

Abstract
The NMDA receptor antagonist phencyclidine (PCP) creates schizophrenia-like symptoms in normal controls. The effect of PCP on non-human primate brain gene expression was examined and compared to changes induced by olanzapine treatment. Experimental studies of PCP and antipsychotic drugs have direct relevance to understanding the pathophysiology and treatment of schizophrenia. Genome-wide changes in prefrontal cortex gene expression revealed alterations of 146 transcripts in the PCP treatment group compared to vehicle controls. Dysregulated genes were enriched in identified classes implicated in neurological and genetic disorders, including schizophrenia genes from the Psychiatric Genomics Consortium 108 loci as well as cell death in PCP-treated primates. Canonical pathway analysis revealed a significant overrepresentation of several groups including synaptic long-term potentiation and calcium signaling. Olanzapine coadministered with PCP normalized 34% of the 146 PCP-induced probe set expression changes, and a network of 17 olanzapine-normalized genes was identified enriched in schizophrenia candidate genes containing RGS4, SYN1 and AKT as nodes. The results of this study support the use of PCP administration in non-human primates as a glutamatergic model of schizophrenia and suggest that a large number of PCP-induced expression differences can be reversed by olanzapine. The results of this study may be informative for identification of potential candidates for pharmacogenetics and biomarker research related to the treatment of schizophrenia.
Key Words: Schizophrenia, Antipsychotics, Gene expression, Phencyclidine, Cynomolgus monkey, Prefrontal cortex
Introduction
Schizophrenia is a highly heritable disorder characterized by positive, negative, and cognitive symptoms. Current antipsychotic treatments for schizophrenia are modifications of serendipitous discoveries and primarily involve antagonism of dopamine and serotonin receptors. The molecular mechanism of action of antipsychotics remains to be elucidated. To date, genome-wide changes in gene expression in response to antipsychotic treatment have not been assessed in a non-human primate model of schizophrenia. The purpose of this study was to examine the effects of antipsychotic administration on whole-genome prefrontal cortex (PFC) gene expression in a primate model of schizophrenia.
One commonly used pharmacological model of schizophrenia involves mimicking glutamate receptor hypofunction, which is hypothesized to play a role in the etiology of schizophrenia [1]. This model is partly based on the knowledge that NMDA receptor antagonists such as phencyclidine (PCP) exacerbate schizophrenia and produce positive and negative symptoms and cognitive deficits in schizophrenia [2,3] as well as symptoms in subjects with no previous psychiatric history [4]. Although there is no complete animal model of schizophrenia, PCP blockade of NMDA receptors has been used in animals to model NMDA receptor hypofunction, which is hypothesized to be causally related to some clinical features of schizophrenia [5]. Other evidence of glutamate dysfunction in schizophrenia includes alterations in glutamate receptor subunits (for a review, see [6]). Moreover, clinical data have shown that an mGlu2/3 agonist is effective in reducing positive and negative symptoms in subjects with schizophrenia [7], while preclinical data have demonstrated that mGlu2/3 agonism decreases presynaptic glutamate activity and is effective in preventing the prefrontal increase in glutamate efflux due to PCP as well as PCP-induced behaviors in rats [8].
In the current study, cynomolgus monkeys were administered the NMDA receptor antagonist PCP with or without OLZ cotreatment to determine if genome-wide level changes could be reversed by antipsychotic administration.
Materials and Methods
Animals and Drug Treatments
Cynomolgus monkeys (n = 42) were randomly assigned to 1 of 6 drug groups and administered vehicle (VEH; saline and 10% acacia), the antipsychotics chronic haloperidol (HAL; 1.5 mg/kg), acute haloperidol (AHAL; 8.0 mg/kg), chronic olanzapine (OLZ; 0.75 mg/kg), or acute olanzapine (AOLZ; 3.0 mg/kg), the NMDA receptor antagonist PCP (1.0-2.0 mg/kg/day), or PCP and OLZ (PCP + OLZ). The duration of the drug administration over a 6-week period is described in table 1. The protocol was reviewed and approved by the Institutional Animal Care and Use Committee at Covance Laboratories Inc. (Madison, Wis., USA).
Table 1.
Study design: cynomolgus monkeys were administered 1 of 6 drug regimens for up to 6 weeks
| Group | Week 1 | Week 2 | Week 3 | Week 4 | Week 5 | Week 6 | Acute |
|---|---|---|---|---|---|---|---|
| VEH | VEH | VEH | VEH | VEH | VEH | VEH | VEH |
| PCP | PCP (1 mg/kg/day) | PCP (1 mg/kg/day) | PCP (2 mg/kg/day) | PCP (2 mg/kg/day) | PCP (2 mg/kg/day) | PCP (2 mg/kg/day) | VEH |
| PCP + OLZ | PCP (1 mg/kg/day) | PCP (1 mg/kg/day) | PCP (2 mg/kg/day)/OLZ (0.75 mg/kg b.i.d.) | PCP (2 mg/kg/day)/OLZ (0.75 mg/kg b.i.d.) | PCP (2 mg/kg/day)/OLZ (0.75 mg/kg b.i.d.) | PCP (2 mg/kg/day)/OLZ (0.75 mg/kg b.i.d.) | VEH |
| OLZ | VEH | VEH | OLZ (0.75 mg/kg b.i.d.) | OLZ (0.75 mg/kg b.i.d.) | OLZ (0.75 mg/kg b.i.d.) | OLZ (0.75 mg/kg b.i.d.) | VEH |
| HAL | VEH | VEH | HAL (1.5 mg/kg b.i.d.) | HAL (1.5 mg/kg b.i.d.) | HAL (1.5 mg/kg b.i.d.) | HAL (1.5 mg/kg b.i.d.) | VEH |
| AOLZ | VEH | VEH | VEH | VEH | VEH | VEH | OLZ (3 mg/kg) |
| AHAL | VEH | VEH | VEH | VEH | VEH | VEH | HAL (8 mg/kg) |
Dose Administration
The PCP hydrochloride or saline was administered via an ALZET Osmotic Pump (Model 2ML2, Lot No. 9910925; Alza Corporation, Palo Alto, Calif., USA). The 10% acacia or OLZ were administered orally after injecting the required volume into dried Calimyrna figs (Mariani Packing Co. Inc., Vacaville, Calif., USA). The method and dose of PCP administration used in the current study was the same as described previously [9], which was shown to produce serum PCP levels in primates similar to the range associated with psychosis-like behavioral consequences of PCP intoxication in humans. Each animal had three surgeries to implant an osmotic pump. Each pump was maintained for at least a 2-week infusion interval, then discarded and replaced with a new pump or discarded after removal at necropsy. Animals were anesthetized by administration of propofol A, and the osmotic pump was implanted subcutaneously in the scapular region. The sterile saline or PCP was administered by continuous subcutaneous infusion 7 days/week for at least 6 weeks via an indwelling osmotic pump. Starting with the pm dose on day 1, 10% acacia was offered in figs to all animals twice daily (approx. 12 h apart). Beginning with the am dose on day 15 and continuing until the day 43 am dose, 10% acacia or OLZ preparations were offered in figs twice daily (approx. 12 h apart). The prepared suspensions were injected into 1 dried fig/animal for each dose. Animals were offered figs twice daily for at least 6 weeks.
Plasma Concentration Results
Mean plasma concentrations of PCP administered via the osmotic pumps were relatively proportional to the dose level. At the nominal dose level of 1 mg/kg/day, the mean PCP plasma concentration was 14.07 ± 1.25 ng/ml for animals in the PCP group and 11.4 ± 1.44 ng/ml for animals in the PCP + OLZ group on day 15. After doubling the nominal dose level to 2 mg/kg/day, the mean PCP plasma concentration was 23.05 ± 2.01 ng/ml for animals in the PCP group and 19.20 ± 1.60 ng/ml for animals in the PCP + OLZ group on day 36. Mean plasma concentrations of OLZ were relatively consistent over the course of the study and within the range of plasma concentrations seen in humans after the administration of efficacious doses of OLZ [10]. The dose for OLZ was chosen to produce plasma concentrations that are within the range of drug concentrations observed after the administration of efficacious doses of OLZ in patients [10]. The mean OLZ plasma concentrations ranged between 3.64 ± 0.71 and 7.39 ± 1.85 ng/ml in the OLZ group and between 6.92 ± 1.74 and 16.20 ± 4.31 ng/ml in the PCP + OLZ group. This is within the range of concentrations observed in schizophrenia patients treated with 1-10 mg (1.47 ± 0.77 to 20.68 ± 17.07 µg/l). The mean HAL plasma concentrations were also relatively consistent over the course of the study and ranged between 0.31 ± 0.02 and 0.74 ± 0.11 ng/ml. The mean AHAL and AOLZ plasma concentrations were 3.71 ± 0.31 and 68.51 ± 9.71 ng/ml, respectively.
Tissue Collection
After 6 weeks of treatment, animals were fasted overnight, then anesthetized with sodium pentobarbital, weighed, exsanguinated, and necropsied. From the right half of the brain, the anterior portion of the frontal cortex including Brodmann areas 9, 10, 11, 32 and 46 was carefully dissected, flash frozen in liquid nitrogen, and then stored in a freezer, set to maintain 60-80°C, until analyzed.
Microarray Detection of Probe Set Levels
Subjects were run in duplicate on two arrays (Affymetrix HU6800 and HG-U95A/HG-U95Av2) for a total of four arrays per animal. Double-stranded cDNA was synthesized from 10 μg of total RNA from each RNA extraction using the SuperScript Choice system (Gibco-BRL) with an oligo(dt) primer containing T7 RNA polymerase promoter (Geneset). The cDNA was purified by phenol/chloroform extraction. Biotin-labeled cRNA was synthesized and purified and hybridized to Affymetrix U95 or HU6800 microarrays. Microarrays were stained and washed on Affymetrix fluidics station 400 according to the manufacturer's instructions.
Data Analysis
Data were analyzed with Partek Genomics Suite statistical software version 6.5 (St. Louis, Mo., USA). Two outliers were removed from both arrays based on visualization of principal components analysis scatter plot. A batch effect related to the scan date was observed by principal components analysis, and the scan date was used in ANCOVA to reduce this effect. The resulting data for each microarray platform were analyzed separately by a three-way ANOVA with array technical replicate, subject and treatment as factors, with subject nested within treatment. Results were corrected for multiple comparisons by Bonferroni correction or false discovery rate step-up correction.
Functional Annotation Analyses
Genes with nominal p values <0.05 on both the U95 and HU6800 arrays and consistent fold change directions between arrays were used as input variables for the dataset to query the Ingenuity Pathways Analysis (IPA) software v7.1 canonical pathway analysis unless otherwise stated [11]. Gene symbols were mapped to the corresponding gene objects in the Ingenuity Pathways Knowledge Base. Each network or pathway was set to have a maximum of 35 focus genes. IPA identified those pathways containing direct and indirect relationships that were most significant to the input dataset. The significance of the association between the dataset and the canonical pathway was determined based on the Benjamini-Hochberg step-down false discovery rate calculated with Fisher's exact test by calculating the probability that the association between the genes in the dataset and the canonical pathway is due to chance alone. Overrepresented related transcription factors were identified using DAVID Bioinformatics Database (http://david.abcc.ncifcrf.gov/).
Results
Antipsychotic Effects on Gene Expression
To assess treatment effects on gene expression, we identified genes with significant treatment effects on both the Affymetrix HU6800 and Affymetrix HG-U95A/HG-U95Av2 arrays. Genes with expression significantly associated with treatment at the p < 0.05 level with consistent fold change directions on both arrays were considered true treatment effect genes. A total of 7,130 probe sets were represented on the HU6800 array and 12,600 on the U95 array. A total of 5,282 unique gene symbols overlapped between arrays. The number of significant findings on each array and the overlap between arrays are outlined in table 2. All probe set data for the overlapping 5,282 genes on the U95 and HU6800 arrays are shown in online supplementary tables 1 and 2, respectively (see www.karger.com/doi/10.1159/000430786 for all online suppl. material).
Table 2.
Number of significant findings (p < 0.05) by microarray platform according to nominal p value, Bonferroni corrected p value, and FDR step-up corrected p value
| Overall treatment | OLZ vs. VEH | PCP vs. VEH | PCP vs. PCP + OLZ | |
|---|---|---|---|---|
| HG6800 | ||||
| Nominal p value | 1,952/1,897 | 198/190 (11↓, 187↑) | 2,504/2,425 (2,502↓, 2↑) | 1,796/1,735 (1,792↓, 4↑) |
| Bonferroni | 54/53 | 3/3 (0↓, 3↑) | 19/19 (19↓, 0↑) | 18/18 (18↓, 0↑) |
| Step-up | 801/785 | 5/5 (0↓, 5↑) | 1,216/1,191 (1,215↓, 1↑) | 295/288 (295↓, 0↑) |
| HG-U95 | ||||
| Nominal p value | 400/361 | 48/45 (19↓, 29↑) | 228/216 (215↓,13↑) | 166/156 (129↓, 37↑) |
| Bonferroni | 2/2 | 0 (0↓, 0↑) | 0 (0↓, 0↑) | 1 (1↓, 0↑) |
| Step-up | 13/13 | 0 (0↓, 0↑) | 0 (0↓, 0↑) | 1 (1↓, 0↑) |
| Overlap | ||||
| Nominal p value | 244 | 12 (0↓, 12↑) | 146 (145↓,1↑) | 85 (84↓, 1↑) |
| Bonferroni | 2 | 0 (0↓, 0↑) | 0 (0↓, 0↑) | 1 (1↓, 0↑) |
| Step-up | 8 | 0 (0↓, 0↑) | 0 (0↓, 0↑) | 1 (1↓, 0↑) |
The number of probesets/gene symbols is shown for each array. The overlap between both platforms is shown for the same comparisons in the last rows.
PCP Effects on PFC Gene Expression
There were a total of 146 overlapping genes with nominally significantly altered gene expression at p < 0.05 in PCP-administered animals compared to VEH controls on the U95 and HU6800 arrays. All but one gene were downregulated in PCP-administered animals compared to VEH controls. Genes that were significant at the p < 0.05 level on both arrays with consistent fold change directions are listed in online supplementary table 3. IPA was used to functionally annotate the 146 genes and identified a significant overrepresentation of genes implicated in neurological and genetic disorders, including schizophrenia (19 genes in total; p = 9.08 × 10-5) as well as cell death (table 3). These 19 significantly dysregulated genes altered by PCP were annotated by IPA as implicated in the etiology of schizophrenia: ATP1A3, ATP2B2, ATP6V1B2, CALR, DHPS, DPYSL2, EGR3, GABRB3, GRIA1, GRIK2, GSN, NEFL, PIK3R1, PLP1, PRKAR1A, RGS4, SNAP25, SOX5, and VDAC1. Pathways associated with PCP effects on PFC identified by canonical pathway analysis included a significant overrepresentation of genes implicated in calcium signaling and synaptic long-term potentiation (table 4). There was a 2.4-fold enrichment of genes from the PGC2 108 loci with PCP-dysregulated genes in the PFC (ATP2A2, GRIA1, MEF2C, PPP4C, and PTN).
Table 3.
IPA was used to functionally annotate 146 genes altered on both microarray platforms in the PCP treatment group compared to the VEH control group
| Category | B-H p value | Genes, n |
|---|---|---|
| Neurological disease | 4.59E-10 | 76 |
| Genetic disorder | 6.95E-10 | 92 |
| Skeletal and muscular disorders | 6.95E-10 | 64 |
| Cell death | 1.03E-09 | 69 |
| Cancer | 5.31E-07 | 32 |
IPA identified a significant over-representation of genes implicated in neurological disorders and genetic disorders, including schizophrenia (19 genes in total; p = 9 × 10−5), after correcting for multiple comparisons with Benjamini-Hochberg (B-H) associated with PCP effects.
Table 4.
IPA was queried with 146 genes altered on both microarray platforms in the PCP treatment group compared to the VEH control group
| Ingenuity canonical pathways | B-H p value | Genes |
|---|---|---|
| Huntington's disease signaling | 4.27 × 10−7 | MAPK1, PIK3R1, UBE2S, SNAP25, CDK5R1, PRKCG, DNM1, BCL2L1, MTOR, PRKCI, PENK, STX16, NCOR2, NAPA, RASA1 |
| Melatonin signaling | 1.78 × 10−5 | PRKACB, CAMK4, PRKCI, MAPK1, RORB, PRKCG, PRKAR1A, CAMK2G |
| Neuropathic pain signaling in dorsal horn neurons | 1.78 × 10−5 | PRKACB, CAMK4, PRKCI, MAPK1, GRIA1, PIK3R1, PRKCG, PRKAR1A, CAMK2G |
| Synaptic long-term potentiation | 2.63 × 10−5 | PRKACB, CAMK4, PRKCI, MAPK1, GRIA1, PPP3CA, PRKCG, PRKAR1A, CAMK2G |
| Calcium signaling | 2.63 × 10−5 | PRKACB, CALR, CAMK4, MAPK1, GRIA1, MEF2C, ATP2A2, PPP3CA, ATP2B2, PRKAR1A, CAMK2G |
IPA canonical pathway analysis identified the following over-represented canonical pathways after correcting for multiple comparisons. B-H = Benjamini-Hochberg.
OLZ Effects on PFC Gene Expression
There were 12 genes with significantly differential expression in OLZ-administered animals compared to VEH controls, overlapping between both arrays (see online suppl. table 4). All 12 were upregulated in OLZ-administered subjects compared to controls. Canonical pathway analysis revealed overrepresentation of genes implicated in glutamate receptor signaling, ERK5 signaling, and calcium signaling (table 5). There was a 12.4-fold enrichment of genes from the PGC2 108 loci with OLZ-dysregulated genes in the PFC (NRGN, MEF2C).
Table 5.
Using a set of 12 genes altered by OLZ, IPA canonical pathway analysis identified the following over-represented canonical pathways after correcting for multiple comparisons
| Ingenuity canonical pathways | B-H p value | Genes |
|---|---|---|
| Melatonin signaling | 0.009 | PRKACB, RORB, PRKCG, CAMK2G |
| Calcium signaling | 0.009 | PRKACB, MEF2C, ATP2A2, ATP2B2, CAMK2G |
| FYN receptor-mediated phagocytosis in macrophages and monocytes | 0.009 | PAK1, PLD3, ACTB, PRKCG |
| G protein-coupled receptor signaling | 0.013 | PRKACB, RGS4, RASA1, PRKCG, CAMK2G |
| Molecular mechanisms of cancer | 0.013 | PRKACB, PAK1, RHOB, RASA1, PRKCG, CAMK2G |
B-H = Benjamini-Hochberg.
Effects of OLZ Coadministration on PCP-Induced Changes in PFC Gene Expression
There were a total of 85 genes with significantly differential expression in OLZ + PCP-administered animals compared to PCP alone, overlapping between both array platforms. Of these 85, 50 were normalized by OLZ + PCP, meaning that these genes fulfilled three criteria: (1) significantly dysregulated in PCP- versus VEH-administered animals, (2) significantly altered in the same direction between PCP- and PCP + OLZ-administered animals, and (3) showed no significant difference in expression between PCP + OLZ- and VEH-administered animals (see fig. 1, 2, 3, 4 for the expression profiles of the representative genes). Because plasma levels for OLZ- and PCP-treated animals varied, we examined the correlation between individual plasma level and gene expression of the 50 OLZ-normalized genes on both arrays. None of the 50 genes showed a significant correlation with gene expression and PCP or OLZ plasma levels on both arrays. IPA was used to functionally annotate the 50 genes normalized by OLZ administration (see tables 6, 7 for HU6800 and U95 results, respectively), which identified a significant overrepresentation of genes implicated in genetic disorders, including schizophrenia (7 genes: ATP1A3, ATP2B2, GABRB3, PLP1, RGS4, SNAP25 and VDAC1; p = 2.33 × 10-2), neurological disorders, and cell death (tables 8, 9). Canonical pathway analysis revealed a significant overrepresentation of genes implicated in melatonin signaling, calcium signaling, FYN receptor-mediated phagocytosis in macrophages and monocytes, and G protein-coupled receptors (table 7). IPA was used to identify biological networks by querying interactions between genes stored in the Ingenuity Pathway Knowledge Base. IPA calculates a significance score for each network, which is the negative log of the p value associated with the network. IPA identified one gene network connecting 17 genes normalized by OLZ with a network score of 39 or a p value of 10-39. This network contained only genes downregulated by PCP and included ACTB, ATP2A2, ATP2B2, CAMK2G, GDI2, MAP2, NF2, PAK1, PRKACB, RAB3A, RAB6A, RASA1, RGS4, RHOB, SLC3A2, SNAP25, and SYN1. Two transcription factors, SOX5 and MEF, were identified by DAVID Bioinformatic Database to regulate a significant number of OLZ-reversed transcripts (see online suppl. tables 5, 6). There was a 2.8-fold enrichment of genes from the PGC2 108 loci with OLZ-normalized, PCP-dysregulated genes in the PFC (ATPA2, MEF2C).
Fig. 1.

Representative gene RGS4 expression was reduced by PCP treatment and normalized by OLZ + PCP. Fold change is shown with respect to control levels.
Fig. 2.

Representative gene NCAM expression was reduced by PCP treatment and normalized by OLZ + PCP.
Fig. 3.

Representative gene RAB3A expression was reduced by PCP treatment and normalized by OLZ + PCP.
Fig. 4.

Representative gene SYN1 expression was reduced by PCP treatment and normalized by OLZ + PCP.
Table 6.
PCP-altered genes were normalized by coadministration of OLZ and fulfilled three criteria: significantly dysregulated in PCP-versus VEH-administered animals, significantly altered in the same direction between PCP- and PCP + OLZ-administered animals, and showed no significant difference in expression between PCP+ OLZ- and VEH-administered animals
| Gene symbol | Gene title | PCP vs. VEH |
PCP vs. PCP/OLZ |
PCP/OLZ vs. VEH |
|||
|---|---|---|---|---|---|---|---|
| p value | FC | p value | FC | p value | FC | ||
| ACTB | actin, beta | 4.5E-04 | −1.24 | 3.3E-06 | −1.36 | 8.8E-02 | 1.10 |
| ACYP1 | acylphosphatase 1, erythrocyte (common) type | 3.0E-02 | −1.16 | 5.0E-02 | −1.15 | 8.4E-01 | −1.01 |
| ATP1A3 | ATPase, Na+/K+ transporting, alpha 3 polypeptide | 8.0E-04 | –1.22 | 5.5E-04 | –1.23 | 8.4E-01 | 1.01 |
| ATP2A2 | ATPase, Ca++ transporting, cardiac muscle, slow twitch 2 | 1.2E-02 | –1.21 | 4.5E-02 | –1.16 | 5.7E-01 | –1.04 |
| ATP2B2 | ATPase, Ca++ transporting, plasma membrane 2 | 1.4E-04 | –1.28 | 6.3E-04 | –1.25 | 6.5E-01 | –1.03 |
| ATP6V1A | ATPase, H+ transporting, lysosomal 70 kDa, V1 subunit A | 1.4E-03 | –1.28 | 2.2E-02 | –1.19 | 3.0E-01 | –1.07 |
| C1orf61 | chromosome 1 open reading frame 61 | 9.2E-03 | –1.19 | 3.6E-02 | –1.15 | 5.8E-01 | –1.03 |
| CACNB4 | calcium channel, voltage-dependent, beta 4 subunit | 1.1E-03 | –1.25 | 1.7E-02 | –1.18 | 3.1E-01 | –1.07 |
| CAMK2G | calcium/calmodulin-dependent protein kinase II gamma | 3.4E-02 | –1.13 | 3.0E-02 | –1.14 | 9.2E-01 | 1.01 |
| CBX3 /// LOC644101 | chromobox homolog 3 (HP1 gamma homolog, Drosophila) /// similar to chromobox homolog | 2.2E-03 | –1.24 | 6.4E-03 | –1.21 | 7.2E-01 | –1.02 |
| CNTN1 | Contactin 1 | 2.2E-05 | –1.38 | 6.5E-04 | –1.29 | 2.8E-01 | –1.07 |
| CSNK1G2 | casein kinase 1, gamma 2 | 3.7E-02 | –1.16 | 4.7E-02 | –1.16 | 9.5E-01 | –1.00 |
| CTBP1 | C-terminal-binding protein 1 | 5.9E-03 | –1.22 | 2.7E-02 | –1.18 | 5.5E-01 | –1.04 |
| CYP2A13 | cytochrome P450, family 2, subfamily A, polypeptide 13 | 4.3E-02 | –1.14 | 4.6E-02 | –1.14 | 1.0E+00 | 1.00 |
| GABRB3 | gamma-aminobutyric acid (GABA) A receptor, beta 3 | 2.8E-04 | –1.29 | 1.6E-02 | –1.17 | 1.4E-01 | –1.09 |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase | 5.2E-03 | –1.19 | 6.2E-05 | –1.32 | 1.0E-01 | 1.10 |
| GDI2 | GDP dissociation inhibitor 2 | 2.4E-03 | –1.21 | 9.7E-03 | –1.18 | 6.2E-01 | –1.03 |
| KCNAB2 | potassium voltage-gated channel, shaker-related subfamily, beta member 2 | 7.4E-03 | –1.17 | 9.3E-02 | –1.10 | 2.7E-01 | –1.06 |
| KLHDC3 | kelch domain containing 3 | 2.5E-03 | –1.23 | 5.2E-03 | –1.22 | 8.2E-01 | –1.01 |
| MAP2 | microtubule-associated protein 2 | 1.1E-02 | –1.24 | 3.0E-03 | –1.30 | 5.7E-01 | 1.05 |
| MAT2A | methionine adenosyltransferase II, alpha | 2.4E-02 | –1.17 | 2.7E-02 | –1.17 | 9.8E-01 | –1.00 |
| MBP | myelin basic protein | 1.3E-03 | –1.28 | 4.3E-02 | –1.17 | 1.7E-01 | –1.10 |
| MEF2C | myocyte enhancer factor 2C | 1.2E-03 | –1.25 | 4.7E-03 | –1.21 | 6.4E-01 | –1.03 |
| NF2 | neurofibromin 2 (merlin) | 4.7E-02 | –1.13 | 1.7E-02 | –1.17 | 6.1E-01 | 1.03 |
| NOMO1 /// NOMO2 /// NOMO3 | NODAL modulator 1 /// NODAL modulator 2 /// NODAL modulator 3 | 1.3E-02 | –1.15 | 1.7E-02 | –1.15 | 9.6E-01 | –1.00 |
| NRCAM | neuronal cell adhesion molecule | 1.2E-03 | –1.26 | 2.0E-02 | –1.18 | 2.8E-01 | –1.07 |
| PAK1 | p21 protein (Cdc42/Rac)-activated kinase 1 | 1.2E-03 | –1.33 | 6.7E-03 | –1.27 | 5.5E-01 | –1.05 |
| PLD3 | phospholipase D family, member 3 | 1.1E-02 | –1.19 | 6.5E-03 | –1.21 | 7.9E-01 | 1.02 |
| PLP1 | proteolipid protein 1 | 7.6E-03 | –1.21 | 9.3E-03 | –1.21 | 9.7E-01 | –1.00 |
| PRKACB | protein kinase, cAMP-dependent, catalytic, beta | 4.2E-03 | –1.19 | 4.6E-03 | –1.19 | 9.9E-01 | 1.00 |
| PRKCG | protein kinase C, gamma | 2.9E-03 | –1.19 | 6.9E-04 | –1.23 | 5.5E-01 | 1.03 |
| PTPRS | protein tyrosine phosphatase, receptor type, S | 2.6E-04 | –1.28 | 1.6E-03 | –1.24 | 5.6E-01 | –1.04 |
| RAB3A | RAB3A, member RAS oncogene family | 3.4E-02 | –1.15 | 2.8E-03 | –1.23 | 2.9E-01 | 1.07 |
| RAB6A | RAB6A, member RAS oncogene family | 1.3E-03 | –1.26 | 9.0E-03 | –1.20 | 4.9E-01 | –1.05 |
| RASA1 | RAS p21 protein activator (GTPase activating protein) 1 | 3.6E-02 | –1.15 | 1.3E-02 | –1.19 | 6.3E-01 | 1.03 |
| RGS4 | regulator of G-protein signaling 4 | 1.5E-04 | –1.29 | 1.0E-04 | –1.31 | 8.4E-01 | 1.01 |
| RHOB | ras homolog gene family, member B | 6.8E-03 | –1.20 | 2.0E-02 | –1.17 | 6.8E-01 | –1.03 |
| RORB | RAR-related orphan receptor B | 5.1E-04 | –1.31 | 1.0E-02 | –1.22 | 2.8E-01 | –1.08 |
| RUNX1T1 | runt-related transcription factor 1; translocated to, 1 (cyclin D-related) | 4.7E-02 | –1.14 | 6.0E-04 | –1.27 | 7.8E-02 | 1.12 |
| SFRS3 | splicing factor, arginine/serine-rich 3 | 8.7E-03 | –1.25 | 3.9E-02 | –1.19 | 5.4E-01 | –1.05 |
| SLC3A2 | solute carrier family 3 (activators of dibasic and neutral amino acid transport) | 4.1E-02 | –1.12 | 2.7E-02 | –1.14 | 8.2E-01 | 1.01 |
| SNAP25 | synaptosomal-associated protein, 25 kDa | 4.6E-03 | –1.22 | 1.8E-02 | –1.18 | 6.1E-01 | –1.03 |
| SYN1 | synapsin I | 7.2E-03 | –1.16 | 5.4E-03 | –1.17 | 8.7E-01 | 1.01 |
| THRA | thyroid hormone receptor, alpha (erythroblastic leukemia viral (v-erb-a) oncogen | 2.3E-02 | –1.13 | 7.0E-03 | –1.17 | 5.8E-01 | 1.03 |
| UBAP2L | ubiquitin-associated protein 2-like | 3.7E-02 | –1.16 | 4.5E-02 | –1.15 | 9.5E-01 | –1.00 |
| UBE2D3 | ubiquitin-conjugating enzyme E2D 3 (UBC4/5 homolog, yeast) | 3.8E-03 | –1.25 | 2.5E-02 | –1.19 | 4.6E-01 | –1.05 |
| UBE2S | ubiquitin-conjugating enzyme E2S | 2.7E-02 | –1.15 | 1.6E-02 | –1.17 | 7.9E-01 | 1.02 |
| VDAC1 | voltage-dependent anion channel 1 | 1.0E-02 | –1.15 | 2.2E-02 | –1.14 | 7.7E-01 | –1.02 |
| YWHAZ | tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, zeta pol | 1.9E-02 | –1.16 | 1.6E-02 | –1.17 | 9.2E-01 | 1.01 |
| ZNF238 | zinc finger protein 238 | 2.0E-02 | –1.18 | 1.0E-02 | –1.21 | 7.6E-01 | 1.02 |
The 50 normalized genes are shown that fulfilled three criteria on the U95 array. FC = Fold change.
Table 7.
PCP-altered genes were normalized by coadministration of OLZ and fulfilled three critieria: significantly dysregulated in PCP-versus VEH-administered animals, significantly altered in the same direction between PCP- and PCP + OLZ-administered animals, and showed no significant difference in expression between PCP + OLZ- and VEH-administered animals
| Gene symbol | Gene title | PCP vs. VEH |
PCP vs. PCP/OLZ |
PCP/OLZ vs. VEH |
|||
|---|---|---|---|---|---|---|---|
| p value | FC | p value | FC | p value | FC | ||
| ACTB | actin, beta | 5.54E-06 | –1.29 | 1.06E-08 | –1.42 | 4.61E-02 | 1.10 |
| ACYP1 | acylphosphatase 1, erythrocyte (common) type | 4.17E-02 | –1.11 | 3.81E-02 | –1.12 | 9.66E-01 | 1.00 |
| ATP1A3 | ATPase, Na+/K+ transporting, alpha 3 polypeptide | 9.48E-04 | –1.18 | 8.75E-05 | –1.23 | 4.07E-01 | 1.04 |
| ATP2A2 | ATPase, Ca++ transporting, cardiac muscle, slow twitch 2 | 1.07E-03 | –1.18 | 2.57E-03 | –1.16 | 7.45E-01 | –1.01 |
| ATP2B2 | ATPase, Ca++ transporting, plasma membrane 2 | 5.89E-04 | –1.27 | 2.92E-03 | –1.22 | 5.57E-01 | 1.04 |
| ATP6V1A | ATPase, H+ transporting, lysosomal 70 kDa, V1 subunit A | 3.35E-07 | –1.39 | 1.76E-07 | –1.41 | 8.34E-01 | 1.01 |
| C1orf61 | chromosome 1 open reading frame 61 | 2.39E-02 | –1.16 | 3.89E-02 | –1.14 | 8.25E-01 | –1.01 |
| CACNB4 | calcium channel, voltage-dependent, beta 4 subunit | 7.64E-09 | –1.40 | 3.97E-06 | –1.28 | 4.60E-02 | –1.09 |
| CAMK2G | calcium/calmodulin-dependent protein kinase II gamma | 2.70E-03 | –1.13 | 6.50E-03 | –1.12 | 7.32E-01 | –1.01 |
| CBX3 /// LOC644101 | chromobox homolog 3 (HP1 gamma homolog, Drosophila) /// similar to chromobox homolog | 3.59E-02 | –1.14 | 2.34E-02 | –1.16 | 8.46E-01 | 1.01 |
| CNTN1 | contactin 1 | 3.62E-03 | –1.18 | 1.17E-02 | –1.15 | 6.40E-01 | –1.02 |
| CSNK1G2 | casein kinase 1, gamma 2 | 1.16E-02 | –1.15 | 9.82E-03 | –1.15 | 9.46E-01 | 1.00 |
| CTBP1 | C-terminal binding protein 1 | 5.07E-03 | –1.17 | 5.08E-03 | –1.17 | 9.99E-01 | –1.00 |
| CYP2A13 | cytochrome P450, family 2, subfamily A, polypeptide 13 | 2.92E-02 | –1.12 | 4.30E-02 | –1.11 | 8.57E-01 | –1.01 |
| GABRB3 | gamma-aminobutyric acid (GABA) A receptor, beta 3 | 3.38E-06 | –1.32 | 8.44E-04 | –1.20 | 6.71E-02 | –1.10 |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase | 1.80E-02 | –1.13 | 2.10E-02 | –1.12 | 9.46E-01 | –1.00 |
| GDI2 | GDP dissociation inhibitor 2 | 2.20E-04 | –1.27 | 8.47E-04 | –1.24 | 6.34E-01 | –1.03 |
| KCNAB2 | potassium voltage-gated channel, shaker-related subfamily, beta member 2 | 7.22E-03 | –1.13 | 1.84E-02 | –1.11 | 6.96E-01 | –1.02 |
| KLHDC3 | kelch domain containing 3 | 2.64E-04 | –1.25 | 4.24E-07 | –1.39 | 3.78E-02 | 1.12 |
| MAP2 | microtubule-associated protein 2 | 1.60E-03 | –1.33 | 5.96E-04 | –1.37 | 7.20E-01 | 1.03 |
| MAT2A | methionine adenosyltransferase II, alpha | 6.02E-05 | –1.26 | 7.90E-05 | –1.25 | 9.27E-01 | –1.00 |
| MBP | myelin basic protein | 2.84E-03 | –1.16 | 1.19E-03 | –1.18 | 7.45E-01 | 1.01 |
| MEF2C | myocyte enhancer factor 2C | 3.59E-03 | –1.21 | 2.12E-04 | –1.29 | 3.07E-01 | 1.06 |
| NF2 | neurofibromin 2 (merlin) | 1.54E-03 | –1.24 | 7.69E-03 | –1.19 | 5.36E-01 | –1.04 |
| NOMO1 /// NOMO2 /// NOMO3 | NODAL modulator 1 /// NODAL modulator 2 /// NODAL modulator 3 | 2.19E-03 | –1.18 | 2.16E-03 | –1.18 | 9.97E-01 | 1.00 |
| NRCAM | neuronal cell adhesion molecule | 1.05E-06 | –1.34 | 3.20E-05 | –1.27 | 2.65E-01 | –1.06 |
| PAK1 | p21 protein (Cdc42/Rac)-activated kinase 1 | 4.56E-04 | –1.22 | 9.67E-03 | –1.15 | 2.53E-01 | –1.06 |
| PLD3 | phospholipase D family, member 3 | 1.21E-03 | –1.15 | 2.60E-03 | –1.14 | 7.76E-01 | –1.01 |
| PLP1 | proteolipid protein 1 | 1.25E-02 | –1.12 | 1.06E-02 | –1.12 | 9.47E-01 | 1.00 |
| PRKACB | protein kinase, cAMP-dependent, catalytic, beta | 4.72E-08 | –1.36 | 7.16E-08 | –1.35 | 8.92E-01 | –1.01 |
| PRKCG | protein kinase C, gamma | 2.15E-03 | –1.17 | 1.28E-05 | –1.26 | 8.14E-02 | 1.08 |
| PTPRS | protein tyrosine phosphatase, receptor type, S | 2.50E-03 | –1.23 | 6.67E-04 | –1.26 | 6.28E-01 | 1.03 |
| RAB3A | RAB3A, member RAS oncogene family | 7.58E-05 | –1.21 | 3.18E-07 | –1.30 | 7.74E-02 | 1.08 |
| RAB6A | RAB6A, member RAS oncogene family | 3.21E-06 | –1.27 | 6.88E-04 | –1.18 | 7.55E-02 | –1.08 |
| RASA1 | RAS p21 protein activator (GTPase activating protein) 1 | 1.26E-03 | –1.18 | 6.84E-04 | –1.19 | 8.24E-01 | 1.01 |
| RGS4 | regulator of G-protein signaling 4 | 8.76E-07 | –1.31 | 1.51E-07 | –1.34 | 5.67E-01 | 1.03 |
| RHOB | ras homolog gene family, member B | 1.02E-06 | –1.36 | 6.86E-07 | –1.37 | 8.99E-01 | 1.01 |
| RORB | RAR-related orphan receptor B | 2.58E-04 | –1.25 | 6.22E-04 | –1.23 | 7.57E-01 | –1.02 |
| RUNX1T1 | runt-related transcription factor 1; translocated to, 1 (cyclin D-related) | 1.83E-04 | –1.26 | 2.64E-06 | –1.36 | 1.63E-01 | 1.08 |
| SFRS3 | splicing factor, arginine/serine-rich 3 | 1.25E-06 | –1.32 | 6.48E-07 | –1.34 | 8.31E-01 | 1.01 |
| SLC3A2 | solute carrier family 3 (activators of dibasic and neutral amino acid transport) | 6.39E-03 | –1.12 | 3.09E-02 | –1.10 | 5.05E-01 | –1.03 |
| SNAP25 | synaptosomal-associated protein, 25 kDa | 4.23E-02 | –1.14 | 2.42E-02 | –1.15 | 7.97E-01 | 1.02 |
| SYN1 | synapsin I | 5.38E-04 | –1.17 | 4.61E-07 | –1.29 | 2.26E-02 | 1.10 |
| THRA | thyroid hormone receptor, alpha (erythroblastic leukemia viral (v-erb-a) oncogen | 2.38E-02 | –1.11 | 2.77E-02 | –1.11 | 9.45E-01 | –1.00 |
| UBAP2L | ubiquitin-associated protein 2-like | 9.94E-04 | –1.22 | 3.54E-04 | –1.25 | 7.13E-01 | 1.02 |
| UBE2D3 | ubiquitin-conjugating enzyme E2D 3 (UBC4/5 homolog, yeast) | 2.40E-04 | –1.21 | 2.45E-04 | –1.21 | 9.94E-01 | –1.00 |
| UBE2S | ubiquitin-conjugating enzyme E2S | 8.75E-03 | –1.14 | 4.48E-02 | –1.10 | 4.76E-01 | –1.03 |
| VDAC1 | voltage-dependent anion channel 1 | 4.28E-02 | –1.11 | 3.18E-02 | –1.12 | 8.91E-01 | 1.01 |
| YWHAZ | tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, zeta pol | 7.41E-04 | –1.20 | 1.60E-02 | –1.14 | 2.38E-01 | –1.06 |
| ZNF238 | zinc finger protein 238 | 4.36E-04 | –1.20 | 4.49E-04 | –1.20 | 9.92E-01 | –1.00 |
This table shows the 50 normalized genes that fulfilled three criteria on the HU6800 array. FC = Fold change.
Table 8.
IPA was used to functionally annotate 50 PCP-dysregulated genes that were normalized by olanzapine coadministration
| Category | B-H p value | Genes, n |
|---|---|---|
| Genetic disorder | 7.13E-05 | 34 |
| Neurological disease | 7.13E-05 | 29 |
| Skeletal and muscular disorders | 7.13E-05 | 19 |
| Cell death | 2.90E-04 | 4 |
| Cell signaling | 2.07E-03 | 3 |
The top five over-represented biological functions are shown. B-H = Benjamini-Hochberg.
Table 9.
Using a set of 50 PCP-dysregulated genes that were normalized by olanzapine coadministration, IPA canonical pathway analysis identified the following over-represented canonical pathways after correcting for multiple comparisons
| Ingenuity canonical pathways | B-H p value | Genes |
|---|---|---|
| Melatonin signaling | 9.12E-03 | PRKACB, RORB, PRKCG, CAMK2G |
| Calcium signaling | 9.33E-03 | PRKACB, MEF2C, ATP2A2, ATP2B2, CAMK2G |
| FYN receptor-mediated phagocytosis in macrophages and monocytes | 9.33E-03 | PAK1, PLD3, ACTB, PRKCG |
| G protein-coupled receptor signaling | 1.35E-02 | PRKACB, RGS4, RASA1, PRKCG, CAMK2G |
| Molecular mechanisms of cancer | 1.35E-02 | PRKACB, PAK1, RHOB, RASA1, PRKCG, CAMK2G |
B-H = Benjamini-Hochberg.
Specificity of OLZ Effects on PCP-Induced Changes in PFC Gene Expression
To test the specificity of PCP-induced gene expression changes reversed by the coadministration of OLZ, we also assessed the effects of AOLZ, AHAL, OLZ, and HAL on probe sets that were altered by PCP treatment and normalized by OLZ + PCP. The results are summarized in Venn diagrams (fig. 5, 6). Of the 50 genes normalized in OLZ + PCP-administered animals (fig. 5), only a small number was changed by AOLZ, OLZ, AHAL, or HAL administration: 2 genes were altered by AOLZ (MEF2C and CACNB4) and 1 gene by OLZ (MEF2C) (fig. 6), while 2 genes were altered by AHAL (CACNB4 and GAPDH) and 3 genes by HAL (CACNB4, GABRB3, and GAPDH).
Fig. 5.
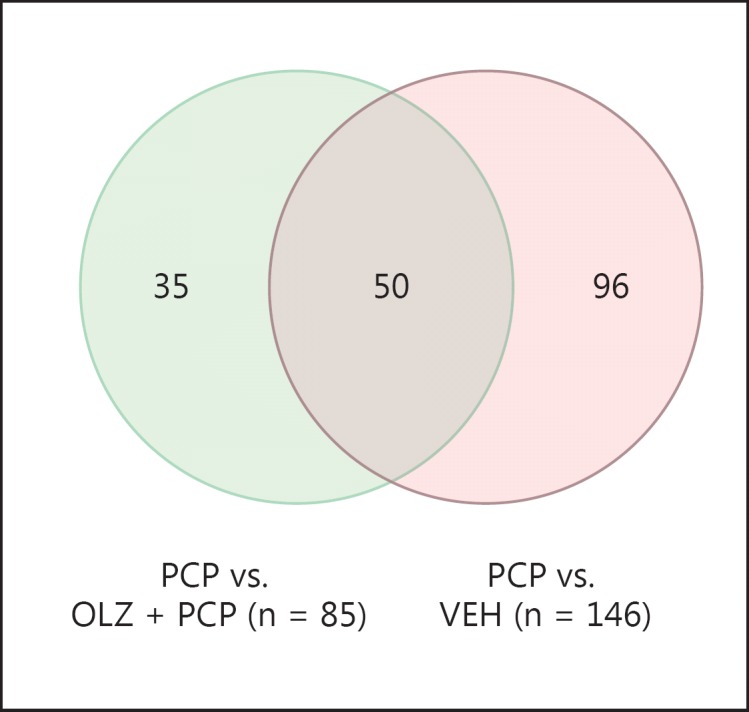
A total of 146 genes were dysregulated in PCP-administered animals compared to VEH controls. Treatment with OLZ reversed 50 of these expression alterations.
Fig. 6.
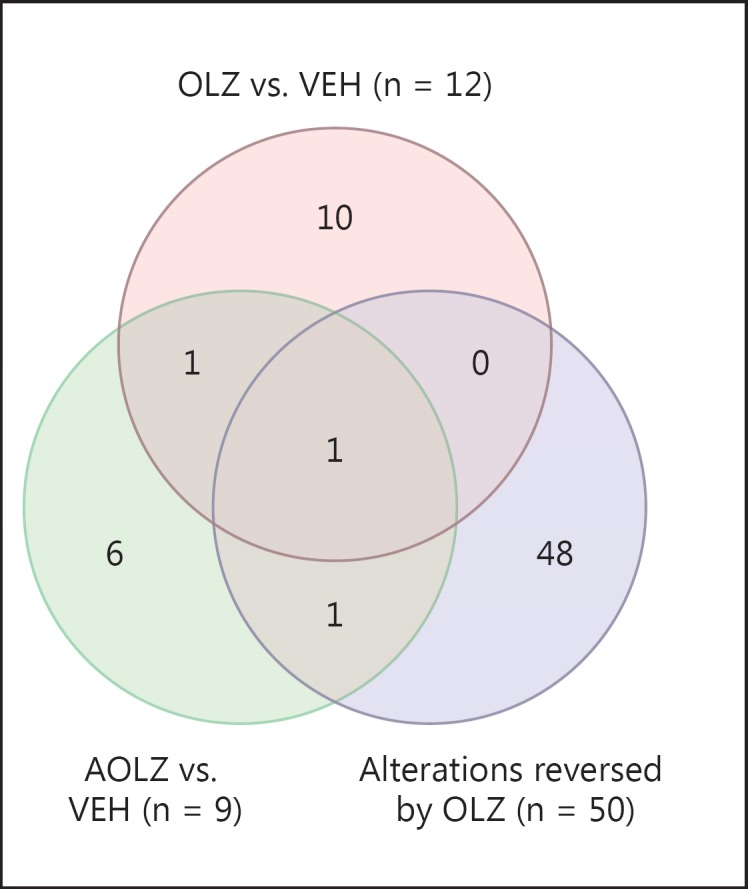
The Venn diagram shows the overlap between probe sets altered in expression by AOLZ and OLZ treatment and 50 PCP-induced expression alterations that were normalized by OLZ (fig. 5).
Discussion
These are the first results of PCP and gene expression in a non-human primate model of schizophrenia. The results demonstrate that a large number of PCP-induced changes in gene expression are reversed by the administration of the antipsychotic OLZ. IPA was run using the list of 146 genes with PCP-induced expression alterations and showed an enrichment of genes previously implicated in schizophrenia and neurological disorders, further supporting the use of PCP treatment as a model system for the study of glutamate dysfunction-related gene expression alterations in schizophrenia. Genes associated with cell death were also significantly altered, which was expected given the previously demonstrated neurotoxic effects of PCP [12]. It was not surprising that genes involved in synaptic long potentiation were altered by the NMDA receptor antagonist PCP given the role of NMDA and long-term potentiation [13]. This is consistent with previous work showing overrepresentation of altered expression of synapse-related genes in a chronic rat PCP model of schizophrenia [14].
Antipsychotic treatment with OLZ reversed the effects of PCP administration for 50 genes. Again, there was an overrepresentation of OLZ + PCP genes with those previously implicated in the etiology of schizophrenia and cell death (n = 24). This is interesting in light of previous work suggesting neuroprotective effects of OLZ in rodents and humans [15,16,17,18,19] and raises the question of whether such changes in gene expression are related to antipsychotic efficacy.
We assessed the effects of AOLZ and OLZ as well as AHAL and HAL administration on gene expression in the absence of PCP and found very little overlap between the genes with expression altered by antipsychotic treatment compared to those normalized by OLZ + PCP, suggesting that antipsychotics have a limited effect on expression of these genes in the absence of glutamate dysfunction. There were 12 significant gene expression alterations in OLZ-administered animals versus those administered VEH (MEF2C, CTNNA2, NRGN, MIF, TYRO3, BRD8, STMN2, YWHAH, ATP1B2, PPIA, GRIA3, and DLG4), including an overrepresentation of genes implicated in glutamate receptor signaling, ERK5 signaling, and calcium signaling, suggesting possible mechanisms by which PCP effects on expression are reversed or prevented by OLZ coadministration. Interestingly, neurogranin (NRGN), which plays a role in long-term potentiation [20], has been implicated in schizophrenia in a clinical association study [21,22] and in an immunoreactivity study in postmortem human brain tissue [23].
IPA analysis identified one gene network connecting 17 genes normalized by OLZ. One gene of interest in this gene network was regulator of G protein signaling 4 (RGS4). RGS4 is a GTPase activator protein that has been shown to interact with opiate, adrenergic, muscarinic, and glutamate receptors, and its expression can influence synaptic function [24]. It has been previously implicated in the etiology of schizophrenia (for reviews, see [25,26,27]), and RGS4 genotypes at markers rs2661319 and rs2842030 have previously been shown to be associated with more severe baseline PANSS total scores and to predict responsiveness to antipsychotic treatment [28]. RSG4 was not significantly altered in OLZ or HAL compared to VEH controls, consistent with previous findings that expression of this gene is not altered by antipsychotic treatment in the absence of glutamate dysfunction [27]. However, RGS4 was one of the most significantly differentially expressed genes in the comparison between PCP- and PCP + OLZ-administered animals and may be of interest for investigation as a target for antipsychotic action. Also of interest within the network were SNAP-25 and SYN1, which have been shown to be decreased in postmortem brains of subjects with schizophrenia and bipolar disorder, respectively [29,30], and YWHAZ, which has been identified as a schizophrenia susceptibility gene [31].
DAVID Bioinformatics Database identified two transcription factors associated with the regulation of a significant number of OLZ normalized genes. These transcription factors were SOX5 and MEF2 (see online suppl. tables 5, 6), which have binding sites to possibly regulate 36 and 41 of the 50 OLZ-reversed genes, respectively. Interestingly, an SNP in the SOX5 gene has been previously associated with metabolic adverse events during treatment with antipsychotics in schizophrenia, further implicating this transcription factor in antipsychotic response.
One shortcoming of the present study was that gene expression was only investigated in one brain region. Future work will be necessary to determine whether the changes in gene expression observed in the current study were specific to the PFC or generalizable across regions. A second shortcoming of this study was that findings were not validated by qPCR or at the protein level. However, the use of two different gene expression platforms and the use of technical duplicates within each platform to find replicable genes strongly support the reliability of the present findings. These follow-up studies were not conducted as tissue from the primates is not available.
It was unclear whether OLZ treatment reversed or prevented gene expression changes induced by PCP. Because animals were treated with PCP alone for 2 weeks prior to 4 weeks of OLZ coadministration, we speculate that PCP-induced changes in gene expression were reversed rather than prevented. However, because the comparison PCP group was administered PCP for 6 weeks, it is not possible to determine which PCP effects that we observed would have occurred by 2 weeks. OLZ treatment alone resulted in an upregulation of transcription factor MEF2C, which again may have mediated the upregulated expression we observed after OLZ treatment. However, the majority of OLZ changes occurred only in the presence of dysregulation, such as in the PCP model. All but one probe set that was significantly differentially expressed between PCP- and VEH-administered animals was downregulated, which is inconsistent with previous microarray findings in rats which found approximately equal numbers of up- and downregulated genes in response to PCP administration [32]. There were six transcription factors downregulated on both arrays: GTF2I, TCEA1, RUNX1T1, ATF6B, E2F4, and MEF2C. MEF2C is a candidate gene for schizophrenia and known to regulate neurogenesis and the excitatory synapse number. This gene is located at 5q14.3, within one of the 108 schizophrenia-associated genetic loci identified by the Psychiatric Genomics Consortium [33]. It is possible that the downregulation of these transcription factors leads to a cascade of downregulation of gene expression in the PFC. In line with this is the observation of NMDA receptor subunit downregulation in the PFC of postmortem subjects with schizophrenia [34]. The current expression data give an understanding of drug effects of OLZ, PCP, and HAL that are otherwise difficult to obtain in the human brain and often reflect the mode of death and environmental consequences of illness. The present gene expression results are not contaminated by agonal factors and have intrinsic value for interpreting other published gene expression datasets on the human brain, since the current results are based on careful quality control of quadruplicate arrays for each sample.
Conclusions
This study is the first to examine genome-wide changes in brain gene expression influenced by PCP and the reversal of effects by antipsychotic treatment in primates. This study identified 146 genes altered in expression by PCP and found an overrepresentation of PCP-induced effects of genes implicated in cell death, neurological disorders, and schizophrenia. Canonical pathway analysis of PCP-altered genes revealed a significant overrepresentation of genes implicated in calcium signaling and synaptic long-term potentiation. Although there is no complete model of schizophrenia, which is a uniquely human disorder, the results of the present study support the use of PCP administration as a pharmacological model of glutamatergic dysfunction in schizophrenia, and we found that 34% of all genes downregulated by PCP administration are normalized by administration of OLZ. Furthermore, novel candidate genes and networks for future pharmacogenetic studies were identified.
Statement of Ethics
None.
Disclosure Statement
This study was funded by Eli Lilly and Company.
References
- 1.Carlsson A, et al. Neurotransmitter aberrations in schizophrenia: new perspectives and therapeutic implications. Life Sci. 1997;61:75–94. doi: 10.1016/s0024-3205(97)00228-2. [DOI] [PubMed] [Google Scholar]
- 2.Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148:1301–1308. doi: 10.1176/ajp.148.10.1301. [DOI] [PubMed] [Google Scholar]
- 3.Javitt DC. Glutamate and schizophrenia: phencyclidine, N-methyl-D-aspartate receptors, and dopamine-glutamate interactions. Int Rev Neurobiol. 2007;78:69–108. doi: 10.1016/S0074-7742(06)78003-5. [DOI] [PubMed] [Google Scholar]
- 4.Allen RM, Young SJ. Phencyclidine-induced psychosis. Am J Psychiatry. 1978;135:1081–1084. doi: 10.1176/ajp.135.9.1081. [DOI] [PubMed] [Google Scholar]
- 5.Olney JW, Farber NB. Glutamate receptor dysfunction and schizophrenia. Arch Gen Psychiatry. 1995;52:998–1007. doi: 10.1001/archpsyc.1995.03950240016004. [DOI] [PubMed] [Google Scholar]
- 6.Kristiansen LV, et al. NMDA receptors and schizophrenia. Curr Opin Pharmacol. 2007;7:48–55. doi: 10.1016/j.coph.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 7.Patil ST, et al. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized phase 2 clinical trial. Nat Med. 2007;13:1102–1107. doi: 10.1038/nm1632. [DOI] [PubMed] [Google Scholar]
- 8.Moghaddam B, Adams BW. Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science. 1998;281:1349–1352. doi: 10.1126/science.281.5381.1349. [DOI] [PubMed] [Google Scholar]
- 9.Linn GS, et al. Behavioral effects of chronic phencyclidine in monkeys. Neuroreport. 1999;10:2789–2793. doi: 10.1097/00001756-199909090-00017. [DOI] [PubMed] [Google Scholar]
- 10.Callaghan JT, et al. Olanzapine. Pharmacokinetic and pharmacodynamic profile. Clin Pharmacokinet. 1999;37:177–193. doi: 10.2165/00003088-199937030-00001. [DOI] [PubMed] [Google Scholar]
- 11.Benes FM, et al. The expression of proapoptosis genes is increased in bipolar disorder, but not in schizophrenia. Mol Psychiatry. 2006;11:241–251. doi: 10.1038/sj.mp.4001758. [DOI] [PubMed] [Google Scholar]
- 12.Olney JW, Labruyere J, Price MT. Pathological changes induced in cerebrocortical neurons by phencyclidine and related drugs. Science. 1989;244:1360–1362. doi: 10.1126/science.2660263. [DOI] [PubMed] [Google Scholar]
- 13.Yashiro K, Philpot BD. Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology. 2008;55:1081–1094. doi: 10.1016/j.neuropharm.2008.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pratt JA, et al. Modelling prefrontal cortex deficits in schizophrenia: implications for treatment. Br J Pharmacol. 2008;153(suppl 1):S465–S470. doi: 10.1038/bjp.2008.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thompson PM, et al. Time-lapse mapping of cortical changes in schizophrenia with different treatments. Cereb Cortex. 2009;19:1107–1123. doi: 10.1093/cercor/bhn152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lieberman JA, Perkins DO, Jarskog LF. Neuroprotection: a therapeutic strategy to prevent deterioration associated with schizophrenia. CNS Spectr. 2007;12(3 suppl 4):1–13. doi: 10.1017/s1092852900025906. quiz 14. [DOI] [PubMed] [Google Scholar]
- 17.Yulug B, et al. Olanzapine attenuates brain damage after focal cerebral ischemia in vivo. Brain Res Bull. 2006;71:296–300. doi: 10.1016/j.brainresbull.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 18.Bonelli RM, Wenning GK. Pharmacological management of Huntington's disease: an evidence-based review. Curr Pharm Des. 2006;12:2701–2720. doi: 10.2174/138161206777698693. [DOI] [PubMed] [Google Scholar]
- 19.Lu XH, Bradley RJ, Dwyer DS. Olanzapine produces trophic effects in vitro and stimulates phosphorylation of Akt/PKB, ERK1/2, and the mitogen-activated protein kinase p38. Brain Res. 2004;1011:58–68. doi: 10.1016/j.brainres.2004.03.018. [DOI] [PubMed] [Google Scholar]
- 20.Zhong L, et al. Neurogranin enhances synaptic strength through its interaction with calmodulin. EMBO J. 2009;28:3027–3039. doi: 10.1038/emboj.2009.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stefansson H, et al. Common variants conferring risk of schizophrenia. Nature. 2009;460:744–747. doi: 10.1038/nature08186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruano D, et al. Association of the gene encoding neurogranin with schizophrenia in males. J Psychiatr Res. 2008;42:125–133. doi: 10.1016/j.jpsychires.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 23.Broadbelt K, Ramprasaud A, Jones LB. Evidence of altered neurogranin immunoreactivity in areas 9 and 32 of schizophrenic prefrontal cortex. Schizophr Res. 2006;87:6–14. doi: 10.1016/j.schres.2006.04.028. [DOI] [PubMed] [Google Scholar]
- 24.Levitt P, et al. Making the case for a candidate vulnerability gene in schizophrenia: convergent evidence for regulator of G-protein signaling 4 (RGS4) Biol Psychiatry. 2006;60:534–537. doi: 10.1016/j.biopsych.2006.04.028. [DOI] [PubMed] [Google Scholar]
- 25.Shi J, Gershon ES, Liu C. Genetic associations with schizophrenia: meta-analyses of 12 candidate genes. Schizophr Res. 2008;104:96–107. doi: 10.1016/j.schres.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Allen NC, et al. Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat Genet. 2008;40:827–834. doi: 10.1038/ng.171. [DOI] [PubMed] [Google Scholar]
- 27.Mirnics K, et al. Disease-specific changes in regulator of G-protein signaling 4 (RGS4) expression in schizophrenia. Mol Psychiatry. 2001;6:293–301. doi: 10.1038/sj.mp.4000866. [DOI] [PubMed] [Google Scholar]
- 28.Campbell DB, et al. Ethnic stratification of the association of RGS4 variants with antipsychotic treatment response in schizophrenia. Biol Psychiatry. 2008;63:32–41. doi: 10.1016/j.biopsych.2007.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thompson PM, Egbufoama S, Vawter MP. SNAP-25 reduction in the hippocampus of patients with schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:411–417. doi: 10.1016/S0278-5846(03)00027-7. [DOI] [PubMed] [Google Scholar]
- 30.Vawter MP, et al. Reduction of synapsin in the hippocampus of patients with bipolar disorder and schizophrenia. Mol Psychiatry. 2002;7:571–578. doi: 10.1038/sj.mp.4001158. [DOI] [PubMed] [Google Scholar]
- 31.Jia Y, Yu X, Zhang B, Yuan Y, Xu Q, Shen Y, Shen Y. An association study between polymorphisms in three genes of 14-3-3 (tyrosine 3-monooxygenase/tryptophan 5-mono- oxygenase activation protein) family and paranoid schizophrenia in northern Chinese population. Eur Psychiatry. 2004;19:377–379. doi: 10.1016/j.eurpsy.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 32.Kaiser S, et al. Phencyclidine-induced changes in rat cortical gene expression identified by microarray analysis: implications for schizophrenia. Neurobiol Dis. 2004;16:220–235. doi: 10.1016/j.nbd.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 33.Schizophrenia Working Group of the Psychiatric Genomics Consortium Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Errico F, et al. Decreased levels of D-aspartate and NMDA in the prefrontal cortex and striatum of patients with schizophrenia. J Psychiatr Res. 2013;47:1432–1437. doi: 10.1016/j.jpsychires.2013.06.013. [DOI] [PubMed] [Google Scholar]