV and +40 mV. Scale bars: 10 pA, 10 ms. (E) Failure rate of eEPSCs recorded at −60 mV and +40 mV from DMSO-, TTX- and RA- treated neurons (***, p < 1 x 10−9). (F) Failure rate of eEPSCs recorded at −60 mV and +40 mV from neurons treated with TTX wash/RA, TTX no wash/RA, TTX wash, and TTX wash/RA+APV (***, p < 0.0005). (G) Top: example traces of mEPSC recordings. Bottom: quantification of amplitude and frequency of mEPSCs recorded from CA1 pyramidal neurons treated with DMSO, TTX and TTX wash/RA (**, p < 0.01; ***, p < 0.001). Scale bars: 15 pA, 1s. (H) Top: example traces of mEPSC recordings. Bottom: quantification of amplitude and frequency of mEPSCs recorded from CA1 pyramidal neurons treated with DMSO, TTX and TTX no wash/RA (**, p < 0.01; ***, p < 1 x 10−6). Scale bars: 15 pA, 1s. All graphs represent average values ± s.e.m.
In slices cultured under control conditions, the failure rate was comparable between the negative and the positive holding potentials (Figures 3B and 3E: −60 mV: 55.73 ± 2.59 %, + 40 mV: 53.82 ± 3.83 %), indicating that most synapses are active and contained both AMPARs and NMDARs at this stage of development. Consistent with previous work (Arendt et al., 2013), prolonged TTX treatment induced formation of silent synapses, indicated by the significantly higher failure rate at −60 mV than at +40 mV holding potential (Figures 3C and 3E: −60 mV: 49.29 ± 2.28 %, + 40 mV: 20.61 ± 2.74 %), which suggests that a larger proportion of synapses contain only NMDARs but not AMPARs. Importantly, acute RA treatment per se did not induce silent synapses (Figures 3D and 3E: −60 mV: 49.18 ± 3.76 %, + 40 mV: 48.57 ± 5.10 %).
We then applied RA to slices that had been treated with TTX for 60 hours. Washing out TTX during RA treatment to restore network activity converted silent synapses to active synapses, as evidenced by the similar failure rates at −60 mV and +40 mV (Figure 3F:TTX 60 hr wash + RA: −60 mV,50.05 ± 2.38 %, + 40 mV: 48.82 ± 3.71 %). This conversion of silent to functional synapses by RA requires intact network activity as well as NMDAR activation because the presence of TTX or APV during RA treatment prevented the activation of silent synapses (Figure 3F: TTX 60 hr no wash + RA: −60 mV,49.16 ± 4.12 %, + 40 mV: 26.76 ± 3.25 %; TTX 60 hr wash + RA + APV: −60 mV,52.80 ± 4.30 %, + 40 mV: 28.51 ± 4.87 %). Importantly, simply washing out TTX without RA treatment was not sufficient to convert silent synapses into active synapses within the four-hour time window used for our experiments (Figure 3F:TTX 60 hr wash: −60 mV,54.22 ± 4.00 %, + 40 mV: 31.68 ± 3.60 %).
In addition to evoked EPSCs, we also examined mEPSCs during silent synapse activation. As expected, homeostatic synaptic plasticity induced by prolonged TTX treatment manifested as an increase in mEPSC amplitude (Figure 3G: DMSO, 11.58 ± 0.27pA; TTX 60 hr, 14.06 ± 0.62pA) without changes in mEPSC frequency (Figure 3G: DMSO, 0.43 ± 0.05 Hz; TTX 60 hr, 0.37 ± 0.05 Hz)(Turrigiano et al., 1998). By contrast, following prolonged TTX treatment, incubation with RA (in the absence of TTX) activated silent synapses as evidenced by a significant increase in the mEPSC frequency (Figure 3G: TTX 60 hr wash + RA: 0.66 ± 0.07 Hz). Interestingly, RA treatment after prolonged TTX treatment also decreased the mEPSC amplitude (Figure 3G: TTX 60 hr wash + RA: 12.06 ± 0.54 pA), presumably because newly activated silent synapses have smaller mEPSC amplitudes. We thus plotted ranked amplitudes of the three groups (Figure S3A). Indeed, while prolonged TTX treatment induced synaptic scaling, responses recorded in neurons that received RA after TTX treatments displayed a large population of small amplitudes that are not apparently potentiated, likely representing the population of newly activated silent synapses (Figure S3A). Similar to the failure rate experiments described above, presence of TTX during RA treatment prevented activation of silent synapses as evidenced by a lack of change in mEPSC frequency (Figure 3H, amplitude: DMSO, 10.93 ± 0.25 pA; TTX 60 hr, 13.70 ± 0.41 pA; TTX 60 hr no wash + RA, 12.66 ± 0.51 pA; frequency: DMSO, 0.37 ± 0.04 Hz; TTX 60 hr, 0.42 ± 0.05 Hz; TTX 60 hr no wash + RA, 0.34 ± 0.07 Hz). Ranked plots of the mEPSC amplitudes confirmed this notion in that the TTX and the TTX no wash+RA groups showed a similar degree of potentiation of mEPSC amplitudes (Figure S3B). Moreover, we previously showed that generation of silent synapses by prolonged TTX treatment leads to a greater potentiation when LTP is induced subsequently (Arendt et al., 2013). This enhancement of LTP was completely reversed to an impairment of LTP if the slices were incubated with RA after TTX washout (Figures S3C and S3D: Control, 224.21 ± 30.24 %; TTX 60 hr,406.67 ± 44.38 %; TTX 60 hr wash + RA, 142.64 ± 17.21 %), which is consistent with RA’s action in converting silent synapses into functional synapses. By contrast, if RA was applied without TTX washout, the greater LTP was maintained (Figures S3C and S3D:TTX 60 hr no wash + RA, 425.93 ± 73.96 %;).
Taken together, the results obtained with both miniature and evoked synaptic responses strongly suggest that RA-induced AMPAR insertion into synapses shares common features with LTP-induced AMPAR insertion in that both processes require neuronal activity and NMDAR activation. Although at first glance this finding may appear to be counterintuitive given that RA-dependent homeostatic plasticity is induced by inactivation of synaptic activity, not by increased synaptic activity, it is remarkable that expression of homeostatic plasticity nevertheless requires a suspension of chronic inactivity – continuous blockade of synaptic activity in fact prevents homeostatic plasticity.
Postsynaptic complexin is not required for RA-mediated AMPAR delivery to the synapse
Intrigued by the common requirement of NMDAR activation for AMPAR insertion in the LTP and RA pathways, we sought to further dissect the steps downstream of NMDAR activation in RA-induced AMPAR insertion. Similar to presynaptic neurotransmitter vesicle release, postsynaptic vesicle exocytosis is thought to involve assembly of SNARE complexes and SNARE-binding proteins. Indeed, complexin (Cpx), an important co-factor for synaptotagmin-triggered presynaptic vesicle fusion (McMahon et al., 1995; Reim et al., 2001), is required for the activity-regulated exocytosis of postsynaptic AMPAR-containing vesicles during LTP, but not for constitutive AMPAR insertion (Ahmad et al., 2012). We therefore investigated the involvement of complexin in the RA-induced increase of excitatory synaptic transmission using an shRNA-mediated knockdown (KD) of complexin (Ahmad et al., 2012; Maximov et al., 2009). Specifically, a multi-promoter lentivirus encoding two shRNAs targeting both Cpx1 and Cpx2 was injected into the CA1 pyramidal cell layer of cultured hippocampal slices to knock down postsynaptic complexin. We found that consistent with previous observations (Ahmad et al., 2012), postsynaptic complexin was not required for maintaining basal synaptic transmission (Figure 4A, mEPSC amplitude: control, 11.04 ± 0.38 pA; Cpx KD, 10.23 ± 0.30 pA). Importantly, acute RA treatment significantly increased the mEPSC amplitude in both control and neighboring Cpx KD neurons (Figure 4A, mEPSC amplitude in RA: control, 12.81 ± 0.35 pA; Cpx KD, 12.77± 0.40 pA). Thus, unlike the LTP pathway, RA-induced AMPAR delivery to the synapse does not require complexin.
Figure 4. Synaptobrevin-2 and SNAP-47, but not complexin, are required for RA-induced increase in excitatory synaptic transmission.

(A) Amplitude and frequency analysis of mEPSCs recorded from CA1 pyramidal neurons infected with lentivirus expressing Cpx KD constructs and treated with DMSO or RA. Neighboring uninfected neurons were recorded as controls (*, p < 0.05; ***, p < 0.001). (B) Amplitude and frequency analysis of mEPSCs recorded from CA1 pyramidal neurons expressing tetanus toxin light chain (tetTox) treated with DMSO or RA. Neighboring uninfected neurons were recorded as controls (*, p < 0.05; ***, p < 1 x 10−5). (C) Amplitude and frequency analysis of mEPSCs recorded from CA1 pyramidal neurons expressing SNAP-47 KD construct treated with DMSO or RA. Neighboring uninfected neurons were recorded as controls (***, p < 1 x 10−4). (D) Amplitude and frequency analysis of mEPSCs recorded from CA1 pyramidal neurons infected with lentivirus expressing both SNAP-47 KD and wild-type SNAP-47 rescue constructs (SNAP-47 Rep) treated with DMSO or RA. Neighboring uninfected neurons were recorded as controls (***, p < 1 x 10−4). All graphs represent average values ± s.e.m.
Postsynaptic Synaptobrevin-2 and SNAP-47 are both required for RA-mediated AMPAR delivery to the synapse
Previous studies using postsynaptic loading of the light chains of botulinum toxin B or tetanus toxin (which cleave the R-SNARE synaptobrevin-2 (Syb-2))(Link et al., 1992; Schiavo et al., 1992), and more recently studies using Syb-2 KO mice and postsynaptic rescue suggested that this R-SNARE protein is critical for the delivery of AMPARs to the plasma membrane during LTP (Jurado et al., 2013; Lledo et al., 1998). To examine the involvement of Syb-2 in RA-mediated AMPAR synaptic delivery, we injected a lentivirus expressing tetanus toxin light chain (tetTox) (Xu et al., 2012) into postsynaptic CA1 neurons in cultured hippocampal slices. RA strongly increased the mEPSC amplitude in neighboring uninfected neurons (Figure 4B, mEPSC amplitude: DMSO, 9.99 ± 0.29 pA; RA, 12.88 ± 0.46 pA), but had no effect on tetTox-expressing neurons (Figure 4B, mEPSC amplitude: DMSO, 11.45 ± 0.49 pA; RA, 10.86 ± 0.34 pA), indicating a critical involvement of postsynaptic Syb-2 in RA-induced AMPAR exocytosis.
Two target Q-SNAREs, syntaxins and SNAP-25 or its homologs, are involved in vesicle exocytosis. Presynaptically, SNAP-25 is specifically required for neurotransmitter release (Sudhof and Rothman, 2009); postsynaptically, SNAP-25 is essential for normal surface expression of NMDARs (Jurado et al., 2013; Lau et al., 2010). However, for activity-dependent surface delivery of AMPARs during LTP, SNAP-47 but not SNAP-25, is critical (Jurado et al., 2013). We therefore examined the involvement of SNAP-47 in RA-mediated AMPAR insertion using the same shRNA lentiviral constructs that were previously characterized (Jurado et al., 2013). Postsynaptic KDof SNAP-47 completely abolished the RA-induced increase in mEPSC amplitude (Figure 4C: SNAP-47 KD, DMSO, 10.83 ± 0.43 pA; RA, 11.10 ± 0.40 pA), whereas neighboring uninfected neurons responded robustly to RA treatment (Figure 4C: DMSO, 10.89 ± 0.30 pA; RA, 13.28 ± 0.45 pA). The deficits observed in the SNAP-47 KD neurons were fully rescued by simultaneous expression of an shRNA-resistant wild-type SNAP-47 (SNAP-47 Rep) (Figure 4D: control/DMSO, 10.52 ± 0.22 pA; control/RA, 12.72± 0.50 pA; SNAP-47 Rep/DMSO, 10.32 ± 0.20 pA; SNAP-47 Rep/RA, 12.00 ± 0.29 pA). Thus, both LTP- and RA- induced exocytosis of AMPAR-containing vesicles requires the R-SNARE Syb-2 and the Q-SNARE SNAP-47.
Postsynaptic Syntaxin-4, but not Syntaxin-3, is required for RA-mediated synaptic delivery of AMPARs
We next focused on syntaxin Q-SNAREs. Syntaxins contain a conserved N-terminal peptide that binds to Sec1/Munc18-like proteins (SM proteins), followed by an Habc domain, a SNARE motif, and a C-terminal transmembrane region (Figure 5A). We systematically examined syntaxin-1, -3 and -4 (Stx-1, Stx-3 and Stx-4) because all of these isoforms are highly expressed in neurons but seem to mediate distinct types of vesicle fusion. Stx-1 forms the SNARE complex with Syb-2 and SNAP-25 that mediates transmitter release (Sudhof and Rothman, 2009), but is not required for postsynaptic constitutive AMPAR exocytosis during basal synaptic transmission or for regulated AMPAR insertion induced by LTP (Jurado et al., 2013). We found that similar to these previous observations, the postsynaptic Stx-1 KD had no effect on the basal mEPSC amplitude or on the RA-mediated increase in mEPSC amplitude (Figure 5A: control/DMSO, 10.94 ± 0.26 pA; control/RA, 12.87 ± 0.56 pA; Stx-1 KD/DMSO, 10.71 ± 0.37 pA; Stx-1 KD/RA, 12.13 ± 0.40 pA). Stx-3, like Stx-1, binds complexin via a specific 12 amino acid stretch within the SNARE motif (Figures 5A and 5B, (Pabst et al., 2000)), but differs from Stx-1 in that Stx-3 is required for AMPAR exocytosis during LTP whereas Stx-1 is not (Jurado et al., 2013). We confirmed that the Stx-3 KD had no effect on basal synaptic transmission, but were surprised to find that the Stx-3 KD, different from its effect on LTP, did not block the RA-induced increase in postsynaptic AMPAR responses (Figure 5B: mEPSC amplitudes, control/DMSO, 10.24 ± 0.21 pA; control/RA, 12.04 ± 0.43 pA; Stx-3 KD/DMSO, 10.36 ± 0.36 pA; Stx-3 KD/RA, 12.33 ± 0.60 pA). In stark contrast, the Stx-4 KD completely abolished the RA-induced enhancement of AMPAR-mediated responses without affecting basal transmission (Figure 5C: control/DMSO, 10.63 ± 0.21 pA; control/RA, 13.39 ± 0.39 pA; Stx-4 KD/DMSO, 10.25 ± 0.28 pA; Stx-4 KD/RA, 10.64 ± 0.17 pA).
Figure 5. Syntaxin-4, but not Syntaxin-1 or Syntaxin-3, is required for RA-induced increase in excitatory synaptic transmission.

(A) Analysis of amplitude and frequency of mEPSCs recorded from control and Stx-1 KD CA1 pyramidal neurons in cultured hippocampal slices treated with DMSO or RA (**, p < 0.01). Top: schematic of Stx-1 showing its functional domains (Habc, Habc domains; SNARE, soluble NSF-attachment protein receptor motif; TMR, transmembrane domain; green indicates complexin-binding sequence). (B) Summary of mEPSC amplitude and frequency analysis from control and Stx-3 KD CA1 pyramidal neurons in cultured hippocampal slices treated with DMSO or RA (**, p < 0.01; ***, p < 0.0005). Top: schematic of Stx-3 showing its functional domains. (C) Summary of mEPSC amplitude and frequency analysis from control and Stx-4 KD CA1 pyramidal neurons in cultured hippocampal slices treated with DMSO or RA (*, p < 0.05; ***, p < 1 x 10−6). Top: schematic of Stx-4 showing its functional domains (red indicates non-complexin-binding sequence). (D) Summary graph (left) and trace examples (right) of LTP recorded from CA1 pyramidal neuron expressing Stx-3 KD or Stx-4 KD constructs. Neighboring uninfected neurons were recorded as controls. Black bar in the summary graph indicates the time window for LTP magnitude quantification. Scale bars: 20 pA, 10 ms. (E) Scatter plots of LTP obtained from individual experiments summarized in (D) with bar graphs representing mean ± s.e.m (**, p < 0.01).
Interestingly, Stx-4 was previously shown to mediate insulin-dependent glucose transport through regulation of membrane GLUT4 trafficking(Dugani and Klip, 2005; Olson et al., 1997). Moreover, a study using hippocampal primary cultures proposed that Stx-4 is critically involved in activity-dependent exocytosis of a transferrin-EGFP fusion protein, used as a proxy for AMPARs, in dendritic spines, and thus suggested that Stx-4 plays a role in AMPAR trafficking in LTP (Kennedy et al., 2010). A more direct investigation of syntaxins in LTP using acute hippocampal slices, however, showed that it was Stx-3 but not Stx-4 that mediates AMPAR-containing vesicle exocytosis in LTP (Jurado et al., 2013). Since we now suggest using a different preparation (cultured hippocampal slices) that Stx-4 is critical for RA-triggered AMPAR exocytosis, we were concerned that differences between previous studies may have been due to differences in experimental approach instead of true differences in biological processes. To address this concern, we repeated the LTP experiments with syntaxin KD in our cultured hippocampal slices with the aim to directly compare the apparently distinct roles of Stx-3 and Stx-4 in two different types of plasticity in the same preparation. We found that as with acute slices(Jurado et al., 2013), Stx-3, but not Stx-4, was essential for hippocampal LTP (Figures 5D and 5E: control, 264.63% ± 24.13%; Stx-3 KD, 142.55 % ± 23.55%; Stx-4 KD, 260.35% ± 48.76%). Taken together, these results thus suggest the intriguing possibility that RA-induced AMPAR exocytosis is mediated by a novel activity-dependent trafficking pathway that is mechanistically distinct from the LTP pathway or from the constitutive ANPAR exocytosis pathway. While the LTP- and RA-induced AMPAR exocytosis pathways share two SNARE proteins, Syb-2 and SNAP-47, they utilize distinct syntaxins, which may play a role in defining these distinct pathways.
Complexin-binding sequence of Syntaxin-3 blocks RA-mediated synaptic delivery of AMPARs
Stx-1 and Stx-3 contain a complexin-binding sequence in the SNARE motif, whereasStx-4does not (Figures 5A, 5B and 5C, (Pabst et al., 2000)). Complexin binding to Stx-1 and Stx-3 is required for their respective function in neurotransmitter release and LTP (Jurado et al., 2013; Sudhof and Rothman, 2009). Although we have shown that complexin is not required for RA-mediated AMPAR delivery to the synapse, we were curious whether Stx-4’s inability to bind complexin is important for its function in RA-mediated AMPAR insertion. We addressed this question by swapping the complexin-binding sequence of Stx-3 (aa 213–224) with the homologous sequence of Stx-4 that does not interact with complexin, resulting in two chimeric proteins – a Stx-4 that now binds complexin (Stx-4/3) and a Stx-3 that no longer binds complexin (Stx-3/4). We then tested the ability of these two chimeras to rescue the effect of the Stx-4 KD on RA-induced AMPAR insertion. In contrast to an shRNA-resistant wild-type Stx-4, which fully rescued the effect of RA on AMPAR-mediated synaptic transmission (Figure 6A: control/DMSO, 10.25 ± 0.22 pA; control/RA, 12.23 ± 0.23 pA; Stx-4 Rep/DMSO, 10.36 ± 0.33 pA; Stx-4 Rep/RA, 11.88 ± 0.26 pA), mutant Stx-4/3 failed to restore the RA-induced increase in AMPAR mEPSC amplitude (Figure 6B: control/DMSO, 10.65 ± 0.21 pA; control/RA, 12.48 ± 0.36 pA; Stx-4/3 Rep/DMSO, 11.06 ± 0.25 pA; Stx-4/3 Rep/RA, 10.22 ± 0.30 pA). This result indicates that lack of complexin binding is indeed an important functional feature of Stx-4 for its role in mediating RA-induced, activity-regulated AMPAR insertion. We also tested the reverse chimera, Stx-3/4, where the Stx-3 complexin-binding sequence is replaced by the homologous sequence in Stx-4 that does not interact with complexin. This rescue construct also failed to reverse the Stx-4 KD phenotype - RA was still unable to increase the mEPSCs amplitude in neurons expressing this chimera (Figure 6C: control/DMSO, 10.37 ± 0.40 pA; control/RA, 12.89 ± 0.52 pA; Stx-3/4 Rep/DMSO, 10.43 ± 0.41 pA; Stx-3/4 Rep/RA, 10.42 ± 0.26 pA). Thus, additional Stx-4-specific functional sequence motifs besides the absence of complexin-binding sequence are required for the RA-mediated activity-dependent synaptic delivery of AMPARs.
Figure 6. Non-complexin-binding sequence of Stx-4 is required for normal Stx-4 function in RA-induced AMPAR-containing vesicle exocytosis.

(A) Analysis of amplitude and frequency of mEPSCs obtained from CA1 pyramidal neurons expressing both Stx-4 KD and wildtype Stx-4 rescue constructs (Stx-4 Rep) (*, p < 0.05; ***, p < 0.001). Top: schematic of Stx-4 showing its functional domains (red indicates non-complexin-binding sequence). (B) Analysis of amplitude and frequency of mEPSCs obtained from CA1 pyramidal neurons expressing both Stx-4 KD and Stx-4/3 rescue constructs (Stx-4/3 Rep) (***, p < 1 x 10−4). Top: schematic of Stx-4/3 showing the non-complexin-binding domain of Stx-4 is replaced by the complexin-binding domain of Stx-3 (green). (C) Analysis of amplitude and frequency of mEPSCs obtained from CA1 pyramidal neurons expressing both Stx-4 KD and Stx-3/4 rescue constructs (Stx-3/4 Rep) (***, p < 0.001). Top: schematic of Stx-3/4 showing the complexin-binding domain of Stx-3 is replaced by the non-complexin-binding domain of Stx-4 (red).
Blocking Stx-4-mediated AMPAR synaptic delivery rescues LTP after RA treatment
Our molecular dissection of the AMPAR exocytosis mechanisms revealed a novel AMPAR insertion pathway that is regulated by synaptic activity. However, it remains undetermined how RA treatment blocks subsequent development of LTP (see Figure 1). One possibility is that because the RA and LTP pathways converge onto the same final common step – an increase in AMPAR content at synapses – these two processes occlude each other either due to depletion of the AMPARs in the reserve pool or to the filling of the synaptic slots that are required for receiving AMPARs at synapses. If one of these hypotheses was correct, selectively preventing RA-induced AMPAR exocytosis but not LTP-induced AMPAR exocytosis should rescue the block of LTP by RA. However, it is also possible that RA has additional unknown effects in a neuron that somehow compromise LTP signaling pathways upstream to AMPAR insertion. If this were the case, preventing AMPAR insertion during RA treatment should not rescue LTP. We directly tested this possibility by examining LTP in Stx-4 KD neurons that were treated with RA. Both DMSO- and RA-treated neurons receiving Stx-4 KD expressed normal LTP (Figures 7A and 7B: Stx-4 KD/DMSO, 349.17% ± 43.90%; Stx-4 KD/RA, 421.29% ± 79.01%). Thus, RA treatment impairs subsequent LTP induction through its effect on the step of AMPAR synaptic delivery.
Figure 7. Stx-4 KD rescues LTP in RA-treated hippocampal slices.
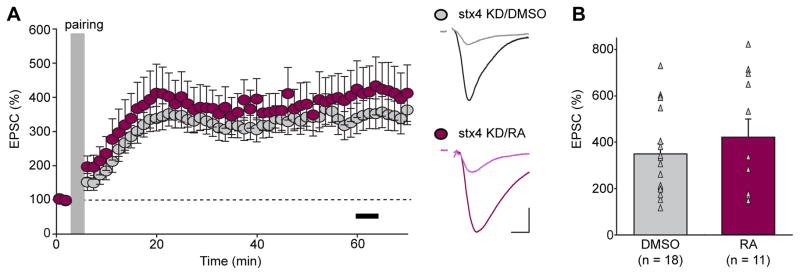
(A) Summary graph (left) and trace examples (right) of LTP recorded from Stx-4 KD CA1 pyramidal neuron treated with DMSO or RA. Black bar in the summary graph represents the time window for LTP magnitude quantification. Scale bars: 50 pA, 10 ms. (B) Scatter plots of LTP obtained from individual experiments summarized in (A) with bar graphs representing mean ± s.e.m (p > 0.05).
DISCUSSION
The connectivity of a neural network can be modified by at least two major forms of long-lasting plasticity – Hebbian and homeostatic plasticity. Although operating under opposite computational mechanisms (positive feedback mechanisms for Hebbian plasticity vs. negative feedback mechanisms for homeostatic plasticity), these two types of plasticity likely interact at multiple points at the biochemical, molecular, and structural level, altering parameters such as neuronal excitability through modulation of expression levels of ion channels (Campanac and Debanne, 2007; Magee and Johnston, 2005), synaptic strength through modulation of release probability and postsynaptic receptor abundance (Davis and Muller, 2014; Grasselli and Hansel, 2014; Huganir and Nicoll, 2013), and – at a longer time scale – number of synaptic contacts through modulation of synapse formation and elimination (Bourne and Harris, 2008; Sala and Segal, 2014; Yu and Zuo, 2011). In the present study, we dissected the AMPAR trafficking mechanisms involved in RA-dependent homeostatic plasticity, and compared these mechanisms to those involved in LTP. Moreover, we examined the intersection of the mechanisms mediating RA- and LTP-induced AMPAR-trafficking, and focused on the interaction between synaptic RA signaling and LTP.
We found that increasing excitatory synaptic strength through synaptic RA signaling impaired subsequent induction of LTP at hippocampal synapses through a mechanism that is mediated by the RA receptor RARα and requires protein synthesis. In further exploring the molecular mechanism of such interaction, we made two unexpected discoveries. First, although RA synthesis in neurons seems to be triggered by reduced synaptic activity (Wang et al., 2011), RA-induced insertion of AMPARs into synapses, paradoxically, requires activity and NMDAR activation. Second, RA-induced exocytosis of AMPAR-containing vesicles requires a SNARE-dependent membrane-fusion machinery (Synaptobrevin-2, SNAP-47, Syntaxin-4, but not complexin) whose components only partially overlap with those involved in LTP-induced exocytosis of AMPAR-containing vesicles (Synaptobrevin-2, SNAP-47, Syntaxin-3, with complexin), thus revealing a novel activity-regulated AMPAR trafficking pathway that is distinct from the LTP pathway.
AMPAR trafficking in RA- and LTP-induced synaptic potentiation requires distinct SNARE-dependent vesicle fusion machineries
The speed and precision required for presynaptic neurotransmitter release makes it an attractive system to study vesicle fusion and to decipher the regulatory mechanisms that gate the fusion event. Indeed, we have a reasonably good understanding of the molecular players required for Ca2+-regulated exocytosis of synaptic vesicles (Rizo and Rosenmund, 2008; Sudhof, 2013; Sudhof and Rothman, 2009). However, although it has been long accepted that Ca2+ is also a key element in the regulation of biochemical processes in postsynaptic compartments, and that activity-regulated postsynaptic exocytosis likely requires SNAREs and SNARE-binding proteins, we are just beginning to identify individual components required for various postsynaptic vesicle exocytosis events. For example, complexin plays multiple roles in neurotransmitter release including vesicle priming (Yang et al., 2010), activation of SNARE complexes before vesicle fusion (Maximov et al., 2009; Reim et al., 2001; Xue et al., 2009), and clamping of SNARE complexes to prevent inappropriate fusion (Giraudo et al., 2006; Huntwork and Littleton, 2007; Maximov et al., 2009; Tang et al., 2006; Xue et al., 2009; Yang et al., 2010). The role of complexin in postsynaptic AMPAR-containing vesicle exocytosis during LTP was recently documented, and is thought to function by binding to the Q-SNARE Stx-3 instead of Stx-1 (Ahmad et al., 2012), thus expanding the repertoire of complexin functions to postsynaptic vesicular trafficking. Interestingly, for RA-mediated AMPAR trafficking during non-Hebbian plasticity, complexin-binding is not only unnecessary, but may even “clamp” the fusion event and prevent it from occurring when a complexin-binding sequence is introduced into Stx-4 (Figure 6B). These results raise the possibility that complexin-binding might help define a subpopulation of postsynaptic AMPAR-containing vesicles that is exclusively involved in LTP but not in homeostatic or other activity-dependent forms of plasticity. This notion is supported in part by our observation that the non-complexin-binding Stx-4 is selectively required for RA-induced AMPAR insertion, instead of Stx-3, which is required for LTP, or of Stx-1, which is required for presynaptic vesicle fusion. Thus, one attractive model for the differential regulation of postsynaptic activity-regulated AMPAR exocytosis is that Stx-3 and Stx-4 define separate vesicle fusion sites on the target plasma membrane for insertion of AMPAR-containing vesicles. During LTP, AMPAR-containing vesicles associate with complexin, dock and exocytose at Stx-3-rich membrane sites, whereas during synaptic RA signaling, non-complexin-associated AMPAR-containing vesicles dock and exocytose at Stx-4-rich membrane sites (Figure 8).
Figure 8. Model of the shared and distinct vesicle fusion machinery for AMPAR-containing vesicle exocytosis during LTP and synaptic RA signaling.

Top panel: R-SNARE Synaptobrevin-2 is present in the membrane of AMPAR-containing vesicles. Syntaxin-3 and Syntaxin-4 may form distinct vesicle fusion micro-domains on the synaptic or perisynaptic membranes and define AMPAR-containing vesicle exocytosis locations for LTP and RA pathways, respectively. Q-SNARE SNAP-47 is present and is required for both pathways.
Bottom panel: AMPAR-containing vesicle fusion occurs at distinct surface membrane locations during LTP or RA signaling. RA-induced vesicle exocytosis occludes LTP by either depleting the AMPAR-containing vesicle pool or by occupying postsynaptic slots necessary for anchoring AMPARs.
Despite the mechanistic similarities between pre- and postsynaptic SNARE-mediated vesicle exocytosis, these processes exhibit important differences that involve distinct molecular mechanisms, possibly for the purpose of differentially regulating these processes. To support presynaptic transmitter release, which is triggered rapidly (< 1ms) by calcium at the active zone, intricate presynaptic scaffold structures are required for docking and priming of synaptic vesicles (Sudhof, 2013). By contrast, ultrastructural studies failed to observe docked AMPAR-containing vesicles on the postsynaptic membranes, either because postsynaptic fusion events are much less frequent than those that occur at presynaptic site, or because calcium-triggered postsynaptic vesicle exocytosis during LTP may occur at a slower time course (Makino and Malinow, 2009; Patterson et al., 2010; Petrini et al., 2009; Yudowski et al., 2007). Findings from the present study add another level of complexity to the picture by revealing a second form of activity-regulated AMPAR-containing vesicle exocytosis, mediated by a different syntaxin than that used by the LTP pathway. Additionally, SNAP-47, the other Q-SNARE required for both postsynaptic vesicle exocytosis processes, may be partly soluble compared to the permanently membrane-localized SNAP-25 at the presynaptic side (Holt et al., 2006). Thus more work is required to determine whether postsynaptic vesicle exocytosis sites for the LTP and RA pathways are permanent or transient.
How does RA-induced AMPAR insertion blocks subsequent development of LTP? One possibility is that the AMPAR-containing vesicle pool is a limiting factor. RA induces synaptic incorporation of GluA2-lacking AMPARs (Aoto et al., 2008; Maghsoodi et al., 2008; Wang et al., 2011). Similarly, LTP has been proposed to induce insertion of GluA2-lacking AMPARs (Guire et al., 2008; Morita et al., 2014; Plant et al., 2006), but see (Adesnik and Nicoll, 2007; Gray et al., 2007). Thus, it is possible that the two processes share and compete for the same GluA2-lacking AMPAR vesicle pool. Local biochemical events specifically associated with each type of plasticity could direct these vesicles for fusion with a complexin/Stx-3-dependent mechanism for the LTP pathway or fusion with a complexin-independent, Stx-4-dependent mechanism for the RA pathway (Figure 8). Alternatively, the AMPAR vesicles used for each pathway may differ. Instead, the synaptic “slots” available for stabilizing newly inserted AMPARs could be limiting. It is then possible that RA-induced AMPAR insertion into synapses occupies these slots and prevents subsequent LTP (Figure 8). Further experiments are required to distinguish between these two possibilities.
Activity-dependence of RA-mediated AMPAR trafficking
An intriguing finding in the current study is the requirement of neuronal activity and NMDAR activation for RA-induced AMPAR insertion – a process that has much similarity with the LTP pathway except in the molecular constituents of the fusion machinery. The function of RA in synaptic signaling was discovered in the context of homeostatic synaptic plasticity, namely, the synaptic inactivity-induced upregulation of synaptic strength (Aoto et al., 2008; Wang et al., 2011). RA synthesis is triggered by reduced dendritic Ca2+(Wang et al., 2011). Our findings here suggest that once RA is made and synthesis of new AMPARs is activated by RA, AMPAR incorporation into the existing synapses is not merely a default step. Instead, activity and NMDAR activation are required to execute the final step of receptor localization to the synapse. Thus, homeostatic synaptic plasticity, at least the form that is mediated by RA signaling, seems to require two major regulatory steps. The first step is the prolonged inactivity-triggered RA production and AMPAR synthesis, a step that “primes” the postsynaptic neurons for rapid strengthening of excitatory synapses. However, AMPARs are not “blindly” inserted into all existing excitatory synapses. The second step is an execution step that inserts AMPARs to functional synapses via a validation process – brief activation of the synapse (mediated by NMDAR activation and a transient postsynaptic Ca2+ rise) that is indicative of a functional synapse. The execution step ensures insertion of AMPARs into synapses that are part of a functional circuit.
What might be the advantage of having such an additional synaptic function “check point” for homeostatic synaptic plasticity? Conceptually the role of homeostatic plasticity in a neural network is to compensate for prolonged changes in the input activity that deviates from its optimal range (too high or too low). In the case of prolonged reduction in input activity, inputs that are experiencing reduced upstream activity (thus representing weaker but still functional input) and those that are experiencing permanent loss of input activity (thus are destined to be eliminated) can both contribute to the activation of homeostatic compensation mechanisms (i.e. RA synthesis). As such, it is perhaps most effective if compensation through an increase in postsynaptic receptor number occurs at synapses where active presynaptic release still occurs (albeit at a reduced level) as opposed to synaptic contacts that are destined to be removed. A validation step using NMDAR activity acts as an ideal switch to permit activity-regulated exocytosis of AMPAR-containing vesicles at functional synapses. Such selective strengthening of all functional connections renormalizes synaptic strength of a network to allow readjustment of an optimal dynamic range of activity for all meaningful connections while leaving the nonfunctional ones out. Furthermore, this validation process might in fact support the phenomenon of “synaptic scaling”– in which synapses of different sizes increase their strength in a multiplicative fashion during homeostatic upregulation – as previously proposed based on observations in cultured cortical neuron networks (Turrigiano et al., 1998). If the AMPAR insertion into functional synapses is regulated by local synaptic activity (and presumably local Ca2+ influx), it is conceivable that bigger synapses with more NMDARs can potentially gain more new AMPARs than smaller ones, thus providing a biochemical mechanism for synaptic scaling.
In summary, our findings suggest that the functional impact of RA at synapses goes beyond homeostatic plasticity, and that despite the divergence in the molecular mechanisms underlying Hebbian and homeostatic plasticity, these two major forms of synaptic plasticity interact functionally (Arendt et al., 2013; Toyoizumi et al., 2014). Additionally, the findings presented here provide concrete molecular tools that can be used for future work to investigate the function of synaptic RA signaling and homeostatic synaptic plasticity in vivo in the context of animal learning.
EXPERIMENTAL PROCEDURES
The RARα floxed mouse is a gift from Drs. Pierre Chambon and Norbert Ghyselinck (IGBMC, Strasbourg, France) (Chapellier et al., 2002). Breeding colonies are maintained in the animal facility at Stanford Medical School following standard procedures approved by the Stanford University APLAC. Organotypic slice cultures were prepared from RARα floxed mice (postnatal day 6–7) and maintained for 5–7 days prior to recording (Aoto et al., 2008; Arendt et al., 2013).
Full experimental procedures and associated references are available in the Supplemental Information.
Supplementary Material
Acknowledgments
We thank members of the Chen, Südhof and Malenka labs for comments and help during the course of the study. The work was supported by NIH grants MH086403 (L.C., R.C.M. and T.C.S.) and MH091193 (L.C.).
Footnotes
AUTHOR CONTRIBUTIONS
K.L.A. performed all the electrophysiology recordings for the study. Y.Z. provided various Stx-3 and Stx-4 chimera rescue constructs. S.J. and Y.Z. prepared lentiviruses expressing various shRNAs for the study. L.C. directed the project and wrote the manuscript. All authors reviewed and edited the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adesnik H, Nicoll RA. Conservation of glutamate receptor 2-containing AMPA receptors during long-term potentiation. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007;27:4598–4602. doi: 10.1523/JNEUROSCI.0325-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad M, Polepalli JS, Goswami D, Yang X, Kaeser-Woo YJ, Sudhof TC, Malenka RC. Postsynaptic complexin controls AMPA receptor exocytosis during LTP. Neuron. 2012;73:260–267. doi: 10.1016/j.neuron.2011.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoto J, Nam CI, Poon MM, Ting P, Chen L. Synaptic signaling by all-trans retinoic acid in homeostatic synaptic plasticity. Neuron. 2008;60:308–320. doi: 10.1016/j.neuron.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt KL, Sarti F, Chen L. Chronic inactivation of a neural circuit enhances LTP by inducing silent synapse formation. J Neurosci. 2013;33:2087–2096. doi: 10.1523/JNEUROSCI.3880-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne JN, Harris KM. Balancing structure and function at hippocampal dendritic spines. Annual review of neuroscience. 2008;31:47–67. doi: 10.1146/annurev.neuro.31.060407.125646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanac E, Debanne D. Plasticity of neuronal excitability: Hebbian rules beyond the synapse. Arch Ital Biol. 2007;145:277–287. [PubMed] [Google Scholar]
- Chapellier B, Mark M, Garnier JM, LeMeur M, Chambon P, Ghyselinck NB. A conditional floxed (loxP-flanked) allele for the retinoic acid receptor alpha (RARalpha) gene. Genesis. 2002;32:87–90. doi: 10.1002/gene.10071. [DOI] [PubMed] [Google Scholar]
- Chen L, Lau AG, Sarti F. Synaptic retinoic acid signaling and homeostatic synaptic plasticity. Neuropharmacology. 2014;78:3–12. doi: 10.1016/j.neuropharm.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang MY, Misner D, Kempermann G, Schikorski T, Giguere V, Sucov HM, Gage FH, Stevens CF, Evans RM. An essential role for retinoid receptors RARbeta and RXRgamma in long-term potentiation and depression. Neuron. 1998;21:1353–1361. doi: 10.1016/s0896-6273(00)80654-6. [DOI] [PubMed] [Google Scholar]
- Cocco S, Diaz G, Stancampiano R, Diana A, Carta M, Curreli R, Sarais L, Fadda F. Vitamin A deficiency produces spatial learning and memory impairment in rats. Neuroscience. 2002;115:475–482. doi: 10.1016/s0306-4522(02)00423-2. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Kehl SJ, McLennan H. Excitatory amino acids in synaptic transmission in the Schaffer collateral-commissural pathway of the rat hippocampus. J Physiol. 1983;334:33–46. doi: 10.1113/jphysiol.1983.sp014478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GW, Muller M. Homeostatic Control of Presynaptic Neurotransmitter Release. Annu Rev Physiol. 2014 doi: 10.1146/annurev-physiol-021014-071740. [DOI] [PubMed] [Google Scholar]
- Dugani CB, Klip A. Glucose transporter 4: cycling, compartments and controversies. EMBO Rep. 2005;6:1137–1142. doi: 10.1038/sj.embor.7400584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraudo CG, Eng WS, Melia TJ, Rothman JE. A clamping mechanism involved in SNARE-dependent exocytosis. Science. 2006;313:676–680. doi: 10.1126/science.1129450. [DOI] [PubMed] [Google Scholar]
- Grasselli G, Hansel C. Cerebellar long-term potentiation: cellular mechanisms and role in learning. Int Rev Neurobiol. 2014;117:39–51. doi: 10.1016/B978-0-12-420247-4.00003-8. [DOI] [PubMed] [Google Scholar]
- Gray EE, Fink AE, Sarinana J, Vissel B, O’Dell TJ. Long-term potentiation in the hippocampal CA1 region does not require insertion and activation of GluR2-lacking AMPA receptors. J Neurophysiol. 2007;98:2488–2492. doi: 10.1152/jn.00473.2007. [DOI] [PubMed] [Google Scholar]
- Guire ES, Oh MC, Soderling TR, Derkach VA. Recruitment of calcium-permeable AMPA receptors during synaptic potentiation is regulated by CaM-kinase I. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2008;28:6000–6009. doi: 10.1523/JNEUROSCI.0384-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt M, Varoqueaux F, Wiederhold K, Takamori S, Urlaub H, Fasshauer D, Jahn R. Identification of SNAP-47, a novel Qbc-SNARE with ubiquitous expression. J Biol Chem. 2006;281:17076–17083. doi: 10.1074/jbc.M513838200. [DOI] [PubMed] [Google Scholar]
- Huganir RL, Nicoll RA. AMPARs and synaptic plasticity: the last 25 years. Neuron. 2013;80:704–717. doi: 10.1016/j.neuron.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntwork S, Littleton JT. A complexin fusion clamp regulates spontaneous neurotransmitter release and synaptic growth. Nat Neurosci. 2007;10:1235–1237. doi: 10.1038/nn1980. [DOI] [PubMed] [Google Scholar]
- Isaac JT, Nicoll RA, Malenka RC. Evidence for silent synapses: implications for the expression of LTP. Neuron. 1995;15:427–434. doi: 10.1016/0896-6273(95)90046-2. [DOI] [PubMed] [Google Scholar]
- Jurado S, Goswami D, Zhang Y, Molina AJ, Sudhof TC, Malenka RC. LTP requires a unique postsynaptic SNARE fusion machinery. Neuron. 2013;77:542–558. doi: 10.1016/j.neuron.2012.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MJ, Davison IG, Robinson CG, Ehlers MD. Syntaxin-4 defines a domain for activity-dependent exocytosis in dendritic spines. Cell. 2010;141:524–535. doi: 10.1016/j.cell.2010.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau CG, Takayasu Y, Rodenas-Ruano A, Paternain AV, Lerma J, Bennett MV, Zukin RS. SNAP-25 is a target of protein kinase C phosphorylation critical to NMDA receptor trafficking. J Neurosci. 2010;30:242–254. doi: 10.1523/JNEUROSCI.4933-08.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao D, Hessler NA, Malinow R. Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature. 1995;375:400–404. doi: 10.1038/375400a0. [DOI] [PubMed] [Google Scholar]
- Link E, Edelmann L, Chou JH, Binz T, Yamasaki S, Eisel U, Baumert M, Sudhof TC, Niemann H, Jahn R. Tetanus toxin action: inhibition of neurotransmitter release linked to synaptobrevin proteolysis. Biochem Biophys Res Commun. 1992;189:1017–1023. doi: 10.1016/0006-291x(92)92305-h. [DOI] [PubMed] [Google Scholar]
- Lledo PM, Zhang X, Sudhof TC, Malenka RC, Nicoll RA. Postsynaptic membrane fusion and long-term potentiation. Science. 1998;279:399–403. doi: 10.1126/science.279.5349.399. [DOI] [PubMed] [Google Scholar]
- Maden M. Retinoic acid in the development, regeneration and maintenance of the nervous system. Nat Rev Neurosci. 2007;8:755–765. doi: 10.1038/nrn2212. [DOI] [PubMed] [Google Scholar]
- Magee JC, Johnston D. Plasticity of dendritic function. Curr Opin Neurobiol. 2005;15:334–342. doi: 10.1016/j.conb.2005.05.013. [DOI] [PubMed] [Google Scholar]
- Maghsoodi B, Poon MM, Nam CI, Aoto J, Ting P, Chen L. Retinoic acid regulates RARalpha-mediated control of translation in dendritic RNA granules during homeostatic synaptic plasticity. Proc Natl Acad Sci U S A. 2008;105:16015–16020. doi: 10.1073/pnas.0804801105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino H, Malinow R. AMPA receptor incorporation into synapses during LTP: the role of lateral movement and exocytosis. Neuron. 2009;64:381–390. doi: 10.1016/j.neuron.2009.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximov A, Tang J, Yang X, Pang ZP, Sudhof TC. Complexin controls the force transfer from SNARE complexes to membranes in fusion. Science. 2009;323:516–521. doi: 10.1126/science.1166505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon HT, Missler M, Li C, Sudhof TC. Complexins: cytosolic proteins that regulate SNAP receptor function. Cell. 1995;83:111–119. doi: 10.1016/0092-8674(95)90239-2. [DOI] [PubMed] [Google Scholar]
- Misner DL, Jacobs S, Shimizu Y, de Urquiza AM, Solomin L, Perlmann T, De Luca LM, Stevens CF, Evans RM. Vitamin A deprivation results in reversible loss of hippocampal long-term synaptic plasticity. Proc Natl Acad Sci U S A. 2001;98:11714–11719. doi: 10.1073/pnas.191369798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita D, Rah JC, Isaac JT. Incorporation of inwardly rectifying AMPA receptors at silent synapses during hippocampal long-term potentiation. Philos Trans R Soc Lond B Biol Sci. 2014;369:20130156. doi: 10.1098/rstb.2013.0156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomoto M, Takeda Y, Uchida S, Mitsuda K, Enomoto H, Saito K, Choi T, Watabe AM, Kobayashi S, Masushige S, et al. Dysfunction of the RAR/RXR signaling pathway in the forebrain impairs hippocampal memory and synaptic plasticity. Mol Brain. 2012;5:8. doi: 10.1186/1756-6606-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson AL, Knight JB, Pessin JE. Syntaxin 4, VAMP2, and/or VAMP3/cellubrevin are functional target membrane and vesicle SNAP receptors for insulin-stimulated GLUT4 translocation in adipocytes. Mol Cell Biol. 1997;17:2425–2435. doi: 10.1128/mcb.17.5.2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabst S, Hazzard JW, Antonin W, Sudhof TC, Jahn R, Rizo J, Fasshauer D. Selective interaction of complexin with the neuronal SNARE complex. Determination of the binding regions. J Biol Chem. 2000;275:19808–19818. doi: 10.1074/jbc.M002571200. [DOI] [PubMed] [Google Scholar]
- Patterson MA, Szatmari EM, Yasuda R. AMPA receptors are exocytosed in stimulated spines and adjacent dendrites in a Ras-ERK-dependent manner during long-term potentiation. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:15951–15956. doi: 10.1073/pnas.0913875107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrini EM, Lu J, Cognet L, Lounis B, Ehlers MD, Choquet D. Endocytic trafficking and recycling maintain a pool of mobile surface AMPA receptors required for synaptic potentiation. Neuron. 2009;63:92–105. doi: 10.1016/j.neuron.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant K, Pelkey KA, Bortolotto ZA, Morita D, Terashima A, McBain CJ, Collingridge GL, Isaac JT. Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nature neuroscience. 2006;9:602–604. doi: 10.1038/nn1678. [DOI] [PubMed] [Google Scholar]
- Poon MM, Chen L. Retinoic acid-gated sequence-specific translational control by RARalpha. Proc Natl Acad Sci U S A. 2008;105:20303–20308. doi: 10.1073/pnas.0807740105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reim K, Mansour M, Varoqueaux F, McMahon HT, Sudhof TC, Brose N, Rosenmund C. Complexins regulate a late step in Ca2+-dependent neurotransmitter release. Cell. 2001;104:71–81. doi: 10.1016/s0092-8674(01)00192-1. [DOI] [PubMed] [Google Scholar]
- Rizo J, Rosenmund C. Synaptic vesicle fusion. Nat Struct Mol Biol. 2008;15:665–674. doi: 10.1038/nsmb.1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala C, Segal M. Dendritic spines: the locus of structural and functional plasticity. Physiol Rev. 2014;94:141–188. doi: 10.1152/physrev.00012.2013. [DOI] [PubMed] [Google Scholar]
- Sarti F, Schroeder J, Aoto J, Chen L. Conditional RARalpha knockout mice reveal acute requirement for retinoic acid and RARalpha in homeostatic plasticity. Front Mol Neurosci. 2012;5:16. doi: 10.3389/fnmol.2012.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarti F, Zhang Z, Schroeder J, Chen L. Rapid suppression of inhibitory synaptic transmission by retinoic acid. J Neurosci. 2013;33:11440–11450. doi: 10.1523/JNEUROSCI.1710-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiavo G, Benfenati F, Poulain B, Rossetto O, Polverino de Laureto P, DasGupta BR, Montecucco C. Tetanus and botulinum-B neurotoxins block neurotransmitter release by proteolytic cleavage of synaptobrevin. Nature. 1992;359:832–835. doi: 10.1038/359832a0. [DOI] [PubMed] [Google Scholar]
- Sudhof TC. Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron. 2013;80:675–690. doi: 10.1016/j.neuron.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC, Rothman JE. Membrane fusion: grappling with SNARE and SM proteins. Science. 2009;323:474–477. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Maximov A, Shin OH, Dai H, Rizo J, Sudhof TC. A complexin/synaptotagmin 1 switch controls fast synaptic vesicle exocytosis. Cell. 2006;126:1175–1187. doi: 10.1016/j.cell.2006.08.030. [DOI] [PubMed] [Google Scholar]
- Toyoizumi T, Kaneko M, Stryker MP, Miller KD. Modeling the dynamic interaction of hebbian and homeostatic plasticity. Neuron. 2014;84:497–510. doi: 10.1016/j.neuron.2014.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391:892–896. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- Wang HL, Zhang Z, Hintze M, Chen L. Decrease in calcium concentration triggers neuronal retinoic acid synthesis during homeostatic synaptic plasticity. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2011;31:17764–17771. doi: 10.1523/JNEUROSCI.3964-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Morishita W, Buckmaster PS, Pang ZP, Malenka RC, Sudhof TC. Distinct neuronal coding schemes in memory revealed by selective erasure of fast synchronous synaptic transmission. Neuron. 2012;73:990–1001. doi: 10.1016/j.neuron.2011.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue M, Lin YQ, Pan H, Reim K, Deng H, Bellen HJ, Rosenmund C. Tilting the balance between facilitatory and inhibitory functions of mammalian and Drosophila Complexins orchestrates synaptic vesicle exocytosis. Neuron. 2009;64:367–380. doi: 10.1016/j.neuron.2009.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Kaeser-Woo YJ, Pang ZP, Xu W, Sudhof TC. Complexin clamps asynchronous release by blocking a secondary Ca(2+) sensor via its accessory alpha helix. Neuron. 2010;68:907–920. doi: 10.1016/j.neuron.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Zuo Y. Spine plasticity in the motor cortex. Curr Opin Neurobiol. 2011;21:169–174. doi: 10.1016/j.conb.2010.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yudowski GA, Puthenveedu MA, Leonoudakis D, Panicker S, Thorn KS, Beattie EC, von Zastrow M. Real-time imaging of discrete exocytic events mediating surface delivery of AMPA receptors. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007;27:11112–11121. doi: 10.1523/JNEUROSCI.2465-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.