

Abstract
The tumor suppressor INPP4B is an important regulator of phosphatidyl-inositol signaling in the cell. Reduced INPP4B expression is associated with poor outcomes for breast, prostate, and ovarian cancer patients. INPP4B contains a CX5R catalytic motif characteristic of dual-specificity phosphatases, such as PTEN. Lipid phosphatase activity of INPP4B has previously been described. In this report we show that INPP4B can dephosphorylate para-nitrophenyl phosphate (pNPP) and 6,8-difluoro-4-methylumbelliferyl (DiFMUP), synthetic phosphotyrosine analogs, suggesting that INPP4B has protein tyrosine phosphatase (PTP) activity. Using mutagenesis, we examined the functional role of specific amino acids within the INPP4B C842KSAKDR catalytic site. The K843M mutant displayed increased pNPP hydrolysis, the K846M mutant lost lipid phosphatase activity with no effect on PTP activity, and the D847E substitution ablated PTP activity and significantly reduced lipid phosphatase activity. Further, we show that INPP4B but not PTEN is able to reduce tyrosine phosphorylation of Akt1 and both the lipid and PTP activity of INPP4B likely contribute to the reduction of Akt1 phosphorylation. Taken together our data identified key residues in the INPP4B catalytic domain associated with lipid and protein phosphatase activities and found a robust downstream target regulated by INPP4B but not PTEN.
Keywords: INPP4B, Phosphatidylinositol phosphatase, Dual specificity phosphoprotein, phosphatase, Lipid metabolism, Akt
1. Introduction
Deregulation of phosphatidyl inositol signaling plays an important role in various disorders. These pathways are stimulated by phosphatidyl 3, 4, and 5 kinases (PI3K, PI4K, PI5K) and inhibited by lipid phosphatases such as PTEN, INPP4B, and SHIP [1]. These phosphatases dephosphorylate the phosphatidyl inositol ring on the 3rd, 4th, and 5th position respectively. Loss of INPP4B is a poor prognostic factor for breast, ovarian, and prostate cancers [2–4]. Similar to PTEN, INPP4B contains a dual specificity phosphatase (DuSP) domain with a characteristic DuSP CX5R motif, C842KSAKDR848. Residue C842 is required for INPP4B enzymatic activity [3,5]. There are three known INPP4B substrates: phosphatidylinositol 3,4-bisphosphate (PI(3,4)P2), phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2), and inositol 1,3,4-trisphosphate (Ins(1,3,4)P3) [5]. Substrates of INPP4B lipid phosphatase activity are important second messengers in pathways regulating cellular proliferation and metastasis and have been implicated in prostate cancer progression [6,7]. PI(4,5)P2 is a substrate for both INPP4B and phospholipase C, an enzyme implicated in cellular motility and tumor dissemination in cancer [8,9]. PI(3,4)P2 is present at low levels on the cell membrane and accumulates at the sites of invadopodia [10], cellular appendages responsible for focal pericellular proteolysis of the extracellular matrix necessary for cellular invasion and metastasis [11]. PI(3,4)P2 binds to the pleckstrin homology domains of Akt and PDK1, recruiting them to the plasma membrane, leading to Akt phosphorylation on T308 and S473, and the activation of Akt. In addition to T308 and S473, tyrosine (Y) phosphorylation of Akt is emerging as an important regulatory mechanism for Akt signaling. Recruitment to the cell membrane and activation of Akt are regulated by its phosphorylation on several Y residues [12–14]. Elevated Y176 phosphorylation promotes Akt recruitment to the plasma membrane and increases phosphorylation of the Akt residues T308 and S473 [12]. In breast cancer patients, increased levels of Y176 phosphorylation correlate with poor overall survival [13].
In this report, we show that INPP4B can dephosphorylate para-nitrophenyl phosphate (pNPP) and 6,8-difluoro-4-methylumbel-liferyl (DifMUP), analogs of phosphotyrosine, suggesting that INPP4B has protein tyrosine phosphatase (PTP) activity. Mutation of four key residues in the active site identified distinct residues that contribute to INPP4B lipid and protein phosphatase activities. Finally, we found that INPP4B reduced Akt1 phospho-Y levels in Hek293T cells. Co-expression of Akt1 with wild-type (WT) and mutant INPP4B proteins revealed that both lipid and PTP activities contribute to downregulation of Akt1 phospho-Y levels. PTEN over-expression did not reduce phosphorylation of Akt1 on Y residues, suggesting distinct downstream signaling for INPP4B and PTEN tumor suppressors.
2. Materials and methods
2.1. Constructs
p3xFLAG-CMV-10-INPP4B-C842S, p3xFLAG-CMV-10-INPP4B-K843M, p3xFLAG-CMV-10-INPP4B-K846M, and p3xFLAG-CMV-10-INPP4B-D847E were generated by site directed mutagenesis of p3xFLAG-CMV-10-INPP4B [4]. Site-directed mutagenesis was conducted using the Stratagene QuikChange II XL Site-Directed Mutagenesis Kit from Agilent Technologies (La Jolla, CA) and the primers used are listed in Table 1. FLAG-HA-Akt1 construct was a kind gift of William Sellers (Addgene plasmid 9021) [15]. FLAG-PTEN was generated by PCR amplification GACAAGCTT GCGGCCGCAACAGCCA, GATGAATTCGCGGC CGCTCAGACTTT) of PTEN from DU145 cells and cloned into p3xFLAG-CMV-10 (Sigma–Aldrich), (St. Louis, MO). Constitutively active Src was obtained by PCR amplification (ACCCAAGCTG GCTAGCACCATGGG TAGCAACAAGA, AAGTTTAAACGCTAGCCTAGTACTGGGGCTCGGT) of Src [16] from PC-3 cells and insertion into pCR3.1. All cloning was performed using the Infusion HD cloning kit from Clontech (Mountain View, CA). All expression constructs were fully sequenced.
Table 1.
INPP4B active site mutations and associated primer sequences used in this study.
| INPP4B mutation | Primers |
|---|---|
| C842S | 5′-tggtattcgtttcacctgtagtaaaagtgccaaagacag-3′ 5′-ctgtctttggcacttttactacaggtgaaacgaatacca-3′ |
| K843M | 5′-ctgaatggtattcgtttcacctgttgtatgagtgccaaagacagg-3′ 5′-cctgtctttggcactcatacaacaggtgaaacgaataccattcag-3′ |
| K846M | 5′-acctgttgtaaaagtgccatggacaggacatcgatgtcag-3′ 5′-ctgacatcgatgtcctgtccatggcacttttacaacaggt-3′ |
| D847E | 5′-acctgttgtaaaagtgccaaagagaggacatcgatg-3′ 5′-catcgatgtcctctctttggcacttttacaacaggt-3′ |
2.2. Cell culture and reagents
Hek293T cell line was purchased from ATCC (Manassas, VA) and maintained in Dulbecco’s Modified Eagle Medium (DMEM), 5% FBS, and 1% penicillin–streptomycin. Media and antibiotics were purchased from Life Technologies (Carlsbad, CA). 3xFLAG peptide was purchased from Sigma–Aldrich. PI(3,4)P2 DiC8, PI(4,5)P2 DiC8, Ins(1,3,4)P3 and Malachite Green were purchased from Echelon Biosciences (Salt Lake City, UT). pNPP was obtained from New England Bio Labs (Ipswich, MA).
2.3. Protein expression and immunoprecipitation
Hek293T cells were transfected with 20 μg of p3xFLAG-CMV10-WT and indicated mutant expression constructs using Lipofectamine 2000 reagent from Life Technologies. Three days later cell lysates were prepared as previously described [4]. Cleared lysates were immunoprecipitated using EZview Red ANTI-FLAG M2 Affinity Gel from Sigma–Aldrich (St. Louis, MO). Affinity agarose gel beads were washed with ice cold TBS and 100 mM Tris–HCl pH 8.0. Proteins were eluted with 100 μl of 3xFLAG elution buffer [100 mM Tris–HCl pH 8.0, 1 mM EDTA, 30 mM NaCl, 0.01% Triton-X 100, 10 mM DTT, 150 ng/μL 3xFLAG peptide]. Eluted protein was used for lipid phosphatase assay, protein phosphatase assay, and western blotting.
2.4. Western blot analysis
Approximately 2% of eluted proteins were resolved on 7.5% SDS–PAGE and transferred using an iBlot Western Blotting transfer device (Invitrogen, Carlsbad, CA). Immunoblotting was performed using the following antibodies: anti-FLAG M2 (1:5000) (Sigma–Aldrich), PTEN (1:1000), phospho-Tyrosine (1:1000) (Cell Signaling Technology, Beverly, MA). Images were visualized with SuperSignal West Pico chemiluminescent substrate from Thermo Scientific (Pittsburg, PA) and Kodak Gel Logic 2200 imaging system and Carestream software (Carestream, Rochester, NY).
2.5. Lipid phosphatase assay
Eighteen μL of eluates were mixed with 2 μL of 1 mM PI(3,4)P2, PI(4,5)P2, or Ins(1,3,4)P3 and incubated for five minutes at 37 °C. One hundred μL of Malachite Green was added to each reaction or lysis buffer for calorimetric detection of free phosphate groups. Reactions were then measured at 660 nm using a FLUOstar Omega plate reader (BMG Labtech, Durham, NC).
2.6. Phosphotyrosine phosphatase assay
Thirty μL of eluates were diluted with 100 mM Tris–HCl pH 8.0 and pNPP added to a final concentration of 100 mM. Reactions were incubated at 25 °C and absorbance at 405 nm measured at two minute intervals using a FLUOstar Omega plate reader. Alternatively, the same amount of eluate was incubated with 200 μM 6,8-difluoro-4-methylumbelliferyl phosphate (DiF-MUP). Excitation/emission of the substrate, DiFMU, was measured every 2 min at 358/452 nm on a FLUOstar Omega plate reader.
For determination of the Michaelis constant, km, 70 μL of WT INPP4B eluate or elution buffer as a background control were incubated at 25 °C with 10 mM, 25 mM, 50 mM, 75 mM, and 100 mM of pNPP in a total reaction volume of 100 μL. Absorbance at 405 nm was recorded with a FLUOstar Omega plate reader. km was calculated from five independent experiments using Michaelis–Menten steady state kinetics.
3. Results
3.1. INPP4B has PTP activity
Since the INPP4B catalytic site is characteristic of DuSP, we tested whether INPP4B has PTP activity. 3xFLAG-WT INPP4B and a phosphatase dead mutant, 3xFLAG-C842A, were expressed in Hek293T cells, purified by immunoprecipitation, and incubated with pNPP. WT-INPP4B could dephosphorylate pNPP, but not the phosphatase dead mutant, C842A, ruling out coimmunoprecipitated phosphatase activity (Fig. 1A). INPP4B protein phosphatase activity was confirmed using 6,8-difluoro-4-methylumbelliferyl (DiFMUP) (Fig. 1B). To evaluate substrate affinity we incubated immunoprecipitated INPP4B with pNPP at 10 mM, 25 mM, 50 mM, 75 mM, and 100 mM and determined Vmax at these concentrations (Fig. 1C). Using Michaelis–Menten steady state kinetics and Lineweaver–Burk calculation we determined the km for INPP4B (Fig. 1D). After five independent measurements the average km for INPP4B at 25 °C was 38.03 mM ± 6.82 SEM. To compare INPP4B and PTEN PTP activities we used 3xFLAG-PTEN and 3xFLAG-INPP4B immunoprecipitated from Hek293T cells in side by side pNPP reactions. Based on FLAG Western blot Analysis the levels of both INPP4B and PTEN were similar while INPP4B activity was higher (Fig. 1E and F). This data suggest that in vitro activity and affinity for pNPP substrate are similar for both INPP4B and PTEN.
Fig. 1.

INPP4B has protein tyrosine phosphatase activity. Hek293T cells were transfected with WT or mutant INPP4B in the p3xFLAG-CMV-10 expression vector. Proteins were expressed for 3 days, immunoprecipitated using M2 affinity gel, and eluted with excess 3xFLAG peptide. (A) Twenty-five percent of indicated eluates were incubated with 100 mM pNPP. (B) INPP4B prepared as in (A) was incubated with 200 μM of DifMUP and fluorescent DiFMU accumulation measured (excitation/emission 358/452 nm). (C) 10 mM, 25 mM, 50 mM, 75 mM, and 100 mM of pNPP were incubated with the same amount of INPP4B as in (A) or with reaction buffer as a blank for each concentration. The maximum velocity was measured and plotted against concentration of pNPP. (D) Values from (C) were used to build a Lineweaver–Burk plot. Michaelis constant, km, was calculated using measurements from (C) and (D). (E) p3xFLAG-CMV-10-INPP4B and p3xFLAG-CMV-10-PTEN were expressed and immunoprecipitated as described in materials and methods. Next, 30% of eluate was used for pNPP hydrolysis and 2% for western blot analysis with 3xFLAG antibody M2 (inset). (F) Slopes of the pNPP enzymatic reaction were divided by the relative protein levels as determined by Western blot analysis. Three independently measured velocities were normalized for protein levels and the average ± SEM calculated.
3.2. Role of K843, K846, and D847 in INPP4B PTP activity
The catalytic site residues of INPP4B, C842KSAKDR848, are evolutionarily conserved from nematodes and drosophila through to vertebrates suggesting their functional significance. Two lysine residues in the catalytic site, K843 and K846, are conserved between INPP4B and PTEN. Substitution of K843 with methionine increased INPP4B PTP activity (Fig. 2A, blue1). The K846M substitution did not alter INPP4B PTP activity (Fig. 2A, green1). In PTEN there is an uncharged G prior to the R residue; in INPP4B the preceding residue is D, which has an acidic side chain. The G129E PTEN mutant was first discovered in patients with Cowden disease, resulting in complete loss of lipid phosphatase activity while retaining PTP activity [17,18]. The conservative D847E substitution in INPP4B significantly reduced INPP4B PTP activity (Fig. 2A, purple1). Active site mutations did not significantly alter steady state protein levels of mutant INPP4B proteins (Fig. 2B). Fig. 2B shows average velocities from four independent experiments that were normalized for protein levels.
Fig. 2.

Differential regulation of INPP4B protein tyrosine phosphatase activity by K843M, K846M, and D847E. (A) Cells were transfected and INPP4B purified as in Fig. 1. Thirty percent of eluate was used in reactions with 100 mM pNPP. Duplicate measurements were taken for each INPP4B expression construct and average ± SEM calculated. (B) Reaction velocities were calculated and normalized for levels of each respective protein. The average velocity of four independent experiments and standard errors are shown. Comparative amounts of protein were analyzed by resolving 2% of eluate on SDS–PAGE and Western blotting with ANTI-FLAG M2 antibody. Representative Western blot is shown.
3.3. K846 and D847 are essential for INPP4B lipid phosphatase activity
The INPP4B K843M mutation did not alter lipid phosphatase activity for the substrate PI(3,4)P2, but reduced INPP4B hydrolysis of Ins(1,3,4)P3 by about 70% (Fig. 3A). The K846M substitution abolished all INPP4B lipid phosphatase activity (Fig. 3B). The D847E mutant of INPP4B could not dephosphorylate PI(3,4)P2 and PI(4,5)P2 but retained approximately 35% of activity towards Ins(1,3,4)P3 (Fig. 3C). We have previously shown that INPP4B can lower Akt phosphorylation of T308 and S473 [4]. Here we tested if phospho-Y levels of Akt1 are regulated by INPP4B and PTEN. WT-INPP4B reduced pospho-Y Akt levels by approximately 67%, whereas PTEN had no effect on pospho-Y levels of Akt1 (Fig. 3D). The catalytic dead C842S INPP4B mutant had no effect on pospho-Y levels, indicating that INPP4B catalytic activity is required. Reduction of pospho-Y Akt1 levels by K843M mutant did not reach statistical significance, while both the K846M and D847E mutants significantly reduced Akt1 pospho-Y levels by approximately 60% and 50% respectively.
Fig. 3.

Role of K843M, K846M, and D847E in INPP4B lipid phosphatase activity. Cells were transfected as in Fig. 1 and indicated INPP4B proteins immunoprecipitated by M2 affinity gel. Fifteen percent of eluate was incubated with 1 mM PI(4,5)P2, PI(3,4)P2, or Ins(1,3,4)P3 for each lipid phosphatase assay. (A) Lipid phosphatase activity of K843M and WT-INPP4B with lipid substrates. (B) Comparison of K846M and WT INPP4B lipid phosphatase activities. (C) Comparison of D847E and WT-INPP4B lipid phosphatase activities. Average absorbance measurements at 660 nm from four individual experiments are shown for each individual mutation (black bars) alongside WT-INPP4B (white bars). (D) Transiently expressed FLAG-tagged Akt1 and WT-INPP4B, indicated mutants of INPP4B, or vector control were immunoprecipitated with M2 anti-FLAG beads from Hek293T cells. Eluted proteins were analyzed by Western blot for FLAG, phospho-Y and PTEN levels by Western blotting. Note that Akt1 and PTEN are of similar size and the bands are indistinguishable on FLAG Western blot. (E) Akt1 phospho-Y levels were normalized to level of total Akt1. Values are shown as % of phospho-Y level of Akt1 in the cells transfected with vector only (V) Data are presented as means ± SEM. /P < 0.05, //P < 0.001, ///P < 0.0001, two-tailed Student’s t test.
While the three dimensional structure of INPP4B is not known, in silico analysis shows that INPP4B has almost no structural similarity to PTEN. INPP4B and PTEN share only 11% sequence similarity [19], with a maximal similarity of 24% around the catalytic site. We compared the phosphatase domain (aa 819–880) of INPP4B against known crystal structures. The highest similarity was found with Mycobacterium tuberculosis PtpB, a bacterial protein tyrosine phosphatase with a CX5R catalytic domain [20]. The phosphatase domain of INPP4B and the region of PtpB used as a template have only 16% sequence similarity but share a high degree of tertiary structure similarity. According to the predicted structure from I-TASSER [21], the active site is positioned in an unstructured loop between two helices (Fig. 4A). As seen in Fig. 4B, mutations introduced to the flexible catalytic loop were not predicted to affect INPP4B tertiary structure.
Fig. 4.
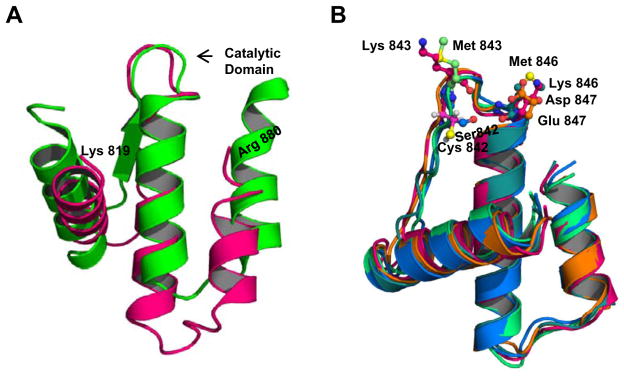
Predicted structure of the INPP4B phosphatase domain does not change with introducing mutations into catalytic loop. (A) Alignment of crystal structure models of INPP4B (pink) and PtpB (green), a protein tyrosine phosphatase from Mycobacterium tuberculosis. (B) Alignment of WT INPP4B and mutant INPP4B proteins; mutations do not alter the general orientation of adjacent helices.
4. Discussion
INPP4B and PTEN are phosphatases with tumor suppressor activity. They are both known to reduce Akt phosphorylation and Akt activity in some cellular milieus [3,4]. In prostate cancer, both phosphatases are lost at similar rates during the transition to metastases [4,22]. In addition to functional similarity and loss during cancer progression, their CX5R motifs are highly conserved. Here we show some striking differences in the functions of individual amino acid residues within the catalytic sites of INPP4B and PTEN.
The first enzymatic activity described for PTEN was protein tyrosine phosphatase activity [23,24]. Potential protein substrates for PTEN include FAK, SMAD3, and PDGFR, proteins involved in the development of metastases [25–27]. In the glioblastoma cell line U87MG, expression of PTEN that lacked either lipid or protein phosphatase activity was able to suppress proliferation, while only PTEN with both of these activities was able to suppress invasion [28] suggesting a functional significance for the PTP activity of PTEN. In vitro, PTEN dephosphorylates pNPP with a km of 4.5 mM [29]. In our experiments we observed that INPP4B was able to use pNPP as a substrate with a somewhat higher km of approximately 38 mM suggesting that structural differences may affect INPP4B affinity for this particular substrate and that optimal phosphoprotein substrates for PTEN and INPP4B likely differ.
INPP4B and PTEN CX5R motifs, C842KSAKDR and C124KAGKR, have 2 lysines, which are highly conserved across species for both proteins suggesting functional significance. In PTEN, mutation of either lysine to methionine alters PTP activity only slightly. While K846M does not alter INPP4B PTP activity significantly, the K843M substitution in INPP4B increases PTP activity by about 60%. According to the reported crystal structure of PTEN, the K125 amino acid residue is positioned next to the D1 phosphate group of Ins(1,3,4,5)P4 and the K125 M substitution causes a decline in PTEN phosphatase activity towards all substrates [30]. The analogous K843M substitution in INPP4B does not alter activity towards its main substrate PI(3,4)P2, but decreases Ins(1,3,4)P3 rate of hydrolysis suggesting a K843–D1 functional interaction. K843 may play a role in orienting the substrate and substrate preference. The K128 residue in PTEN is predicted to interact with the phosphate at position D5 of the inositol ring and consequentially in PTEN K128 M mutant hydrolysis of PI(3,4,5)P3 is significantly diminished but not for PI(3,4)P2 and PI(3)P [30]. Analogous mutation in INPP4B, K846M, completely abolishes phosphatase activity towards phospholipids, raising the possibility that the positively charged lysine residue interacts with the phosphate at position D4 of the inositol ring present in each substrate.
The PTEN G129E mutation was found repeatedly in Cowden disease patients [17]. This mutation completely abolishes PTEN lipid phosphatase activity leaving PTP activity unaltered [17,18]. PTEN G129E changes both the charge and bulk of the residue in this position. A more conservative D847E substitution in INPP4B that only slightly increases the length of the side chain without altering its charge, almost completely abolishes INPP4B activity with pNPP. However, some lipid phosphatase activity for D847E INPP4B remains. This subtle substitution may induce enough of a change in the active site that the smaller PI(3,4)P2, PI(4,5)P2 and pNPP molecules are no longer able to make adequate contacts with the active site leading to loss of hydrolysis. The larger Ins(1,3,4)P3 head group may therefore be able to maintain at least some contact that allows for some remaining degree of hydrolysis. Alternatively, this residue may also play a critical role in interactions with position D4 and, although subtle, this substitution is enough to abolish soluble substrate interactions.
Several tyrosine kinases phosphorylate and activate Akt [12–14]. In this study we show that INPP4B, but not PTEN is able to reduce tyrosine phosphorylation of Akt1 in Hek293t cells, which is dependent on INPP4B catalytic activity. Interestingly, modifications within the catalytic site of INPP4B modified this activity. While K843M retained both lipid phosphatase and PTP activities in vitro, in this cell based assay it was unable to reduce phospho-Y on Akt1 to the same extent as WT-INPP4B. INPP4B K846M mutation caused complete loss of in vitro lipid phosphatase activity. However, this mutant was as efficient in reducing phospho-Y Akt1 levels as WT-INPP4B, suggesting that lipid phosphatase activity is dispensable for this function. The D847E substitution that abolished PI(3,4)P2 and PI(4,5)P2 hydrolysis and significantly reduced PTP activity was able to significantly reduce phospho-Y on Akt1. It is possible that residual PTP activity of D847E mutant was sufficient to reduce phospho-Y Akt1 levels in the course of 48 h coexpression. These data suggest that INPP4B suppression of Y phosphorylation of Akt1 may not be mediated through phospho-lipid signaling. It is also possible that INPP4B lipid phosphatase and PTP activities differ in vitro and in vivo.
The INPP4B and PTEN phosphatase domains are located within the C-terminus and N-terminus respectively. Amino acid alignment reveals maximal identity of 24% around the active site. It is not possible to overlay tertiary structures of these DuSPs; the PTEN phosphatase domain is composed of intermittent helices and β-strands, while INPP4B is predicted to have mostly α-helices and overlaps best with Mycobacterium tuberculosis PtpB, whose protein tyrosine phosphatase domain is composed exclusively of α-helices. As in PTEN, the INPP4B catalytic motif is predicted to be located in a flexible loop and mutations in that loop are not predicted to alter INPP4B conformation.
In conclusion, we have shown that INPP4B has protein phosphatase activity. While both PTEN and INPP4B are dual specificity phosphatases and tumor suppressors, different residues within the catalytic site of INPP4B are responsible for activity with lipid and protein substrates and for substrate specificity. Using Akt1 pY levels, we demonstrated existence of unique downstream signaling pathways for INPP4B.
Abbreviations
- PI3K
by phosphatidyl 3-kinase
- PI4K
phosphatidyl 4-kinase
- PI5K
phosphatidyl 5-kinase
- INPP4B
inositol polyphosphate 4-phosphatase type II
- PTP
protein tyrosine phosphatase
- pNPP
para-nitrophenyl phosphate
- DifMUP
6,8-difluoro-4-methylumbelliferyl phosphate
- PTEN
phosphatase and tensin homolog
- DuSP
dual specificity phosphatase
- PI(34)P2
phosphatidylinositol 3,4-bisphosphate
- PI(45)P2
phosphatidylinositol 4,5-bisphosphate
- Ins(13,4)P3
inositol 1,3,4-trisphosphate
Footnotes
For interpretation of color in Fig. 2, the reader is referred to the web version of this article.
References
- 1.Bunney TD, Katan M. Phosphoinositide signalling in cancer: beyond PI3K and PTEN. Nat Rev Cancer. 2010;10:342–352. doi: 10.1038/nrc2842. [DOI] [PubMed] [Google Scholar]
- 2.Fedele CG, Ooms LM, Ho M, et al. Inositol polyphosphate 4-phosphatase II regulates PI3K/Akt signaling and is lost in human basal-like breast cancers. Proc Natl Acad Sci USA. 2010;107:22231–22236. doi: 10.1073/pnas.1015245107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gewinner C, Wang ZC, Richardson A, et al. Evidence that inositol polyphosphate 4-phosphatase type II is a tumor suppressor that inhibits PI3K signaling. Cancer Cell. 2009;16:115–125. doi: 10.1016/j.ccr.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hodgson MC, Shao LJ, Frolov A, et al. Decreased expression and androgen regulation of the tumor suppressor gene INPP4B in prostate cancer. Cancer Res. 2011;71:572–582. doi: 10.1158/0008-5472.CAN-10-2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferron M, Boudiffa M, Arsenault M, et al. Inositol polyphosphate 4-phosphatase B as a regulator of bone mass in mice and humans. Cell Metab. 2011;14:466–477. doi: 10.1016/j.cmet.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Agoulnik IU, Hodgson MC, Bowden WA, et al. INPP4B: the new kid on the PI3K block. Oncotarget. 2011;2:321–328. doi: 10.18632/oncotarget.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He H, Watanabe T, Zhan X, et al. Role of phosphatidylinositol 4,5-bisphosphate in Ras/Rac-induced disruption of the cortactin-actomyosin II complex and malignant transformation. Mol Cell Biol. 1998;18:3829–3837. doi: 10.1128/mcb.18.7.3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kassis J, Moellinger J, Lo H, et al. A role for phospholipase C-gamma-mediated signaling in tumor cell invasion. Clin Cancer Res. 1999;5:2251–2260. [PubMed] [Google Scholar]
- 9.Turner T, Epps-Fung MV, Kassis J, et al. Molecular inhibition of phospholipase cgamma signaling abrogates DU-145 prostate tumor cell invasion. Clin Cancer Res. 1997;3:2275–2282. [PubMed] [Google Scholar]
- 10.Oikawa T, Itoh T, Takenawa T. Sequential signals toward podosome formation in NIH-src cells. J Cell Biol. 2008;182:157–169. doi: 10.1083/jcb.200801042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weaver AM. Invadopodia: specialized cell structures for cancer invasion. Clin Exp Metastasis. 2006;23:97–105. doi: 10.1007/s10585-006-9014-1. [DOI] [PubMed] [Google Scholar]
- 12.Conus NM, Hannan KM, Cristiano BE, et al. Direct identification of tyrosine 474 as a regulatory phosphorylation site for the Akt protein kinase. J Biol Chem. 2002;277:38021–38028. doi: 10.1074/jbc.M203387200. [DOI] [PubMed] [Google Scholar]
- 13.Mahajan K, Coppola D, Challa S, et al. Ack1 mediated AKT/PKB tyrosine 176 phosphorylation regulates its activation. PLoS One. 2010;5:e9646. doi: 10.1371/journal.pone.0009646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zheng Y, Peng M, Wang Z, et al. Protein tyrosine kinase 6 directly phosphorylates AKT and promotes AKT activation in response to epidermal growth factor. Mol Cell Biol. 2010;30:4280–4292. doi: 10.1128/MCB.00024-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsieh AC, Bo R, Manola J, et al. A library of siRNA duplexes targeting the phosphoinositide 3-kinase pathway: determinants of gene silencing for use in cell-based screens. Nucleic Acids Res. 2004;32:893–901. doi: 10.1093/nar/gkh238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Irby RB, Mao W, Coppola D, et al. Activating SRC mutation in a subset of advanced human colon cancers. Nat Genet. 1999;21:187–190. doi: 10.1038/5971. [DOI] [PubMed] [Google Scholar]
- 17.Myers MP, Pass I, Batty IH, et al. The lipid phosphatase activity of PTEN is critical for its tumor supressor function. Proc Natl Acad Sci USA. 1998;95:13513–13518. doi: 10.1073/pnas.95.23.13513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Furnari FB, Huang HJ, Cavenee WK. The phosphoinositol phosphatase activity of PTEN mediates a serum-sensitive G1 growth arrest in glioma cells. Cancer Res. 1998;58:5002–5008. [PubMed] [Google Scholar]
- 19.Thompson JD, Gibson TJ, Plewniak F, et al. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grundner C, Ng HL, Alber T. Mycobacterium tuberculosis protein tyrosine phosphatase PtpB structure reveals a diverged fold and a buried active site. Structure. 2005;13:1625–1634. doi: 10.1016/j.str.2005.07.017. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taylor BS, Schultz N, Hieronymus H, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li DM, Sun H. TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor beta. Cancer Res. 1997;57:2124–2129. [PubMed] [Google Scholar]
- 24.Myers MP, Stolarov JP, Eng C, et al. P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. Proc Natl Acad Sci USA. 1997;94:9052–9057. doi: 10.1073/pnas.94.17.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hjelmeland AB, Hjelmeland MD, Shi Q, et al. Loss of phosphatase and tensin homologue increases transforming growth factor beta-mediated invasion with enhanced SMAD3 transcriptional activity. Cancer Res. 2005;65:11276–11281. doi: 10.1158/0008-5472.CAN-05-3016. [DOI] [PubMed] [Google Scholar]
- 26.Mahimainathan L, Choudhury GG. Inactivation of platelet-derived growth factor receptor by the tumor suppressor PTEN provides a novel mechanism of action of the phosphatase. J Biol Chem. 2004;279:15258–15268. doi: 10.1074/jbc.M314328200. [DOI] [PubMed] [Google Scholar]
- 27.Tamura M, Gu J, Matsumoto K, et al. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science. 1998;280:1614–1617. doi: 10.1126/science.280.5369.1614. [DOI] [PubMed] [Google Scholar]
- 28.Davidson L, Maccario H, Perera NM, et al. Suppression of cellular proliferation and invasion by the concerted lipid and protein phosphatase activities of PTEN. Oncogene. 2010;29:687–697. doi: 10.1038/onc.2009.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li L, Ernsting BR, Wishart MJ, et al. A family of putative tumor suppressors is structurally and functionally conserved in humans and yeast. J Biol Chem. 1997;272:29403–29406. doi: 10.1074/jbc.272.47.29403. [DOI] [PubMed] [Google Scholar]
- 30.Lee JO, Yang H, Georgescu MM, et al. Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell. 1999;99:323–334. doi: 10.1016/s0092-8674(00)81663-3. [DOI] [PubMed] [Google Scholar]