

Abstract
The distal nephron plays a critical role in the renal control of homeostasis. Until very recently most studies focused on the control of Na+, K+, and water balance by principal cells of the collecting duct and the regulation of solute and water by hormones from the renin-angiotensin-aldosterone system and by antidiuretic hormone. However, recent studies have revealed the unexpected importance of renal intercalated cells, a subtype of cells present in the connecting tubule and collecting ducts. Such cells were thought initially to be involved exclusively in acid-base regulation. However, it is clear now that intercalated cells absorb NaCl and K+ and hence may participate in the regulation of blood pressure and potassium balance. The second paradigm-challenging concept we highlight is the emerging importance of local paracrine factors that play a critical role in the renal control of water and electrolyte balance.
Keywords: pendrin, purinergic signaling, serine protease, hypertension, potassium, sodium, acid-base
INTRODUCTION
The composition of the body’s different compartments is constant as a result of the dynamic and controlled exchange of solute and water between body cells and our surrounding environment. The kidney plays a central role in this function, termed electrolyte and water homeostasis. This equilibrium is achieved by controlling the amount of water and ions excreted into urine to exactly match the daily input of various substances due either to dietary intake or to metabolic production. Renal homeostasis is the result of the combination of two primary processes: glomerular filtration and tubular ion transport. The glomeruli filter daily very large amounts (~180 liters) of water and of ions (e.g., 25,000 mmol of Na+, 4,300 mmol of HCO3−, 720 mmol of K+). These quantities easily exceed those in the body. Accordingly, the renal tubule must absorb most substances filtered at the glomerulus to avoid their loss into urine. However, as indicated above, a small fraction of water and ions that exactly matches the daily input from diet and metabolism must also be excreted.
The nephron can be functionally divided into three different portions: the proximal part, which corresponds approximately to the proximal tubule; the loop of Henle; and the distal nephron. The first portion achieves massive absorption of water and solute. The second portion creates the cortico-papillary gradient of solute and osmoles in the interstitial space and hence plays a critical role in the control of transport of water and ions by the distal nephron. The distal tubule and the collecting ducts (CDs) are the final nephron segments that are sensitive to systemic hormones like aldosterone and vasopressin (AVP). Depending on these hormones’ actions, these final nephron segments can reabsorb 10–15% of filtered salt and water. These tubule segments enable fine-tuning of systemic salt, water, K+, and acid-base balance.
The distal nephron features cellular heterogeneity. The distal nephron begins after the macula densa and is composed of five successive segments: the distal convoluted tubule (DCT), the connecting tubule (CNT), the cortical collecting duct (CCD), the outer medullary collecting duct (OMCD), and the inner medullary collecting duct (IMCD) (1). Even though the CNT, the CCD, and the OMCD exhibit some functional differences and hence might be viewed as different nephron segments, they also share many characteristics because they are composed of very similar cell types (2–4). Thus, for the sake of simplicity, we hereafter refer to these nephron segments as the distal nephron. Several different cell types are identified along the distal nephron: DCT cells, principal cells (PCs), intercalated cells (ICs), and IMCD cells. Additional cell subtypes exist. For example, CNT cells and PCs differ with regard to Ca2+ transport mechanisms, which are present in the former but are absent in the latter. In mice, rats, and humans, but not in rabbits, a cell type with features in common with both DCT and CNT cells exists in the latter portion of the DCT. However, because all these three cell subtypes are characterized by the presence of the epithelial Na+ channel ENaC, they are considered to be PCs in this review. ICs undergo transdifferentiation from an acid-secreting phenotype termed α-intercalated cells (α-ICs) to a base-secreting phenotype termed β-intercalated cells (β-ICs) (5–7). The mechanisms underlying this functional plasticity have recently been reviewed in detail (8). α-ICs secrete H+ through an apical vacuolar H+-ATPase (vH+-ATPase) and reclaim HCO3− via the basolateral Cl−/HCO3− exchanger AE1 (SLC4A1). β-ICs have the opposite polarity: They extrude H+ through a basolateral vH+-ATPase and secrete HCO3− into urine via the apical Cl−/HCO3− exchanger pendrin (9–13). However, even if they represent a unique cell type, α-ICs and β-ICs represent two states of differentiation with completely opposite functions and express different Cl−/HCO3− exchangers. Therefore, in this review we consider α-ICs and β-ICs to be two different cell types. There is at least one other subtype of ICs, termed the non-α, non-β IC, which expresses both the H+ pump and the Cl−/HCO3− exchanger pendrin at the apical membrane (11, 14). Whether these cells represent a transitional form of β-ICs to α-ICs or have a specific function is not known. For simplicity, we here use the term β-IC to refer to both canonical β-ICs and non-α, non-β ICs.
Our classical view of the distal nephron assumes that this cellular heterogeneity reflects a very high degree of functional specialization. For example, DCT cells mediate net reabsorption of NaCl and are impermeable to water. Therefore, DCT cells are critical for extracellular fluid volume and blood pressure regulation, as well as for urine dilution. In contrast, PCs mediate Na+/K+ exchange and have highly regulated water absorption. Thus, they are important for K+ homeostasis and urine concentration. Finally, ICs can excrete either acid or base, depending on the acid-base status. Recent evidence challenges the view that hormone-regulatory loops activate or inhibit only one specific cell type and thus one specific function. For example, the role of ICs is not restricted to acid-base regulation because they can also transport NaCl and absorb K+ and thus participate in both blood pressure and plasma K+ regulation. Also, a large number of studies have examined the regulation of homeostasis by the classical hormones AVP and aldosterone, whereas important local autocrine and paracrine regulatory mechanisms that are based on the special tissue environment, diverse cell types, and unique cell metabolism of distal nephron epithelia have received much less attention.
In this article we highlight recently identified, emerging functions of ICs and paracrine mechanisms in the distal nephron that may play paradigm-shifting roles in renal physiology, body electrolyte and water homeostasis, and blood pressure control. Our mission is to depict the central role of ICs and to give the distal nephron a new face, depicting it as a complex, environmentally sensitive, semiautonomous nephron segment fully loaded with local sensory and regulatory machinery for the ultimate control of kidney functions and body homeostasis.
INTERCALATED CELLS ABSORB CHLORIDE AND PARTICIPATE IN BLOOD PRESSURE REGULATION
Two different molecular mechanisms of NaCl transport have been identified along the distal nephron. In the DCT, Na+ and Cl− are reabsorbed via the NaCl cotransporter NCC. First cloned by Gamba et al. (15) from the flounder bladder, NCC belongs to the superfamily of cation Cl− cotransporters (16) and is encoded by the gene SLC12A3 (17, 18). The importance of NCC in blood pressure regulation is attested by observations that inactivating mutations of SLC12A3 in humans (19), or targeted disruption of Slc12a3 in mice (20), cause Gitelman’s syndrome, an inherited recessive disease characterized by low blood pressure. More recently, researchers showed that even a mild decrease in NCC activity observed in individuals harboring heterozygous inactivating mutations in the SLC12A3 locus can significantly lower blood pressure (21). Moreover, NCC is the canonical target of thiazide diuretics, the oldest (22) but still one of the most effective therapies for human hypertension (23, 24).
In the CNT and the CD, NCC is not expressed, and Na+ reabsorption occurs through ENaC, which is located in the PCs (3, 25). ENaC is a heteromultimeric channel (26) that belongs to the degenerin gene superfamily, which also includes channels involved in mechanotransduction or neurotransmission and the H+-gated cation channel ASIC (27). In the mammalian kidney, ENaC has been characterized as a target for the diuretic amiloride (28, 29). Because ENaC is electrogenic, Na+ absorption by PCs leads to the development of a lumen-negative transepithelial voltage, which is a critical determinant of K+ secretion. Thus, the function of PCs is generally viewed as that of Na+/K+ exchange, even though both ion fluxes are not directly coupled by a single membrane transport protein (30–32). Because significant permeability of the paracellular pathway for Cl− has been measured in isolated CCDs (32–35), until recently researchers believed (36) that Cl− absorption by the CNT and CD is passive, occurring through the paracellular pathway, and is driven by the lumen-negative transepithelial voltage. However, several studies indicate that significant transcellular Cl− absorption also exists (33). The primary mechanism for Cl− transport in the CCD involves an electroneutral anion exchanger that can operate in either a Cl−/Cl− self-exchange or a Cl−/HCO3− exchange mode (37–39). This transport activity has been detected exclusively in β-ICs (37, 40, 41) and appears to be directly linked to HCO3− secretion (38, 42). The transepithelial movement of Cl− is effected by an apical Cl−/HCO3− exchanger, operating in series with basolateral Cl− conductance, that exhibits the same functional properties as does the voltage-gated chloride channel ClC-Kb (43).
A considerable advance in our understanding of Cl− transport by the CD originated from the identification of pendrin (SLC26A4) as the apical Cl−/HCO3− exchanger in β-ICs (12). Pendrin is not restricted to the kidney but is also expressed in the inner ear and thyroid. Pendrin is responsible for Pendred’s syndrome, which is characterized by deafness associated with goiter (44). Initially described as a sulfate transporter on the basis of sequence homologies with SAT-1, a prototype of sulfate transporter (45), pendrin is not a sulfate transporter, but rather a Cl−/anion exchanger (46). Pendrin has a very broad substrate specificity and can accept iodide, formate, or HCO3− (47–49). In the inner ear, pendrin mediates Cl−/HCO3− exchange. Pendrin mediates HCO3− secretion into the lumen of the cochlea, the vestibular labyrinth, and the endolymphatic sac of the inner ear (50–53). During embryonic development of the inner ear, pendrin participates in the net absorption of fluid in the endolymphatic sac (53). Loss of pendrin during the embryonic phase of development leads to an enlargement of the inner ear, which in the cochlea causes failure to develop a robust hearing phenotype, leading to deafness in children and in mouse models (52, 54, 55). The role of pendrin in the thyroid is less well understood. However, because pendrin is expressed in the apical membrane of thyrocytes and can exchange iodide for Cl−, it may be important for the secretion of iodide into the colloid and for thyroid hormone organification (56, 57).
A study by Royaux et al. (12) elucidated the role of pendrin in the kidney. These authors showed that pendrin is confined to the apical domain of β-ICs. They further clarified that pendrin represents the long-sought molecular basis for apical Cl−/HCO3− exchange in β-ICs. Thus, disruption of Slc26a4 abolishes HCO3− secretion in CDs isolated from alkalotic animals (12). In addition, Wall et al. (58) subsequently reported that pendrin disruption not only impairs HCO3− secretion but also totally abolishes Cl− reabsorption. They demonstrated that pendrin-mediated Cl− transport is the most important, if not the only, pathway for Cl− absorption in the collecting system (58).
That Cl absorption is not passive but rather occurs through β-ICs and involves a protein-mediated mechanism raises several important questions. (a) Is Cl− transport specifically regulated by changes in extracellular fluid volume or in Cl− balance? (b) Is Cl− transport an independent determinant of vascular fluid and blood pressure? (c) How are Cl− and Na+ fluxes synchronized to yield net NaCl absorption because they are not operated by the same cell type?
We designed a study in which rats were treated by different challenges to manipulate the amount of Cl− excreted into urine, and we measured renal pendrin protein abundance by semiquantitative Western blot analyses. Rats administered 0.28 M NaCl, NH4Cl, NaHCO3, KCl, or KHCO3 solution for 6 days or fed a low-NaCl diet exhibited an inverse relationship between pendrin expression and changes in Cl− excretion independently of the administered cation or of acid-base disturbances (59). Other investigators observed a decrease in pendrin in response to high renal Cl− delivery (60) or to dietary Cl− restriction (58, 61) in mice. Pendrin expression is also increased by angiotensin II (62) and by aldosterone (63).
Because the total NaCl body content is the critical determinant of the extracellular fluid volume and is under the tight control of the renin-angiotensin-aldosterone system (RAAS), the two latter studies suggested that pendrin is adapted to control vascular volume and thus blood pressure. In line with this hypothesis, pendrin knockout mice were found to be protected against deoxycorticosterone pivalate–induced hypertension (63). Conversely, following NaCl restriction, Cl− excretion was excessive in pendrin knockout mice, leading to negative balance and hypotension (58). Taken together, the aforementioned studies demonstrate that pendrin is critically involved in maintaining Cl− balance, responds to prohypertensive agents, and plays a critical role in blood pressure regulation by controlling distal Cl− transport.
INTERCALATED CELLS MEDIATE THIAZIDE-SENSITIVE NaCl ABSORPTION
Prior to our studies of the role of ICs in extracellular fluid volume regulation, the only Na+ transporter identified in the CD was ENaC. Because extracellular fluid volume is determined by NaCl and not only by Na+, we expected ENaC and pendrin to function in parallel to mediate net NaCl absorption. However, we were puzzled by several observations. Using a model of acute thiazide administration to create a state of volume depletion with secondary hyperaldosteronism together with high urinary Cl− delivery, we observed the expected increase in ENaC abundance. However, in this setting, we detected not an increase but rather a decrease in pendrin abundance (64). Using a mouse model of Liddle syndrome (65), we also did not detect any change in pendrin expression (64), even though ENaC-mediated Na+ absorption was markedly increased in this model (66). Finally, targeted deletion of α-ENaC in the CD disrupts ENaC activity but does not impair the kidney’s ability to maintain normal Na+ balance (67).
However, previous studies indicated that an additional mechanism for Na+ absorption might be present in the CD. Thus, bradykinin inhibits net Na+ absorption by 50% without affecting K+ secretion or the transepithelial voltage, suggesting that this hormone acts on an electroneutral Na+ transporter rather than on ENaC (31, 68). Furthermore, pharmacological studies on isolated CCD segments demonstrated that two distinct Na+ transporters are present in this nephron segment. The first transporter, mediated by ENaC, is highly amiloride sensitive, is electrogenic, and accounts for the transepithelial voltage that drives K+ secretion. In contrast, the second transporter is electroneutral, is not affected by amiloride, and is thiazide sensitive (69). However, because other researchers did not confirm these observations (70), and because careful localization studies did not confirm the expression of NCC, the only target known to date for thiazide diuretics in CCD cells (71–73), the identity of this electroneutral, thiazide-sensitive Na+ transporter remained unknown. Because of the availability of many genetically engineered mouse models with specific deletion of transporters, we reexamined the presence of this electroneutral NaCl transport pathway in isolated CCD segments (74).
When we simultaneously measured the transepithelial Na+ (JNa), K+ (JK ), and Cl− (JCl) fluxes and the transepithelial voltage (Vte) in isolated mouse CCDs microperfused in vitro, we detected two components of Na+ absorption. The first component was abolished either by amiloride or by ENaC genetic deletion and accounted for both the negative Vte and K+ secretion. In contrast, the second component was electroneutral and thiazide sensitive. Importantly, thiazide-sensitive NaCl absorption was present and even stimulated in NCC−/− mice (20). This finding indicates that such NaCl transport is operated by a mechanism that differs from NCC and that it may be one of the compensatory mechanisms that limit the effects of NCC deletion.
In many epithelia, Na+-dependent Cl− transport occurs through the functional coupling of a Cl−/HCO3− exchanger and a Na+/H+ exchanger. Because genetic ablation of pendrin eliminates Cl− absorption in the CCD, we hypothesized that thiazide-sensitive NaCl transport results from the coupling of pendrin-mediated Cl− reabsorption with a H+/HCO3−-dependent Na+ transporter. Indeed, by an approach combining intracellular pH measurements on isolated CCDs and different ion substitutions, we identified a thiazide-sensitive, Na+-driven Cl−/HCO3− exchanger in the apical membrane of ICs. Many HCO3− transporters have been reported, but only NDCBE/Slc4a8 and Slc4a10 have been reported to mediate Na+- and Cl−-dependent HCO3− transport in mammals (75–77). However, we did not find any evidence for the presence of Slc4a10 in the mouse CCD, whereas we detected both Slc4a8 mRNA and protein in mouse CCD. Furthermore, NDCBE deletion in mice abrogated amiloride-resistant electroneutral NaCl absorption in isolated CCD segments. This confirms that NDCBE operates in tandem with pendrin to mediate NaCl absorption (74). Thus, we identified a specific novel NaCl transport system along the nephron that is expressed by ICs (Figure 1). This finding indicates that the role of these cells is not limited to acid-base transport but also encompasses NaCl reabsorption and, accordingly, blood pressure regulation.
Figure 1.

Schematic model of Na+ transport in the collecting duct. (a) Cell models with the different transporters of the principal cells and intercalated cells. Two cycles of pendrin for one cycle of NDCBE are predicted to mediate net NaCl uptake with the recycling of one Cl− and two HCO3− ions. Dashed lines indicate ion recycling. (b) Cell model summarizing the characteristic features of Na+ transport in principal cells and intercalated cells. Abbreviations: BK, large-conductance, Ca2+-activated potassium channel; ClC-Kb, voltage-gated chloride channel Kb; ENaC, epithelial Na+ channel; NDCBE, Na+-driven Cl−/HCO3− exchanger; PDS, pendrin; ROMK, renal outer medullary K+ channel.
Pendrin is critically required to mediate net NaCl uptake by ICs. In vivo experiments in which we administrated thiazide to NCC−/− mice demonstrated that this novel system mediates a significant fraction of renal NaCl absorption (see figure 4 in Reference 74). Moreover, pendrin has emerged as an important player in renal NaCl conservation and in blood pressure regulation. Recent data suggest that this is not the only function of pendrin. In fact, pendrin disruption not only affects IC function but also impairs ENaC function and molecular regulation in the adjacent PC (78, 79). This finding indicates strong functional cooperation between the different cell types within the CCD.
Figure 4.

Schematic model depicting how tissue kallikrein (TK) participates in the response to dietary K+ loading. Dietary K+ loading stimulates TK production by connecting tubule cells. TK is then released into the urinary fluid and reaches the cortical collecting duct. There, TK may favor K+ secretion by principal cells through TK’s stimulating action on epithelial Na+ channel (ENaC)-mediated Na+ absorption, an effect that occurs through proteolytic processing of the γ-ENaC subunit (161). TK also inhibits K+ absorption by intercalated cells by decreasing colonic H+,K+-ATPase expression and activity. Moreover, TK upregulates ENaC-independent electroneutral NaCl absorption, presumably through a decrease in the local production of bradykinin (68). Other abbreviations: BK, large-conductance, Ca2+-activated potassium channel; NDCBE, Na+-driven Cl−/HCO3− exchanger; PDS, pendrin; ROMK, renal outer medullary K+ channel. From Reference 132.
PHYSIOLOGY OF ATP-RELEASING MEMBRANE (HEMI)CHANNELS
The aforementioned study of Pech et al. (78) proposed that ICs can modulate PC function through changes in HCO3− secretion. However, this is not the only paracrine signaling pathway that links IC and PC activities. One of the emerging new topics in renal transport physiology is the role of purinergic signaling in the regulation of tubular salt and water reabsorption and K+ secretion. In particular, the field of P2 purinergic receptors that bind extracellular (released) ATP has advanced significantly. According to the current view, a local purinergic system intrinsic to the distal nephron provides a powerful control of ENaC- and aquaporin 2 (AQP2)-mediated Na+ and water excretion. This purinergic system involves ectonucleotidases (ATP-degrading enzymes) and purinergic receptors. Due to the availability of P2Y2 knockout mice, P2Y2 receptors in the luminal plasma membrane of PCs provide most of our knowledge on purinergic receptors in this context. Mice lacking the P2Y2 receptor have impaired Na+ excretion, increased blood pressure, and consequently reduced plasma renin, aldosterone, and K+ (80). The renal physiological role of the P2Y2 receptor and the purinergic regulation of renal salt and water transport were extensively studied and reviewed recently (81–90). Here we focus on a new development in this purinergic system, namely, the mechanisms by which ATP, the ligand, is released from renal epithelial cells into the tubular lumen.
Studies in several organs suggest that connexin (Cx) proteins, the building blocks of gap junctions, can form hemichannels, or connexons, which allow ATP release into the extracellular fluid (91, 92). Inspired by these results, our laboratories first localized Cx30 (93) and then Cx30.3 (94) to the apical, nonjunctional plasma membrane of specific cells in the distal nephron. A detailed review of the localization and function of connexins in the kidney is available (95). The initial studies found that both Cx30 and Cx30.3 immunolabeling in the mouse kidney was particularly strong and limited to the apical membrane of ICs in the CNT and CCD. A high-salt diet caused upregulation of Cx30 expression (93). The distinct, continuous labeling of the IC luminal plasma membrane and upregulation by high salt suggest that Cx30 may function as a hemichannel involved in the paracrine regulation of salt reabsorption in the distal nephron.
Subsequent work by Sipos et al. (96) used an ATP biosensor approach to demonstrate that functional Cx30 hemichannels mediate luminal ATP release in the intact, microperfused CCD and control salt and water transport by purinergic signaling. Cx30 hemichannel opening at the luminal cell membrane of the CCD required mechanical stimulation by either increased tubular fluid flow rate or an interstitium-to-lumen osmotic pressure gradient and resulted in significant amounts of released ATP (up to 50 μM) in the luminal microenvironment. Consistent with preferential Cx30 localization in ICs, ATP biosensor responses were threefold larger in ICs versus PCs. Confirming the physiological significance of Cx30, genetic deletion of Cx30 markedly reduced flow-induced luminal ATP release and impaired salt and water excretion associated with pressure natriuresis, an important mechanism that maintains body fluid and electrolyte balance and blood pressure (96). Thus, Cx30−/− mice express a salt retention phenotype due to the hyperactive Cx30-expressing CD. Benzamil, a specific inhibitor of ENaC, prevents the salt-sensitive hypertension observed in Cx30−/− mice (96).
Additional whole-animal studies using Cx30−/− mice confirmed the importance of Cx30-mediated ATP release in the control of renal salt excretion by testing the effects of changes in dietary salt intake. Studies by Mironova et al. (97) established that urinary ATP levels increase with high Na+ intake in wild-type mice. In contrast, urinary ATP in Cx30−/− mice was lower and less dependent on dietary salt. Loss of inhibitory ATP regulation in Cx30−/− mice also caused high ENaC open probability, particularly with high Na+ intake. The level of ENaC expression in Cx30−/− mice was not altered (96). Together with the observed increase in ENaC open probability rather than in the number of Na+ channels (97), this last finding suggests that ATP functions in a paracrine manner. Importantly, the loss of paracrine ATP feedback regulation of ENaC in Cx30−/− mice disrupts normal responses to changes in Na+ intake. Thus, Cx30−/− mice have lower Na+ excretion in states of positive Na+ balance. Moreover, the loss of the ability of ENaC to respond to changes in Na+ levels causes salt-sensitive hypertension in Cx30−/− mice (97).
The finding that reduced salt excretion in Cx30−/− mice is most apparent in high-salt conditions is consistent with several observations. First, in normal mice, a high-salt diet upregulates Cx30 expression (93), and the greatest feedback inhibition of ENaC by Cx30-mediated ATP release is observed in animals receiving a high-salt diet (97). Moreover, clamping mineralocorticoids high in Cx30−/− mice fed a high-Na+ diet caused a marked decline in renal Na+ excretion. Such responses are in contrast to normal physiological responses in wild-type mice that are capable of undergoing aldosterone escape, which refers to increased salt excretion during high Na+ intake despite inappropriately increased levels of mineralocorticoids (98). These studies not only provide novel insights into aldosterone research but further support the key importance of the local CD purinergic system, including the role of Cx30 in renal control of salt transport.
On the basis of these data, the disruption of Cx30 function seems to be equivalent to the loss of the P2Y2 receptor (80, 84, 85) in terms of ENaC control. This conclusion is not surprising because the P2Y2 receptor is localized downstream of Cx30-mediated ATP release, linking the inhibitory ATP signal to ENaC. Thus, several lines of evidence point to Cx30 as an integral component of a local purinergic signaling system within the distal nephron. It provides insights into a dietary salt–sensitive negative feedback mechanism for the control of renal salt excretion via ATP release from ICs and ATP’s paracrine purinergic actions on PCs targeting ENaC.
Purinergic regulation of K+ secretion in the distal nephron is quite complex, with different types of K+ channels involved. Extracellular ATP is a potent inhibitor of the small-conductance K+ (SK) channel located in the apical plasma membrane of PCs, suggesting that ATP in the tubular fluid inhibits net K+ secretion (99). However, the large-conductance, Ca2+-activated K+ (BK) channel, which is localized predominantly in the luminal membrane of ICs, seems also to be regulated by ATP released via connexin hemichannels. Holtzclaw et al. (100) found recently that pharmacological inhibition of connexins or P2 receptors in ICs diminished flow-induced K+ efflux through BK channels as well as ATP secretion. These investigators also showed that K+ efflux and ATP release from ICs are interdependent because downregulation of the BK-β4 subunit using siRNA decreased ATP secretion and both ATP-dependent and flow-induced K+ efflux. In mice with high distal nephron flows induced by a high-K+ diet, the genetic disruption of BK channel function resulted in significantly reduced urinary ATP excretion. The observed Na+ and water retention as well as decreased K+ secretion in BK-β4−/− mice are consistent with reduced purinergic tone (101). Because ICs have not traditionally been considered to play a major role in K+ secretion, further studies are needed to clarify the autocrine/paracrine actions of ATP on ICs and PCs and their possible interactions.
The physiological importance of ATP–BK channel interaction may be related to the need to maintain high distal flow rates; maximal K+ secretion and minimal Na+ reabsorption occur when animals consume a high-K+ diet. High flow may be augmented by the release of ATP, which inhibits Na+ reabsorption (via ENaC) and water reabsorption (via AQP2) yet at the same time stimulates K+ secretion. This newly described BK channel function may help us to understand the so-called aldosterone paradox. This term refers to the apparently opposite effects of aldosterone, salt reabsorption (e.g., in hypovolemia), and K+ secretion (e.g., in hyperkalemia). Investigators recently reviewed the molecular mechanisms mediating the aldosterone paradox, that is, the interdependent but differential regulation of renal Na+ and K+ transport, including the role of the serine/threonine kinases WNK1, WNK4, SPAK, and OSR1 (102). The local purinergic system in the distal nephron may also be involved via modulation of tubular flow. In volume depletion, aldosterone favors salt retention, with no change in K+ secretion (low tubular flow, low ATP levels, inactive purinergic inhibition, and high ENaC but low BK channel activity). In contrast, in hyperkalemia aldosterone helps to excrete K+ without affecting salt reabsorption (high tubular flow, high levels of ATP release, powerful purinergic inhibition of ENaC, high BK-mediated K+ secretion). The precise mechanism by which ATP affects the activity of ROMK and maxi-K+ channels as well as purinergically mediated activity needs further investigation.
On the basis of these findings, a more complete picture of the local intratubular purinergic system has emerged (Figure 2). Mechanically induced opening of Cx30 hemichannels in ICs (e.g., by tubular flow, interstitial pressure, cell volume changes) allows ATP to enter the tubular fluid and to bind to local P2 receptors in both ICs and PCs in an autocrine fashion and a paracrine fashion, respectively. Activation of these receptors inhibits salt and water reabsorption in PCs via ENaC and AQP2 and facilitates K+ secretion via BK channels. Sustained activity of this purinergic system appears to participate in the homeostasis of body fluid and electrolytes and blood pressure in a variety of physiological conditions, including pressure natriuresis, high-salt diet, aldosterone escape, the aldosterone paradox, and high-K+ diet.
Figure 2.
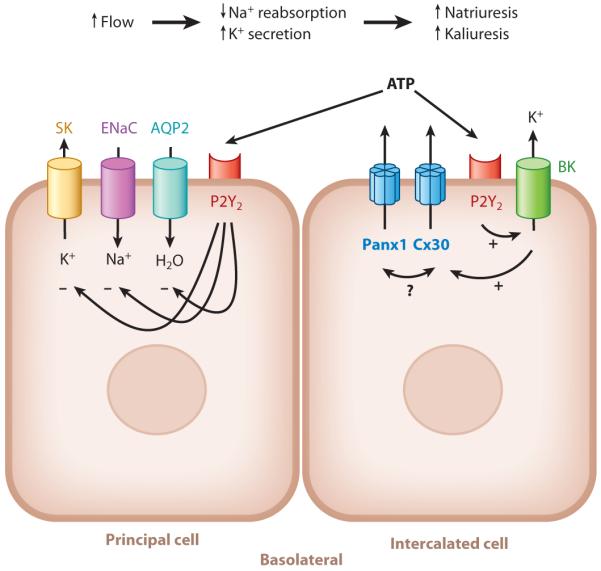
Novel elements and proposed function of the intratubular autocrine/paracrine purinergic system in the collecting duct. Abbreviations: AQP2, aquaporin 2 water channel; BK, large-conductance, Ca2+-activated potassium channel; Cx30, connexin 30 hemichannel; ENaC, epithelial Na+ channel; P2Y2, ATP receptor subtype P2Y2; Panx1, pannexin 1 channel; SK, small-conductance K+ channel.
However, the idea that functional hemichannels are formed solely by connexins under physiological conditions is controversial (103). Then how does Cx30 mediate ATP release from ICs in the CD? Such release may occur due to the close association of Cx30 with the recently identified plasma membrane ATP channel pannexin 1 (Panx1) (Figure 2). Pannexins are a class of proteins that also form hexameric transmembrane channels that are similar to those formed by connexins (104). Despite having similar structures, connexins and pannexins have no sequence homology. In several cell types, including epithelial cells, Panx1 forms functional, mechanosensitive ATP-releasing channels under physiological conditions (104, 105). Interestingly, a preliminary study from our laboratory (106) found strong Panx1 expression in the apical membrane of the CD system, raising a question about its role alongside that of Cx30. Future work will address the exciting possibility of Cx30-Panx1 interactions in the CD.
Cx30−/− mice are deaf due to inner-ear dysfunction (107). Similarities in regulatory mechanisms and common ion transporters in the renal CD and the inner ear are well known (108). Consistent with this, these deaf animals also possess a renal phenotype (i.e., salt retention), as detailed above, on the basis of the dysfunction of Cx30 in the renal CD system. A potential interaction between Cx30 and pendrin may also be part of the local purinergic system, as suggested by their colocalization in non-α ICs (93) and the similar phenotype of pendrin knockout mice (Pendred syndrome, deafness) (12). Recently, a genetic link between Bartter syndrome (which causes renal salt wasting) and sensorineural deafness in humans was established (109). Unlike in the mouse kidney, in other species Cx30 is expressed in the thick ascending limb and distal tubule (93). The findings suggest that Cx30-mediated ATP release is an important physiological regulatory mechanism in many organs and species, including humans. ATP release mediated by connexin hemichannels (and other associated proteins) and the complex regulation of water and electrolyte transport mechanisms by a local purinergic signaling system appear to be a well-preserved mechanosensory and regulatory machinery in various organs and cell types.
THE COLLECTING DUCT RENIN-ANGIOTENSIN SYSTEM AND NOVEL METABOLIC REGULATORS
The RAAS is a key regulator of renal salt and water reabsorption whose molecular and cellular effects in various nephron segments have been extensively studied. Here we briefly emphasize two important developments concerning paracrine regulation of distal nephron function. First, a local, distal nephron’s RAAS has been established and characterized (110, 111). This local system is now recognized as a key anatomical site of the overall RAAS in pathological states including diabetes and as the major source of systemic RAAS components, for example, (pro)renin (110, 112). The local generation of angiotensin peptides in the tubular fluid of the CD and their stimulatory effects on salt reabsorption via ENaC have been demonstrated (113, 114). The importance of angiotensin II generated de novo by this local intratubular RAAS in overall renal salt and water excretion as well as its interaction with the systemic RAAS (e.g., aldosterone) need to be further studied.
Another emerging field is the direct metabolic control of distal nephron function by metabolic intermediates in the tubular fluid, for example, succinate and α-ketoglutarate. The special cell metabolism (high anaerobic glycolysis) and tissue environment (hypoxia) under which some cells of the distal nephron operate are highly consistent with the local accumulation of these dicarboxylates (115). The G protein–coupled receptors GPR91 and GPR99, newly identified specific cell membrane receptors for succinate and α-ketoglutarate, respectively, are localized predominantly in the luminal cell membrane of the distal nephron–CD system and have been linked to the activation of RAAS (116–119). GPR91 signaling involves the generation of prostaglandins (e.g., PGE2), which is highly relevant to the control of renal salt and water transport (117–119). In addition, elements of an olfactory signaling system along the apical surface of the distal nephron have been discovered (120). Although the activating ligand is not known, the idea that cells of the distal nephron and CD can “smell” certain chemical signals in the tubular fluid that may directly regulate electrolyte transport is exciting and will undoubtedly open new areas in kidney research.
ROLE OF TISSUE KALLIKREIN IN POTASSIUM HOMEOSTASIS
Another example of paracrine regulation is the modulation of K+ transport in the distal nephron by tissue kallikrein (TK, klk1 gene; number NM_010639), a serine protease involved in kinin generation. In the kidney, TK is synthesized in large amounts by CNT cells and, to a lesser extent, by the DCT and CCD (121, 122). Although TK can be released into the circulation, active TK is found predominantly in the urine (123). TK release leads to kinin formation. TK cleaves locally available kininogen to yield kallidin (lysyl-bradykinin), which is converted through the action of an N-aminopeptidase to bradykinin. Bradykinin in turn activates the bradykinin type 2 (B2) receptor. B2 receptors have been localized to both the luminal side and the basal side of CD cells (124). Kinins have potent vasodilatory properties, and intrarenal infusions of kinins increase renal blood flow and produce diuresis and natriuresis (125). This response is probably due both to kinin-induced redistribution of medullary renal blood flow (medullary washout of solute) and to direct effects on Na+ and water transport across the CD. In fact, kinins (bradykinin and kallidin) inhibit Na+ and water transport when applied to the basolateral surface but not to the luminal surface of the CD (31, 68, 126). Although a kinin-dependent effect of TK on renal Na+ and water transport is well established, the role of the large amount of TK that is secreted into the urinary fluid remains poorly understood.
MECHANISMS FOR THE RELEASE OF RENAL TISSUE KALLIKREIN: EFFECT OF POTASSIUM
A recent study performed in humans by Azizi et al. (127) confirmed earlier observations on the influence of dietary K+ intake on urinary TK activity. A diet with high K+ and low Na+ content increased whereas a diet with low K+ and high Na+ content decreased TK activity and excretion (127). Thus, the distal nephron segments responsible for regulation of K+ excretion (i.e., late distal tubules, CNTs, and CCDs) are exposed to variable amounts of TK. Because maneuvers modulating urinary kallikrein activity alter K+ balance, it is tempting to postulate that the controlled upstream release of TK from the apical membranes of CNT cells into the tubular fluid by K+ may lead to autocrine and paracrine action of TK on distal tubular K+ transport. Early studies indicate that K+ intake stimulates TK synthesis and excretion directly and/or through secondary mechanisms (125, 128, 129). Aldosterone may also influence the effect of K+ on the urinary excretion of renal TK because high K+ intake stimulates the secretion of aldosterone, which has a stimulatory effect on TK synthesis and excretion. However, distinct effects of mineralocorticoids on TK synthesis and excretion have been reported (130, 131). In fact, acute administration of aldosterone does not induce the synthesis of renal TK, whereas chronic treatment with deoxycorticosterone or adrenalectomy increases and decreases, respectively, TK synthesis in the CNTs and TK secretion in the urine.
The concept that K+ regulates renal TK has been reconsidered. In a recent study, we showed that a high-K+ diet increases to a similar extent renal TK secretion in both control and aldosterone synthase–deficient mice, indicating that aldosterone is not required for the stimulatory effect of K+ intake on TK secretion (132). We also showed that a single meal, which represents an acute K+ load, increases renal K+ and TK excretion in parallel, whereas aldosterone secretion is not significantly modified. An early increase in renal TK secretion, which occurs after intravenous infusion of K+, was also reported in rats (133, 134). Administration of ATP-sensitive K+ channel (KATP) blockers also increased renal kallikrein secretion in superfused slices of kidney cortex (134). Thus, K+ and KATP channel blockers may increase renal kallikrein secretion through the same mechanism. A decrease in membrane potential caused by high concentrations of extracellular K+ has been reported for renal tubular cells (135), and K+ channel blockers such as barium Cl− produced the same effect (135).
El Moghrabi et al. (132) showed that acute K+ loading rendered TK-deficient mice hyperkalemic, whereas this maneuver did not lead to any significant change in plasma [K+] in wild-type mice. The authors concluded that TK is regulated by extracellular K+ independently of aldosterone and in turn protects the organism against an acute K+ load. A typical meal of a Western-style diet represents a K+ load equivalent to half of the total amount of K+ in the extracellular fluid. As a consequence, every meal represents an acute and massive K+ load with which the body has to cope (136). Renal TK represents a new factor that protects the body against postfeeding hyperkalemia besides insulin or β-adrenergic agonists, both of which stimulate the rapid transfer of K+ from the extracellular space to the intracellular space (137). Although K+ balance is under the tight control of the mineralocorticoid hormone aldosterone, which activates and fine-tunes K+ secretion by the CD, aldosterone, whose effects are mostly transcriptional, is not suitable for such an acute adaptation.
TISSUE KALLIKREIN AND RENAL POTASSIUM HANDLING
K+ is freely filtered at the glomerulus and is almost totally reabsorbed by the proximal tubule and the loop of Henle. The amount of K+ excreted by the kidney is determined by events beyond the early distal tubule, where either reabsorption or secretion of K+ can occur. Generally, a large amount of K+ has to be eliminated to maintain balance. K+ secretion occurs mainly through the PCs and involves the following: (a) cellular K+ entry across the basolateral membrane via the Na+,K+-ATPase pump and (b) K+ exit across the apical membrane via K+ channels, allowing K+ secretion into the lumen. In PCs, Na+ reabsorption is energized by the basolateral Na+,K+-ATPase, which exchanges three Na+ for two K+. The resulting electrochemical gradient for Na+ is dissipated mainly by apical Na+ entry via ENaC, which depolarizes the apical membrane and facilitates apical K+ exit via the K+ channel ROMK. Na+ transport via ENaC is thus an important determinant for K+ secretion through ROMK. Accordingly, amiloride administration decreased urinary K+ excretion. Under conditions in which the kidney must retain K+, such as a situation of K+ depletion, the CD reverses the transepithelial net flux of K+ to yield net K+ reabsorption (136). K+ absorption is localized to the ICs in the CCD and is mediated by H+,K+-ATPases, which reabsorb K+ in exchange for H+ ions (138). Net secretion may be the result of simultaneous bidirectional secretory and reabsorptive K+ fluxes.
If one does not consider a situation of K+ depletion in which net K+ absorption occurs, the CCD is a site of net K+ secretion. Remarkably, microperfused CCDs isolated from TK-deficient mice (139) exhibit net transepithelial K+ absorption (132). TK may also modulate K+ transport by favoring ENaC-mediated Na+ absorption that, in turn, is expected to favor renal K+ secretion and/or by inhibiting K+ absorption through H+,K+-ATPase.
TISSUE KALLIKREIN AND PROTEOLYTIC ACTIVATION OF THE EPITHELIAL SODIUM CHANNEL
In 1997, the channel-activating protease 1 (CAP1, also referred as prostasin), a membrane-bound serine protease, was identified in Xenopus kidney A6 cells on the basis of its ability to activate ENaC when coexpressed in Xenopus oocytes (140). From this study emerged the concept that a variety of serine proteases can activate ENaC. Such proteases include (a) extracellular serin proteases [e.g., trypsin; plasmin; and elastase, which is a serine protease produced by neutrophils (141–144)], (b) intracellular serin proteases [e.g., furin, a serine protease located in the trans-Golgi network (145)], and (c) membrane-bound serine proteases [e.g., members of the CAP family (146–148)], all of which act through their catalytic activity to increase the open probability of ENaC (141–143, 147).
ENaC is a heteromultimeric protein composed of three related subunits: α, β, and γ. All subunits have an intracytoplasmic N terminus and C terminus and two membrane-spanning domains connected by a large extracellular loop. Proteolytic cleavage of ENaC occurs for the α subunit and the γ subunit (γ-ENaC) in vitro. Hughey et al. (149, 150) demonstrated that furin activates ENaC by cleaving the channel in the ectodomain at two sites in the α subunit and at a single site within γ-ENaC. There is increasing in vitro evidence that ENaC activation by serine proteases is related to proteolytic cleavage of the extracellular domain of γ-ENaC at sites distal to the furin cleavage site (for review, see References 144 and 151–155). Masilamani et al. (156) suggest that endogenous serine proteases may cleave γ-ENaC in vivo. However, the serine protease(s) that mediates this process in vivo has not been identified, including in the kidney.
Endogenous membrane-bound and/or secreted serine proteases such as CAPs are likely candidates for protease-mediated regulation of Na+ transport in the lung (157, 158). Accordingly, gene disruption of CAP1 specifically in the alveolar epithelium using conditional Cre-loxP-mediated recombination revealed that CAP1 plays a crucial role in the regulation of ENaC-mediated alveolar Na+ and water transport and in mouse lung fluid balance (158). CAP1 is coexpressed with ENaC in Na+-transporting epithelia, including the kidney (140), where CAP1 is present in native and cultured CCD cells (148). CAP1 is detectable in mouse exosomes, a preparation that reflects the pool of protein apically expressed in renal tubular cells (159), suggesting that CAP1 is present at the apical surface of renal cells (R. Chambrey & B. Rossier, unpublished data). Activation of ENaC by aldosterone has been consistently associated with a shift in the molecular weight of renal γ-ENaC from 85 to 70 kDa, consistent with physiological cleavage of the extracellular loop of that subunit (156, 160, 161). Aldosterone increases the expression of prostasin, the human ortholog for CAP1, in vitro and in vivo (162). Narikiyo et al. (162) showed that urinary excretion of prostasin is inversely correlated with the urinary Na+:K+ ratio in patients with primary aldosteronism, suggesting that prostasin may be responsible for aldosterone-dependent proteolytic ENaC activation. Gene disruption of CAP1 specifically in the CD will be necessary for direct assessment of its physiological role in ENaC regulation.
Because CNT cells secrete a large amount of TK into the tubular fluid, the luminal membranes of renal tubular cells starting from the CNT are highly exposed to TK. Thus, TK, through its catalytic activity, may act directly on ENaC in the apical membranes of tubular cells located downstream of the site of TK secretion to modulate ENaC activity.
Picard et al. (161) recently reported that TK-deficient mice have decreased ENaC activity in the kidney and the distal colon, two organs that exhibit ENaC-mediated Na+ transport and produce abundant amounts of TK, whereas in lung alveolae, an organ devoid of TK expression, TK disruption does not impair ENaC activity. In this study, reduced ENaC activity in TK-deficient mice correlated with a dramatic decrease in expression of the 70-kDa truncated form of γ-ENaC in both renal cortical membrane and urinary exosome preparations. In contrast, in the lung, TK disruption did not impair ENaC processing, confirming that in lung alveolae a protease other than TK, such as CAP1, regulates ENaC, as recently described by Planès et al. (158).
When applied to the luminal surface of CCDs isolated and microperfused in vitro, TK has a marked stimulatory effect on ENaC-dependent Na+ entry (161). In vitro experiments using either commercially available TK or mouse TK from desalted urine demonstrated that TK promotes the cleavage of γ-ENaC (Figure 3) (161). Thus, urinary TK promotes in vivo and in vitro γ-ENaC cleavage, which is associated with ENaC activation.
Figure 3.

Tissue kallikrein (TK) promotes cleavage of the epithelial Na+ channel (ENaC) γ subunit (γ-ENaC) in vitro. (a) Renal membrane fractions from TK-deficient (TK−/−) mice were incubated with TK, and appearance of the 70-kDa form of γ-ENaC was studied by immunoblotting. Membrane proteins were mixed with either 0, 30, or 100 μg ml−1 TK purified from porcine pancreas and were incubated for 15 min at 37°C. (b) Densitometric analyses of data showing the appearance of the 70-kDa form after treatment with TK. (c) Renal membrane fractions from TK−/− mice were incubated with desalted and concentrated urine from wild-type (WT) or TK−/− mice for 1.5 h at 37°C. The appearance of the 70-kDa form of γ-ENaC was studied by immunoblotting. Modified from Reference 161.
Interestingly, we also observed that elevated circulating aldosterone levels did not interfere with ENaC processing and activation in the absence of TK (161). Thus, serine proteases other than TK, presumably CAPs (see above), may promote the cleavage and activation of γ-ENaC in the kidneys of TK-deficient mice with elevated circulating aldosterone. These data suggest that TK is not critical for protease-mediated regulation of Na+ absorption in the CD.
Although ENaC activity is reduced in the absence of TK, CCDs from TK-deficient mice absorb Na+ without affecting the transepithelial potential difference, indicating that Na+ absorption occurs through an electroneutral process (74, 132). Early studies showed that bradykinin inhibits Na+ absorption without affecting net K+ transport or the transepithelial potential difference in isolated and microperfused CDs through the activation of the basolateral B2 receptor (68). TK gene inactivation in the mouse not only disrupts TK production but also impairs local kinin production (139). Thus, the absence of TK upregulates ENaC-independent electroneutral NaCl absorption, presumably through a decrease in the local production of bradykinin, and prevents proteolytic activation of ENaC (161). The combination of these two opposite effects on Na+ transport explains why TK-deficient mice do not have a significantly altered Na+ balance (139, 161). In conclusion, TK-dependent ENaC activation is not critical for the regulation of renal Na+ homeostasis but rather plays an important role in maintaining K+ balance.
TISSUE KALLIKREIN AND THE COLONIC H+,K+-ATPASE
As discussed above, CCDs isolated from TK-deficient mice exhibit net transepithelial K+ absorption (132). Because TK-deficient mice display decreased ENaC activity, a shutdown of K+ secretion likely contributes to net K+ absorption in the CDs of these mice. However, these data do not rule out the possibility that H+,K+-ATPase activity is also involved by further reducing urinary K+ losses.
We refer the reader to the review by Gumz et al. (163) for background on H+,K+-ATPases. Briefly, the H+,K+-ATPases consist of a catalytic α subunit and a regulatory β subunit. Researchers have identified two renal H+,K+-ATPases that contain the catalytic subunit HKα1 or HKα2. The HKα1 H+,K+-ATPase, or gastric H+,K+-ATPase, has a role in acidifying stomach content. The HKα2 H+,K+-ATPase, termed the colonic H+,K+-ATPase, is abundant in and cloned from the colon. Using HKα2-deficient mice, Meneton et al. (164) demonstrated that under K+ deprivation HKα2 plays a critical role in the maintenance of K+ homeostasis in vivo.
K+ depletion, which causes renal K+ absorption, is associated with an increase in kidney HKα2 mRNA, suggesting that the colonic type of the H+,K+-ATPase plays an important role in K+ conservation by the kidney. Accordingly, the colonic type of the H+,K+-ATPase mediates increased HCO3− absorption in the CD during K+ depletion (165, 166). H+,K+-ATPase activity estimated by the initial recovery rate of intracellular pH after an acute intracellular acid load was higher in CD ICs from TK-deficient mice. Acid extrusion in ICs from TK-deficient mice was inhibited either by 30 μM SCH28080, a specific inhibitor of H+,K+-ATPases, or by 1 mM ouabain. These pharmacological properties correspond to those described for the colonic type of the H+,K+-ATPase (167). Consistently, mRNA for the HKα2 subunit but not that for the HKα1 subunit of the H+,K+-ATPase was markedly increased in CNTs and CCDs of TK-deficient mice (132). Conversely, in CCDs isolated from TK-deficient mice and microperfused in vitro, the addition of TK to the luminal surface but not to the basolateral surface of the tubule caused a 70% inhibition of H+,K+-ATPase activity. These results demonstrate that the absence of TK stimulates the expression and activity of the colonic H+,K+-ATPase and results in net K+ absorption by the CCD. Under conditions of K+ excretion, TK exerts a negative braking effect on K+ absorption by inhibiting the H+,K+-ATPase. The mechanism or signaling pathway by which luminal TK controls H+,K+-ATPase remains unknown. As mentioned above, serine proteases act directly on ENaC gating by enhancing the open probability of ENaC. Recent studies show that TK can also directly activate the B2 receptor independently of bradykinin release (168). TK, which colocalizes with the epithelial Ca2+ channel TRPV5 in the distal part of the nephron, activates TRPV5 from the luminal side (169). Using primary cultures of CNT/CCD, Gkika et al. (169) showed that TK, like bradykinin, stimulates TRPV5 indirectly via activation of the B2 receptor. This induces the phosphorylation of TRPV5 through a protein kinase C–dependent mechanism, with consequent redistribution of TRPV5 channels toward the plasma membrane.
The experimental data summarized in this part indicate that TK regulates K+ transport by two mechanisms: (a) by favoring ENaC-mediated Na+ transport that, in turn, is expected to favor renal K+ secretion and (b) by inhibiting K+ absorption through the colonic H+,K+-ATPase (Figure 4). Thus, TK is a kaliuretic factor that provides rapid and aldosterone-independent protection against hyperkalemia after a dietary K+ load. Importantly, these data also suggest that K+ absorption through the colonic H+,K+-ATPase is under the negative control of TK in the distal nephron.
CONCLUSIONS
There has been significant progress in our understanding of the molecular mechanisms involved in the renal control of homeostasis. Recent studies in particular have highlighted the emerging importance of paracrine regulation of ion transport and cross talk between nephron segments and different cell types within a single nephron segment. The identification of several novel ion transport mechanisms and local signaling pathways in the distal nephron that we summarize in this review will help us to further understand the pathophysiology of several diseases. Various past studies that focused on the classical hormonal regulation of the distal nephron and its abnormalities repeatedly failed to identify a unifying molecular mechanism. This failure is particularly evident for disorders that affect Na+ balance, such as hypertension and nephrotic syndrome. Another significant development that will challenge the current concepts in renal homeostasis is the emerging importance of ICs in Na+ and K+ balance. This is highlighted by the strong modifier effects of pendrin disruption on blood pressure regulation (61, 63) and by the identification of a novel therapeutic target of thiazide in these cells (74). Another perhaps paradigm-shifting finding is the identification of ICs as an important source of paracrine factors that regulate diverse functions of the CD, particularly those of the adjacent PCs. Therefore, future studies should reevaluate the precise role and involvement of ICs in disorders that were initially thought to originate from PC dysfunction.
SUMMARY POINTS.
In contrast to the existing paradigm, intercalated cells not only are involved in acid-base transport but also are important for NaCl and K+ absorption. Accordingly, these cells participate in the regulation of extracellular fluid volume and blood K+ concentration.
Cl− transport in the collecting duct occurs through intercalated cells rather than through the paracellular pathway and is an important determinant of blood pressure.
Intercalated cells are the primary source of ATP that is released into the tubular fluid (urine), and hence intercalated cells play a central role in purinergic signaling in the distal nephron.
Disruption of connexin30 impairs ATP release and purinergic signaling within the distal nephron, leading to the loss of negative feedback control of ENaC that results in excessive salt retention and ultimately salt-sensitive hypertension.
Tissue kallikrein is a serine protease abundantly released into urine by connecting tubule cells. It stimulates K+ secretion by principal cells indirectly through proteolytic activation of ENaC and inhibits K+ absorption by the H+,K+-ATPase in intercalated cells.
FUTURE ISSUES.
In light of the new ion transport and paracrine signaling mechanisms that have been identified in the distal nephron, the role of intercalated cells and principal cells and their interactions in the regulation of NaCl and K+ transport need to be further investigated.
The involvement of intercalated cells in disorders characterized by abnormal regulation of blood pressure (particularly Cl−-dependent hypertension like pseudohypoaldosteronism type II) should be reevaluated.
The physiological importance of the local purinergic regulation of collecting duct K+ secretion (via BK and SK channels) and water reabsorption (via aquaporin 2) needs to be determined in vivo (for example, by using connexin30 knockout mice).
On the basis of the functional similarities in molecular mechanisms between the renal collecting duct and the inner ear, future clinical studies may identify the diagnostic potential of newborn hearing tests as a predictor of kidney disease and hypertension.
Whether tissue kallikrein modulates H+,K+-ATPase activity through proteolysis or through another mechanism remains to be determined.
ACKNOWLEDGMENTS
Dominique Eladari and Régine Chambrey are funded by INSERM, by CNRS, by the Transatlantic Network for Hypertension (TNH) from the Fondation Leducq, and by grants ANR PHYSIO 2007-RPV07084 and ANR BLANC 2010-R10164DD to Dr. Eladari from l’Agence Nationale de la Recherche (ANR). Janos Peti-Peterdi is funded by NIH grants DK64324 and DK74754, by American Diabetes Association Research grant 1-11-BS-121, and by American Heart Association Established Investigator award 0640056N.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Kriz W, Kaissling B. Structural organization of the distal nephron. In: Seldin DW, Giebisch G, editors. The Kidney: Physiology and Pathophysiology. Philadelphia, PA: Lippincott: 2000. pp. 587–654. [Google Scholar]
- 2.Bachmann S, Bostanjoglo M, Schmitt R, Ellison DH. Sodium transport-related proteins in the mammalian distal nephron—distribution, ontogeny and functional aspects. Anat. Embryol. 1999;200:447–68. doi: 10.1007/s004290050294. [DOI] [PubMed] [Google Scholar]
- 3.Loffing J, Kaissling B. Sodium and calcium transport pathways along the mammalian distal nephron: from rabbit to human. Am. J. Physiol. Ren. Physiol. 2003;284:628–43. doi: 10.1152/ajprenal.00217.2002. [DOI] [PubMed] [Google Scholar]
- 4.Meneton P, Loffing J, Warnock DG. Sodium and potassium handling by the aldosterone-sensitive distal nephron: the pivotal role of the distal and connecting tubule. Am. J. Physiol. Ren. Physiol. 2004;287:593–601. doi: 10.1152/ajprenal.00454.2003. [DOI] [PubMed] [Google Scholar]
- 5.Schwartz GJ, Tsuruoka S, Vijayakumar S, Petrovic S, Mian A, Al-Awqati Q. Acid incubation reverses the polarity of intercalated cell transporters, an effect mediated by hensin. J. Clin. Investig. 2002;109:89–99. doi: 10.1172/JCI13292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schwartz GJ, Barasch J, Al-Awqati Q. Plasticity of functional epithelial polarity. Nature. 1985;318:368–71. doi: 10.1038/318368a0. [DOI] [PubMed] [Google Scholar]
- 7.Gao X, Eladari D, Leviel F, Tew BY, Miro-Julia C, et al. Deletion of hensin/DMBT1 blocks conversion of β- to α-intercalated cells and induces distal renal tubular acidosis. Proc. Natl. Acad. Sci. USA. 2010;107:21872–77. doi: 10.1073/pnas.1010364107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Al-Awqati Q. Terminal differentiation in epithelia: the role of integrins in hensin polymerization. Annu. Rev. Physiol. 2011;73:401–12. doi: 10.1146/annurev-physiol-012110-142253. [DOI] [PubMed] [Google Scholar]
- 9.Brown D, Hirsch S, Gluck S. An H+-ATPase in opposite plasma membrane domains in kidney epithelial cell subpopulations. Nature. 1988;331:622–24. doi: 10.1038/331622a0. [DOI] [PubMed] [Google Scholar]
- 10.Alper SL, Natale J, Gluck S, Lodish HF, Brown D. Subtypes of intercalated cells in rat kidney collecting duct defined by antibodies against erythroid band 3 and renal vacuolar H+-ATPase. Proc. Natl. Acad. Sci. USA. 1989;86:5429–33. doi: 10.1073/pnas.86.14.5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim J, Kim YH, Cha JH, Tisher CC, Madsen KM. Intercalated cell subtypes in connecting tubule and cortical collecting duct of rat and mouse. J. Am. Soc. Nephrol. 1999;10:1–12. doi: 10.1681/ASN.V1011. [DOI] [PubMed] [Google Scholar]
- 12.Royaux IE, Wall SM, Karniski LP, Everett LA, Suzuki K, et al. Pendrin, encoded by the Pendred syndrome gene, resides in the apical region of renal intercalated cells and mediates bicarbonate secretion. Proc. Natl. Acad. Sci. USA. 2001;98:4221–26. doi: 10.1073/pnas.071516798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wall SM, Hassell KA, Royaux IE, Green ED, Chang JY, et al. Localization of pendrin in mouse kidney. Am. J. Physiol. Ren. Physiol. 2003;284:229–41. doi: 10.1152/ajprenal.00147.2002. [DOI] [PubMed] [Google Scholar]
- 14.Teng-umnuay P, Verlander JW, Yuan W, Tisher CC, Madsen KM. Identification of distinct subpopulations of intercalated cells in the mouse collecting duct. J. Am. Soc. Nephrol. 1996;7:260–74. doi: 10.1681/ASN.V72260. [DOI] [PubMed] [Google Scholar]
- 15.Gamba G, Saltzberg SN, Lombardi M, Miyanoshita A, Lytton J, et al. Primary structure and functional expression of a cDNA encoding the thiazide-sensitive, electroneutral sodium-chloride cotransporter. Proc. Natl. Acad. Sci. USA. 1993;90:2749–53. doi: 10.1073/pnas.90.7.2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gamba G. Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol. Rev. 2005;85:423–93. doi: 10.1152/physrev.00011.2004. [DOI] [PubMed] [Google Scholar]
- 17.Gamba G, Miyanoshita A, Lombardi M, Lytton J, Lee WS, et al. Molecular cloning, primary structure, and characterization of two members of the mammalian electroneutral sodium-(potassium)-chloride cotransporter family expressed in kidney. J. Biol. Chem. 1994;269:17713–22. [PubMed] [Google Scholar]
- 18.Hebert SC, Gamba G. Molecular cloning and characterization of the renal diuretic-sensitive electroneutral sodium-(potassium)-chloride cotransporters. Clin. Investig. 1994;72:692–94. doi: 10.1007/BF00212991. [DOI] [PubMed] [Google Scholar]
- 19.Simon DB, Nelson-Williams C, Bia MJ, Ellison D, Karet FE, et al. Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat. Genet. 1996;12:24–30. doi: 10.1038/ng0196-24. [DOI] [PubMed] [Google Scholar]
- 20.Schultheis PJ, Lorenz JN, Meneton P, Nieman ML, Riddle TM, et al. Phenotype resembling Gitelman’s syndrome in mice lacking the apical Na+-Cl− cotransporter of the distal convoluted tubule. J. Biol. Chem. 1998;273:29150–55. doi: 10.1074/jbc.273.44.29150. [DOI] [PubMed] [Google Scholar]
- 21.Ji W, Foo JN, O’Roak BJ, Zhao H, Larson MG, et al. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat. Genet. 2008;40:592–99. doi: 10.1038/ng.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beyer KH, Jr, Baer JE, Russo HF, Noll R. Electrolyte excretion as influenced by chlorothiazide. Science. 1958;127:146–47. doi: 10.1126/science.127.3290.146. [DOI] [PubMed] [Google Scholar]
- 23.ALLHAT Off. Coord. ALLHAT Collab. Res. Group Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker versus diuretic: the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT) JAMA. 2002;288:2981–97. doi: 10.1001/jama.288.23.2981. [DOI] [PubMed] [Google Scholar]
- 24.Appel LJ. The verdict from ALLHAT–thiazide diuretics are the preferred initial therapy for hypertension. JAMA. 2002;288:3039–42. doi: 10.1001/jama.288.23.3039. [DOI] [PubMed] [Google Scholar]
- 25.Chang SS, Grunder S, Hanukoglu A, Rosler A, Mathew PM, et al. Mutations in subunits of the epithelial sodium channel cause salt wasting with hyperkalaemic acidosis, pseudohypoaldosteronism type 1. Nat. Genet. 1996;12:248–53. doi: 10.1038/ng0396-248. [DOI] [PubMed] [Google Scholar]
- 26.Canessa CM, Schild L, Buell G, Thorens B, Gautschi I, et al. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature. 1994;367:463–67. doi: 10.1038/367463a0. [DOI] [PubMed] [Google Scholar]
- 27.Kellenberger S, Schild L. Epithelial sodium channel/degenerin family of ion channels: a variety of functions for a shared structure. Physiol. Rev. 2002;82:735–67. doi: 10.1152/physrev.00007.2002. [DOI] [PubMed] [Google Scholar]
- 28.Qadri YJ, Song Y, Fuller CM, Benos DJ. Amiloride docking to acid-sensing ion channel-1. J. Biol. Chem. 2010;285:9627–35. doi: 10.1074/jbc.M109.082735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bicking JB, Mason JW, Woltersdorf OW, Jr, Jones JH, Kwong SF, et al. Pyrazine diuretics. I. N-Amidino-3-amino-6-halopyrazinecarboxamides. J. Med. Chem. 1965:638–42. doi: 10.1021/jm00313a014. [DOI] [PubMed] [Google Scholar]
- 30.Frindt G, Burg MB. Effect of vasopressin on sodium transport in renal cortical collecting tubules. Kidney Int. 1972;1:224–31. doi: 10.1038/ki.1972.32. [DOI] [PubMed] [Google Scholar]
- 31.Tomita K, Pisano JJ, Knepper MA. Control of sodium and potassium transport in the cortical collecting duct of the rat. Effects of bradykinin, vasopressin, and deoxycorticosterone. J. Clin. Investig. 1985;76:132–36. doi: 10.1172/JCI111935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O’Neil RG, Helman SI. Transport characteristics of renal collecting tubules: influences of DOCA and diet. Am. J. Physiol. Ren. Physiol. 1977;233:544–58. doi: 10.1152/ajprenal.1977.233.6.F544. [DOI] [PubMed] [Google Scholar]
- 33.Sansom SC, Weinman EJ, O’Neil RG. Microelectrode assessment of chloride-conductive properties of cortical collecting duct. Am. J. Physiol. Ren. Physiol. 1984;247:291–302. doi: 10.1152/ajprenal.1984.247.2.F291. [DOI] [PubMed] [Google Scholar]
- 34.Warden DH, Schuster VL, Stokes JB. Characteristics of the paracellular pathway of rabbit cortical collecting duct. Am. J. Physiol. Ren. Physiol. 1988;255:720–27. doi: 10.1152/ajprenal.1988.255.4.F720. [DOI] [PubMed] [Google Scholar]
- 35.O’Neil RG, Boulpaep EL. Ionic conductive properties and electrophysiology of the rabbit cortical collecting tubule. Am. J. Physiol. Ren. Physiol. 1982;243:81–95. doi: 10.1152/ajprenal.1982.243.1.F81. [DOI] [PubMed] [Google Scholar]
- 36.Hou J, Renigunta A, Yang J, Waldegger S. Claudin-4 forms paracellular chloride channel in the kidney and requires claudin-8 for tight junction localization. Proc. Natl. Acad. Sci. USA. 2010;107:18010–15. doi: 10.1073/pnas.1009399107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schuster VL, Stokes JB. Chloride transport by the cortical and outer medullary collecting duct. Am. J. Physiol. Ren. Physiol. 1987;253:203–12. doi: 10.1152/ajprenal.1987.253.2.F203. [DOI] [PubMed] [Google Scholar]
- 38.Tago K, Schuster VL, Stokes JB. Stimulation of chloride transport by HCO3−-CO2 in rabbit cortical collecting tubule. Am. J. Physiol. Ren. Physiol. 1986;251:49–56. doi: 10.1152/ajprenal.1986.251.1.F49. [DOI] [PubMed] [Google Scholar]
- 39.Star RA, Burg MB, Knepper MA. Bicarbonate secretion and chloride absorption by rabbit cortical collecting ducts. Role of chloride/bicarbonate exchange. J. Clin. Investig. 1985;76:1123–30. doi: 10.1172/JCI112067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schuster VL, Bonsib SM, Jennings ML. Two types of collecting duct mitochondria-rich (intercalated) cells: lectin and band 3 cytochemistry. Am. J. Physiol. Cell Physiol. 1986;251:347–55. doi: 10.1152/ajpcell.1986.251.3.C347. [DOI] [PubMed] [Google Scholar]
- 41.Emmons C, Kurtz I. Functional characterization of three intercalated cell subtypes in the rabbit outer cortical collecting duct. J. Clin. Investig. 1994;93:417–23. doi: 10.1172/JCI116976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Matsuzaki K, Stokes JB, Schuster VL. Stimulation of Cl− self exchange by intracellular HCO3− in rabbit cortical collecting duct. Am. J. Physiol. Cell Physiol. 1989;257:94–101. doi: 10.1152/ajpcell.1989.257.1.C94. [DOI] [PubMed] [Google Scholar]
- 43.Nissant A, Paulais M, Lachheb S, Lourdel S, Teulon J. Similar chloride channels in the connecting tubule and cortical collecting duct of the mouse kidney. Am. J. Physiol. Ren. Physiol. 2006;290:1421–29. doi: 10.1152/ajprenal.00274.2005. [DOI] [PubMed] [Google Scholar]
- 44.Pendred V. Deaf-mutism and goitre. Lancet. 1896;2:532. [Google Scholar]
- 45.Everett LA, Glaser B, Beck JC, Idol JR, Buchs A, et al. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS) Nat. Genet. 1997;17:411–22. doi: 10.1038/ng1297-411. [DOI] [PubMed] [Google Scholar]
- 46.Kraiem Z, Heinrich R, Sadeh O, Shiloni E, Nassir E, et al. Sulfate transport is not impaired in Pendred syndrome thyrocytes. J. Clin. Endocrinol. Metab. 1999;84:2574–76. doi: 10.1210/jcem.84.7.5973. [DOI] [PubMed] [Google Scholar]
- 47.Scott DA, Karniski LP. Human pendrin expressed in Xenopus laevis oocytes mediates chloride/formate exchange. Am. J. Physiol. Cell Physiol. 2000;278:207–11. doi: 10.1152/ajpcell.2000.278.1.C207. [DOI] [PubMed] [Google Scholar]
- 48.Scott DA, Wang R, Kreman TM, Sheffield VC, Karniski LP. The Pendred syndrome gene encodes a chloride-iodide transport protein. Nat. Genet. 1999;21:440–43. doi: 10.1038/7783. [DOI] [PubMed] [Google Scholar]
- 49.Soleimani M, Greeley T, Petrovic S, Wang Z, Amlal H, et al. Pendrin: an apical Cl−/OH−/HCO3− exchanger in the kidney cortex. Am. J. Physiol. Ren. Physiol. 2001;280:356–64. doi: 10.1152/ajprenal.2001.280.2.F356. [DOI] [PubMed] [Google Scholar]
- 50.Wangemann P, Nakaya K, Wu T, Maganti RJ, Itza EM, et al. Loss of cochlear HCO3− secretion causes deafness via endolymphatic acidification and inhibition of Ca2+ reabsorption in a Pendred syndrome mouse model. Am. J. Physiol. Ren. Physiol. 2007;292:1345–53. doi: 10.1152/ajprenal.00487.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nakaya K, Harbidge DG, Wangemann P, Schultz BD, Green ED, et al. Lack of pendrin HCO3− transport elevates vestibular endolymphatic [Ca2+] by inhibition of acid-sensitive TRPV5 and TRPV6 channels. Am. J. Physiol. Ren. Physiol. 2007;292:1314–21. doi: 10.1152/ajprenal.00432.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim HM, Wangemann P. Epithelial cell stretching and luminal acidification lead to a retarded development of stria vascularis and deafness in mice lacking pendrin. PLoS ONE. 2011;6:e17949. doi: 10.1371/journal.pone.0017949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim HM, Wangemann P. Failure of fluid absorption in the endolymphatic sac initiates cochlear enlargement that leads to deafness in mice lacking pendrin expression. PLoS ONE. 2010;5:e14041. doi: 10.1371/journal.pone.0014041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wangemann P, Kim HM, Billings S, Nakaya K, Li X, et al. Developmental delays consistent with cochlear hypothyroidism contribute to failure to develop hearing in mice lacking Slc26a4/pendrin expression. Am. J. Physiol. Ren. Physiol. 2009;297:1435–47. doi: 10.1152/ajprenal.00011.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Everett LA, Belyantseva IA, Noben-Trauth K, Cantos R, Chen A, et al. Targeted disruption of mouse Pds provides insight about the inner-ear defects encountered in Pendred syndrome. Hum. Mol. Genet. 2001;10:153–61. doi: 10.1093/hmg/10.2.153. [DOI] [PubMed] [Google Scholar]
- 56.Bizhanova A, Kopp P. Minireview: the sodium-iodide symporter NIS and pendrin in iodide homeostasis of the thyroid. Endocrinology. 2009;150:1084–90. doi: 10.1210/en.2008-1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bizhanova A, Kopp P. Genetics and phenomics of Pendred syndrome. Mol. Cell Endocrinol. 2010;322:83–90. doi: 10.1016/j.mce.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 58.Wall SM, Kim YH, Stanley L, Glapion DM, Everett LA, et al. NaCl restriction upregulates renal Slc26a4 through subcellular redistribution: role in Cl− conservation. Hypertension. 2004;44:982–87. doi: 10.1161/01.HYP.0000145863.96091.89. [DOI] [PubMed] [Google Scholar]
- 59.Quentin F, Chambrey R, Trinh-Trang-Tan MM, Fysekidis M, Cambillau M, et al. The Cl−/HCO3− exchanger pendrin in the rat kidney is regulated in response to chronic alterations in chloride balance. Am. J. Physiol. Ren. Physiol. 2004;287:1179–88. doi: 10.1152/ajprenal.00211.2004. [DOI] [PubMed] [Google Scholar]
- 60.Hafner P, Grimaldi R, Capuano P, Capasso G, Wagner CA. Pendrin in the mouse kidney is primarily regulated by Cl− excretion but also by systemic metabolic acidosis. Am. J. Physiol. Cell Physiol. 2008;295:1658–67. doi: 10.1152/ajpcell.00419.2008. [DOI] [PubMed] [Google Scholar]
- 61.Verlander JW, Kim YH, Shin W, Pham TD, Hassell KA, et al. Dietary Cl− restriction upregulates pendrin expression within the apical plasma membrane of type B intercalated cells. Am. J. Physiol. Ren. Physiol. 2006;291:833–39. doi: 10.1152/ajprenal.00474.2005. [DOI] [PubMed] [Google Scholar]
- 62.Pech V, Zheng W, Pham TD, Verlander JW, Wall SM. Angiotensin II activates H+-ATPase in type A intercalated cells. J. Am. Soc. Nephrol. 2008;19:84–91. doi: 10.1681/ASN.2007030277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Verlander JW, Hassell KA, Royaux IE, Glapion DM, Wang ME, et al. Deoxycorticosterone upregulates PDS (Slc26a4) in mouse kidney: role of pendrin in mineralocorticoid-induced hypertension. Hypertension. 2003;42:356–62. doi: 10.1161/01.HYP.0000088321.67254.B7. [DOI] [PubMed] [Google Scholar]
- 64.Vallet M, Picard N, Loffing-Cueni D, Fysekidis M, Bloch-Faure M, et al. Pendrin regulation in mouse kidney primarily is chloride-dependent. J. Am. Soc. Nephrol. 2006;17:2153–63. doi: 10.1681/ASN.2005101054. [DOI] [PubMed] [Google Scholar]
- 65.Pradervand S, Wang Q, Burnier M, Beermann F, Horisberger JD, et al. A mouse model for Liddle’s syndrome. J. Am. Soc. Nephrol. 1999;10:2527–33. doi: 10.1681/ASN.V10122527. [DOI] [PubMed] [Google Scholar]
- 66.Pradervand S, Vandewalle A, Bens M, Gautschi I, Loffing J, et al. Dysfunction of the epithelial sodium channel expressed in the kidney of a mouse model for Liddle syndrome. J. Am. Soc. Nephrol. 2003;14:2219–28. doi: 10.1097/01.asn.0000080204.65527.e6. [DOI] [PubMed] [Google Scholar]
- 67.Rubera I, Loffing J, Palmer LG, Frindt G, Fowler-Jaeger N, et al. Collecting duct-specific gene inactivation of αENaC in the mouse kidney does not impair sodium and potassium balance. J. Clin. Investig. 2003;112:554–65. doi: 10.1172/JCI16956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tomita K, Pisano JJ, Burg MB, Knepper MA. Effects of vasopressin and bradykinin on anion transport by the rat cortical collecting duct. Evidence for an electroneutral sodium chloride transport pathway. J. Clin. Investig. 1986;77:136–41. doi: 10.1172/JCI112268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Terada Y, Knepper MA. Thiazide-sensitive NaCl absorption in rat cortical collecting duct. Am. J. Physiol. Ren. Physiol. 1990;259:519–28. doi: 10.1152/ajprenal.1990.259.3.F519. [DOI] [PubMed] [Google Scholar]
- 70.Rouch AJ, Chen L, Troutman SL, Schafer JA. Na+ transport in isolated rat CCD: effects of bradykinin, ANP, clonidine, and hydrochlorothiazide. Am. J. Physiol. Ren. Physiol. 1991;260:86–95. doi: 10.1152/ajprenal.1991.260.1.F86. [DOI] [PubMed] [Google Scholar]
- 71.Plotkin MD, Kaplan MR, Verlander JW, Lee WS, Brown D, et al. Localization of the thiazide sensitive Na-Cl cotransporter, rTSC1, in the rat kidney. Kidney Int. 1996;50:174–83. doi: 10.1038/ki.1996.300. [DOI] [PubMed] [Google Scholar]
- 72.Bachmann S, Velazquez H, Obermuller N, Reilly RF, Moser D, Ellison DH. Expression of the thiazide-sensitive Na-Cl cotransporter by rabbit distal convoluted tubule cells. J. Clin. Investig. 1995;96:2510–14. doi: 10.1172/JCI118311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Obermuller N, Bernstein P, Velazquez H, Reilly R, Moser D, et al. Expression of the thiazide-sensitive Na-Cl cotransporter in rat and human kidney. Am. J. Physiol. Ren. Physiol. 1995;269:900–10. doi: 10.1152/ajprenal.1995.269.6.F900. [DOI] [PubMed] [Google Scholar]
- 74.Leviel F, Hubner CA, Houillier P, Morla L, El Moghrabi S, et al. The Na+-dependent chloride-bicarbonate exchanger SLC4A8 mediates an electroneutral Na+ reabsorption process in the renal cortical collecting ducts of mice. J. Clin. Investig. 2010;120:1627–35. doi: 10.1172/JCI40145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Damkier HH, Aalkjaer C, Praetorius J. Na+-dependent HCO3− import by the slc4a10 gene product involves Cl− export. J. Biol. Chem. 2010;285:26998–7007. doi: 10.1074/jbc.M110.108712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Grichtchenko II, Choi I, Zhong X, Bray-Ward P, Russell JM, Boron WF. Cloning, characterization, and chromosomal mapping of a human electroneutral Na+-driven Cl-HCO3 exchanger. J. Biol. Chem. 2001;276:8358–63. doi: 10.1074/jbc.C000716200. [DOI] [PubMed] [Google Scholar]
- 77.Parker MD, Musa-Aziz R, Rojas JD, Choi I, Daly CM, Boron WF. Characterization of human SLC4A10 as an electroneutral Na/HCO3 cotransporter (NBCn2) with Cl− self-exchange activity. J. Biol. Chem. 2008;283:12777–88. doi: 10.1074/jbc.M707829200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pech V, Pham TD, Hong S, Weinstein AM, Spencer KB, et al. Pendrin modulates ENaC function by changing luminal HCO3. J. Am. Soc. Nephrol. 2010;21:1928–41. doi: 10.1681/ASN.2009121257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kim YH, Pech V, Spencer KB, Beierwaltes WH, Everett LA, et al. Reduced ENaC protein abundance contributes to the lower blood pressure observed in pendrin-null mice. Am. J. Physiol. Ren. Physiol. 2007;293:1314–24. doi: 10.1152/ajprenal.00155.2007. [DOI] [PubMed] [Google Scholar]
- 80.Rieg T, Bundey RA, Chen Y, Deschenes G, Junger W, et al. Mice lacking P2Y2 receptors have salt-resistant hypertension and facilitated renal Na+ and water reabsorption. FASEB J. 2007;21:3717–26. doi: 10.1096/fj.07-8807com. [DOI] [PubMed] [Google Scholar]
- 81.Bailey M, Shirley D. Effects of extracellular nucleotides on renal tubular solute transport. Purinergic Signal. 2009;5:473–80. doi: 10.1007/s11302-009-9149-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Garvin JL, Herrera M, Ortiz PA. Regulation of renal NaCl transport by nitric oxide, endothelin, and ATP: clinical implications. Annu. Rev. Physiol. 2011;73:359–76. doi: 10.1146/annurev-physiol-012110-142247. [DOI] [PubMed] [Google Scholar]
- 83.Kishore BK, Chou CL, Knepper MA. Extracellular nucleotide receptor inhibits AVP-stimulated water permeability in inner medullary collecting duct. Am. J. Physiol. Ren. Physiol. 1995;269:863–69. doi: 10.1152/ajprenal.1995.269.6.F863. [DOI] [PubMed] [Google Scholar]
- 84.Pochynyuk O, Bugaj V, Rieg T, Insel PA, Mironova E, et al. Paracrine regulation of the epithelial Na+ channel in the mammalian collecting duct by purinergic P2Y2 receptor tone. J. Biol. Chem. 2008;283:36599–607. doi: 10.1074/jbc.M807129200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pochynyuk O, Rieg T, Bugaj V, Schroth J, Fridman A, et al. Dietary Na+ inhibits the open probability of the epithelial sodium channel in the kidney by enhancing apical P2Y2-receptor tone. FASEB J. 2010;24:2056–65. doi: 10.1096/fj.09-151506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rieg T, Vallon V. ATP and adenosine in the local regulation of water transport and homeostasis by the kidney. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009;296:419–27. doi: 10.1152/ajpregu.90784.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vallon V. P2 receptors in the regulation of renal transport mechanisms. Am. J. Physiol. Ren. Physiol. 2008;294:10–27. doi: 10.1152/ajprenal.00432.2007. [DOI] [PubMed] [Google Scholar]
- 88.Wildman SSP, Boone M, Peppiatt-Wildman CM, Contreras-Sanz A, King BF, et al. Nucleotides downregulate aquaporin 2 via activation of apical P2 receptors. J. Am. Soc. Nephrol. 2009;20:1480–90. doi: 10.1681/ASN.2008070686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang Y, Kohan DE, Nelson RD, Carlson NG, Kishore BK. Potential involvement of P2Y2 receptor in diuresis of postobstructive uropathy in rats. Am. J. Physiol. Ren. Physiol. 2010;298:634–42. doi: 10.1152/ajprenal.00382.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang Y, Nelson RD, Carlson NG, Kamerath CD, Kohan DE, Kishore BK. Potential role of purinergic signaling in lithium-induced nephrogenic diabetes insipidus. Am. J. Physiol. Ren. Physiol. 2009;296:1194–201. doi: 10.1152/ajprenal.90774.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ebihara L. New roles for connexons. Physiology. 2003;18:100–3. doi: 10.1152/nips.01431.2002. [DOI] [PubMed] [Google Scholar]
- 92.Evans WH, De Vuyst E, Leybaert L. The gap junction cellular internet: Connexin hemichannels enter the signalling limelight. Biochem. J. 2006;397:1–14. doi: 10.1042/BJ20060175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.McCulloch F, Chambrey R, Eladari D, Peti-Peterdi J. Localization of connexin 30 in the luminal membrane of cells in the distal nephron. Am. J. Physiol. Ren. Physiol. 2005;289:1304–12. doi: 10.1152/ajprenal.00203.2005. [DOI] [PubMed] [Google Scholar]
- 94.Hanner F, Schnichels M, Zheng-Fischhofer Q, Yang LE, Toma I, et al. Connexin 30.3 is expressed in the kidney but not regulated by dietary salt or high blood pressure. Cell Commun. Adhes. 2008;15:219–30. doi: 10.1080/15419060802013836. [DOI] [PubMed] [Google Scholar]
- 95.Hanner F, Sorensen CM, Holstein-Rathlou N-H, Peti-Peterdi J. Connexins and the kidney. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010;298:1143–55. doi: 10.1152/ajpregu.00808.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sipos A, Vargas SL, Toma I, Hanner F, Willecke K, Peti-Peterdi J. Connexin 30 deficiency impairs renal tubular ATP release and pressure natriuresis. J. Am. Soc. Nephrol. 2009;20:1724–32. doi: 10.1681/ASN.2008101099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mironova E, Peti-Peterdi J, Bugaj V, Stockand JD. Diminished paracrine regulation of the epithelial Na+ channel by purinergic signaling in mice lacking connexin 30. J. Biol. Chem. 2011;286:1054–60. doi: 10.1074/jbc.M110.176552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Stockand JD, Mironova E, Bugaj V, Rieg T, Insel PA, et al. Purinergic inhibition of ENaC produces aldosterone escape. J. Am. Soc. Nephrol. 2010;21:1903–11. doi: 10.1681/ASN.2010040377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lu M, MacGregor GG, Wang W, Giebisch G. Extracellular ATP inhibits the small-conductance K channel on the apical membrane of the cortical collecting duct from mouse kidney. J. Gen. Physiol. 2000;116:299–310. doi: 10.1085/jgp.116.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Holtzclaw JD, Cornelius RB, Hatcher LI, Sansom SC. Coupled ATP and potassium efflux from intercalated cells. Am. J. Physiol. Ren. Physiol. 2011;300:1319–26. doi: 10.1152/ajprenal.00112.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Holtzclaw JD, Grimm PR, Sansom SC. Intercalated cell BK-α/β4 channels modulate sodium and potassium handling during potassium adaptation. J. Am. Soc. Nephrol. 2010;21:634–45. doi: 10.1681/ASN.2009080817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Arroyo JP, Ronzaud C, Lagnaz D, Staub O, Gamba G. Aldosterone paradox: differential regulation of ion transport in distal nephron. Physiology. 2011;26:115–23. doi: 10.1152/physiol.00049.2010. [DOI] [PubMed] [Google Scholar]
- 103.Spray DC, Ye Z-C, Ransom BR. Functional connexin “hemichannels”: a critical appraisal. Glia. 2006;54:758–73. doi: 10.1002/glia.20429. [DOI] [PubMed] [Google Scholar]
- 104.Barbe MT, Monyer H, Bruzzone R. Cell-cell communication beyond connexins: the pannexin channels. Physiology. 2006;21:103–14. doi: 10.1152/physiol.00048.2005. [DOI] [PubMed] [Google Scholar]
- 105.Huang Y-J, Maruyama Y, Dvoryanchikov G, Pereira E, Chaudhari N, Roper SD. The role of pannexin 1 hemichannels in ATP release and cell–cell communication in mouse taste buds. Proc. Natl. Acad. Sci. 2007;104:6436–41. doi: 10.1073/pnas.0611280104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hanner F, Peti-Peterdi J. Pannexin1 is a novel renal ATP release mechanism. FASEB J. 2010;24 606.27 (Meet. Abstr. Suppl.) [Google Scholar]
- 107.Teubner B, Michel V, Pesch J, Lautermann J, Cohen-Salmon M, et al. Connexin30 (Gjb6)-deficiency causes severe hearing impairment and lack of endocochlear potential. Hum. Mol. Genet. 2003;12:13–21. doi: 10.1093/hmg/ddg001. [DOI] [PubMed] [Google Scholar]
- 108.Lang F, Vallon V, Knipper M, Wangemann P. Functional significance of channels and transporters expressed in the inner ear and kidney. Am. J. Physiol. Cell Physiol. 2007;293:1187–208. doi: 10.1152/ajpcell.00024.2007. [DOI] [PubMed] [Google Scholar]
- 109.Nozu K, Inagaki T, Fu XJ, Nozu Y, Kaito H, et al. Molecular analysis of digenic inheritance in Bartter syndrome with sensorineural deafness. J. Med. Genet. 2008;45:182–86. doi: 10.1136/jmg.2007.052944. [DOI] [PubMed] [Google Scholar]
- 110.Prieto-Carrasquero MC, Botros FT, Kobori H, Navar LG. Collecting duct renin: a major player in angiotensin II-dependent hypertension. J. Am. Soc. Hypertens. 3:96–104. doi: 10.1016/j.jash.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rohrwasser A, Morgan T, Dillon HF, Zhao L, Callaway CW, et al. Elements of a paracrine tubular renin-angiotensin system along the entire nephron. Hypertension. 1999;34:1265–74. doi: 10.1161/01.hyp.34.6.1265. [DOI] [PubMed] [Google Scholar]
- 112.Kang JJ, Toma I, Sipos A, Meer EJ, Vargas SL, Peti-Peterdi J. The collecting duct is the major source of prorenin in diabetes. Hypertension. 2008;51:1597–604. doi: 10.1161/HYPERTENSIONAHA.107.107268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Komlosi P, Fuson AL, Fintha A, Peti-Peterdi J, Rosivall L, et al. Angiotensin I conversion to angiotensin II stimulates cortical collecting duct sodium transport. Hypertension. 2003;42:195–99. doi: 10.1161/01.HYP.0000081221.36703.01. [DOI] [PubMed] [Google Scholar]
- 114.Peti-Peterdi J, Warnock DG, Bell PD. Angiotensin II directly stimulates ENaC activity in the cortical collecting duct via AT1 receptors. J. Am. Soc. Nephrol. 2002;13:1131–35. doi: 10.1097/01.asn.0000013292.78621.fd. [DOI] [PubMed] [Google Scholar]
- 115.Peti-Peterdi J. High glucose and renin release: the role of succinate and GPR91. Kidney Int. 2010;78:1214–17. doi: 10.1038/ki.2010.333. [DOI] [PubMed] [Google Scholar]
- 116.He W, Miao FJP, Lin DCH, Schwandner RT, Wang Z, et al. Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature. 2004;429:188–93. doi: 10.1038/nature02488. [DOI] [PubMed] [Google Scholar]
- 117.Robben JH, Fenton RA, Vargas SL, Schweer H, Peti-Peterdi J, et al. Localization of the succinate receptor in the distal nephron and its signaling in polarized MDCK cells. Kidney Int. 2009;76:1258–67. doi: 10.1038/ki.2009.360. [DOI] [PubMed] [Google Scholar]
- 118.Toma I, Kang JJ, Sipos A, Vargas S, Bansal E, et al. Succinate receptor GPR91 provides a direct link between high glucose levels and renin release in murine and rabbit kidney. J. Clin. Investig. 2008;118:2526–34. doi: 10.1172/JCI33293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Vargas SL, Toma I, Kang JJ, Meer EJ, Peti-Peterdi J. Activation of the succinate receptor GPR91 in macula densa cells causes renin release. J. Am. Soc. Nephrol. 2009;20:1002–11. doi: 10.1681/ASN.2008070740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Pluznick JL, Zou D-J, Zhang X, Yan Q, Rodriguez-Gil DJ, et al. Functional expression of the olfactory signaling system in the kidney. Proc. Natl. Acad. Sci. USA. 2009;106:2059–64. doi: 10.1073/pnas.0812859106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Figueroa CD, MacIver AG, Mackenzie JC, Bhoola KD. Localisation of immunoreactive kininogen and tissue kallikrein in the human nephron. Histochemistry. 1988;89:437–42. doi: 10.1007/BF00492599. [DOI] [PubMed] [Google Scholar]
- 122.Proud D, Vio CP. Localization of immunoreactive tissue kallikrein in human trachea. Am. J. Resp. Cell Mol. Biol. 1993;8:16–19. doi: 10.1165/ajrcmb/8.1.16. [DOI] [PubMed] [Google Scholar]
- 123.van Leeuwen BH, Grinblat SM, Johnston CI. Release of active and inactive kallikrein from the isolated perfused rat kidney. Clin. Sci. 1984;66:207–15. doi: 10.1042/cs0660207. [DOI] [PubMed] [Google Scholar]
- 124.Figueroa CD, Gonzalez CB, Grigoriev S, Abd Alla SA, Haasemann M, et al. Probing for the bradykinin B2 receptor in rat kidney by anti-peptide and anti-ligand antibodies. J. Histochem. Cytochem. 1995;43:137–48. doi: 10.1177/43.2.7822771. [DOI] [PubMed] [Google Scholar]
- 125.Bhoola KD, Figueroa CD, Worthy K. Bioregulation of kinins: kallikreins, kininogens, and kininases. Pharmacol. Rev. 1992;44:1–80. [PubMed] [Google Scholar]
- 126.Schuster VL, Kokko JP, Jacobson HR. Interactions of lysyl-bradykinin and antidiuretic hormone in the rabbit cortical collecting tubule. J. Clin. Investig. 1984;73:1659–67. doi: 10.1172/JCI111372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Azizi M, Boutouyrie P, Bissery A, Agharazii M, Verbeke F, et al. Arterial and renal consequences of partial genetic deficiency in tissue kallikrein activity in humans. J. Clin. Investig. 2005;115:780–87. doi: 10.1172/JCI23669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Horwitz D, Margolius HS, Keiser HR. Effects of dietary potassium and race on urinary excretion of kallikrein and aldosterone in man. J. Clin. Endocrinol. Metab. 1978;47:296–99. doi: 10.1210/jcem-47-2-296. [DOI] [PubMed] [Google Scholar]
- 129.Vio CP, Figueroa CD. Evidence for a stimulatory effect of high potassium diet on renal kallikrein. Kidney Int. 1987;31:1327–34. doi: 10.1038/ki.1987.146. [DOI] [PubMed] [Google Scholar]
- 130.Marchetti J, Imbert-Teboul M, Alhenc-Gelas F, Allegrini J, Menard J, Morel F. Kallikrein along the rabbit microdissected nephron: a micromethod for its measurement. Effect of adrenalectomy and DOCA treatment. Pflüg. Arch. 1984;401:27–33. doi: 10.1007/BF00581529. [DOI] [PubMed] [Google Scholar]
- 131.Miller DH, Lindley JG, Margolius HS. Tissue kallikrein levels and synthesis rates are not changed by an acute physiological dose of aldosterone. Proc. Soc. Exp. Biol. Med. 1985;180:121–25. doi: 10.3181/00379727-180-42152. [DOI] [PubMed] [Google Scholar]
- 132.El Moghrabi S, Houillier P, Picard N, Sohet F, Wootla B, et al. Tissue kallikrein permits early renal adaptation to potassium load. Proc. Natl. Acad. Sci. USA. 2010;107:13526–31. doi: 10.1073/pnas.0913070107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Fujita T, Hayashi I, Kumagai Y, Inamura N, Majima M. K+ loading, but not Na+ loading, and blockade of ATP-sensitive K+ channels augment renal kallikrein secretion. Immunopharmacology. 1999;44:169–75. doi: 10.1016/s0162-3109(99)00088-0. [DOI] [PubMed] [Google Scholar]
- 134.Fujita T, Hayashi I, Kumagai Y, Inamura N, Majima M. Early increases in renal kallikrein secretion on administration of potassium or ATP-sensitive potassium channel blockers in rats. Br. J. Pharmacol. 1999;128:1275–83. doi: 10.1038/sj.bjp.0702899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Stanton BA. Characterization of apical and basolateral membrane conductances of rat inner medullary collecting duct. Am. J. Physiol. Ren. Physiol. 1989;256:862–68. doi: 10.1152/ajprenal.1989.256.5.F862. [DOI] [PubMed] [Google Scholar]
- 136.Wang W. Regulation of renal K transport by dietary K intake. Annu. Rev. Physiol. 2004;66:547–69. doi: 10.1146/annurev.physiol.66.032102.112025. [DOI] [PubMed] [Google Scholar]
- 137.Youn JH, McDonough AA. Recent advances in understanding integrative control of potassium homeostasis. Annu. Rev. Physiol. 2009;71:381–401. doi: 10.1146/annurev.physiol.010908.163241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Giebisch G, Malnic G, Berliner RW. Control of renal potassium excretion. In: Brenner BM, editor. The Kidney. Vol. 1. W.B. Saunders; Philadelphia: 1996. pp. 371–407. [Google Scholar]
- 139.Meneton P, Bloch-Faure M, Hagege AA, Ruetten H, Huang W, et al. Cardiovascular abnormalities with normal blood pressure in tissue kallikrein-deficient mice. Proc. Natl. Acad. Sci. USA. 2001;98:2634–39. doi: 10.1073/pnas.051619598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Vallet V, Chraibi A, Gaeggeler HP, Horisberger JD, Rossier BC. An epithelial serine protease activates the amiloride-sensitive sodium channel. Nature. 1997;389:607–10. doi: 10.1038/39329. [DOI] [PubMed] [Google Scholar]
- 141.Caldwell RA, Boucher RC, Stutts MJ. Serine protease activation of near-silent epithelial Na+ channels. Am. J. Physiol. Cell Physiol. 2004;286:190–94. doi: 10.1152/ajpcell.00342.2003. [DOI] [PubMed] [Google Scholar]
- 142.Caldwell RA, Boucher RC, Stutts MJ. Neutrophil elastase activates near-silent epithelial Na+ channels and increases airway epithelial Na+ transport. Am. J. Physiol. Lung Cell Mol. Physiol. 2005;288:813–19. doi: 10.1152/ajplung.00435.2004. [DOI] [PubMed] [Google Scholar]
- 143.Chraibi A, Vallet V, Firsov D, Hess SK, Horisberger JD. Protease modulation of the activity of the epithelial sodium channel expressed in Xenopus oocytes. J. Gen. Physiol. 1998;111:127–38. doi: 10.1085/jgp.111.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Passero CJ, Mueller GM, Rondon-Berrios H, Tofovic SP, Hughey RP, Kleyman TR. Plasmin activates epithelial Na+ channels by cleaving the γ subunit. J. Biol. Chem. 2008;283:36586–91. doi: 10.1074/jbc.M805676200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hughey RP, Bruns JB, Kinlough CL, Harkleroad KL, Tong Q, et al. Epithelial sodium channels are activated by furin-dependent proteolysis. J. Biol. Chem. 2004;279:18111–14. doi: 10.1074/jbc.C400080200. [DOI] [PubMed] [Google Scholar]
- 146.Guipponi M, Vuagniaux G, Wattenhofer M, Shibuya K, Vazquez M, et al. The transmembrane serine protease (TMPRSS3) mutated in deafness DFNB8/10 activates the epithelial sodium channel (ENaC) in vitro. Hum. Mol. Genet. 2002;11:2829–36. doi: 10.1093/hmg/11.23.2829. [DOI] [PubMed] [Google Scholar]
- 147.Vuagniaux G, Vallet V, Jaeger NF, Hummler E, Rossier BC. Synergistic activation of ENaC by three membrane-bound channel-activating serine proteases (mCAP1, mCAP2, and mCAP3) and serum- and glucocorticoid-regulated kinase (Sgk1) in Xenopus oocytes. J. Gen. Physiol. 2002;120:191–201. doi: 10.1085/jgp.20028598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Vuagniaux G, Vallet V, Jaeger NF, Pfister C, Bens M, et al. Activation of the amiloride-sensitive epithelial sodium channel by the serine protease mCAP1 expressed in a mouse cortical collecting duct cell line. J. Am. Soc. Nephrol. 2000;11:828–34. doi: 10.1681/ASN.V115828. [DOI] [PubMed] [Google Scholar]
- 149.Hughey RP, Bruns JB, Kinlough CL, Kleyman TR. Distinct pools of epithelial sodium channels are expressed at the plasma membrane. J. Biol. Chem. 2004;279:48491–94. doi: 10.1074/jbc.C400460200. [DOI] [PubMed] [Google Scholar]
- 150.Hughey RP, Mueller GM, Bruns JB, Kinlough CL, Poland PA, et al. Maturation of the epithelial Na+ channel involves proteolytic processing of the α- and γ-subunits. J. Biol. Chem. 2003;278:37073–82. doi: 10.1074/jbc.M307003200. [DOI] [PubMed] [Google Scholar]
- 151.Adebamiro A, Cheng Y, Rao US, Danahay H, Bridges RJ. A segment of gamma ENaC mediates elastase activation of Na+ transport. J. Gen. Physiol. 2007;130:611–29. doi: 10.1085/jgp.200709781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Bruns JB, Carattino MD, Sheng S, Maarouf AB, Weisz OA, et al. Epithelial Na+ channels are fully activated by furin- and prostasin-dependent release of an inhibitory peptide from the gamma-subunit. J. Biol. Chem. 2007;282:6153–60. doi: 10.1074/jbc.M610636200. [DOI] [PubMed] [Google Scholar]
- 153.Diakov A, Bera K, Mokrushina M, Krueger B, Korbmacher C. Cleavage in the γ-subunit of the epithelial sodium channel (ENaC) plays an important role in the proteolytic activation of near-silent channels. J. Physiol. 2008;586:4587–608. doi: 10.1113/jphysiol.2008.154435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Harris M, Firsov D, Vuagniaux G, Stutts MJ, Rossier BC. A novel neutrophil elastase inhibitor prevents elastase activation and surface cleavage of the epithelial sodium channel expressed in Xenopus laevis oocytes. J. Biol. Chem. 2007;282:58–64. doi: 10.1074/jbc.M605125200. [DOI] [PubMed] [Google Scholar]
- 155.Kleyman TR, Carattino MD, Hughey RP. ENaC at the cutting edge: regulation of epithelial sodium channels by proteases. J. Biol. Chem. 2009;284:20447–51. doi: 10.1074/jbc.R800083200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Masilamani S, Kim GH, Mitchell C, Wade JB, Knepper MA. Aldosterone-mediated regulation of ENaC α, β, and γ subunit proteins in rat kidney. J. Clin. Investig. 1999;104:R19–23. doi: 10.1172/JCI7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Donaldson SH, Hirsh A, Li DC, Holloway G, Chao J, et al. Regulation of the epithelial sodium channel by serine proteases in human airways. J. Biol. Chem. 2002;277:8338–45. doi: 10.1074/jbc.M105044200. [DOI] [PubMed] [Google Scholar]
- 158.Planés C, Randrianarison NH, Charles RP, Frateschi S, Cluzeaud F, et al. ENaC-mediated alveolar fluid clearance and lung fluid balance depend on the channel-activating protease 1. EMBO Mol. Med. 2010;2:26–37. doi: 10.1002/emmm.200900050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Pisitkun T, Shen RF, Knepper MA. Identification and proteomic profiling of exosomes in human urine. Proc. Natl. Acad. Sci. USA. 2004;101:13368–73. doi: 10.1073/pnas.0403453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ergonul Z, Frindt G, Palmer LG. Regulation of maturation and processing of ENaC subunits in the rat kidney. Am. J. Physiol. Ren. Physiol. 2006;291:683–93. doi: 10.1152/ajprenal.00422.2005. [DOI] [PubMed] [Google Scholar]
- 161.Picard N, Eladari D, El Moghrabi S, Planés C, Bourgeois S, et al. Defective ENaC processing and function in tissue kallikrein-deficient mice. J. Biol. Chem. 2008;283:4602–11. doi: 10.1074/jbc.M705664200. [DOI] [PubMed] [Google Scholar]
- 162.Narikiyo T, Kitamura K, Adachi M, Miyoshi T, Iwashita K, et al. Regulation of prostasin by aldosterone in the kidney. J. Clin. Investig. 2002;109:401–8. doi: 10.1172/JCI13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Gumz ML, Lynch IJ, Greenlee MM, Cain BD, Wingo CS. The renal H+-K+-ATPases: physiology, regulation, and structure. Am. J. Physiol. Ren. Physiol. 2010;298:12–21. doi: 10.1152/ajprenal.90723.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Meneton P, Schultheis PJ, Greeb J, Nieman ML, Liu LH, et al. Increased sensitivity to K+ deprivation in colonic H,K-ATPase-deficient mice. J. Clin. Investig. 1998;101:536–42. doi: 10.1172/JCI1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Nakamura S, Amlal H, Galla JH, Soleimani M. Colonic H+-K+-ATPase is induced and mediates increased HCO3− reabsorption in inner medullary collecting duct in potassium depletion. Kidney Int. 1998;54:1233–39. doi: 10.1046/j.1523-1755.1998.00105.x. [DOI] [PubMed] [Google Scholar]
- 166.Wingo CS. Active proton secretion and potassium absorption in the rabbit outer medullary collecting duct. Functional evidence for proton-potassium-activated adenosine triphosphatase. J. Clin. Investig. 1989;84:361–65. doi: 10.1172/JCI114165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Dherbecourt O, Cheval L, Bloch-Faure M, Meneton P, Doucet A. Molecular identification of Sch28080-sensitive K-ATPase activities in the mouse kidney. Pflüg. Arch. 2006;451:769–75. doi: 10.1007/s00424-005-1508-1. [DOI] [PubMed] [Google Scholar]
- 168.Hecquet C, Tan F, Marcic BM, Erdos EG. Human bradykinin B2 receptor is activated by kallikrein and other serine proteases. Mol. Pharmacol. 2000;58:828–36. doi: 10.1124/mol.58.4.828. [DOI] [PubMed] [Google Scholar]
- 169.Gkika D, Topala CN, Chang Q, Picard N, Thebault S, et al. Tissue kallikrein stimulates Ca2+ reabsorption via PKC-dependent plasma membrane accumulation of TRPV5. EMBO J. 2006;25:4707–16. doi: 10.1038/sj.emboj.7601357. [DOI] [PMC free article] [PubMed] [Google Scholar]