

Abstract
Introduction
Toll-like receptors (TLRs) are a family of pattern recognition receptors that are expressed on cells of the innate immune system. The ligands can be pathogen derived (pathogen associated molecular patterns; PAMPs) or endogenous (damage associated molecular patters; DAMPs) that when bound induces activation of nuclear factor kappa B (NF-κB) and transcription of pro-inflammatory genes. TLRs have also been discovered in various malignant cell types, but with unknown function.
Methods
In this study we performed a detailed analysis of TLR and co-receptor expression pattern and function in breast cancer. Expression patterns were examined using real-time quantitative polymerase chain reaction (RT-qPCR) and immunohistochemistry (IHC) on three estrogen receptor-positive (ER+) and four estrogen receptor/progesterone receptor-negative (ER−/PR−; ER/PR-negative) breast cancer cell lines, and a breast cancer cohort consisting of 144 primary breast cancer samples. The function was investigated using in vitro assays comprising PAMP/DAMP-stimulation, downstream signaling and TLR-silencing experiments.
Results
We found that TLR4 was expressed in a biologically active form and responded to both PAMPs and DAMPs primarily in ER/PR-negative breast cancers. Stimulation of TLR2/4 in vitro induced expression of pro-inflammatory genes and a gene expression analysis of primary breast cancers showed a strong correlation between TLR4 expression and expression of pro-inflammatory mediators. In line with this, TLR4 protein expression correlated with a decreased survival.
Conclusions
These findings suggest that TLR4 is expressed in a functional form in ER/PR-negative breast cancers. Studies regarding TLR4-antagonist therapies should be focusing on ER/PR-negative breast cancer particularly.
Electronic supplementary material
The online version of this article (doi:10.1186/s13058-015-0640-x) contains supplementary material, which is available to authorized users.
Introduction
Breast cancer is the most common form of cancer among women today [1]. The prognosis of breast cancer patients varies depending on the breast cancer subtype. Clinical breast cancer classification is based on expression of various immunohistochemical markers, with the hormone receptors being the most important. One of the worst prognosis subtypes is the triple-negative (TN) breast cancer subtype, where the malignant cells lack expression of the hormone receptors, estrogen receptor (ER) and progesterone receptor (PR), and human epidermal growth factor receptor 2 (Her2) (ER−PR−Her2−). The treatment options are few for patients with TN breast cancer [2–4].
Toll-like receptors (TLRs) are a family of receptors that are expressed on innate immune cells [5]. They are part of the pattern recognition receptor (PRR) family and recognize molecular patterns from pathogens (pathogen-associated molecular patterns; PAMPs) or from endogenous stress-induced proteins (damage-associated molecular patterns; DAMPs) [6–9]. Signaling via TLRs leads to activation of nuclear factor kappa B (NFκB) and a subsequent expression of pro-inflammatory genes [10]. There are 10 different TLRs (TLR1-10) in humans, and these are divided into two subgroups depending on cellular localization; on the surface of the cell (TLR1, TLR2, TLR4, TLR5 and TLR6), or in vesicles such as endoplasmic reticulum, endosomes or lysosomes (TLR3, TLR7, TLR8 and TLR9). Lately, expression of different TLRs has been described in various malignancies, although their function is as yet unclear [5, 11, 12].
TLR2 and TLR4 respond to the typical PAMP from Gram-negative bacteria, lipopolysaccharide (LPS). Different variants of LPS (from Escherichia coli and Salmonella typhimurium) induce different TLR-intracellular signals [13]. DAMPs can also bind to and activate TLR2 or TLR4, and two endogenous ligands that are well-described are HMGB1 and S100A9 [14–19]. To signal via TLR2 or TLR4, different ligands may also require the co-receptors CD14 or MD2 [20–23]. All TLR ligands initiate activation of NFκB, but also mitogen-activated protein kinase (MAPK) pathways that affect protein translation and processing rather than transcription can be activated [24]. TLR4 has previously been shown to be expressed in breast cancer [25, 26].
The transcriptional factors ERα and NFκB are synergistically interrelated, although their exact interactions are unknown [10, 27–31]. NFκB is a transcriptional factor that induces a wide array of pro-inflammatory mediators and is also related to several oncogenic processes [32]. Both ER and NFκB have previously been shown to attenuate each other in different ways. In line with this observation, ER− breast cancers have a stronger pro-inflammatory phenotype and microenvironment. NFκB has even been shown to downregulate ERα expression in breast cancer cells [29], but there is no direct proof that constitutive NFκB would generate ER− breast cancers in general. On the other hand, a recent positive synergy between ER and NFκB was published, where TNFα and estrogen were shown to remodulate the ERα-promoter landscape in an NFκB and FoxA1 dependent manner resulting in an altered gene expression pattern [33].
In this study we performed an analysis of TLR expression patterns and function in breast cancer. Using a carefully validated TLR4-specific antibody for immunohistochemistry (IHC), we found that TLR4 protein expression was primarily present in breast cancers of ER/PR-negative phenotype. Using three cell lines of ER+ phenotype and four cell lines of the TN phenotype, we further showed that the expressed TLR4 was biologically active and hence responding to both PAMPs and DAMPs, primarily in the TN breast cancer cell lines. Finally, TLR4 protein expression correlated with a decreased survival in a cohort of 144 primary breast cancer patients. We propose that novel therapies targeting TLR4 may be of value, in particular in ER/PR-negative breast cancers.
Methods
Cell culture
The human breast cancer cell lines MCF-7, T47D, MDA-MB-231 and MDA-MB-468 were purchased from ATCC and were cultured in RPMI 1640 medium supplemented with 10 % fetal bovine serum (FBS) (Biosera, Boussens, France), 1 % sodium pyruvate, 1 % HEPES and penicillin/streptomycin (100 U/ml and 100 μg/ml respectively); CAMA-1 (also purchased from ATCC) was cultured in MEM/EBSS supplemented with 10 % FBS and penicillin/streptomycin, and SUM-149 and SUM-159 were cultured in F-12 HAM’S medium supplemented with 5 % FBS, 1 mM L-Glutamine, 1 μg/ml hydrocortisone (BD BioScience, San Diego, CA, USA) and 5 μg/ml insulin (Novo Nordisk A/S, Måløv, Denmark). The SUM-149 and SUM-159 cell lines were produced by Professor S Ethier. Media and supplements were purchased from Thermo Scientific HyClone (South Logan, UT, USA) unless otherwise stated.
Compounds and cytokine analysis
LPS was purchased from Sigma Aldrich (St Louis, MO, USA) and originated from S. Typhimurium (LPS1) and E. Coli (LPS2), respectively. All stimulations were performed for a total of 6 h except for rhS100A9 (20 h). IL-1β and HMGB1 was from R&D Systems. Recombinant human S100A9 (rhS100A9) was a gift from Active Biotech AB and a detailed description on endotoxin-free S100A9 generation and purification has been published previously [15] and was used in the presence of calcium and zinc (Ca2+ ≥200 μM; 10 μM ZnCl2 [34, 35]). Supernatants from stimulated or siRNA transfected cells were harvested and analyzed using human inflammatory cytokine cytometric bead array (CBA; BD Biosciences, San Diego, CA, USA) according to the manufacturer’s instructions or using IL-6 and IL-8 Quantikine ELISA (R&D Systems, Minneapolis, MN, USA). Annexin V-allophycocyanin (APC) and propium iodide (PI) staining was performed according to the manufacturer’s instructions (BD Biosciences). The cycloheximide (CHX) experiments (Sigma Aldrich) where performed by adding 10 μg/ml CHX, with or without 100 ng/ml LPS for 6 h.
Preparation of necrotic cell supernatant (NCS)
Confluent monolayers of MDA-MB-231 cells were harvested by trypsinization and 3.2 × 106 cells were resuspended in 2 ml serum-free RPMI-1640 medium. Necrosis was induced by performing three freeze-thaw cycles and NCS was separated from the necrotic cell pellet by centrifugation.
Tissue microarray (TMA) and immunohistochemistry
The breast cancer cohort analyzed in this study consists of 144 patients diagnosed with invasive breast cancer at Skåne University Hospital, Malmö, Sweden, between 2001 and 2002. The cohort and TMA have previously been described in detail [36–38] and [39]. TMA sections of 4 μm thickness were mounted onto glass slides and deparaffinized followed by antigen retrieval using the PT-link system (DAKO, Glostrup, Denmark) and stained in an Autostainer Plus (DAKO) with the EnVisionFlex High pH-kit (DAKO). Antibody used for TLR4 IHC was anti-TLR4 NB100-56566 at 1:250 (Novus Biologicals, Littleton, CO, USA). TLR4 expression in TMA tumor samples was estimated as cytoplasmic staining intensity (0 = negative, 1 = weak, 2 = moderate, 3 = strong intensity and 4 = very strong intensity).
Ethical considerations
Ethical permit was obtained from the regional ethical committee at Lund University (Dnr 447/07), waiving the requirement for signed informed consent. Patients were offered to opt out of research. Ethical permission for using blood from healthy blood donors was obtained from the regional ethical committee at Lund University (Dnr 2012/689).
Gene expression profile array
The publicly available database R2: microarray analysis and visualization platform [40]; Tumor breast EXPO-351 was used for gene expression profile analysis.
Quantitative real-time PCR (RT-qPCR)
RNeasy Plus kit was used to extract total RNA according to the manufacturer’s instructions (Qiagen, Hilden, MD, USA). Random hexamers and the M-MuLV reverse transcriptase enzyme (Thermo Scientific) was used and quantitative real-time PCR (RT-qPCR) were performed in triplicates for the genes analyzed using Maxima SYBR Green/Rox (Thermo Scientific) according to the manufacturer’s instructions. RT-qPCR analysis was performed on the Mx3005P QPCR system (Agilent Technologies, Santa Clara, CA, USA) and the relative mRNA expression was normalized to YWHAZ, UBC and SDHA and calculated using the comparative cycle threshold (Ct) method [41]. For primers see Additional file 1: Table S1.
Transient transfections
siRNA transfections were performed using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA): 2 μM of the following silencer select siRNA oligonucleotides from Ambion (Carlsbad, CA, USA) were used; Silencer Select Negative Control #2: 4390846, siTLR2 #1: s168, siTLR2 #2: s170, siTLR4 #1: s14194, siTLR4 #2: s14195. Analyses were performed 48 h and 72 h post transfection. For luciferase assays, breast cancer cells were co-transfected using Lipofectamine 2000 with a total of 0.6 μg pNFκB-luciferase (BD Biosciences) and 0.06 μg TK-renilla-luciferase (Promega, Madison, WI, USA) plasmids and was subsequently analyzed using Dual-Luciferase Reporter System (Promega). For TLR4 transfections breast cancer cells were transfected using Lipofectamine 2000 with a total of 1.0 μg pDUO-MD2/hTLR4 or pUNOI-hTLR4-GFP (Invivogen, San Diego, CA, USA) per 24 wells for 72 h or 48 h, respectively, and was subsequently analyzed using immunofluorescence (×40 magnification) or ELISA as described in the figure legends.
Statistical analyses
Graph Pad Prism software was used to perform analysis of variance (ANOVA) or Students t test for the in vitro experiments as indicated. Spearman's Rho and the chi-square (χ2) test was used for correlation analysis and Kaplan-Meier analysis with the log-rank test was used to illustrate differences in survival. All statistical tests were two sided and P ≤0.05 was considered significant. Calculations were performed with IBM SPSS Statistics version 19.0 (SPSS Inc).
Results
TLR and co-receptor mRNA expression pattern in breast cancer cell lines
Most studies of TLRs in breast cancer have been performed using the ER+ cell line MCF-7 and the TN cell line MDA-MB-231 [5]. To our knowledge, a detailed comparison between ER+ and TN cell lines or cancers has not been published. We initially performed a broad analysis on TLR and TLR2/4 co-receptor (CD14 and MD2) mRNA expression patterns in various breast cancer cell lines. We used three cell lines with an ER+PR+ phenotype (MCF-7, T47D and CAMA-1) and four with an ER−PR−Her2− (TN) phenotype (MDA-MB-231; MDA-MB-468, SUM-149 and SUM-159). As shown in Fig. 1a-c, TLR2, TLR3 and TLR4 were preferentially expressed in the TN cell lines while TLR9 was more generally expressed (Fig. 1d). Only MDA-MB-468 had low/absent mRNA expression levels of TLR2 and TLR4 of the TN cell lines. Similarly, the TLR4 co-receptors CD14 and MD2 were expressed primarily in the TN cells lines (Fig. 1e, f). Again, the TN cell line MDA-MB-468 stood out with high CD14 mRNA expression levels, but low MD2 levels (Fig. 1e, f). This means that three out of the four TN breast cancer cell lines had the necessary proteins for a functional TLR4 signal to occur.
Fig. 1.
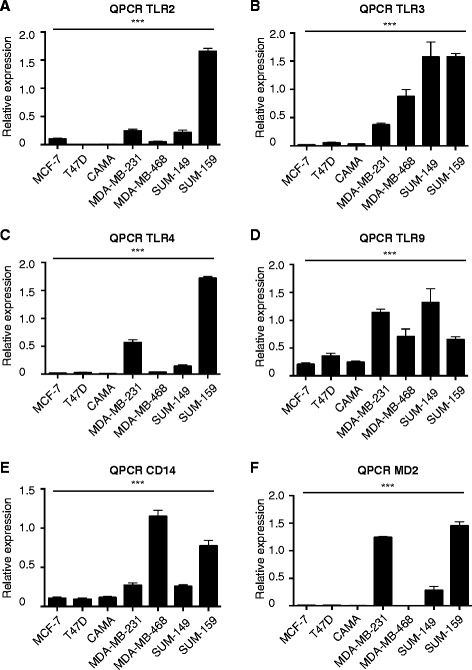
Breast cancer cell line mRNA expression levels of Toll-like receptor 2 (TLR2), TLR3, TLR4, TLR9 and co-receptors CD14 and MD2. a-f The relative expression of indicated mRNA using quantitative real-time PCR (QPCR) on mRNA from the cell lines indicated. Error bars standard error of the mean: n = 6 − 9; ***P <0.001 (analysis of variance)
The TLRs are functional and activation promotes expression of pro-inflammatory genes
To investigate whether the expressed TLRs were functional in the breast cancer cells following LPS stimulation, we analyzed the expression levels of some pro-inflammatory genes that are known targets for NFκB. The pro-inflammatory cytokines IL-6 and IL-8 were expressed at both protein (Fig. 2a and b) and mRNA (Fig. 2c and d) levels and only in the TN breast cancer cells but not the ER+ breast cancer cells. The TLR2/4-ligand LPS induces different TLR downstream signaling pathways when originating from different bacterial strains [13]. When the breast cancer cells were stimulated with LPS for 6 h (LPS1 from S. Typhimurium and LPS2 from E. Coli), we could see that IL-6, IL-8 and TNFα were induced by both LPS1 and LPS2 in the MDA-MB-231, SUM-149 and SUM-159 cell lines, but not the MDA-MB-468 cells with inherent low expression of TLR4 (Fig. 2b). A slight effect of LPS2 was seen in the TLR2/4-negative cell line, CAMA-1, which might represent unspecific binding to other receptors. This was supported by the finding that the mRNA levels of IL-6 and IL-8 increased in a similar manner in all TN cell lines except MDA-MB-468, and not in the CAMA cell line (Fig. 3a and data not shown). Interestingly, TLR signaling affected not only the transcription of IL-6 and IL-8, but also the protein translation as judged by cycloheximide (CHX) experiments showing a decreased release of both IL-6 and IL-8 after LPS1 stimulation upon simultaneous treatment with LPS and CHX (Fig. 3b).
Fig. 2.
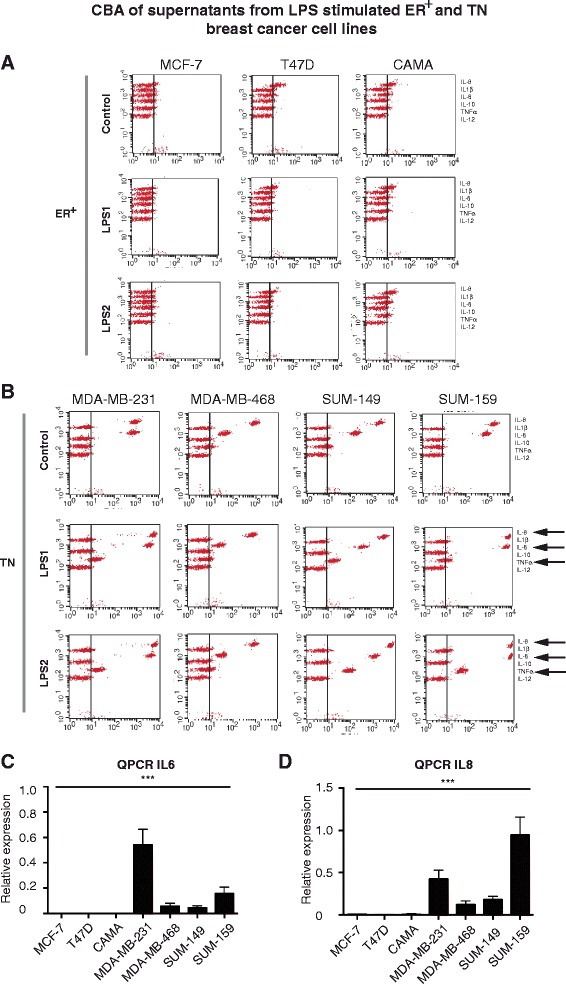
Lipolysaccharide (LPS) induced cytokine release in human breast cancer cells in vitro. Release of cytokines by breast cancer cells of estrogen receptor-positive (ER +) origin (a) and triple-negative (TN) origin (b) was analyzed using cytokine bead array (CBA). Unstimulated breast cancer cells of TN origin (b) produce IL-6 and IL-8 at high levels. LPS1 (from S. Typhimurium) and LPS2 (from E. Coli) stimulation for 6 h induced release of IL-8, IL-6 and TNFα from indicated breast cancer cell lines. At least three experiments were performed for each cell line. The relative mRNA expression of IL-6 (c) and IL-8 (d) in unstimulated cells was measured using quantitative real-time PCR (QPCR). At least five experiments were performed for each cell line. Error bars standard error of the mean; ***P <0.001 (analysis of variance)
Fig. 3.
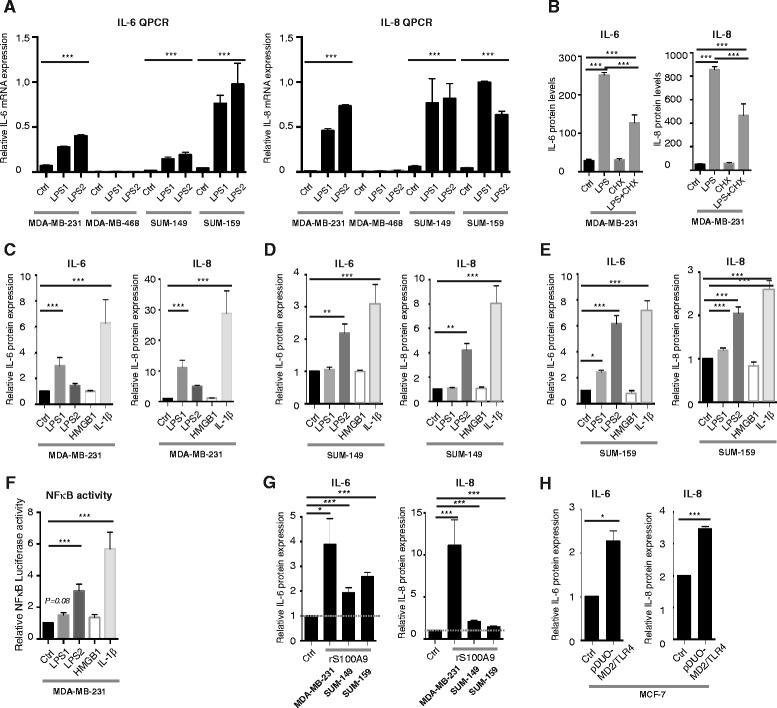
Pathogen-associated molecular patterns (PAMPs) and damage-associated molecular pattern (DAMPs) induce cytokine expression in human breast cancer cells in vitro. a Relative expression of IL-6 and IL-8 mRNAs after 6 h stimulation with lipopolysaccharide 1 (LPS1) and LPS2, using quantitative real-time PCR (QPCR) on mRNA from the cell lines indicated; n = 6. Error bars standard error of the mean (SEM); ***P <0.001 (analysis of variance (ANOVA)). b Release of IL-6 and IL-8 after stimulation with LPS1 and cycloheximide for 6 h, using ELISA on supernatants from stimulated MDA-MB-231 cells; n = 3. c-e Relative release of IL-6 and IL-8 after stimulation with LPS1, LPS2, the DAMP HMGB1 or IL-1β for 6 h, using ELISA on supernatants from stimulated MDA-MB-231 cells (c), SUM-149 cells (d) and SUM-159 cells (e); n = 10. Error bars SEM; *P <0.05, ***P <0.001 (ANOVA). f Dual luciferase reporter assays of MDA-MB-231 cells transfected with an NFκB reporter. TK-Renilla was co-transfected as control (Ctrl). LPS1, LPS2, HMGB1 or IL-1β was added to stimulate NFκB activity as described in “Methods”; n = 14. Error bars SEM; ***P <0.001 (ANOVA). g The relative release of IL-6 and IL-8 after stimulation with the DAMP S100A9 for 20 h, using ELISA on supernatants from stimulated MDA-MB-231, SUM-149 and SUM-159 cells; n = 6. Error bars SEM. *P <0.05, ***P <0.001 (ANOVA). h Transient transfection of pDUO-MD2/hTLR4 for 72 h in MCF-7 cells induces release of both IL-6 and IL-8 measured by ELISA; n = 6. Error bars SEM; *P <0.05, ***P <0.001 (Students t test)
A TLR4-specific DAMP induces pro-inflammatory cytokines in breast cancer cells
We further investigated whether DAMPs could induce TLR2/4-signaling in TN cell lines (MDA-MB-231, SUM-149 and SUM-159) and found that LPS (LPS1 and LPS2), but not the endogenous DAMP HMGB1, significantly induced IL-6 and IL-8 release in MDA-MB-231 cells and SUM-159 cells (Fig. 3c and e), whereas in SUM-149 cells LPS2 induced IL-6 and IL-8 release primarily (Fig. 3d). This finding might reflect that TLR2/4-induced transcription v/s translation might be differentially regulated in breast cancer cells. IL-1β was used as a positive control. Using dual luciferase assays and an NFκB reporter, we confirmed that LPS stimulation of MDA-MB-231 cells induced activation of NFκB but HMGB1 did not (Fig. 3f). We continued with another cancer-related DAMP reported to be a TLR4 ligand, the S100A9 protein [35]. Indeed, stimulating MDA-MB-231, SUM-149 and SUM-159 cells with rS100A9 for 20 h induced a significant increase in both IL-6 and IL-8 release (Fig. 3g). We also tested whether stimulating with HMGB1 for 20 h would induce cytokine release but with negative results (data not shown). Finally, by introducing the MD2/TLR4 complex (pDUO-MD2/TLR4) in otherwise negative MCF-7 cells, we could see a significant expression of both IL-6 and IL-8 as compared to control MCF-7 cells (Fig. 3h). pDUO-MD2/TLR4 is an expression vector that is designed to co-express the MD2 and TLR4 genes needed to interact with each other for functional signaling to occur upon ligand binding [42].
Constitutive expression of IL-6 and IL-8 is inhibited by silencing of TLR4
The impact of TLR4 signaling (possibly by endogenous DAMPs) on the constitutive expression of IL-6 and IL-8 seen in the MDA-MB-231 cells was analyzed. To this end we used negative control (nc) siRNA or siRNA specific for TLR2 (siTLR2#1 and #2) and TLR4 (siTLR4#1 and #2) (Fig. 4a), and analyzed the IL-6 and IL-8 levels 72 h post transfection. Both siTLR2 and siTLR4 slightly decreased the endogenous levels of IL-6 and IL-8 (Fig. 4b).
Fig. 4.
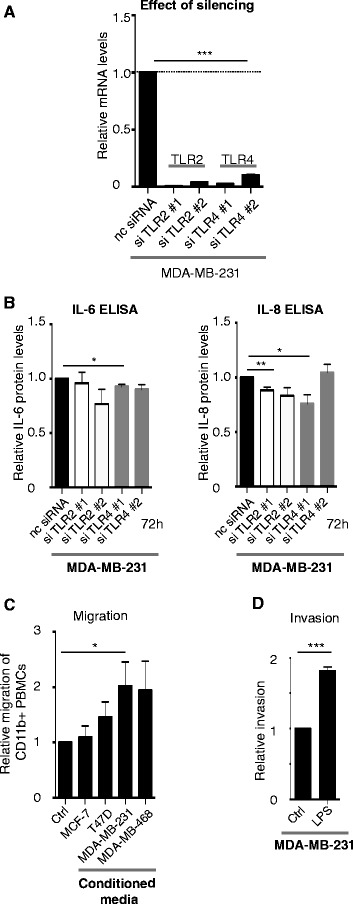
Toll-like receptor 4 (TLR4) silencing decreases endogenous levels of pro-inflammatory cytokines. a Effect of TLR2/4 silencing in breast cancer cells transfected with negative control (nc) siRNA, or siRNA directed against TLR2 mRNA (si#1 and si#2) or TLR4 mRNA (si#1 and si#2) was analyzed using quantitative real-time PCR; ***P <0.001 (analysis of variance (ANOVA)). b IL-6 (left) and IL-8 (right) ELISA on supernatants from MDA-MB-231 breast cancer cells transfected with nc siRNA, or siRNA directed against TLR2 mRNA (si#1 and si#2) or TLR4 mRNA (si#1 and si#2); n = 4. Error bars standard error of the mean (SEM); *P <0.05, **P <0.01, ***P <0.001 (ANOVA). c Boyden chamber migration assays. Migration of primary human myeloid cells towards supernatants from different cell lines indicated. Human primary peripheral blood mononuclear cells were isolated as previously described [48] and allowed to migrate through a Costar Transwell® Permeable Support 8.0-μm 24-well plate (Corning) to the supernatants of breast cancer supernatants cultured under serum-free conditions. Percentage of migrated CD11b+ cells was analyzed using a flow cytometer and CD11b-APC antibodies (BD Sciences); n = 4. Error bars SEM; *P <0.05, **P <0.01, ***P <0.001 (ANOVA). d Matrigel invasion assays. Invasion of lipopolysaccharide (LPS)-stimulated/un-stimulated MDA-MB-231 cells into matrigel invasion chambers (BD Sciences) as indicated: 25 × 103 MDA-MB-231 cells were stimulated or not with LPS and allowed to invade from 72 h. Amount of invaded cells was analyzed using crystal violet staining and manual counting in four separate experiments; n = 4. Error bars SEM; *P <0.05, **P <0.01, ***P <0.001 (Student’s t test)
TLR2/4 expression affects migration and invasion
The TLR2/4-induced pro-inflammatory cytokines can be chemoattractants for myeloid cells. We therefore next investigated whether primary human CD11b+ myeloid cells would migrate toward supernatants collected from breast cancer cells with a TN phenotype as compared to ER+ breast cancer cells. Indeed, primary human myeloid cells migrated significantly more to supernatants collected from MDA-MB-231 cells as compared to from ER+ MCF-7 or T47D cells, but as expected also to the TLR4-negative, but pro-inflammatory cytokine-secreting, MDA-MB-468 cells (Fig. 4c).
Other parameters that might be affected by TLR4 expression in breast cancer cells were also investigated; invasion, apoptosis and proliferation (Fig. 4d and Additional file 2: Figure S1A-B). In summary, invasion into matrigel invasion chambers by MDA-MB-231 breast cancer cells was increased when the TLR2/4 ligand LPS was added to the invading cells (Fig. 4d). Apoptosis in MDA-MB-231 breast cancer cells where TLR2 or TLR4 was silenced gave either conflicting results (TLR2) or was not affected (TLR4) (Additional file 2: Figure S1A), and finally proliferation using 3H-incorporation assays of MDA-MB-231 breast cancer cells where TLR4 was silenced was not affected as compared to control (Additional file 2: Figure S1B).
To finally evaluate if other relevant TLRs were functional in the TN breast cancer cells we also performed stimulations of MDA-MB-231 cells with necrotic cell supernatant (NCS; TLR3 ligands [43]). Release of IL-8 but not IL-6 was affected by addition of NCS in a concentration-dependent manner (see Additional file 2: Figure S1C).
TLR4 is expressed in ER/PR-negative breast cancers and correlates with poor survival
TLR proteins are difficult to analyze because the antibody specificity is generally poor. We carefully evaluated several antibodies and found one to be highly specific. This antibody was confirmed first by using human tonsil tissue as positive control, showing the typical pattern of TLR4-expressing cells surrounding the follicles (Fig. 5a). Having optimized IHC, we subsequently stained formalin-fixed and paraffin-embedded cell pellets of the cell lines used in this study. All ER+ cell lines were negative for TLR4, whereas three out of four TN cell lines displayed marked cytoplasmic positivity, corroborating our mRNA results (Fig. 5b). The cytoplasmic localization of TLR4 in breast cancer cells was supported by transfection of breast cancer cells using a green fluorescent protein (GFP)-tagged TLR4 plasmid (pUNOI-hTLR4-GFP) (Fig. 5c). We found that in cells expressing both the TLR4 co-receptors MD2 and CD14 (MDA-MB-231 cells; Fig. 5c left) TLR4-GFP was expressed in a vesicular pattern in the cytosol, whereas in breast cancer cells lacking both MD2 and CD14 (MCF-7 cells; Fig. 5c right), TLR4-GFP was expressed evenly in the cytoplasm.
Fig. 5.
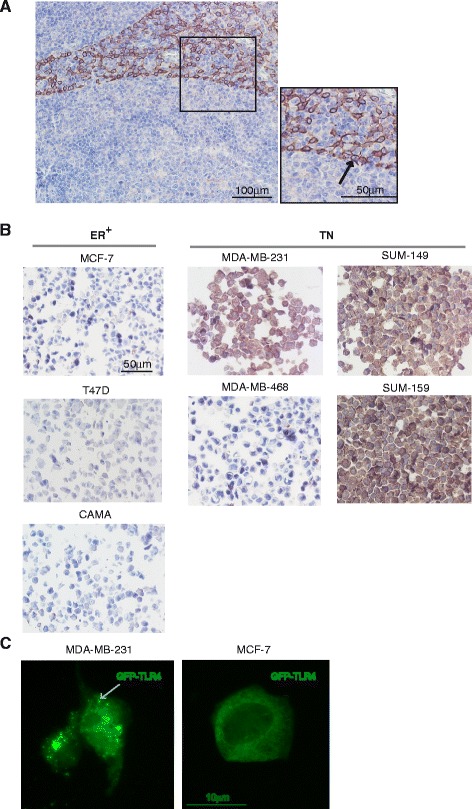
Analysis of Toll-like receptor 4 (TLR4) protein expression in tonsil and breast cancer cells lines. Immunohistochemical analysis using an anti-human TLR4-specific antibody on paraffin-embedded tonsil (a) or cell pellets from the cell lines indicated (b). Arrow indicates the membranous staining. c MDA-MB-231 cells expressing the TLR co-receptors MD2 and CD14 but not ERα left), and MCF-7 cells expressing ERα but not MD2 or CD14 (right) were transfected with GFP-tagged hTLR4 (pUNOI-hTLR4-GFP; Invivogen) for 48 h. Localization was investigated using immunofluorescence microscopy. The green fluorescent protein (GFP)-tagged hTLR4 was expressed in a vesicular pattern in the cytoplasm of MDA-MB-231 cells and evenly in the cytoplasm of MCF-7 cells. Arrow indicates the vesicular pattern. ER+ estrogen receptor-positive, TN triple-negative
A TMA of 144 breast cancer patients was subsequently stained and analyzed for correlation with other histological and clinical parameters. The staining was judged as cytoplasmic staining of intensities 0, 1, 2, 3 and 4 (see “Methods”) (Fig. 6a). When the analysis was performed these cytosolic scoring parameters were grouped into 0–2 (0) and 3–4 (1) critical cutoffs. Table 1 shows the clinical parameters and correlations found with the TLR4-specific antibody. The intensity groups 3–4 (1) correlated significantly with the ER/PR-negative patient group and the basal-like status marker CK5. It did not correlate to Her2 expression (or lack of expression), however (Tables 1, 2 and 3). Tables 2 and 3 specifically show the correlation between Her2 and TLR4. Survival curves using the two cutoff groups indicate that as expected, the group with high TLR4 expression had significantly worse recurrence-free survival (P <0.029) (Fig. 6b). Membranous staining was also scored (0, 1) but there was no strong correlation (data not shown).
Fig. 6.
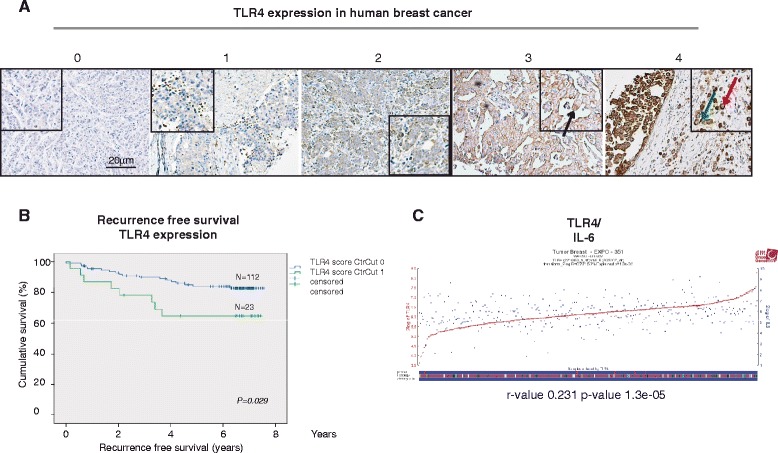
Toll-like receptor 4 (TLR4) expression in breast tumors and survival curves. a Immunohistochemical TLR4 expression sample images from breast cancer tissue microarray cores, from left to right: negative (0), weak (1), moderate (2), strong (3) and very strong (4). Boxes with magnification and arrows to indicate localization (black arrow membranous, green arrow leukocyte, red arrow cytoplasmic staining. b Kaplan-Meier curves illustrating differences in recurrence-free survival according to TLR4 cytoplasmic expression in breast tumors. Cytosolic scoring parameters were grouped into 0–2 (0) and 3–4 (1) critical cutoffs. c Gene expression profile analysis of TLR4 mRNA in relation to IL-6 in (R2: microarray analysis and visualization platform [40]; Tumor breast EXPO-351)
Table 1.
Correlation between TLR4 expression and clinicopathologic features in primary breast cancer (n = 144 patients)
| Toll-like receptor 4 | |||
|---|---|---|---|
| Clinicopathologic features | Correlation coefficient | P value (two-tailed) | Number |
| Age | −0,042 | 0,629 | 135 |
| Nodal stage | 0,042 | 0,646 | 122 |
| Tumor size | −0,006 | 0,944 | 135 |
| Ki67 | 0,003 | 0,971 | 117 |
| nhg | 0,094 | 0,276 | 135 |
| Her2 subtype | 0,158 | 0,072 | 131 |
| ER status | −0,170 | 0,049* | 135 |
| PR status | −0,206 | 0,016* | 135 |
| CK5 | 0,184 | 0,037* | 129 |
*P <0.05 using SPSS and Spearman’s Rho test. Her2 human epidermal growth factor receptor 2, ER estrogen receptor, PR progesterone receptor, nhg Nottingham histological grade
Table 2.
Crosstab over Toll-like receptor 4 (TLR4) (0, 1) and Her2 (0, 1)
| TLR4 (0) | TLR4 (1) | Total | ||
|---|---|---|---|---|
| Her2 | (0) | 83 | 33 | 116 |
| (1) | 6 | 6 | 12 |
Chi square test value 2.366, P = 0.124. Her2 human epidermal growth factor receptor 2
Table 3.
Crosstab over Toll-like receptor 4 (TLR4) (0,1) and ER/PR-negative/positive Her2+ tumors
| TLR4 (0) | TLR4 (1) | Total | |
|---|---|---|---|
| ER/PR-negative Her2+ | 1 | 3 | 4 |
| ER/PR-positive Her2+ | 5 | 3 | 8 |
Chi square test value 1.375, P = 0.241. Her2 human epidermal growth factor receptor 2, ER estrogen receptor, PR progesterone receptor
Finally, using a publicly available data site (R2: microarray analysis and visualization platform [40]; Tumor breast EXPO-351) with gene expression profiles of 351 primary breast cancers, we found positive correlation between expression of TLR4 mRNA and IL-6 (r value 0.231, P = 1.3e-05) (Fig. 6c).
Discussion
Breast cancers with an ER-negative phenotype have previously been shown to promote a strong pro-inflammatory microenvironment [44]. Furthermore, historically there is a negative relationship between ERα and NFκB that has previously been described in depth [10, 27–30]. Despite the fact that ER signaling can inhibit NFκB activity and vice versa, there is no evidence that the development of ER-negative breast tumors are caused by constitutive NFκB activity. Rather, it may be a result of the typical molecular gene landscapes found in luminal A compared to basal breast cancers, respectively. A link between PRR, e.g., TLR-induced activation of NFκB in breast cancer and its relation to expression of ER, has not been described. Both IL-6 and IL-8 can be highly expressed in TN breast cancers and this has partly been attributed to constitutively active NFκB [44]. In order to investigate whether TLRs, which are known to induce strong activation of NFκB, are expressed primarily in TN breast cancers and if this might affect the expression of pro-inflammatory genes in the same, we investigated the functional role of TLRs and co-receptors in breast cancer.
In immune cells, TLR expression is generally inhibited by prolonged activation of NFκB [45]. In contrast, our findings show that TLRs (TLR2, TLR 3, TLR 4) are preferentially expressed in TN breast cancer cell lines with constitutive NFκB activity, suggesting that the TLRs may be responsible for the NFκB activation pathway rather than induced by the same. Although introduction of a functional MD2/TLR4 complex in an ER+ cell line has been shown to induce expression of pro-inflammatory cytokines, silencing of TLR4 in TN cells only caused a slight decrease in pro-inflammatory mediator release, indicating that the constitutive NFκB activation seen in TN cells in general is caused by another mechanism [44]. Apart from MDA-MB-468, the TN breast cancer cells were also demonstrated to express the co-receptors CD14 and MD2 meaning that they harbor the necessary proteins for a functional TLR4 signal to occur [20–22]. The exception, MDA-MB-468, only expressed CD14 and in line with this showed no biological TLR function. In the patient cohort we found correlation between TLR4 expression and ER/PR-negative tumors, but not TN tumors. This strengthens the interrelationship between TLR4, ER and NFκB activity, as expression of HER2 was not correlated in the TLR4-expressing primary tumors. We did not perform our in vitro analyses on any Her2+ breast cancer cell line. Interestingly, the typical membrane staining seen in immune cells was not as obvious in the malignant cells, indicating that different regulation of TLR expression and signaling could be possible in cancers. This was previously described in neuroblastoma cells [46] and is also supported by our finding that a GFP-tagged hTLR4 primarily showed a vesicular cytoplasmic localization in breast cancer cells. Furthermore and supporting this observation, it was recently reported that the TLR4-specific DAMP, S100A9, needs to be internalized to be able to signal via TLR4 [15]. Indeed, scoring of membrane TLR4 expression in breast cancer lesions did not reveal as much as that of cytoplasmic staining, and both TLR2 and TLR4 have been reported to be expressed intracellularly as well [47].
The DAMP, HMGB1, has previously been shown to signal via TLR4 in myeloid cells [6, 19]. Although we have also previously shown this in primary myeloid cells [48], we did not see an effect of HMGB1 on breast cancer cells in vitro. This could be due to different culture conditions, or to receptor expression patterns in myeloid as compared to cancer cells which might also reflect the fact that different sources of LPS generate different signals in the different cell lines in this study. Instead, we could show that the DAMP, S100A9, also induced pro-inflammatory proteins in breast cancer cells expressing TLR4.
It has previously been shown that NFκB [29] and targets (IL-6) [49] can downregulate ERα. We also investigated whether overexpression of TLR4 would affect ERα expression per se in ER+ MCF-7 cells. We did see a slight although non-significant decrease of ERα after 72 h (data not shown), a finding that is probably explained by the significantly increased levels of IL-6 we observed in these experiments (Fig. 3h). In spite of this, we suggest that the ER/PR-negative breast cancer subtype probably is not caused by expression of TLRs and their downstream mediators, but rather further affected by them. Perhaps the expression of TLRs is even affected by the ERα/FoxA1/GATA3 network [50]. We show that both PAMPs and DAMPs induced release of pro-inflammatory mediators in ER/PR-negative breast cancer cells in vitro, a process that was regulated both at the transcriptional and post-transcriptional level. This means that although ER-negative breast cancer cells express high endogenous levels of pro-inflammatory mediators, a functional TLR4 is still likely to enhance their phenotype and surrounding inflammatory microenvironment, and this is also reflected by the decreased recurrence-free survival seen in the patients with tumors expressing TLR4 at high levels. In support of this, previous studies have shown that TLR4 expression promotes metastasis in a breast cancer model, an effect that was even enhanced by Paclitaxel [25, 26].
Conclusion
The findings presented in this study suggest that TLR4 is expressed in a functional form in ER/PR-negative breast cancers primarily. We suggest that TLR4 should be viewed as a possible therapeutic target in ER/PR-negative breast cancers to decrease the pro-inflammatory environment and hence the metastatic spread.
Acknowledgements
The authors thank Ms Elise Nilsson for professional technical skills in preparation of TMAs and IHC. This work was supported by grants from the Swedish Research Council, The Swedish Cancer Society, Kocks Foundation, Österlunds Foundation, Gunnar Nilsson Cancer Foundation, MAS Cancer Foundation, and Åke Wibergs Foundation.
Abbreviations
- ANOVA
analysis of variance
- CBA
cytokine bead array
- CHX
cycloheximide
- DAMP
damage-associated molecular pattern
- ELISA
enzyme-linked immunosorbent assay
- FBS
fetal bovine serum
- ER
estrogen receptor
- GFP
green fluorescent protein
- Her2
human epidermal growth factor receptor 2
- HMGB1
High mobility group protein B1
- IL
interleukin
- LPS
lipopolysaccharide
- MAPK
mitogen-activated protein kinase
- NCS
necrotic cell supernatant
- NFκB
nuclear factor kappa B
- PAMP
pathogen associated molecular pattern
- PR
progesterone receptor
- PRR
pattern recognition receptor
- RT-qPCR
quantitative real-time PCR
- TLR
toll-like receptor
- TMA
tissue microarray
- TN
triple-negative (ER−PR−Her2−)
- TNF
tumor necrosis factor
Additional files
Primer sequences. (PDF 94 kb)
A Annexin V staining of MDA-MB-231 cells using flow cytometry to investigate apoptosis of MDA-MB-231 cells transfected with negative control (nc) siRNA, or siRNA directed against TLR2 mRNA (si#1 and si#2) or TLR4 mRNA (si#1 and si#2). TLR2 (si#1 and #2) gave contradicting results while TLR4 si#1 and #2 gave no effect (n = 3). Error bars indicate standard error of the mean (SEM); *P <0.05, **P <0.01, ***P <0.001 (analysis of variance (ANOVA)). B 3H-incorporation assay using previously published methods [48] to investigate proliferation of MDA-MB-231 cells transfected with negative control (nc) siRNA, or siRNA directed against TLR2 mRNA (si#1 and si#2) or TLR4 mRNA (si#1 and si#2) (n = 6). Error bars indicate SEM; *P <0.05 ** P < 0.01, ***P <0.001 (ANOVA). C IL-6 (left) and IL-8 (right) ELISA performed on supernatants from MDA-MB-231 breast cancer cells stimulated with increasing amounts of necrotic cell supernatants (NCS): 100 μl = 1:1, 50 μl = 1:4, 25 μl = 1:8 (n = 4). Error bars indicate SEM; *P <0.05, **P <0.01, ***P <0.001 (ANOVA). (PDF 176 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MM performed the majority of experiments and analyzed data. RA and CB performed experiments and analyzed data. LHS was involved in revising the manuscript critically for important intellectual content, contributed to analysis and interpretation of data and also was responsible for linguistic correction. SPE produced and provided the TN breast cancer cell lines SUM149 and SUM159, contributed to analysis and interpretation of data and was involved in revising the manuscript critically for important intellectual content. As a clinical pathologist MEJ verified the IHC staining, and helped and mentored RA in scoring of the IHC. KJ was responsible for the breast cancer clinical samples and verified the IHC staining and TMA scoring. KL designed the experiments, wrote the manuscript and interpreted and analyzed the data. All authors read and approved the manuscript and were involved in revising the manuscript critically for important intellectual content.
Contributor Information
Meliha Mehmeti, Email: Meliha.Mehmeti@med.lu.se.
Roni Allaoui, Email: Roni.Allaoui@med.lu.se.
Caroline Bergenfelz, Email: Caroline.Bergenfelz@med.lu.se.
Lao H. Saal, Email: lao.saal@med.lu.se
Stephen P. Ethier, Email: ethier@musc.edu
Martin E. Johansson, Email: Martin.Johansson@med.lu.se
Karin Jirström, Email: Karin.Jirstrom@med.lu.se.
Karin Leandersson, Phone: +4640333154, Email: Karin.Leandersson@med.lu.se.
References
- 1.Kamangar F, Dores GM, Anderson WF. Patterns of cancer incidence, mortality, and prevalence across five continents: defining priorities to reduce cancer disparities in different geographic regions of the world. J Clin Oncol. 2006;24:2137–50. doi: 10.1200/JCO.2005.05.2308. [DOI] [PubMed] [Google Scholar]
- 2.Schnitt SJ. Classification and prognosis of invasive breast cancer: from morphology to molecular taxonomy. Mod Pathol. 2010;23:S60–4. doi: 10.1038/modpathol.2010.33. [DOI] [PubMed] [Google Scholar]
- 3.Allred DC, Brown P, Medina D. The origins of estrogen receptor alpha-positive and estrogen receptor alpha-negative human breast cancer. Breast Cancer Res. 2004;6:240–5. doi: 10.1186/bcr938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98:10869–74. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhatelia K, Singh K, Singh R. TLRs: linking inflammation and breast cancer. Cell Signal. 2014;26:2350–7. doi: 10.1016/j.cellsig.2014.07.035. [DOI] [PubMed] [Google Scholar]
- 6.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 7.Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev. 2009;22:240–73. doi: 10.1128/CMR.00046-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Piccinini AM, Midwood KS. DAMPening inflammation by modulating TLR signalling. Mediators Inflamm. 2010. doi:10.1155/2010/672395. [DOI] [PMC free article] [PubMed]
- 9.Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev. 2012;249:158–75. doi: 10.1111/j.1600-065X.2012.01146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blasius AL, Beutler B. Intracellular toll-like receptors. Immunity. 2010;32:305–15. doi: 10.1016/j.immuni.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 11.Lu H. TLR Agonists for Cancer Immunotherapy: Tipping the balance between the immune stimulatory and inhibitory effects. Front Immunol. 2014;5:83. doi: 10.3389/fimmu.2014.00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang JQ, Jeelall YS, Ferguson LL, Horikawa K. Toll-Like Receptors and Cancer: MYD88 Mutation and Inflammation. Front Immunol. 2014;5:367. doi: 10.3389/fimmu.2014.00367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zughaier SM, Zimmer SM, Datta A, Carlson RW, Stephens DS. Differential induction of the toll-like receptor 4-MyD88-dependent and -independent signaling pathways by endotoxins. Infect Immun. 2005;73:2940–50. doi: 10.1128/IAI.73.5.2940-2950.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benedyk M, Sopalla C, Nacken W, Bode G, Melkonyan H, Banfi B, et al. HaCaT keratinocytes overexpressing the S100 proteins S100A8 and S100A9 show increased NADPH oxidase and NF-kappaB activities. J Invest Dermatol. 2007;127:2001–11. doi: 10.1038/sj.jid.5700820. [DOI] [PubMed] [Google Scholar]
- 15.Riva M, Kallberg E, Bjork P, Hancz D, Vogl T, Roth J, et al. Induction of nuclear factor-kappaB responses by the S100A9 protein is Toll-like receptor-4-dependent. Immunology. 2012;137:172–82. doi: 10.1111/j.1365-2567.2012.03619.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roth J, Vogl T, Sorg C, Sunderkotter C. Phagocyte-specific S100 proteins: a novel group of proinflammatory molecules. Trends Immunol. 2003;24:155–8. doi: 10.1016/S1471-4906(03)00062-0. [DOI] [PubMed] [Google Scholar]
- 17.Sinha P, Okoro C, Foell D, Freeze HH, Ostrand-Rosenberg S, Srikrishna G. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol. 2008;181:4666–75. doi: 10.4049/jimmunol.181.7.4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sunahori K, Yamamura M, Yamana J, Takasugi K, Kawashima M, Yamamoto H, et al. The S100A8/A9 heterodimer amplifies proinflammatory cytokine production by macrophages via activation of nuclear factor kappa B and p38 mitogen-activated protein kinase in rheumatoid arthritis. Arthritis Res Ther. 2006;8:R69. doi: 10.1186/ar1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol. 2011;29:139–62. doi: 10.1146/annurev-immunol-030409-101323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.da da Silva CJ, Soldau K, Christen U, Tobias PS, Ulevitch RJ. Lipopolysaccharide is in close proximity to each of the proteins in its membrane receptor complex. transfer from CD14 to TLR4 and MD-2. J Biol Chem. 2001;276:21129–35. doi: 10.1074/jbc.M009164200. [DOI] [PubMed] [Google Scholar]
- 21.Nagai Y, Akashi S, Nagafuku M, Ogata M, Iwakura Y, Akira S, et al. Essential role of MD-2 in LPS responsiveness and TLR4 distribution. Nat Immunol. 2002;3:667–72. doi: 10.1038/ni809. [DOI] [PubMed] [Google Scholar]
- 22.Shimazu R, Akashi S, Ogata H, Nagai Y, Fukudome K, Miyake K, et al. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J Exp Med. 1999;189:1777–82. doi: 10.1084/jem.189.11.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miyake K. Roles for accessory molecules in microbial recognition by Toll-like receptors. J Endotoxin Res. 2006;12:195–204. doi: 10.1179/096805106X118807. [DOI] [PubMed] [Google Scholar]
- 24.Troutman TD, Bazan JF, Pasare C. Toll-like receptors, signaling adapters and regulation of the pro-inflammatory response by PI3K. Cell Cycle. 2012;11:3559–67. doi: 10.4161/cc.21572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rajput S, Volk-Draper LD, Ran S. TLR4 is a novel determinant of the response to paclitaxel in breast cancer. Mol Cancer Ther. 2013;12:1676–87. doi: 10.1158/1535-7163.MCT-12-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Volk-Draper L, Hall K, Griggs C, Rajput S, Kohio P, DeNardo D, et al. Paclitaxel therapy promotes breast cancer metastasis in a TLR4-dependent manner. Cancer Res. 2014;74:5421–34. doi: 10.1158/0008-5472.CAN-14-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Biswas DK, Dai SC, Cruz A, Weiser B, Graner E, Pardee AB. The nuclear factor kappa B (NF-kappa B): a potential therapeutic target for estrogen receptor negative breast cancers. Proc Natl Acad Sci U S A. 2001;98:10386–91. doi: 10.1073/pnas.151257998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Biswas DK, Shi Q, Baily S, Strickland I, Ghosh S, Pardee AB, et al. NF-kappa B activation in human breast cancer specimens and its role in cell proliferation and apoptosis. Proc Natl Acad Sci U S A. 2004;101:10137–42. doi: 10.1073/pnas.0403621101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Van Laere SJ, Van der Auwera I, Van den Eynden GG, van Dam P, Van Marck EA, Vermeulen PB, et al. NF-kappaB activation in inflammatory breast cancer is associated with oestrogen receptor downregulation, secondary to EGFR and/or ErbB2 overexpression and MAPK hyperactivation. Br J Cancer. 2007;97:659–69. doi: 10.1038/sj.bjc.6603906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou Y, Eppenberger-Castori S, Marx C, Yau C, Scott GK, Eppenberger U, et al. Activation of nuclear factor-kappaB (NFkappaB) identifies a high-risk subset of hormone-dependent breast cancers. Int J Biochem Cell Biol. 2005;37:1130–44. doi: 10.1016/j.biocel.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 31.Sas L, Lardon F, Vermeulen PB, Hauspy J, Van Dam P, Pauwels P, et al. The interaction between ER and NFkappaB in resistance to endocrine therapy. Breast Cancer Res. 2012;14:212. doi: 10.1186/bcr3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mantovani A. Molecular pathways linking inflammation and cancer. Curr Mol Med. 2010;10:369–73. doi: 10.2174/156652410791316968. [DOI] [PubMed] [Google Scholar]
- 33.Franco HL, Nagari A, Kraus WL. TNFalpha signaling exposes latent estrogen receptor binding sites to alter the breast cancer cell transcriptome. Mol Cell. 2015. doi:10.1016/j.molcel.2015.02.001. [DOI] [PMC free article] [PubMed]
- 34.Markowitz J, Carson WE., 3rd Review of S100A9 biology and its role in cancer. Biochim Biophys Acta. 1835;2013:100–9. doi: 10.1016/j.bbcan.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bjork P, Bjork A, Vogl T, Stenstrom M, Liberg D, Olsson A, et al. Identification of human S100A9 as a novel target for treatment of autoimmune disease via binding to quinoline-3-carboxamides. PLoS Biol. 2009;7:e97. doi: 10.1371/journal.pbio.1000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Elkabets M, Gifford AM, Scheel C, Nilsson B, Reinhardt F, Bray MA, et al. Human tumors instigate granulin-expressing hematopoietic cells that promote malignancy by activating stromal fibroblasts in mice. J Clin Invest. 2011;121:784–99. doi: 10.1172/JCI43757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Svensson KJ, Christianson HC, Kucharzewska P, Fagerstrom V, Lundstedt L, Borgquist S, et al. Chondroitin sulfate expression predicts poor outcome in breast cancer. Int J Oncol. 2011;39:1421–8. doi: 10.3892/ijo.2011.1164. [DOI] [PubMed] [Google Scholar]
- 38.Gronberg M, Fjallskog ML, Jirstrom K, Janson ET. Expression of ghrelin is correlated to a favorable outcome in invasive breast cancer. Acta Oncol. 2011. doi:10.3109/0284186X.2011.631576. [DOI] [PubMed]
- 39.Medrek C, Ponten F, Jirstrom K, Leandersson K. The presence of tumor associated macrophages in tumor stroma as a prognostic marker for breast cancer patients. BMC Cancer. 2012;12:306. doi: 10.1186/1471-2407-12-306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.http://r2.amc.nl.
- 41.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:RESEARCH0034. [DOI] [PMC free article] [PubMed]
- 42.Needham BD, Trent MS. Fortifying the barrier: the impact of lipid A remodelling on bacterial pathogenesis. Nat Rev Microbiol. 2013;11:467–81. doi: 10.1038/nrmicro3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lim DM, Wang ML. Toll-like receptor 3 signaling enables human esophageal epithelial cells to sense endogenous danger signals released by necrotic cells. Am J Physiol Gastrointest Liver Physiol. 2011;301:G91–9. doi: 10.1152/ajpgi.00471.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yamaguchi N, Ito T, Azuma S, Ito E, Honma R, Yanagisawa Y, et al. Constitutive activation of nuclear factor-kappaB is preferentially involved in the proliferation of basal-like subtype breast cancer cell lines. Cancer Sci. 2009;100:1668–74. doi: 10.1111/j.1349-7006.2009.01228.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Biswas SK, Lopez-Collazo E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. 2009;30:475–87. doi: 10.1016/j.it.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 46.Hassan F, Islam S, Tumurkhuu G, Naiki Y, Koide N, Mori I, et al. Intracellular expression of toll-like receptor 4 in neuroblastoma cells and their unresponsiveness to lipopolysaccharide. BMC Cancer. 2006;6:281. doi: 10.1186/1471-2407-6-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Uronen-Hansson H, Allen J, Osman M, Squires G, Klein N, Callard RE. Toll-like receptor 2 (TLR2) and TLR4 are present inside human dendritic cells, associated with microtubules and the Golgi apparatus but are not detectable on the cell surface: integrity of microtubules is required for interleukin-12 production in response to internalized bacteria. Immunology. 2004;111:173–8. doi: 10.1111/j.0019-2805.2003.01803.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bergenfelz C, Medrek C, Ekstrom E, Jirstrom K, Janols H, Wullt M, et al. Wnt5a induces a tolerogenic phenotype of macrophages in sepsis and breast cancer patients. J Immunol. 2012;188:5448–58. doi: 10.4049/jimmunol.1103378. [DOI] [PubMed] [Google Scholar]
- 49.D'Anello L, Sansone P, Storci G, Mitrugno V, D'Uva G, Chieco P, et al. Epigenetic control of the basal-like gene expression profile via Interleukin-6 in breast cancer cells. Mol Cancer. 2010;9:300. doi: 10.1186/1476-4598-9-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nakshatri H, Badve S. FOXA1 in breast cancer. Expert Rev Mol Med. 2009;11:e8. doi: 10.1017/S1462399409001008. [DOI] [PubMed] [Google Scholar]