

Abstract
Peptides with diverse amino acid sequences, structures and functions are essential players in biological systems. The construction of well-annotated databases not only facilitates effective information management, search and mining, but also lays the foundation for developing and testing new peptide algorithms and machines. The antimicrobial peptide database (APD) is an original construction in terms of both database design and peptide entries. The host defense antimicrobial peptides (AMPs) registered in the APD cover the five kingdoms (bacteria, protists, fungi, plants, and animals) or three domains of life (bacteria, archaea, and eukaryota). This comprehensive database (http://aps.unmc.edu/AP) provides useful information on peptide discovery timeline, nomenclature, classification, glossary, calculation tools, and statistics. The APD enables effective search, prediction, and design of peptides with antibacterial, antiviral, antifungal, antiparasitic, insecticidal, spermicidal, anticancer activities, chemotactic, immune modulation, or anti-oxidative properties. A universal classification scheme is proposed herein to unify innate immunity peptides from a variety of biological sources. As an improvement, the upgraded APD makes predictions based on the database-defined parameter space and provides a list of the sequences most similar to natural AMPs. In addition, the powerful pipeline design of the database search engine laid a solid basis for designing novel antimicrobials to combat resistant superbugs, viruses, fungi or parasites. This comprehensive AMP database is a useful tool for both research and education.
Keywords: ab initio design, database filtering tech, database screen, peptide design, peptide prediction, universal peptide classification
1. Introduction
There are at least two good reasons for our current focus on host defense antimicrobial peptides (AMPs). First, AMPs have remained potent for millions of years. Therefore, AMPs constitute useful templates for developing a new generation of antimicrobials to meet the growing antibiotic resistance problem worldwide. Second, AMPs are key components of the innate immune system universally required for the survival of both invertebrates and vertebrates. Thus, research in this direction improves our understanding of innate immunity and its relationships with the adaptive immune system in vertebrates [1-6].
Lysozyme, discovered by Alexander Fleming in 1922, is now recognized as the first antimicrobial peptide. However, there was little research on AMPs until the discoveries of cecropins, defensins, and magainins in the 1980s [7-9]. Since then, AMPs have been identified from a variety of living species. Select AMPs identified during 1922-2012 are listed in the discovery timeline page of the antimicrobial peptide database (APD) [10, 11]. In earlier days when the number of AMPs was limited, these peptides were handled in review articles. With a rapid increase in the number of such peptides, it became impractical to continue to manage them manually. As a consequence, several databases have been established to categorize these peptides [10-31]. AMSDb appears to be the first such database available online in 1998 [12]. The information format of this database is identical to the SWISS-Prot (UniProt) [32]. It contains 895 antimicrobial peptides, proteins, and their precursors from plants and animals. Unfortunately, AMSDb is no longer updated. To meet the need of better databases with a broad scope, two general databases were published side by side in 2004. ANTIMIC reported more than 1700 entries [13], while a new version of ANTIMIC called DAMPD [14] contains 1232 entries. In 2004, the first version of the APD [10] reported 525 peptide entries. These peptides were manually collected from the literature with the aid of public search engines such as Pub-Med, Swiss-Prot, and PDB [32-34]. The peptide number reached 1228 entries in the second version of the APD [11] and there are 2329 peptide entries in the current version.
Since the publication of APD and ANTIMIC, several specialized databases have been established to emphasize certain aspects of natural, synthetic, or recombinant AMPs from a special peptide family (circular peptides, defensins, and thiopeptides) or source (e.g., bacteria, plants, shrimps, amphibians) [15-28]. For example, defensin knowledgebase is dedicated to defensins only, while DADP contains only polypeptides from frogs. More recently, the CAMP [29], YADAMP [30], and LAMP [31] were also built. Table 1 lists major databases dedicated to AMPs. Among these databases, the APD [10, 11] stands out. This article highlights the unique aspects of the APD as well as new developments since the publication of the second version in 2009.
Table 1.
A chronological list of the databases for antimicrobial peptidesa
| Year | Database | URL (http://) | Scope | Country | Ref |
|---|---|---|---|---|---|
| 1998 | AMSDb | www.bbcm.univ.trieste.it/~tossi/amsdb.html | Plant/animal AMPs |
Italy | [12] |
| 2002 | SAPD | oma.terkko.helsinki.fi:808 0/~SAPD/ |
Synthetic AMPs |
Finland | [25] |
| 2003, 2004 |
Peptaibols | www.cryst.bbk.ac.uk/peptaibol/home.shtml | Fungal peptaibols |
England | [26] |
| 2004, 2009 |
APD | aps.unmc.edu/AP/ | AMPs | USA | [10- 11] |
| 2004, 2012 |
DAMPD | apps.sanbi.ac.za/dampd/ | AMPs | South Africa/Sau di Arabia |
[13- 14] |
| 2006 | PenBase | penbase.immunaqua.com | Shrimp AMPs |
France | [15] |
| 2006, 2008 |
Cybase | research1t.imb.uq.edu.au/ cybase/ |
Circular proteins |
Australia | [18] |
| 2006, 2010 |
BAGLE | bioinformatics.biol.rug.nl/ websoftware/bagel/bagel_ start.php |
Bacterial AMPs |
Netherland | [21] |
| 2007 | AMPer | marray.cmdr.ubc.ca/cgi- bin/amp.pl |
Like AMSDb |
Canada | [24] |
| 2007, 2010 |
BACTIBAS E |
bactibase.pfba-lab- tun.org/main.php |
Bacteriocins | Canada/Tu nisie |
[17] |
| 2007 | Defensins | defensins.bii.a-star.edu.sg/ | Defensins | Singapore | [16] |
| 2008 | RAPD | faculty.ist.unomaha.edu/chen/rapd/index.php | Recombinan t AMPs |
USA | [20] |
| 2009 | PhytAMP | phytamp.pfba-lab- tun.org/main.php |
Plant AMPs | Tunisie/Ca nada |
[19] |
| 2010 | CAMP | www.bicnirrh.res.in/antimicrobial | AMPs | India | [29] |
| 2012 | YADAMD | yadamp.unisa.it/ | AMPs | Italy | [30] |
| 2012 | DADP | split4.pmfst.hr/dadp/ | Amphibian AMPs |
Croatia | [22] |
| 2012 | THIOBASE | db- mml.sjtu.edu.cn/THIOBA SE/ |
Bacteria thiopeptides |
China | [23] |
| 2012 | EnzyBase | biotechlab.fudan.edu.cn/d atabase/EnzyBase/home.p hp |
Cleaving enzymes |
China | [27] |
| 2013 | LAMP | biotechlab.fudan.edu.cn/d atabase/lamp/guide.php |
AMPs | China | [31] |
| 2013 | MilkAMP | /milkampdb.org | Milk AMPs | Canada | [28] |
Adapted from the APD website (http://aps.unmc.edu/AP/links.php) [10, 11].
2. Database design and search functions
2.1. Criteria for peptide collections
In terms of peptide registration, the APD database [11] follows a set of self-defined criteria. First, the peptide must have a known amino acid sequence, at least partially. Second, the peptide should have demonstrated antimicrobial activity. Third, the peptide contains less than 100 amino acids (this has recently been expanded to 200 amino acids so that some important antimicrobial proteins could be collected). Fourth, the peptide originates primarily from natural sources, including bacteria, protozoa, fungi, plants, and animals. Only a small set of synthetic peptides of general interest was collected. Also, the APD emphasizes unique sequences. Therefore, peptides from different species currently occupy the same entry in this database if they share the same amino acid sequence. At present, there are 46 such entries in the APD, which were “found in multiple species” (the quoted phrase can be searched in the additional information field). Since in silico-predicted peptides may not be truly antimicrobial peptides, they are not registered into the APD at this stage. By following the above criteria, the APD database provides a well-defined set of peptides to the research community. Indeed, the APD is a well-recognized resource in the field of AMPs. For example, the web hits were ~15,000 per year prior to 2008 [11]. Since the publication of the second version in 2009, there is a dramatic increase in database use. For example, the web hits reached 86,000 in 2012 alone.
2.2. A flexible database design
The design of any database is to facilitate information search. Users can conduct a simple search by using peptide name and amino acid sequence in single-letter code. Different from other databases in Table 1, the power of the APD search engine can be ascribed to two important features. First, the search engine is composed of a pipeline of search functions. Second, the modular design of the APD enables continued expansion and development. These features greatly facilitate information search at an advanced level. For example, we obtained 268 defensins using the word “defensin” as a search term. The number of defensins rapidly reduced to 19 when the word “monkey” is also used. Only seven peptides were found when a combination of “defensin”, “monkey” and “theta” are used.
2.3. Database search functions
To make it easier for the APD users, Table 2 lists major search functions, peptide information and examples. Most of these search functions are self-explanatory. The name field of the APD, however, has been substantially expanded and deserves some description. It consists of the following elements:
Table 2.
Search functions of the antimicrobial peptide database
| Search Function |
Peptide Information | Examples |
|---|---|---|
| APD ID | A unique 5-digit number for each database entry |
AP00310 |
| Name | Peptide name or synonyms | LL-37 (LL37, FALL-39) |
| AMP sequence |
Amino acid sequence in single-letter code |
LLGDFFRKSKEKIGKEFKRI VQRIKDFLRNLVPRTES |
| Name | Life kingdoms | Bacteria, plants, fungi, protists, animals |
| Name | Life domains | Bacteria, archaea |
| Name | Classes | Fish, reptiles, amphibians, birds, insects, |
| Name | Peptide family | Defensins, cathelicidins, histatins, cecropins, magainins, |
| Source species |
Location where the peptide is found | Neutrophils; Homo sapiens |
| Length | The number of amino acids | 37 (for LL-37) |
| Net Charge | At pH 7 | +6 (for LL-37) |
| Hydrophobi c% |
Sum of L, I, V. M, A, F, W, C divided by peptide length |
35% (for LL-37) |
| Name | Chemical modification type | See Table 5 |
| Structure | (1) Known 3D (α, β, αβ, non-αβ); (2) Partial known (bridged, rich); (3) unknown |
Helix for LL-37 |
| Structural method |
X-ray; NMR; CD | NMR (for LL-37) |
| PDB ID | Self explained | 2K6O (for LL-37) |
| Activity | Known antimicrobial activity | Gram+/Gram−; Gram+; Gram−; viruses; HIV-1; fungi |
| Name | Binding target | See Table 6 |
| Additional info |
Mechanism of action | Magainin: forming pores |
| Additional info |
Synergy | LL-37 and lysozyme |
| Additional info |
Animal model | Mouse |
| Author or Pub year |
Search author or publication year separately |
Any |
Peptide name + family name + peptide source kingdom + post-translational modification + peptide binding molecules.
In the beginning, it gives peptide name, including synonyms and even the outdated names. In the case of human cathelicidin LL-37, the word LL37 is also used in the literature and FALL-39 is an outdated name. To help users to understand the AMP nomenclature, the major methods used to name AMPs are summarized in the APD website (aps.unmc.edu/AP/naming.php). These include the peptide property-based method, the source-based method, and a third method that uses both peptide features and source information. For examples, please visit the APD website.
After the peptide name, the peptide family name is also given in the NAME field. Selected AMP families are tabulated in Table 3. Using the peptide family name, one can obtain a list of AMPs from the same family. For example, there are 268 defensins from a variety of sources and 185 brevinins from amphibians.
Table 3.
Select antimicrobial peptide families in the APDa
| Peptide family | Count | Peptide family | Count |
|---|---|---|---|
| Defensins | 268 | Aureins | 12 |
| Cathelicidins | 78 | Maximins | 30 |
| Histatins | 12 | Brevinins | 185 |
| Neuropeptides | 20 | Temporins | 105 |
| Chemokines | 26 | Ranatuerins | 49 |
| Ribonucleases | 6 | Dermaseptins | 55 |
| Caerins | 29 | ||
| Cyclotides | 151 | Maculatins | 7 |
| Uperins | 12 | ||
| Lantibiotics | 51 | Magainins | 5 |
| Microcins | 13 | Cecropins | 24 |
Peptide counts in this and subsequent tables were obtained from the APD on November 30, 2013.
Following the family name, the peptide is further annotated in the NAME field based on the source domains or kingdoms. The five kingdoms of life are bacteria, protists (protozoa + algea), fungi, plants, and animals [35], while the three domains of life are bacteria, archaea, and eukaryote [36]. The peptide counts in each kingdom are listed in Table 4. Selected classes in each life domain are also given in the NAME field, allowing users to focus only on the AMPs of their interest.
Table 4.
Antimicrobial peptides from the three domains and five kingdoms of lifea
| Domain | Peptide count | Class | Peptide count |
|---|---|---|---|
| Bacteria | 209 | Insects | 216 |
| Archeae | 2 | Spiders | 33 |
| Eukaryota | 2082 | Molluscs | 27 |
| Worms | 14 | ||
| Kingdom | Peptide count | Crustaceans | 32 |
| Bacteria | 208 | Birds | 36 |
| Protists | 7 | Reptiles | 10 |
| Fungi | 12 | Fish | 79 |
| Plants | 301 | Amphibians | 929 |
| Animals | 1761 | Ruminants | 44 |
| Humans | 102 |
The importance of post-translational modifications (PTMs) is only secondary to the peptide sequence itself [37]. Because PTMs could influence both structure and function of the peptide, it is necessary to annotate sequence modification information in the same location. Table 5 contains 23 types of PTMs in the APD. To our knowledge, the APD is the only AMP database that contains extensive information on peptide chemical modifications. In addition, the effect of chemical modification on a peptide net charge is considered in the APD.
Table 5.
Post-translational modifications of natural antimicrobial peptides
| Search key | Post-translational modification |
Peptide count |
|---|---|---|
| XXA | Amidation | 448 |
| XXB | Chromophore/ion-binding moieties |
4 |
| XXC | Backbone cyclization | 176 |
| XXD | D-amino acids | 17 |
| XXE | Acetylation | 11 |
| XXF | Carboxylic-acid-containing unit |
8 |
| XXG | Glycosylation | 12 |
| XXH | Halogenation (Cl, Br) | 8 |
| XXJ | Sidechain-backbone cyclization |
15 |
| XXK | Hydroxylation | 9 |
| XXL | Lipidation | 9 |
| XXM | Methylation | 3 |
| XXN | Nitrolation | 0 |
| XXO | Oxidation | 10 |
| XXP | Phosphorylation | 3 |
| XXQ | N-terminal cyclic glutamate | 15 |
| XXR | Reduction | 2 |
| XXS | Sulfation | 1 |
| XXT | Thioether bridge | 46 |
| XXU | Rana Box via a single S-S bond |
269 |
| XXW | Dehydration | 21 |
| XXY | Citrullination | 1 |
| Structure searcha |
Disulfide bridges | 551 |
This number was obtained by searching for disulfide bond-containing AMPs classified as “Bridge”, “β structure”, and “αβ structure” families, respectively. The “bridged” AMPs are known to have disulfide bonds but unknown 3D structure. Beta structures without disulfide bonds were excluded by including “c” as a sequence search term. For the αβ structures, only the AMPs with a packed 3D fold were counted.
How AMPs kill pathogens is an important question to ask. The information for binding targets of AMPs is also annotated in the APD (Table 6). In addition to membranes, AMPs can bind to DNA, heat shock proteins, carbohydrates, and lipid II [1-6].
Table 6.
Binding targets of antimicrobial peptides
| Search keya | Binding target | Count |
|---|---|---|
| BBBh2o | Self aggregation in water | 15 |
| BBBm | Oligomers in membranes | 4 |
| BBII | Ions | 16 |
| BBW | Lipid II | 17 |
| BBL | LPS | 54 |
| BBr | Receptors | 3 |
| BBMm | Membranes | 81 |
| BBN | Nucleic acids | 11 |
| BBS | Sugars/carbohydrates | 44 |
3. Classification of AMPs based on peptide activity, 3D structure and chain bonding pattern
There are a variety of approaches for classifying AMPs. Some of these methods are summarized on the classification page of the APD website (aps.unmc.edu/AP/class.php). For example, the peptides may be classified based on the biosynthesis machinery. Some peptides are synthesized by a multiple enzyme system, while the majority of AMPs are gene-coded. The expression and degradation of gene-coded AMPs are elegantly regulated because either over or under expression of AMPs could cause problems [1-5]. AMPs can also be classified based on molecular targets (e.g., membrane targeting and cell-penetrating peptides) [6]. In the following, we first describe structure and activity-based classification schemes in the APD and then introduce a universal classification scheme for antimicrobial peptides.
3.1. Antimicrobial activity
As key effector molecules of innate immunity, AMPs are able to control invading pathogenic microbes, including bacteria, viruses, fungi, and parasites [1-4]. It is natural to classify these host defense peptides based on their functions, including antibacterial, antiviral, antifungal, insecticidal, and spermicidal activities. In addition, some AMPs also possess other functional roles such as anticancer, wound healing and immune modulation [4]. The APD database has annotated 17 types of peptide activities or functions (Table 7). Several newly annotated activity types are unique in this database, making the APD most comprehensive in terms of activity annotation.
Table 7.
Biological activities of host defense antimicrobial peptides
| Year created | Activitya | Count |
|---|---|---|
| 2003 | Antibacterial (G+/G−) | 1909 |
| 2003 | Antifungal | 850 |
| 2003 | Antiviral | 138 |
| 2003 | Anticancer | 158 |
| 2003 | Hemolytic | 284 |
| 2008 | Anti-HIV | 92 |
| 2009 | Anti-G+ | 360 |
| 2009 | Anti-G− | 172 |
| 2009 | Antiparasitic | 59 |
| 2009 | Insecticidal | 22 |
| 2009 | Spermicidal | 9 |
| 2011 | Chemotactic | 47 |
| 2012 | Anti-protist | 4 |
| 2013 | Antioxidant | 10 |
| 2013 | Anti-inflammatory | 2 |
| 2013 | Wound healing | 7 |
| 2013 | Enzyme inhibitor | 5 |
Some newly defined search functions can be searched in the “additional information” field of the APD by entering the words in the table. These include antioxidant, anti-inflammatory, and wound healing, and enzyme inhibitor.
3.2. Three-dimensional structure of AMPs
According to the APD, only a small population of AMPs (13%) has a known 3D structure, primarily determined by solution nuclear magnetic resonance (NMR) spectroscopy [10]. In addition, X-ray diffraction was also used to solve the structures of some AMPs with a folded structure in water. The structural information is well annotated in the APD database, including structural class, method for structural determination, structural regions, key residues, and membrane-mimetic models for structural determination. In addition, users can directly view the 3D structure via the link to the PDB [33]. The AMP structures are usually classified into α-helical, β-sheet, and extended structures [4, 38]. A more general classification approach has been proposed recently [6]. In this approach, the AMP structures are classified into four families: α, β, αβ, and non-αβ based on the types of secondary structures. Peptides in the α family contain α-helical structure (Figure 1A) as the major secondary structure. In contrast, AMPs in the β family are characterized by at least a pair of two β-strands in the structure (Figure 1B). The αβ family contains both α and β structures (Figure 1C), whereas the non-αβ family has neither α nor β structure (Figure 1D). This structural classification scheme is now executed in the APD. Typical examples and peptide counts from different families are provided in Table 8. While the α-helical family is the largest with 328 entries, the non-αβ family is the smallest with merely 9 entries. Table 8 also shows that the lysine/arginine (K/R) ratios in these structural families differ. While lysines are dominant in the α-helical family, arginines are preferred in the β-family as well as the non-αβ family. Not surprisingly, AMPs with both α and β structures have a moderate K/R ratio of ~1.2. These ratios might become useful as indicators for classifying a newly discovered peptide into a particular structural family.
Figure 1. Classification of the 3D structures of antimicrobial peptides into four families [6].
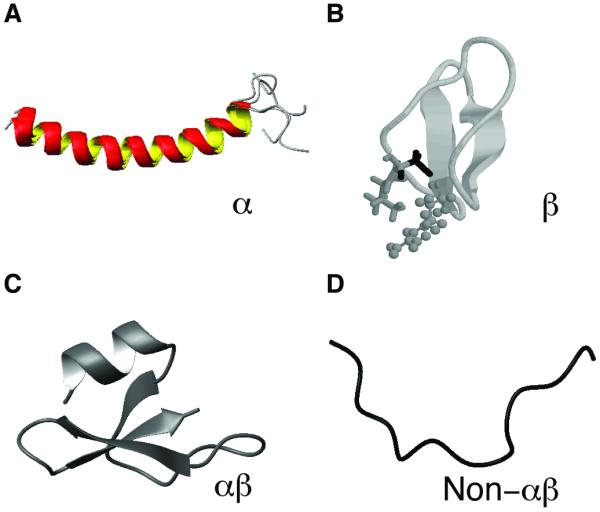
Shown are representatives from each family: (A) α-helical structure of human cathelicidin LL-37 (PDB entry: 2K6O) [55]; (B) the β-sheet structure of plant kalata B1 (PDB entry: 1JNU) [56]; (C) the αβ structure of human β-defensin-1 (HBD-1) (PDB entry: 1IJV) [57]; and (D) the non-αβ structure of cattle indolicidin (PDB entry: 1G89) [58].
Table 8.
Classification of 3D structures of antimicrobial peptides
| Structurea | K/R ratio | Peptide count | Examples |
|---|---|---|---|
| α | 13.65/5.26=2.59 | 329 | Cecropin, dermcidin, LL-37, magainin |
| β | 5.63/10.7=0.53 | 97 | Human alpha defensins (HNP-1, HNP- 4, and HD-5), plant kalata B1 |
| α β | 8.47/7.05=1.2 | 81 | Drosomycin, Human beta defensins (HBD-1, HBD-4), PhD1 |
| Non-αβ | 4.85/10.19=0.48 | 9 | Indolicidin, tritrpticin, drosocin, nisin A |
For AMPs without 3D structures, additional annotations were made in the APD: (1) unknown, no 3D structure; (2) bridge, disulfide-linked, usually beta-structure; (3) rich, rich in certain amino acids.
3.3. A universal classification of AMPs based on peptide bonding patterns
Because only a small number of AMPs has a 3D structure, we herein propose a systematic classification approach that is independent of 3D structure, peptide source, or activity. This classification is framed based on the connection mode of polypeptide chains. Class I includes linear AMPs (Figure 2A), which may be chemically modified (amidation, sulfate, phosphate, bromide, or glycosylation) at side chains or even backbones. However, such modifications (Table 5) for class I AMPs do not lead to chain connections between different amino acids. Class II covers all AMPs with chemical bonds between different peptide side chains (Figure 2B). These include lantibiotics (thioether rings) and the defensin family (disulfide bonds). Broadly, it can be any type of chemical connections between two amino acids. When two or more peptides work together, they belong to this class as long as any of the polypeptide chain contains a sidechain-sidechain connection. Class III AMPs must possess a chemical bond between peptide side chain and backbone (Figure 2C). The typical members are lassos where the carboxyl group of residue E8 or D9 is covalently linked to the N-terminal amine group. It can be any type of chemical bonding between the side chain of one amino acid and the backbone of another amino acid (see Table 9). Lastly, class IV is composed of circular peptides where a peptide bond is formed between the amino and carboxylic ends of the peptide backbone (Figure 2D). These circular peptides may (or may not) contain additional modifications such as disulfide bonds. Examples are enterocin AS-48 from bacteria, cyclotides from plants and θ-defensins from primates [37].
Figure 2. Classification of antimicrobial peptides based on the connection patterns of the polypeptide chain.
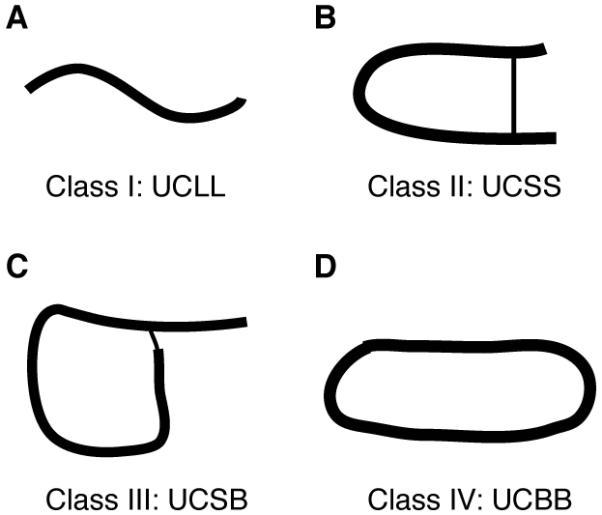
(A) linear polypeptide chains (e.g. LL-37 and magainins); (B) sidechain-linked peptides such as defensins and lantibiotics; (C) polypeptide chains with side chain to backbone connection (e.g. lassos); and (D) circular peptides with a seamless backbone (e.g. cyclotides).
Table 9.
A universal classification of antimicrobial peptides
| Class | Chain linkage |
Subclass | Link type | Class symbol |
Examples |
|---|---|---|---|---|---|
| I | Linear & open chainsa |
1. One chain | None | UCLL1a | LL-37, magainins |
| 2. Two chains | None | UCLL2 | Enterocin L50 | ||
| II | Sidechain- Sidechain |
1. One chain | Cβ-S-S-Cβ (Disulfide- bond) |
UCSS1aa | Defensin-like |
| Cβ-S-Cβ (thioether) |
UCSS1ba | lantibiotics | |||
| 2. Two chains | Inter-chain Cβ-S-S-Cβ |
UCSS2a | Distinctin, halocidin, centrocin |
||
| Intra-chain Cβ-S-Cβ |
UCSS2b | Lacticin-3147, Smb | |||
| III | Sidechain- Backbone |
One chain | CO-NH amide |
UCSB1a | Microcin J25, Lariatins |
| CO-O ester | UCSB1b | Fusaricidin A | |||
| Cβ-S-Cα | UCSB1c | Thuricin CD | |||
| IV | Backbone- Backbone |
One chain | CO-NH amide |
UCBB1aa | AS-48, subtilosin A, cyclotides, θ- defenins |
Each class of AMPs can be further classified. For class I peptides, they can be classified into two subclasses based on the number of polypeptide chains (Table 9). Single-chain linear AMPs are further classified based on chemical modifications. Unmodified AMPs include “amino acid rich” and “not amino acid rich” families. Modified peptides are further divided into two types based on modification sites (side chain or backbone). These systematic classifications for class 1 AMPs are summarized in Table 10 with examples. Likewise, class II AMPs with connections between side chains can be further classified based on the number of polypeptide chains as well as the type of chemical bonds (Table 9). A further classification of the single-chain disulfide-bonded AMPs (e.g. defensins or defensin-like) based on the number of S-S bonds is provided in Table 11. It is also possible to further classify single-chain lantibiotics based on the number of thioether bonds (Table 12). A new type of sidechain-sidechain connection will constitute a new subclass. In the same vein, class III AMPs can be further separated into different types based on the bond type (Table 9). This chemical bond-based classification is also extended to class IV. Circular AMPs are classified based on the additional types and number of chemical bonds in the polypeptide chain (Table 13). This systematic classification system covers all AMPs and should complement with the existing classification systems proposed for AMPs from different life domains [39-43].
Table 10.
Classification of class 1 linear antimicrobial peptides (UCLL1)
| Subclass | Modification sitea |
Modification type |
Sub-type | Peptide examples |
|---|---|---|---|---|
| UCLL1A | None | None | Not-AA-richb | LL-37 |
| AA-Rich (25%) | Pro-rich; Arg-rich PR-39 | |||
| UCLL1B | Sidechain | Group attachment |
Hydroxylation; halogenation; phosphorylation ; glycosylation; lipidation; sulfation |
Piscidin 4 (hydroxylated Trp); datucin, MccC7 |
| Sidechain cyclization |
cyclic glutamate | Heliocin | ||
| UCLL1C | Backbone | End capping | Amidation; acetylation; other attachments |
Aurein 1.2; temproin A |
| Configuration change |
D-amino acids, | Gramicidin; bombinin H4 | ||
| Backbone transformed |
Dehydrated; | Cypemycin (Linaridins) | ||
| Heterocyclic rings |
Thiopeptides in ThioBase |
Post-translational modification (PTM) is a broad concept that includes all types of functional groups attached to the peptide chain via covalent bond formation. A detailed list of PTMs is provided in Table 5. Some common examples are N-terminal acetylation, C-terminal amidation, phosphorylation, glycosylation, aromatic halogenation, and sulfation. In the extreme case, even the peptide backbone is modified, leading to dehydrated or heterocycles. However, all these modifications are limited to a single amino acid and do not lead to a polypeptide chain connection between different amino acids as observed in the other three major classes of AMPs (Table 9).
AA = Amino acids.
Table 11.
Sidechain-sidechain connected antimicrobial peptides: further classification of single-chain peptides containing disulfide bonds (UCSS1a)
| Type | S-S bond count | Sub-typea | Examples |
|---|---|---|---|
| I | 1 | A | Brevinin, esculentin (Rana box) |
| B | Thanatin | ||
| C | Bactenecin | ||
| II | 2 | A | Ec-AMP1, lasiocepsin, Glycocin F |
| B | Protegrin, polyphemusin, CXCL1, LEAP-2 |
||
| III | 3 | A | NK-lysin, caenopore-5 |
| B | HNP-1, HBD-1, big defensins | ||
| IV | 4 | B | ASABF, NaD1, drosomycin |
| V | 5 | B | PhD1, WAMP-1a, Ec-CBP |
| VI | 6 | B | Copsin |
The peptides can further be classified into sub-types based on 3D structure (A: α-helical; B: β-sheet-containing (β and αβ families); C: non-αβ; D: unclassified due to an unknown 3D structure).
Table 12.
Sidechain-sidechain connected antimicrobial peptides: further classification of single-chain lantibiotics containing thioether bonds (UCSS1b)
| Type | Number of linkage | Examples |
|---|---|---|
| I | 1 | Not found |
| II | 2 | Bovicin HJ50 |
| III | 3 | Epilancin 15X, Lacticin 481 |
| IV | 4 | Cinnamycin, Actagardine A |
| V | 5 | Nisin, Microbisporicin, Subtilin, Ericin A, Paenibacillin |
| VI | 6 | Paenicidin A |
| VII | 7 | Geobacillin I |
Table 13.
Classification of circular antimicrobial peptides (UCBB1a)
| Type | Additional Links | Examples |
|---|---|---|
| A | None | Bacterial enterocin AS-48 |
| B | Sidechain-sidechain (Cβ-S-S-Cβ) | Plant cyclotides, primate θ-defensins |
| C | Sidechain-backbone (Cβ-S-Cα) | Bacterial subtilosin |
4. Peptide Prediction
Based on the information content used in the prediction programs, the prediction methods of AMPs have been classified into five types [6]. The first type uses only mature peptide sequences, while the second method involves only the precursor sequences. The third prediction type considers both mature and precursor sequences. The fourth method employs the sequence similarity of the modifying enzymes. Finally, the fifth prediction uses genomic information. It is possible that each prediction above can be achieved in different ways. For example, based on the mature AMP sequences in the APD [10, 11], numerous prediction methods have been developed. In the Lata method [44], two data sets were utilized: antimicrobial and non-antimicrobial. While it is easy to download the positive data set from the APD, it is difficult to get a true negative data set because the activities of the sequences in the negative data set have not been validated by experiments. Yet, the program is set up with a good predictive ability. A recent prediction method iAMP-2L [45] considers multiple functions of AMPs annotated in the APD. Different from all other prediction protocols (reviewed in ref. [6]), a unique prediction method is programmed in the APD. This method does not require a negative data set, but is coupled with the database. In the following, we describe an upgraded version of this APD method.
The original prediction method in the APD made predictions based on some known rules [10]. Hence, the method was referred to as knowledge-based prediction. For example, AMPs are usually cationic. A peptide with a negative net charge was predicted as “less likely to be an antibacterial peptide”. This simple prediction has its limitations because the database does contain anionic AMPs. To overcome this shortcoming, we have updated the prediction interface based on the parameter space defined by the whole peptide set in the APD. The parameters for antimicrobial peptides are better defined due to a four-fold increase in peptide number from the original 525 to the current 2329. Peptide parameters such as length, net charge, hydrophobic percentage, and amino acid composition can all be calculated. These parameters constitute the parameter space of natural AMPs.
In terms of net charge, the known AMPs occupy a very broad range. The AMP with the most negative net charge is chrombacin (net charge −12). Two AMPs, sheep cathelicidin OaBac11 and fish histone-derived Oncorhyncin II, possess the highest net charge of +30. Thus, the boundary conditions for net charge are defined as
The above boundary condition can be incorporated into the APD program to make database-based predictions. This expansion enables the prediction of a broader range of peptide sequences. Because the majority of the AMPs (97.4%) have a net charge between −5 and +10, it may be useful to define this range as the core region. The small number of AMPs outside the core region may be called the minor region. This core region may be used as an alternative condition for prediction.
The hydrophobic content (i.e., the sum of hydrophobic amino acids divided by the total number of amino acids in a peptide) is another important parameter that determines peptide properties. In the APD, hydrophobic amino acids include alanines (Ala), valines (Val), leucines (Leu), isoleucines (Ile), methionines (Met), phenylalanines (Phe), tryptophans (Trp), and cysteins (Cys) [10]. Based on the database sorting function, we identified the AMPs with the lowest and highest hydrophobic contents. Sheep anionic peptide SAAP (sequence: DDDDDD) contains no hydrophobic residues in the sequence, leading to a hydrophobic content of 0%, while gramicidins have the highest hydrophobic content of 93%. Thus, the boundary conditions for peptide hydrophobic contents are defined as
The peak of this hydrophobic distribution is located between 40-50% [46]. This leads to another set of boundary conditions for our database-based prediction. We can also define the core region based on the hydrophobic content. The AMPs in the core region (98.6%) possess a hydrophobic content between 10% and 80%.
The length of the peptides in the current APD ranges from 5 to 174. The lower limit is real, while the upper limit is arbitrary since it is defined by the scope of peptides collected into the database (<200 amino acids). However, the majority of AMPs (92.9%) are less than 60 amino acids in length, leading to a definition of the core length region of 5-60. We can anticipate that these boundary conditions will be fully determined when a sufficient number of representative natural AMPs have been identified and registered into the APD.
During this study, we have executed these new database-derived boundary conditions in the prediction interface of the APD (Figure 3). This interface makes predictions based on sequence similarity. In the first step, the prediction program will calculate the peptide parameters based on the input sequence. The calculated peptide parameters will then be compared with the APD parameter space. If one or more calculated parameters fall outside the database-defined parameter space, the users will be informed that “your input is less likely to be an antibacterial peptide”. If all the parameters fall within the defined parameter space, the database will conduct a second tier of prediction by broadly classifying input peptides into several classes: rich in amino acids (>25% for any amino acid), helical, and disulfide-linked. With the execution of the universal classification proposed in Table 9, a more accurate prediction will be realized. As the third tier of our prediction, the database compares the input sequence with all the peptides in the database by performing sequence alignment. Five peptides with most similar sequences will be provided in the output. Because we use database-derived parameters for prediction, we refer to this upgraded method as the APD-based prediction (the November 2013 version). Compared to the original prediction [10], the upgraded version is able to handle a broader range of peptide sequences. In addition, the chance of identifying the most similar sequences in the APD also increases substantially as a consequence of a four-fold increase in natural compounds.
Figure 3. Prediction of antimicrobial peptides based on the antimicrobial peptide database.
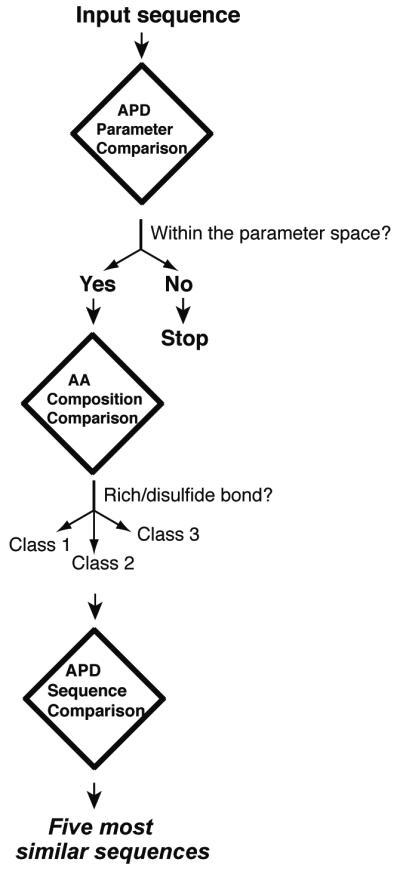
The prediction consists of three steps. As the first step, the program will determine whether the input sequence is in the database-defined parameter space (such as charge and hydrophobic content). If identical, the users will be informed. If one or more calculated parameters of the input peptide are out of the boundaries, it is predicted as “your sequence is less likely to be an antibacterial peptide”. Second, the input sequence will be classified into three families: rich in amino acids such as histatins and tryptophans, disulfide-linked peptides, and linear. Third, sequence alignments will be conducted to find five peptides that are most similar to the query sequence.
The identification of most similar AMPs is a useful feature. For example, O’Shea did not find similar sequences by searching the BLAST database [47], but were able to do so using the APD. Based on the sequence similarity of a novel bacteriocin with plant Ib-AMP3, these authors named the new bacteriocin as bactofensin. The similarity also inspired the authors to test possible antimicrobial activities listed for Ib-AMP3. In addition, the authors can also check whether the new peptide has a similar 3D structure. Thus, the output from the APD prediction programs can guide users to design new experiments to test the structure and activity of the newly identified peptide based on the knowledge annotated for the most similar candidates in the database. Such a prediction of sequence, structure and activity at multiple levels requires careful annotation of AMP information in the APD.
5. Peptide design
The APD [10, 11] also provides a useful platform for identification of useful antimicrobials to combat difficult-to-kill pathogens such as human immune-deficiency virus (HIV) and methicillin-resistant Staphylococcus aureus (MRSA) [46]. Both database screening and database-guided design have been conducted. By screening a representative set of AMPs selected from the APD, we found several potent anti-HIV or anti-MRSA peptides [48, 49]. New peptides were also obtained by modifying, shuffling, or hybriding natural sequences. Mathematically, a known peptide sequence can be shuffled into multiple sequences. Experimentally, we found that sequence shuffling could lead to all the possibilities: less active, equally active, and more potent sequences [49]. An MIT group developed a large-scale hybrid approach by combining sequence segments of 10 residues (i.e., grammars). This grammar approach can generate new sequences, which may, or may not, be bactericidal [50]. A complete different approach in the form of combinatorial libraries can also be pursued [51]. In principle, the amino acid at each position of the peptide sequence can be changed into other amino acids. In practice, it is necessary to bias the choice of amino acids in order to obtain active peptides [52]. This is because the amino acid use in natural AMPs is biased. The APD enabled us to identify the frequently occurring amino acids for AMPs from a variety of life domains [10, 53]. For example, the frequently occurring residues (≥8.5%) are leucines, glycines, and lysines based on the average percentages of all the 2329 peptides in the current APD. We demonstrated previously that these three amino acids contained sufficient information for designing antibacterial peptides [11].
Another important approach is de novo design (reviewed in ref. [6]). We have recently developed a novel database approach [54]. A flow chart for this approach is provided as Figure 4. This flow chart contains two major tiers of information filters. The first tier consists of an activity filter that enables one to obtain a set of peptides with desired activity. Table 3 lists 17 types of peptide activities, each of which contains a set of model peptides. In our design, we selected a group of peptides with activity against Gram-positive bacteria. This set of peptides formed the templates for extracting useful parameters for designing anti-MRSA peptides. The second tier contains numerous filters (F1, F2, to Fn), each defines one parameter for the peptide (P1, P2, to Pn). In determining these parameters, we followed the most probable principle, which projected the maximum for each parameter. Because the most probable parameters were used, the peptides assembled in this manner had a good chance to be antimicrobial. This is indeed the case. The designed peptide DFTamP1 rapidly killed MRSA USA300, a community-associated staphylococcal pathogen. It also showed some bacterial selectivity since DFTamP1 did not kill Gram-negative bacteria E. coli, P. aeruginosa, or Gram-positive B. subtilis. This success opens a new avenue to designing peptides with various types of activities (Table 7). Because this new method differs from all existing de novo approaches, it was referred to as ab initio design [54].
Figure 4. Ab initio peptide design based on the database filtering technology (DFT).

The DFT tech developed recently [54] is composed of two layers of filters. The first layer filter enables the identification of a set of antimicrobial peptides with the desired activity from the antimicrobial peptide database (see Table 7). This set of peptides is then used as templates to extract useful parameters for peptide design by utilizing the second layer filters (F1, F2, F3, …, to Fn). These peptide parameters (P1, P2, P3, …, to Pn) are combined to generate a single or limited number of peptides.
6. Concluding Remarks and Future Studies
The antimicrobial peptide database was constructed in 10 years ago. It is an original construction in terms of both database design and peptide entries. Each peptide entry in the APD was manually collected from the literature using the public search engines such as PubMed, PDB, and Swiss-Prot. By following a set of rules for data registration, the APD presents a well-defined set of natural AMPs. To achieve a more complete sampling of natural AMPs, the database is extensively annotated and regularly updated. In addition, the pipeline design led to a powerful search engine. This unique database, therefore, constitutes the basis for developing new methods for peptide classification, prediction and design. The APD is the first to adopt both five-kingdom and three-domain classifications, allowing users to search the AMP information from any kingdom (bacteria, protists, fungi, plants, and animals) or classes (e.g. insects, spiders, molluscs, crustaceans, reptiles, amphibians, fish, and birds) (Table 4). Once a domain is defined in the NAME field, the APD behaves like a specialized database (e.g., plant AMPs, bacteriocins, and amphibian peptides). The APD also executed a new structure classification scheme based on the types of secondary structures (α, β, αβ, and non-αβ) in a variety of 3D structures of AMPs (Figure 1) [6]. Needless to say, the structures in each family can be further grouped based on the number of secondary structures (e.g., α-helix and β-strand). Due to a limited number of known 3D structures, we have proposed a universal classification scheme here based on peptide chain bonding patterns (Figure 2). Since the information on peptide source, activity, and 3D structure is not required, this systematic classification (Tables 9-13) complements to the existing classification methods for AMPs in a defined life kingdom such as bacteria and plants [39-43]. It also offers an approach to unifying the classification of antimicrobial peptides. This classification is general and can be applied to other biologically active peptides.
There are various prediction methods for AMPs (reviewed in ref. [6]). The APD is unique in that the prediction is highly coupled with the database. The upgraded version of the APD makes predictions in three steps by following the similarity principle. Each step deals with a specific question. The first tier asks whether the peptide parameters of the input sequence fall within the database parameter space. Based on the amino acid composition analysis, the second tier asks which peptide class the input sequence belongs to. The third tier determines five most similar sequences based on sequence alignment with all the peptides in the database. It is clear why we have been strict in following a set of rules in registering AMPs. Our practice allows us to more accurately map the parameter space for natural AMPs. When a large number of predicted or artificial sequences are included, such parameters could deviate from nature’s parameters, thereby influencing the prediction quality. In addition, users can get an idea of the structural type and functional space of the input sequence by viewing the similar sequences already in the APD. For example, the input sequence is most likely to form a helix-bundle structure stabilized by three disulfide bonds if the best match is a saposin-like protein. If the sequence matches human cathelicidin LL-37, it is likely to have multiple functions, ranging from antimicrobial, wound healing, to immune modulation. Like LL-37, the peptide may also have a broad-spectrum activity to kill bacteria, fungi, viruses, and parasites. This information will guide the users to validate both structure and activity of a new peptide.
Finally and importantly, the construction of this well-annotated database also enabled us to develop novel approaches for designing peptides with desired properties. Based on the database, we have tested two general approaches: peptide screening [48,49] and database-guided design [46, 54]. In particular, we demonstrated the first ab initio design based on the database by developing the database filtering technology [54]. This approach is not limited to the development of anti-MRSA peptides and can be applied to the design of peptides with other types of activities (Table 7) as well. It is also desirable that the designed peptides only kill a specific species. Our detailed annotations of AMP targeting organisms into the database set the stage for this effort. In addition, other database filters such as peptide selectivity and stability to proteases can be created as well. Taken together, the APD is a powerful engine for research and education in the field of innate immunity and drug discovery.
Acknowledgements
This study was supported by grants from the NIH (1R56AI105147-01 and 1R01AI105147-01A1) and the State of Nebraska. The author thanks Zhe Wang for programming the original database and Biswajit Mishra for conducting the ab initio design of novel antimicrobials.
References
- 1.Zasloff M. Antimicrobial peptides of multicellullar organisms. Nature. 2002;415:359–365. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- 2.Lehrer RI. Multispecific myeloid defensins. Curr Opin Hematol. 2007;14:16–21. doi: 10.1097/00062752-200701000-00005. [DOI] [PubMed] [Google Scholar]
- 3.Boman HG. Antibacterial peptides: basic facts and emerging concepts. J Inter Med. 2003;254:197–215. doi: 10.1046/j.1365-2796.2003.01228.x. [DOI] [PubMed] [Google Scholar]
- 4.Hancock RE, Sahl HG. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat Biotechnol. 2006;24:1551–1557. doi: 10.1038/nbt1267. [DOI] [PubMed] [Google Scholar]
- 5.Lai Y, Gallo RL. AMPed up immunity: how antimicrobial peptides have multiple roles in immune defense. Trends Immunol. 2009;30:131–141. doi: 10.1016/j.it.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang G, editor. Antimicrobial Peptides: Discovery, Design and Novel Therapeutic Strategies. CABI; England: 2010. [Google Scholar]
- 7.Steiner H, Hultmark D, Engström Å , Bennich H, Boman HG. Sequence and specificity of two antibacterial proteins involved in insect immunity. Nature. 1981;292:246–248. doi: 10.1038/292246a0. [DOI] [PubMed] [Google Scholar]
- 8.Selsted ME, Harwig SS, Ganz T, Schilling JW, Lehrer RI. Primary structures of three human neutrophil defensins. J Clin Invest. 1985;76:1436–1439. doi: 10.1172/JCI112121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zasloff M. Magainins, a class of antimicrobial peptides from Xenopus skin: isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc Natl Acad Sci USA. 1987;84:5449–5453. doi: 10.1073/pnas.84.15.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Z, Wang G. APD: the antimicrobial peptide database. Nucleic Acids Res. 2004;32:D590–D592. doi: 10.1093/nar/gkh025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang G, Li X, Wang Z. The updated antimicrobial peptide database and its application in peptide design. Nucleic Acids Res. 2009;37:D933–D937. doi: 10.1093/nar/gkn823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tossi A, Sandri L. Molecular diversity in gene-coded, cationic antimicrobial polypeptides. Curr Pharm Des. 2002;8:743–761. doi: 10.2174/1381612023395475. [DOI] [PubMed] [Google Scholar]
- 13.Brahmachary M, Krishnan SP, Koh JL, Khan AM, Seah SH, Tan TW, Brusic V, Bajic VB. ANTIMIC: a database of antimicrobial sequences. Nucleic Acids Res. 2004;32:D586–D589. doi: 10.1093/nar/gkh032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seshadri Sundararajan V, Gabere MN, Pretorius A, Adam S, Christoffels A, Lehväslaiho M, Archer JA, Bajic VB. DAMPD: a manually curated antimicrobial peptide database. Nucleic Acids Res. 2012;40:D1108–D1112. doi: 10.1093/nar/gkr1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gueguen Y, Garnier J, Robert L, Lefranc MP, Mougenot I, de Lorgeril J, Janech M, Gross PS, Warr GW, Cuthbertson B, Barracco MA, Bulet P, Aumelas A, Yang Y, Bo D, Xiang J, Tassanakajon A, Piquemal D, Bachère E. PenBase, the shrimp antimicrobial peptide penaeidin database: sequence-based classification and recommended nomenclature. Dev Comp Immunol. 2006;30:283–288. doi: 10.1016/j.dci.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 16.Seebah S, Suresh A, Zhou S, Choong YH, Chua H, Chuon D, Beuerman R, Verma C. Defensins knowledgebase: a manually curated database and information source focused on the defensins family of antimicrobial peptides. Nucleic Acids Res. 2007;35:D265–D268. doi: 10.1093/nar/gkl866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hammami R, Zouhir A, Ben Hamida J, Fliss I. BACTIBASE: a new web-accessible database for bacteriocin characterization. BMC Microbiol. 2007;7:89. doi: 10.1186/1471-2180-7-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang CK, Kaas Q, Chiche L, Craik DJ. Cybase: a database of cyclic protein sequences and structures, with applications in protein discovery and engineering. Nucleic Acids Res. 2008;36:D206–D210. doi: 10.1093/nar/gkm953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hammami R, Ben Hamida J, Vergoten G, Fliss I. PhytAMP: a database dedicated to antimicrobial plant peptides. Nucleic Acids Res. 2009;37:D963–D968. doi: 10.1093/nar/gkn655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Y, Chen Z. RAPD: a database of recombinantly-produced antimicrobial peptides. FEMS Microbiol Lett. 2008;289:126–129. doi: 10.1111/j.1574-6968.2008.01357.x. [DOI] [PubMed] [Google Scholar]
- 21.de Jong A, van Heel AJ, Kok J, Kuipers OP. BAGEL2: mining for bacteriocins in genomic data. Nucleic Acids Res. 2010;38:W647–W651. doi: 10.1093/nar/gkq365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Novković M, Simunić J, Bojović V, Tossi A, Juretić D. DADP: the database of anuran defense peptides. Bioinformatics. 2012;28:1406–1407. doi: 10.1093/bioinformatics/bts141. [DOI] [PubMed] [Google Scholar]
- 23.Li J, Qu X, He X, Duan L, Wu G, Bi D, Deng Z, Liu W, Ou HY. ThioFinder: a web-based tool for the identification of thiopeptide gene clusters in DNA sequences. PLoS One. 2012;7:e45878. doi: 10.1371/journal.pone.0045878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fjell CD, Hancock RE, Cherkasov A. AMPer: a database and an automated discovery tool for antimicrobial peptides. Bioinformatics. 2007;23:1148–55. doi: 10.1093/bioinformatics/btm068. [DOI] [PubMed] [Google Scholar]
- 25.Wade D, Englund J. Synthetic antibiotic peptides database. Protein Pept Lett. 2002;9:53–57. doi: 10.2174/0929866023408986. [DOI] [PubMed] [Google Scholar]
- 26.Whitmore L, Wallace BA. The Peptaibol Database: a database for sequences and structures of naturally occurring peptaibols. Nucleic Acids Res. 2004;32:D593–D594. doi: 10.1093/nar/gkh077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu H, Lu H, Huang J, Li G, Huang Q. EnzyBase: a novel database for enzybiotic studies. BMC Microbiol. 2012;12:54. doi: 10.1186/1471-2180-12-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Theolier J, Fliss I, Jean J, Hammami R. MilkAMP: a comprehensive database of antimicrobial peptides of dairy origin. Dairy Sci Technol. 2013 DOI 10.1007/s13594-013-0153-2. [Google Scholar]
- 29.Thomas S, Karnik S, Barai RS, Jayaraman VK, Idicula-Thomas S. CAMP: a useful resource for research on antimicrobial peptides. Nucleic Acids Res. 2010;38:D774–D780. doi: 10.1093/nar/gkp1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Piotto SP, Sessa L, Concilio S, Iannelli P. YADAMP: yet another database of antimicrobial peptides. Int J Antimicrob Agents. 2012;39:346–351. doi: 10.1016/j.ijantimicag.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 31.Zhao X, Wu H, Lu H, Li G, Huang Q. LAMP: A Database Linking Antimicrobial Peptides. PLoS One. 2013;8:e66557. doi: 10.1371/journal.pone.0066557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu CH, Apweiler R, Bairoch A, Natale DA, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Mazumder R, O’Donovan C, Redaschi N, Suzek B. The Universal Protein Resource (UniProt): an expanding universe of protein information. Nucleic Acids Res. 2006;34:D187–D191. doi: 10.1093/nar/gkj161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rose PW, Bi C, Bluhm WF, Christie CH, Dimitropoulos D, Dutta S, Green RK, Goodsell DS, Prlic A, Quesada M, Quinn GB, Ramos AG, Westbrook JD, Young J, Zardecki C, Berman HM, Bourne PE. The RCSB Protein Data Bank: new resources for research and education. Nucleic Acids Res. 2013;41:D475–D482. doi: 10.1093/nar/gks1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wheeler DL, Church DM, Lash AE, Leipe DD, Madden TL, Pontius JU, Schuler GD, Schriml LM, Tatusova TA, Wagner L, Rapp BA. Database resources of the National Center for Biotechnology Information: 2002 update. Nucleic Acids Res. 2002;30:13–16. doi: 10.1093/nar/30.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Whittaker RH. New concepts of kingdoms of organisms. Science. 1969;163:150–160. doi: 10.1126/science.163.3863.150. [DOI] [PubMed] [Google Scholar]
- 36.Woese CR, Fox GE. Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proc Natl Acad Sci USA. 1977;74:5088–5090. doi: 10.1073/pnas.74.11.5088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang G. Chemical modifications of natural antimicrobial peptides and strategies for peptide engineering. Curr Biotechnol. 2012;1:72–79. doi: 10.2174/2211550111201010072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Epand RM, Vogel HJ. Diversity of antimicrobial peptides and their mechanisms of action. Biochim Biophys Acta. 1999;1462:11–28. doi: 10.1016/s0005-2736(99)00198-4. [DOI] [PubMed] [Google Scholar]
- 39.Klaenhammer TR. Genetics of bacteriocins produced by lactic acid bacteria. FEMS Microbiol Rev. 1993;12:39–85. doi: 10.1111/j.1574-6976.1993.tb00012.x. [DOI] [PubMed] [Google Scholar]
- 40.Duquesne S, Destoumieux-Garzón D, Peduzzi J, Rebuffat S. Microcins, gene-encoded antibacterial peptides from enterobacteria. Nat Prod Rep. 2007;24:708–734. doi: 10.1039/b516237h. [DOI] [PubMed] [Google Scholar]
- 41.Egorov TA, Odintsova TI, Pukhalsky VA, Grishin EV. Diversity of wheat antimicrobial peptides. Peptides. 2005;26:2064–2073. doi: 10.1016/j.peptides.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 42.Bulet P, Stocklin R. Insect antimicrobial peptides: structures, properties and gene regulation. Protein Pept Lett. 2005;12:3–11. doi: 10.2174/0929866053406011. [DOI] [PubMed] [Google Scholar]
- 43.Conlon JM. Reflections on a systematic nomenclature for antimicrobial peptides from the skins of frogs of the family Ranidae. Peptides. 2008;29:1815–1819. doi: 10.1016/j.peptides.2008.05.029. [DOI] [PubMed] [Google Scholar]
- 44.Lata S, Sharma BK, Raghava GP. Analysis and prediction of antibacterial peptides. BMC bioinformatics. 2007;8:263. doi: 10.1186/1471-2105-8-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xiao X, Wang P, Lin WZ, Jia JH, Chou KC. iAMP-2L: A two-level multi-label classifier for identifying antimicrobial peptides and their functional types. Anal Biochem. 2013;436:168–177. doi: 10.1016/j.ab.2013.01.019. [DOI] [PubMed] [Google Scholar]
- 46.Wang G. Database-guided discovery of potent peptides to combat HIV-1 or superbugs. Pharmaceuticals. 2013;6:728–758. doi: 10.3390/ph6060728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.O’Shea EF, O’Connor PM, O’Sullivan O, Cotter PD, Ross RP, Hill C. Bactofencin a, a new type of cationic bacteriocin with unusual immunity. MBio. 2013;4:pii: e00498-13. doi: 10.1128/mBio.00498-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Menousek J, Mishra B, Hanke ML, Heim CE, Kielian T, Wang G. Database screening and in vivo efficacy of antimicrobial peptides against methicillin-resistant Staphylococcus aureus USA300. Int J Antimicrob Agents. 2012;39:402–406. doi: 10.1016/j.ijantimicag.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang G, Watson KM, Peterkofsky A, Buckheit RW., Jr Identification of novel human immunodeficiency virus type 1 inhibitory peptides based on the antimicrobial peptide database. Antimicrob Agents Chemother. 2010;54:1343–1346. doi: 10.1128/AAC.01448-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Loose C, Jensen K, Rigoutsos I, Stephanopoulos G. A linguistic model for the rational design of antimicrobial peptides. Nature. 2006;443:867–869. doi: 10.1038/nature05233. [DOI] [PubMed] [Google Scholar]
- 51.Lam KS, Salmon SE, Hersh EM, Hruby VJ, Kazmierski WM, Knapp RJ. A new type of synthetic peptide library for identifying ligand-binding activity. Nature. 1991;354:82–84. doi: 10.1038/354082a0. [DOI] [PubMed] [Google Scholar]
- 52.Cherkasov A, Hilpert K, Jenssen H, Fjell CD, Waldbrook M, Mullaly SC, Volkmer R, Hancock RE. Use of artificial intelligence in the design of small peptide antibiotics effective against a broad spectrum of highly antibiotic-resistant superbugs. ACS Chemical Biology. 2008;4:65–74. doi: 10.1021/cb800240j. [DOI] [PubMed] [Google Scholar]
- 53.Mishra B, Wang G. The importance of amino acid composition in natural AMPs: an evolutional, structural, and functional perspective. Frontier in Immunology. 2012;3:221. doi: 10.3389/fimmu.2012.00221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mishra B, Wang G. Ab initio design of potent anti-MRSA peptides based on database filtering technology. J Am Chem Soc. 2012;134:12426–12429. doi: 10.1021/ja305644e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang G. Structures of human host defense cathelicidin LL-37 and its smallest antimicrobial peptide KR-12 in lipid micelles. J Biol Chem. 2008;283:32637–32643. doi: 10.1074/jbc.M805533200. [DOI] [PubMed] [Google Scholar]
- 56.Saether O, Craik DJ, Campbell ID, Sletten K, Juul J, Norman DG. Elucidation of the primary and three-dimensional structure of the uterotonic polypeptide kalata B1. Biochemistry. 1995;34:4147–4158. doi: 10.1021/bi00013a002. [DOI] [PubMed] [Google Scholar]
- 57.Bauer F, Schweimer K, Klüver E, Conejo-Garcia JR, Forssmann WG, Rösch P, Adermann K, Sticht H. Structure determination of human and murine beta-defensins reveals structural conservation in the absence of significant sequence similarity. Protein Sci. 2001;10:2470–2479. doi: 10.1110/ps.24401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rozek A, Friedrich CL, Hancock RE. Structure of the bovine antimicrobial peptide indolicidin bound to dodecylphosphocholine and sodium dodecyl sulfate micelles. Biochemistry. 2000;39:15765–15774. [PubMed] [Google Scholar]