

Abstract
Background
Protein functional diversity at the post-transcriptional level is regulated through spliceosome mediated pre-mRNA alternative splicing (AS) events and that has been widely demonstrated to be a key player in regulating the functional diversity in plants. Identification and analysis of AS genes in cereal crop plants are critical for crop improvement and understanding regulatory mechanisms.
Results
We carried out the comparative analyses of the functional landscapes of the AS using the consensus assembly of expressed sequence tags and available mRNA sequences in four cereal plants. We identified a total of 8,734 in Oryza sativa subspecies (ssp) japonica, 2,657 in O. sativa ssp indica, 3,971 in Sorghum bicolor, and 10,687 in Zea mays AS genes. Among the identified AS events, intron retention remains to be the dominant type accounting for 23.5 % in S. bicolor, and up to 55.8 % in O. sativa ssp indica. We identified a total of 887 AS genes that were conserved among Z. mays, S. bicolor, and O. sativa ssp japonica; and 248 AS genes were found to be conserved among all four studied species or ssp. Furthermore, we identified 53 AS genes conserved with Brachypodium distachyon. Gene Ontology classification of AS genes revealed functional assignment of these genes in many biological processes with diverse molecular functions.
Conclusions
AS is common in cereal plants. The AS genes identified in four cereal crops in this work provide the foundation for further studying the roles of AS in regulation of cereal plant growth and development. The data can be accessed at Plant Alternative Splicing Database (http://proteomics.ysu.edu/altsplice/).
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-1914-5) contains supplementary material, which is available to authorized users.
Keywords: Alternative splicing, Cereal crops, Expressed sequence tags, mRNA
Background
Spliceosome mediated post-transcriptional modifications are the biggest challenges in understanding and predicting the degree of certainty and complexity of the proteome diversity [1, 2]. One of the most important mechanisms that contribute to the diversity in the protein isoforms is alternative splicing (AS), thus modulating the protein function as a consequence of the linking of the functional units (exons and introns) in a ubiquitous manner [3]. In addition, to the observed alternative splicing sub-types such as exon skipping (ES), alternative donor (AltD) or acceptor (AltA) site, and intron retention (IR), various complex types can be formed by combination of basic events [4, 5]. Apart from the four basic events, alternative transcripts may arise as a consequence of the alternative transcription initiation, alternative transcription termination, and alternative polyadenylation [2]. AS isoforms might encode distinct functional proteins, or might be nonfunctional, which harbor a premature termination codon. These nonfunctional isoforms generated through the process called “regulated unproductive splicing and translation” are degraded by a process known as nonsense-mediated decay [6].
Previous reports estimated around 90 % of human genes containing multiple exons are alternatively spliced [7, 8]. In line with the observed reports in humans, alternative splicing has been shown to be a major player in generation of the plant proteome diversity with 60 % of Arabidopsis thaliana multi-exon genes undergoing alternative splicing [9]. Genome-wide identification and physiological implications of AS have been reported in a number of model and non-model plant species including A. thaliana [10–13], Oryza sativa [14], Nelumbo nucifera (sacred lotus) [15], Vitis vinifera [16], Brachypodium distachyon [5, 17]. AS transcripts are generally generated through three pathways: (1) IR in the mature mRNA; (2) alternative exon usage (AEU), resulting in ES; and (3) the use of cryptic splice sites that may elongate or shorten an exon that generates AltD or AltA site or both [14, 17]. Approximately 60–75 % of AS events occur within the protein coding regions of mRNAs, resulting changes in binding properties, intracellular localization, protein stability, enzymatic, and signaling activities [18]. In plants, IR has been shown to be the most dominant form with reports suggesting the proportions of intron containing genes undergoing AS in plants ranged from ~30 % to >60 % depending the depth of available transcriptome data [4, 5]. On contrast, recent reports suggest the down-regulation of the IR events and up-regulation of the alternative donor/acceptor site (AltDA) and ES under heat stress in model Physcomitrella patens [19]. With the advent of the Next Generation Sequencing (NGS) based approaches, fine scale physiological implications revealed alternative splicing as the prominent mechanism, which regulates the microRNA- mediated gene regulation by increasing the complexity of the alternative mRNA processing in Arabidopsis [20]. Complex networks of regulation of gene expression and variation in AS has played a major role in the adaptation of plants to their corresponding environment and additionally in coping with environmental stresses [13].
Rice (O. sativa ssp japonica and indica), maize (Zea mays), and sorghum (Sorghum bicolor) are important cereal crops as major sources of food in many countries. Previously several approaches have widely demonstrated the identification of the quantitative trait loci, genes and proteins linked to the functional grain content in these species [21]. However, a major portion of the gene functional diversity is controlled by a spliceosomal regulated AS. AS has been shown to be a critical regulator in grass clade, demonstrating several of the genes involved in flowering and abiotic stress depicting alternative splicing [4, 17, 22]. Identifying alternative splicing genes in these cereal plants is the first step toward understanding the functions and regulations of these genes in plant development and abiotic or biotic stress resistance. Previously, using the homology based mapping approach and expressed sequence tags (ESTs) representing the functional transcripts, we identified a total of 941 AS genes in B. distachyon, a model temperate grass [5, 17]. Previous and recent reports on the identification and prevalence of the alternative splicing events in O. sativa [4, 23], S. bicolor [24], and Z. mays [25] have shown the functional diversity changes through EST/RNA-seq approaches. Previous report by Ner-Gaon et al. suggested a 3.7-fold difference in AS rates between O. sativa and S. bicolor using EST pairs gapped alignment [26]. The lack of the identification of the comparative AS events in cereal plants and realizing the importance of these functional foods in climate changes, we attempted to carry out the large scale analysis using the so far currently ESTs and mRNA based information in cereal plants to identify species specific and conserved AS events across cereal plants. In this work, we compared the AS event landscape and the AS gene functional diversity in cereal plants, which includes O. sativa ssp japonica and indica, S. bicolor and Z. mays, with a much deeper coverage of the identified AS events and also comparatively analyzed these AS genes with AS genes identified from B. distachyon to reveal conserved patterns of the AS across the grass species. Identified AS events will allow for the experimental characterization of the AS genes involved in important physiological processes. Investigation of the genome-wide conserved AS events across different species will shed light on the understanding of the evolution of the functional diversity in cereal plant for crop improvement.
Methods
Sequence datasets and sequence assembly
To identify the putative functional transcriptional changes across the Panicoideae lineage, we systematically queried and downloaded expressed sequence tags (ESTs) and mRNA sequences of O. sativa ssp japonica and indica, S. bicolor, and Z. mays from the dbEST and nucleotide repository of National Center for Biotechnology Information (NCBI; www.ncbi.nlm.nih.gov). Prior to aligning the ESTs/mRNAs to the corresponding genomic sequence, we applied stringent cleaning procedure using the strategy outlined below: 1) ESTs and mRNA sequences were subsequently cleaned using EMBOSS “trim” tool for trimming of the polyA or polyT ends; 2) Cleaned and trimmed ESTs and mRNA sequences were blasted using the BLASTN against UniVec and E. coli database for removal of vector and E. coli contaminants; 3) BLASTN searches against the plant repeat database which was built with TIGR gramineae repeat data and species specific repeat data including sorghum, maize, and rice available from ftp://ftp.plantbiology.msu.edu/pub/data/TIGR_Plant_Repeats/. Following stringent cleaning procedure, we assembled rice and sorghum cleaned EST and mRNA sequences using CAP3 with the following parameters: −p 95 –o 50 –g 3 –y 50 –t 1000 [27]. In case of the maize data, owing to the large number of available ESTs for this species, which is difficult to assemble, we followed an alternative way of assembling those ESTs. We first mapped ESTs and mRNA sequences to each individual chromosome of the maize genome using GMAP with default settings [28], and then chromosome specific-mapped ESTs and mRNAs were assembled individually using CAP3 with the parameters as mentioned above. The unmapped data and all assembled data from each individual assembly were combined and then re-assembled using CAP3 to generate a final consensus assembly for the further identification of the AS events. The raw data and assembled data for each organism were summarized in Table 1. For the prediction of the AS events, genome sequences, predicted protein coding DNA sequences (CDS), and related GFF data of O. sativa ssp japonica, Z. mays, and S. bicolor were downloaded from Phytozome database (http://www.phytozome.net/) [29–32]. The genome sequences and CDS data of O. sativa ssp indica (strain 93–11) were downloaded from BGI database (http://rise2.genomics.org.cn/page/rice/index.jsp) [33].
Table 1.
Summary of raw sequence data and assembled data in each organism
| Species | ESTs | mRNAs | Total Sequences | Cleaned Sequences | Total PUTs | Average Length (bp) |
|---|---|---|---|---|---|---|
| O. sativa ssp japonica | 987327 | 82451 | 1069868 | 1053842 | 163778 | 783 |
| O. sativa ssp indica | 207012 | 11953 | 219065 | 212768 | 102424 | 751 |
| S. bicolor | 209835 | 33248 | 243083 | 241690 | 60189 | 1002 |
| Z. mays | 2019524 | 91990 | 2111514 | 1822653 | 488243 | 466 |
PUTs putative unique transcripts
Putative unique transcripts to genome mapping, identification and functional annotation of AS isoforms
In the present study, taking into the account the genome duplication events in Z. mays and S. bicolor, accurate prediction of the alternative splicing events is a major concern over the decades. In our study, calling and predicting alternative splicing events is taken into account by mapping of EST and mRNA assemblies, i.e. putative unique transcripts (hereafter simply referred them as PUTs), to the corresponding genomic sequences were carried out using in-house developed algorithm, ASFinder (http://proteomics.ysu.edu/tools/ASFinder.html/) [34], which uses SIM4 program [35] to map PUTs to the corresponding genome and then subsequently identifies those PUTs that are mapped to the same genomic location but have variable exon-intron boundaries as AS isoforms. To avoid the call of the spurious alternative splicing events, we applied a threshold of minimum of 95 % identity of aligned PUT with a genomic sequence, a minimum of 80 bp aligned length, and >75 % of a PUT sequence aligned to the genome [17]. Application of the above identity percentage and the aligned length removes the chance of the false positive AS events calling as a result of genome duplication events. The output file (AS.gtf) of ASFinder was then subsequently submitted to AStalavista server (http://genome.crg.es/astalavista/) for AS event analysis [36]. The percentage of alternative splicing genes was estimated using the genome predicted gene models having alternative splicing PUT isoforms among total genes models having at least one PUT, the results were presented in Table 2.
Table 2.
Percentage of alternative splicing genes
| Total mapped PUTs (%) | PUT match to gene model | Total unique genes | AS genes | AS (%) | |
|---|---|---|---|---|---|
| O. sativa ssp japonica | 104447 (63.8) | 71830 | 26191 | 7883 | 30.1 |
| O. sativa ssp indica | 47843 (46.7) | 36467 | 17402 | 2414 | 13.9 |
| S. bicolor | 50224 (83.4) | 38654 | 26540 | 3580 | 13.5 |
| Z. mays | 207332 (42.5) | 119418 | 28698 | 9689 | 33.8 |
AS Alternative splicing
We further queried the coding potential and corresponding coding frame of each PUT using the ORFPredictor [37], and to assess the full–length transcript coverage using TargetIdentifier [38] as previously described. Functional classification was assigned to the PUTs by performing BLASTX searches with an E-value threshold of 1E-5 against UniProtKB/Swiss-Prot. Predicted protein sequences from ORFPredictor were further annotated using rpsBLAST against the PFAM database (http://pfam.xfam.org/). Gene Ontologies (GOs) were assigned on the basis of the functional homology obtained by the BLASTX searching algorithm against the UniProtKB/Swiss-Prot. The GO categories were further analyzed using GO SlimViewer using plant specific GO terms [39]. To assess the functional coverage of the assembled PUTs, we further compared PUTs against the predicted gene primary transcripts using BLASTN with a cut off E-value of 1E-10, ≥ 95 % identity and minimum aligned length of 80 bp.
Conserved alternatively spliced genes in cereal plants and visualization of AS
For the identification of the potentially conserved AS genes among O. sativa ssp japonica and indica, Z. mays and S. bicolor, reciprocal BLASTP (cutoff E-value 1E-10) were done using the longest (or longer) ORF of the AS PUT isoforms for classifying the conserved AS pairs between species or sub-species. Venn graphical visualization for conserved AS pairs were obtained using R programming language (http://www.r-project.org/). Visualization of the alternative splicing events with genome tracks is critically important from two points of views: (1) To have a graphic look at the corresponding genomic coordinate and associated genic functional changes; and (2) To extract the corresponding spliced region of interest for functional primer designing of putative AS events. Keeping in view the above points, AS events identified in this study along with the integrated genomic tracks are available from Plant Alternative Splicing Database (http://proteomics.ysu.edu/altsplice/) [15, 17]. The specific pages associated with the cereal plants offer several end-users functionalities such as querying using the PUT ID, gene ID, keywords in functional annotation, PFAM, or AS event types as “query fields”. Additionally, the identified AS events can be visualized and compared with predicted gene models using GBrowse for comparative assessment. Nevertheless, we also deployed BLASTN functionality to search for the PUTs and AS isoforms. The data analyzed along with the GO and PFAM annotations in the present research are publicly available at: http://proteomics.ysu.edu/publication/data/.
Results and discussion
EST assembly and annotation
Optimization of the assembly parameters and mapping functionally annotated PUTs is a key parameter to provide a robust identification and classification of the AS events. Table 1 represents the assembly information, including the final cleaned reads for the assembly, mRNA count for each species, assembled consensus sequence and average length of assembled consensus. In the present research, we assembled and generated consensus PUTs accounting for a total of 163,778 PUTs in O. sativa ssp japonica, 102,424 PUTs in O. sativa ssp indica, 60,189 PUTs in S. bicolor, and 488,243 PUTs in Z. mays. The average length (N50) of assembled PUTs was 783 bp in O. sativa ssp japonica, 751 bp in O. sativa ssp indica, 1,002 bp in S. bicolor, and 466 bp in Z. mays. To check for the coverage of the assembled functional transcriptome, we further checked for the functional assignments and all the assembled PUTs were structurally and functionally annotated including putative open reading frame (ORF) prediction, coding region full-length prediction, a putative function and PFAM prediction, which ensures the reliability of the assembly strategies in case of large complex ploidy genomes underwent whole genome duplication events. PUTs were mapped to their corresponding genomes and predicted gene models were also visualized using GBrowse.
Gapped alignments of PUTs to genome, detection and classification of alternative splicing events
Following the sequence assembly, resulting unique PUTs were mapped onto their corresponding genomic sequences using gapped alignments as implemented in SIM4 method that was integrated as part of ASFinder [34]. The numbers of mapped PUTs and matched gene models, as well as the number of the observed AS genes are presented in Table 2. We observed that a relatively larger proportion of PUTs in S. bicolor (83.4 %) and O. sativa ssp japonica (63.8 %) aligned to their genomes as compared to the other cereal plants. We identified a total of 8,734 in Oryza sativa subspecies (ssp) japonica, 2,657 in O. sativa ssp indica, 3,971 in Sorghum bicolor, and 10,687 in Zea mays AS genes (Table 3). The percentage of AS genes was estimated based on the proportion of predicted gene models having AS PUT isoforms over the total gene models having an EST (PUT) evidence (Table 2). The percentages of AS genes vary in different cereal plants, up to 30.1 % in O. sativa ssp japonica and 33.8 % in Z. mays, and relatively low in O. sativa ssp indica (13.9 %) and in S. bicolor (13.5 %). The difference in the mapping rate and AS rate might be due to the difference in the number of ESTs available for respective species. Previous reports on AS in B. distachyon clearly illustrates the fact that availability of the more ESTs/mRNAs reflects the prediction of the AS landscape [5, 17].
Table 3.
Alternative splicing events in different cereal species
| Species | IR(%) | AltD(%) | AltA (%) | ES (%) | Complex event (%) | Total events | Total AS genes |
|---|---|---|---|---|---|---|---|
| O. sativa ssp japonica | 8288 (42.0) | 1245 (6.3) | 1950 (9.9) | 762 (3.9) | 7447 (37.8) | 19692 | 8734 |
| O. sativa ssp indica | 2193 (55.8) | 332 (8.5) | 576 (14.7) | 161 (4.1) | 665 (16.9) | 3927 | 2657 |
| S. bicolor | 4448 (23.5) | 1072 (5.7) | 1230 (6.5) | 507 (2.6) | 11681 (61.7) | 18938 | 3971 |
| Z. mays | 11048 (40.4) | 2080 (7.6) | 3314 (11.4) | 1568 (5.7) | 5576 (20.4) | 23386 | 10687 |
IR Intron Retention, AltD Alternative donor, AltA Alternative acceptor, ES exon skipping
Recent reports using the RNA-seq technology revealed that AS is common in plants—around 61 % of multi-exonic genes in A. thaliana are alternatively spliced under normal growth conditions [12], and ~40 % of intron containing genes that undergo AS in maize [25]. Classification of the AS events observed in the cereal plants are listed in Table 3 showing the prevalence of the IR as the major splicing type showing frequency as high as 55.8 % in O. sativa ssp indica and as low as 23.5 % in S. bicolor (Table 3). The high frequency of the IR in the mature mRNA is perfectly in line with the previously observed frequencies of IR (30–50 %) in AS landscape in A. thaliana and O. sativa [14]. It is worthwhile to mention that plant spliceosomal machinery supports the intron definition model, thus identifies the introns for pre-mRNA splicing as oppose to the abundant exon-spliceosome model observed in case of mammals. Previous arguments have clearly justified the cause and benefits of retaining the introns as potential cytoplasmic translatable transcripts [26] or as mediators of increasing the gene expression, a process widely described as intron-mediated enhancement (IME) of gene expression [40]. The abundance of IR as a major AS event is consistent with previous reports including Medicago tuncatula (39 %), Populus trichocarpa (34 %), A. thaliana (56 %), O. sativa (54 %), Chlamydomonas reinhardtii (50 %), Z. mays (58–62 %) and B. distachyon (55.5 %) [14, 17, 25, 41, 42]. In contrast, recently IR has been found remarkably repressed under elevated temperature in P. patens [19].
Alternative acceptor (AltA) and donor (AltD) represent the second most abundant and classified functional class of observed AS events with AltA showing a relatively higher frequency as compared to AltD (Table 3). Although ES events have been described as the rarest events in plants, which are in line with the observed results in this study, recently they have been proposed as the candidates of the transgene regulation using the conditional splicing [43]. We noted that 61.7 % events are complex events in sorghum, which have more than one basic event in compared paired PUTs. This is clearly related to the relative longer lengths of the PUTs in sorghum assembly. Recent reports suggest the differential up-regulation of the alternative donor/acceptor site (AltDA) and ES elucidating the importance of these events as indicators of early heat stress [19].
Our data in this work clearly showed that the number of AS genes and the percentage of genes with AS are different in different crops (Tables 2 and 3). However, this observation only reflects the current state in these plants based on the available data. Our previous analysis on AS in B. distachyon clearly demonstrated that more AS genes were identified with more available ESTs/mRNA data [5, 17]. This is also consistent with the finding of increasing frequency of occurrence of AS in Arabidopsis with time—a reflection of an accumulation of available transcriptome data, for example, only 1.2 % of the genes in Arabidopsis were reported undergo AS in 2003 and now it was estimated over 60 % of intron-containing genes undergo AS [13].
Features of exons and introns in protein coding genes: indicators of gene evolution
Understanding the patterns of gene evolution and identifying signatures of convergent and divergent evolution is of paramount importance, especially when we are addressing the genome complexity in terms of gene evolution. Exon-intron framework properties such as length distribution and GC content evolution have been previously used to demonstrate the gene evolution [44]. Additionally, longer introns as compared to short introns have been shown to play an important role in the gene expression [40, 45]. However, reports by Yang [46] demonstrate the negative correlation of the long introns with the levels of the expression in A. thaliana and O. sativa. Realizing the importance of the features of exon-intron in evolution and physiological responses, we extracted and plotted the length distribution of all internal exons and introns from each plant and the results are summarized (Table 4; Fig. 1; Fig. 2). Interestingly, we observed that the average internal exon lengths in O. sativa ssp indica and Z. mays are almost similar, and are relatively much shorter than the internal exon lengths in O. sativa ssp japonica and S. bicolor. On the other hand, Z. mays had the longer intron length (554 bp) and showed a wide variation in intron lengths as compared to the observed range of intron lengths (422–440 bp) in other three cereal plants in this study. We further analyzed deeply the exon size and intron size distribution frequencies demonstrating that Z. mays and O. sativa ssp indica had a relatively much higher proportion of internal exons of a smaller size (<120 bp) (Fig. 1). The observed frequency of internal exon lengths below 300 bp was 0.93 in Z. mays, 0.95 in O. sativa ssp indica, 0.89 in S. bicolor, and 0.90 in O. sativa ssp japonica. S. bicolor and O. sativa ssp japonica displayed more exons of relatively large size, whereas Z. mays displayed a higher number of long introns (Fig. 2). Prevalence of the introns richness and specifically long introns have been previously been shown to be widely associated with the increased expression of Adh1, Sh1, Bz1, Hsp82, actin, and GapA1 genes in Z. mays [47–51] and salT, Act1, and tpi genes in rice [52, 53]. Additionally, a relative higher proportion of introns having a shorter length were observed in S. bicolor. We also observed ~2 % introns in maize and a small number of introns (<0.5 %) in other plants having a size >10 kb. However, taking into account the possible errors in PUT and genome assembly, these long introns were not included in the calculation of the average intron size. It is worthwhile to mention that the average internal exon size (180 bp) and intron size (440 bp) in O. sativa ssp japonica obtained in this work were close to the exon (193 bp) and intron (433 bp) size obtained previously in O. sativa, which presents the robustness of the implemented approach [14].
Table 4.
Exon and intron size in cereal plants
| Exon | Intron | |||||
|---|---|---|---|---|---|---|
| Sample size | Average size (bp) | SD | Sample Size | Average size (bp) | SD | |
| O. sativa ssp japonica | 127627 | 180 | 261 | 180575 | 440 | 695 |
| O. sativa ssp indica | 52330 | 133 | 113 | 79735 | 434 | 703 |
| S. bicolor | 106753 | 179 | 222 | 144860 | 422 | 747 |
| Z. mays | 137020 | 142 | 133 | 209139 | 554 | 1057 |
SD Standard deviation
Fig. 1.
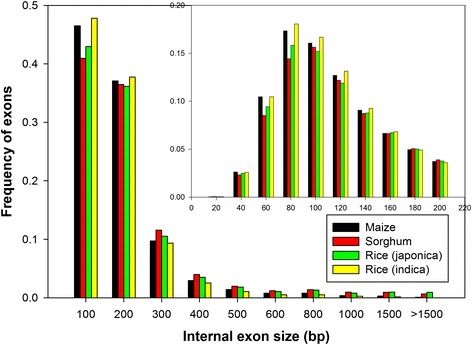
Distribution of internal exon size: The x-axis indicates the size of internal exons. Bin sizes are right inclusive (e.g., bin 100 comprises sequences of lengths 1–100 bp). The y-axis indicates the frequency of internal exons. The inset shows a detailed distribution of small internal exons
Fig. 2.
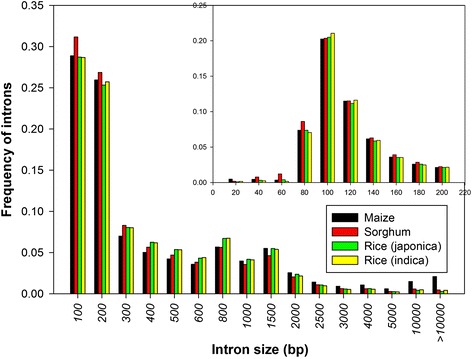
Distribution of intron size: The x-axis indicates the size of introns. Bin sizes are right inclusive (e.g., bin 100 comprises sequences of lengths 1 –100 bp). The y-axis indicates the frequency of introns. The inset shows a detailed distribution of small introns
Functional classification of AS genes
AS and gene regulation can be observed at almost all levels of biological interactions [54]. The AS transcripts identified in the present study were functionally annotated for the Gene Ontologies (GOs) and for putative protein domains association by performing a BLASTX search of all PUTs against UniProt/Swiss-Prot database. The ORFs of PUTs were identified using ORFPredictor webserver [37]. The protein families of the AS genes, using the longest ORF of each AS gene, were predicted using rpsBLAST searching PFAM database. Among predicted ORFs of these AS genes, 6,900 in Z. mays, 4,939 in O. sativa ssp japonica, 1,362 in O. sativa ssp indica, and 2,890 in S. bicolor were classified with a putative protein family (Table 5, Additional file 1: Table S1). We further classified AS gene functional products into 2,030 unique protein families in Z. mays, 1,708 unique protein families in O. sativa ssp japonica, 757 unique protein families in O. sativa ssp indica, and 1,194 unique protein families in S. bicolor. Among the protein functions, encoded by these AS genes, widely includes protein kinase domain, RNA recognition motif, protein tyrosine kinase, ring finger domain, cytochrome P450, Myb-like DNA-binding domain, WRKY DNA-binding domain, Thioredoxin and protein phosphatase 2C (Table 5). A complete list of all the protein families encoded by AS genes is shown in Additional file 1: Table S1. Our analysis demonstrated that AS genes in cereal plants encode diverse protein families that play important roles in various biological processes. A classical example can be WRKY- DNA binding domains, which represents the largest and functionally diverse transcription factors in plants playing a major role in developmental and physiological processes. Previous studies have widely demonstrated the presence of the alternative ORF in the WRKY genes [55, 56]. Yang et al. [57] and Feng et al. [58] have clearly highlighted the role of the alternative splicing and WRKY in plant immunity. Previous functional studies have shown the presence of the splicing of the R-type intron and V-type intron in O. sativa WRKY genes and functionally correlated them to plant immunity [59]. MYB-domains play an important role in plant defense mechanism and are transcriptionally regulated by alterative splicing in A. thaliana and O. sativa and encode MYB- or MYB-related proteins [60]. Alternative splicing of MYB related genes MYR1 and MYR2 have clearly demonstrated the change in protein dimerization and folding as a consequence of alternative splicing thus affecting the transcriptional sensitivity in light mediated responses [61].
Table 5.
Protein family classification of alternative genes in cereal plants
| PFAM | Domain | Z. mays | O. sativa ssp japnoica | O. sativa ssp indica | S. bicolor | Putative Functions |
|---|---|---|---|---|---|---|
| pfam00069 | Pkinase | 205 | 228 | 55 | 74 | Protein kinase domain |
| pfam00076 | RRM_1 | 112 | 61 | 32 | 43 | RNA recognition motif |
| pfam07714 | Pkinase_Tyr | 88 | 79 | 12 | 25 | Protein tyrosine kinase |
| pfam13639 | zf-RING_2 | 53 | 28 | 8 | 15 | Ring finger domain |
| pfam00067 | p450 | 45 | 56 | 4 | 37 | Cytochrome P450 |
| pfam00481 | PP2C | 45 | 25 | 11 | 11 | Protein phosphatase 2C |
| pfam00249 | Myb_DNA-binding | 44 | 14 | 7 | 22 | Myb-like DNA-binding domain |
| pfam00179 | UQ_con | 43 | 17 | 10 | 7 | Ubiquitin-conjugating enzyme |
| pfam00010 | HLH | 41 | 5 | 1 | 7 | Helix-loop-helix DNA-binding domain |
| pfam00071 | Ras | 38 | 20 | 11 | 7 | Ras family |
| pfam00141 | peroxidase | 37 | 30 | 11 | 31 | Peroxidase |
| pfam00153 | Mito_carr | 35 | 24 | 7 | 14 | Mitochondrial carrier protein |
| pfam01559 | Zein | 35 | 0 | 0 | 0 | Zein seed storage protein |
| pfam01490 | Aa_trans | 33 | 12 | 1 | 7 | Transmembrane amino acid transporter protein |
| pfam02365 | NAM | 33 | 33 | 8 | 14 | No apical meristem (NAM) protein |
| pfam00125 | Histone | 31 | 9 | 3 | 5 | Core histone H2A/H2B/H3/H4 |
| pfam01370 | Epimerase | 31 | 26 | 8 | 22 | NAD dependent epimerase/dehydratase family |
| pfam00083 | Sugar_tr | 30 | 22 | 7 | 10 | Sugar (and other) transporter |
| pfam00847 | AP2 | 30 | 9 | 3 | 9 | AP2 domain |
| pfam00106 | adh_short | 29 | 25 | 10 | 15 | short chain dehydrogenase |
| pfam00657 | Lipase_GDSL | 29 | 5 | 1 | 16 | GDSL-like Lipase/Acylhydrolase |
| pfam00085 | Thioredoxin | 28 | 14 | 6 | 11 | Thioredoxin |
| pfam00226 | DnaJ | 28 | 18 | 9 | 9 | DnaJ domain |
| pfam03151 | TPT | 27 | 9 | 2 | 6 | Triose-phosphate Transporter family |
| pfam00004 | AAA | 26 | 18 | 6 | 14 | ATPase family associated with various cellular |
| pfam00270 | DEAD | 24 | 21 | 5 | 8 | DEAD/DEAH box helicase |
| pfam00504 | Chloroa_b-bind | 24 | 19 | 11 | 20 | Chlorophyll A-B binding protein |
| pfam02309 | AUX_IAA | 24 | 13 | 5 | 5 | AUX/IAA family |
| pfam00149 | Metallophos | 23 | 9 | 3 | 13 | Calcineurin-like phosphoesterase |
| pfam00134 | Cyclin_N | 22 | 9 | 2 | 4 | Cyclin |
| pfam00450 | Peptidase_S10 | 21 | 18 | 6 | 18 | Serine carboxypeptidase |
| pfam03106 | WRKY | 21 | 22 | 7 | 7 | WRKY DNA -binding domain |
| pfam13041 | PPR_2 | 21 | 30 | 1 | 12 | PPR repeat family |
| Total | 6900 | 4939 | 1362 | 2890 |
Note: a complete list is shown in Additional file 1: Table S1
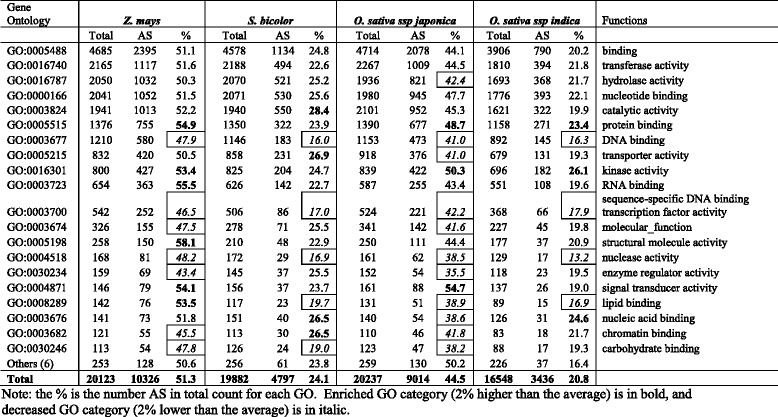
GO analysis according to biological and molecular function revealed a wide visibility in all the major biological and molecular functions (Table 6; Table 7). Interestingly, even the data we collected are from pooled data in the public domain, i.e., not from a strictly controlled experiment, our GO analysis revealed that relative to the average of AS percentage, a higher percentage of genes involved in response to abiotic stimulus, photosynthesis, carbohydrate metabolic process, and cell death are involved in AS in cereal plants. In contrast, the genes involved in multicellular organismal development and reproduction had a lower percentage of AS (Table 6). GO molecular function analysis revealed that genes encoding proteins having DNA binding, sequence-specific DNA binding transcription factor activity, nuclease activity had a lower percentage of AS, and the gene coding proteins for protein binding and having kinase activity had a higher percentage of AS in the majority of plants (Table 7). Our observed results are consistent with literature reviewed recently by Reddy et al. [4] and Staiger and Brown [22] that AS is involved in most plant processes and plays regulated roles in plant development and stress responses.
Table 6.
Classification of biological processes based on Gene Ontology (GO)
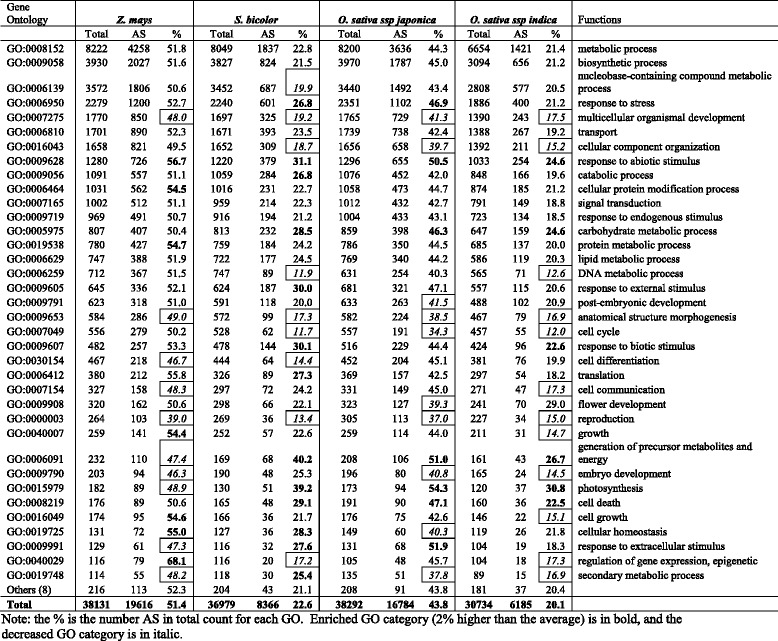
Table 7.
Classification of molecular functions based on Gene Ontology (GO)
Conserved alternatively spliced genes
Classification of the conserved alternative splicing events provides a framework for understanding the evolution of the functional genes and their genic-regulation at the transcriptional level, which may initiate the cross-talks between the evolution of the genes under AS and between the transcriptional environment and the ecological adaptation. For the identification of the conserved AS pairs, longest ORFs of AS genes in each studied species were compared using the BLASTP (cutoff E-value 1E-10) to identify the best-reciprocal top hit as the conserved pairs. In total, we identified 1558 AS genes conserved between O. sativa ssp japonica and indica, 3,246 AS genes conserved between O. sativa ssp japonica and Z. mays, and 1,967 AS genes between S. bicolor and Z. mays (Additional file 2: Table S3). A total of 887 AS genes are conserved among Z. mays, S. bicolor, and O. sativa ssp japonica. More importantly, we identified 248 AS genes conserved among all four plants (Fig. 3). Furthermore, using the same approach, we identified a total of 53 AS genes conserved with B. distachyon belonging to BEP-clade of grass evolution. The co-orthologous conserved 53 AS genes are listed in Table 8. The set of co-orthologs 248 AS genes conserved in the four plants, with 53 of them conserved to B. distachyon, are provided in Additional file 3: Table S2 (can be downloaded at http://proteomics.ysu.edu/altsplice/). Interestingly, one of the candidates among the conserved gene is Drought-induced protein (Di19). It has been previously suggested that the presence of the retained intron within the coding sequence may give rise to the non-sense mediated decay (NMD) [62]. Recent studies highlight the role of cycloheximide in introducing pre-mature termination codons (PTCs) and NMD in A. thaliana Di19, indicating the splicing mechanism in Di19 [63]. Identification of the Di19 mediated splicing will be of critical importance in increasing the drought resistance or increasing the captive yield of the cereal plants, which are acting as major suppliers of food in climate change. As current analysis were based on the pooled EST/mRNA sequences available in the public domain, more biologically functionally conserved AS genes will be identified when more transcriptome data are collected with improved technologies, various environmental conditions, developmental stages and tissues in these cereal crops. The present data is of immense potential for experimental validation and highlights the role of the AS and biological significance in plant, growth development and environmental regulation, which is a standing challenge in climate change.
Fig. 3.
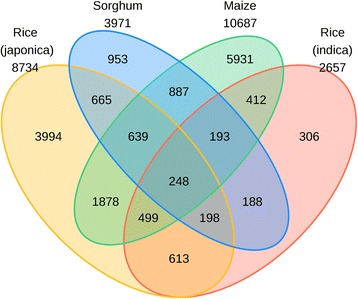
Conserved alternative splicing genes in rice (Oryza sativa) ssp japonica, rice ssp indica, sorghum (Sorghum bicolor), and maize (Zea mays) plants
Table 8.
Conserved alternative splicing genes among five monocot plants
| O. sativa ssp indica | O. sativa ssp japonica | Z. mays | S. bicolor | B. distachyon | CDD/Pfam | ||
|---|---|---|---|---|---|---|---|
| Osi19962 | Osj954 | Zm92934 | Sb6267 | Bd2565 | pfam03171 | 2OG-FeII_Oxy | 2OG-Fe(II) oxygenase superfamily |
| Osi18787 | Osj44013 | Zm40020 | Sb12294 | Bd28385 | pfam00004 | AAA | ATPase family associated with various cellular |
| Osi6875 | Osj22392 | Zm88316 | 45969421 | Bd7352 | pfam00248 | Aldo_ket_red | Aldo/keto reductase family |
| Osi9356 | Osj41340 | Zm162 | Sb17314 | Bd6214 | pfam00248 | Aldo_ket_red | Aldo/keto reductase family |
| CX100091 | Osj15328 | Zm35072 | Sb10885 | Bd29210 | pfam00439 | Bromodomain | Bromodomain |
| Osi12568 | Osj24409 | Zm100060 | Sb8817 | Bd24009 | pfam05042 | Caleosin | Caleosin related protein |
| CT843009 | Osj14649 | Zm32705 | Sb6709 | Bd10918 | pfam00571 | CBS | CBS domain |
| Osi524 | CT828785.1 | Zm73067 | Sb4586 | Bd10523 | pfam04733 | Coatomer_E | Coatomer epsilon subunit |
| Osi21096 | Osj16673 | FL103380 | 2.42E + 08 | Bd4031 | pfam07876 | Dabb | Stress responsive A/B Barrel Domain |
| Osi8549 | Osj47391 | Zm69871 | Sb334 | Bd7166 | pfam05605 | Di19 | Drought induced 19 protein (Di19) |
| CT833644.1 | CI258157 | Zm20082 | Sb10226 | Bd7036 | pfam05057 | DUF676 | Putative serine esterase (DUF676) |
| Osi21136 | Osj16693 | Zm46142 | Sb13903 | Bd7810 | pfam05623 | DUF789 | Protein of unknown function (DUF789) |
| Osi19974 | Osj14932 | Zm70017 | Sb10575 | Bd3731595 | pfam00676 | E1_dh | Dehydrogenase E1 component |
| Osi1759 | Osj22934 | Zm35625 | Sb15873 | Bd7027 | pfam01370 | Epimerase | NAD dependent epimerase/dehydratase family |
| CT842225 | Osj27697 | Zm91971 | Sb4930 | Bd268 | pfam00316 | FBPase | Fructose-1-6-bisphosphatase |
| Osi20900 | Osj16392 | Zm58947 | Sb3303 | Bd7531597 | pfam00210 | Ferritin | Ferritin-like domain |
| Osi339 | Osj20205 | Zm20714 | Sb12056 | Bd6374 | pfam00762 | Ferrochelatase | Ferrochelatase |
| Osi11082 | Osj491 | Zm59942 | Sb3313 | Bd27405 | pfam00125 | Histone | Core histone H2A/H2B/H3/H4 |
| Osi13655 | Osj36042 | Zm81325 | Sb15256 | Bd28446 | pfam00403 | HMA | Heavy-metal-associated domain |
| Osi11360 | Osj36865 | Zm38497 | Sb20674 | Bd9583 | pfam00447 | HSF_DNA-bind | HSF-type DNA-binding |
| Osi17520 | Osj35947 | Zm27347 | Sb9471 | Bd7833 | pfam01156 | IU_nuc_hydro | Inosine-uridine preferring nucleoside |
| Osi13902 | Osj25885 | Zm23750 | Sb12436 | Bd28318 | pfam00013 | KH_1 | KH domain |
| Osi11280 | Osj28328 | Zm35841 | Sb9907 | Bd13744 | cd00116 | LRR_RI | Leucine-rich repeats (LRRs) |
| CT844279 | CB642464 | Zm3338 | Sb7337 | Bd28467 | pfam01717 | Meth_synt_2 | Cobalamin-independent synthase |
| Osi1437 | Osj37916 | Zm4695 | Sb5119 | Bd7994 | pfam00635 | Motile_Sperm | MSP (Major sperm protein) domain |
| Osi231 | Osj25397 | Zm37411 | Sb10332 | Bd28960 | pfam14360 | PAP2_C | PAP2 superfamily C-terminal |
| Osi8815 | Osj32580 | Zm61468 | Sb11226 | Bd6619 | pfam01195 | Pept_tRNA_hydro | Peptidyl-tRNA hydrolase |
| Osi16666 | Osj19199 | Zm104454 | Sb12015 | Bd16056 | pfam00450 | Peptidase_S10 | Serine carboxypeptidase |
| Osi12736 | Osj14309 | Zm22618 | Sb7927 | Bd8683 | pfam00141 | peroxidase | Peroxidase |
| Osi833 | Osj39350 | Zm92939 | Sb19533 | Bd5931597 | pfam00069 | Pkinase | Protein kinase domain |
| Osi3301 | Osj17780 | Zm29726 | Sb14730 | Bd29285 | pfam00069 | Pkinase | Protein kinase domain |
| Osi6061 | Osj15126 | Zm59883 | Sb673 | Bd15932 | PLN02756 | PLN02756 | S-methyl-5-thioribose kinase |
| Osi6187 | Osj42201 | Zm39790 | Sb2138 | Bd8363 | pfam00348 | polyprenyl_synt | Polyprenyl synthetase |
| Osi13092 | Osj21144 | Zm33939 | Sb5787 | Bd23758 | pfam14299 | PP2 | Phloem protein 2 |
| Osi11891 | NM_001070568.2 | Zm87952 | Sb2001 | Bd2595 | pfam00854 | PTR2 | POT family |
| Osi20788 | Osj19691 | Zm39384 | 30944654 | Bd10083 | pfam07992 | Pyr_redox_2 | Pyridine nucleotide-disulphide |
| CT837906.1 | Osj7689 | Zm101865 | Sb5520 | Bd25885 | pfam00719 | Pyrophosphatase | Inorganic pyrophosphatase |
| Osi21504 | Osj17274 | Zm6058 | Sb11340 | Bd21664 | pfam00072 | Response_reg | Response regulator receiver domain |
| Osi15366 | Osj24220 | Zm5068 | Sb10671 | Bd23705 | pfam02453 | Reticulon | Reticulon |
| Osi9029 | Osj47510 | Zm24118 | Sb11323 | Bd8231593 | pfam03214 | RGP | Reversibly glycosylated polypeptide |
| Osi5643 | Osj25267 | Zm80771 | Sb227 | Bd11010 | pfam01246 | Ribosomal_L24e | Ribosomal protein L24e |
| Osi8310 | Osj36859 | Zm101179 | Sb11303 | Bd6311 | pfam00076 | RRM_1 | RNA recognition motif |
| Osi1456 | Osj43479 | Zm371 | Sb12579 | Bd15819 | pfam00076 | RRM_1 | RNA recognition motif |
| Osi773 | Osj43052 | Zm24001 | Sb2305 | Bd20070 | pfam00464 | SHMT | Serine hydroxymethyltransferase |
| Osi9812 | Osj14203 | Zm39491 | 2.42E + 08 | Bd28258 | pfam01406 | tRNA-synt_1e | tRNA synthetases class I (C) catalytic |
| Osi9653 | Osj44577 | Zm33457 | Sb5144 | Bd6360 | pfam00443 | UCH | Ubiquitin carboxyl-terminal hydrolase |
| Osi2251 | Osj35805 | Zm98577 | 2.42E + 08 | Bd20683 | pfam12076 | Wax2_C | WAX2 C-terminal domain |
| Osi15508 | Osj14495 | Zm95 | Sb10474 | Bd4536 | pfam05495 | zf-CHY | CHY zinc finger |
| Osi21052 | Osj4519 | Zm39479 | Sb9831 | Bd24331 | No Pfam predicted | ||
| Osi8778 | Osj195 | Zm100142 | Sb1070 | Bd2265 | No Pfam predicted | ||
| Osi20728 | Osj16408 | Zm34294 | 57806619 | Bd19455 | No Pfam predicted | ||
| Osi17233 | Osj20996 | Zm100171 | Sb1504 | Bd16477 | No Pfam predicted | ||
| CT830510.1 | Osj3010 | Zm96019 | Sb2210 | Bd12504 | No Pfam predicted | ||
Conclusions
In the present work, we investigated the functional landscape of the four most important cereal plants O. sativa ssp indica and japonica, S. bicolor and Z. mays using the updated EST and mRNA sequences available in NCBI thus bridging the knowledge gap and updating the conserved AS catalog with functional elucidation. The availability of the conserved AS genes among the four cereal plants will facilitate to understand the regulation of the alternative physiological processes in global climate change biology and their subsequent impact on the genic-environmental interactions.
Availability of supporting data
The data described in the work can be searched or downloaded at the Plant Alternative Splicing Database (http://proteomics.ysu.edu/altsplice/). Other detailed analysis data can be downloaded at http://proteomics.ysu.edu/publication/data/CerealAS/.
Acknowledgements
The work was funded by the Ohio Plant Biotechnology Consortium (Grant 2013-003) through Ohio State University, Ohio Agricultural Research and Development Center to XJM. XJM was also supported by the College of Science, Technology, Engineering, and Mathematics Dean’s reassigned time for research. JM was supported with a graduate research assistantship by the Center for Applied Chemical Biology, Youngstown State University.
Abbreviations
- AltA
Alternative acceptor site
- AltD
Alternative donor site
- AS
Alternative splicing
- CDS
Coding DNA sequence
- ES
Exon skipping
- IR
Intron retention
- PUT
Putative unique transcript
- ssp
Subspecies
Additional files
Protein family classification of alternative genes in cereal plants. (XLS 402 kb)
Number of conserved alternative splicing genes in cereal crops. (XLS 31 kb)
Conserved alternative splicing gene list in rice, sorghum, corn, and Brachypodium distachyon. (XLS 75 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
XJM conceived the study. BP, JB, and JM contributed to the database construction, XJM, GS, and FY contributed to the experiment design, data analysis, and preparation of the manuscript. All authors have read and approved the final version of the manuscript.
References
- 1.Graveley BR. Alternative splicing: increasing diversity in the proteomic world. Trends Genet. 2001;17:100–107. doi: 10.1016/S0168-9525(00)02176-4. [DOI] [PubMed] [Google Scholar]
- 2.Roberts GC, Smith CW. Alternative splicing: combinatorial output from the genome. Curr Opin Chem Biol. 2002;6:375–383. doi: 10.1016/S1367-5931(02)00320-4. [DOI] [PubMed] [Google Scholar]
- 3.Hiller M, Huse K, Platzer M, Backofen R. Creation and disruption of protein features by alternative splicing - a novel mechanism to modulate function. Genome Biol. 2005;6:R58. doi: 10.1186/gb-2005-6-7-r58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reddy AS, Marquez Y, Kalyna M, Barta A. Complexity of the alternative splicing landscape in plants. Plant Cell. 2013;25:3657–3683. doi: 10.1105/tpc.113.117523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sablok G, Gupta PK, Baek JM, Vazquez F, Min XJ. Genome-wide survey of alternative splicing in the grass Brachypodium distachyon: an emerging model biosystem for plant functional genomics. Biotechnol Lett. 2011;33:629–636. doi: 10.1007/s10529-010-0475-6. [DOI] [PubMed] [Google Scholar]
- 6.Lewis BP, Green RE, Brenner SE. Evidence for the widespread coupling of alternative splicing and nonsense-mediated mRNA decay in humans. Proc Natl Acad Sci U S A. 2003;100:189–192. doi: 10.1073/pnas.0136770100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen L, Tovar-Corona J M, Urrutia AO. Alternative splicing: a potential source of functional innovation in the eukaryotic genome. Int J Evol Biol. 2012, doi:10.1155/2012/596274 [DOI] [PMC free article] [PubMed]
- 9.Carvalho RF, Feijão CV, Duque P. On the physiological significance of alternative splicing events in higher plants. Protoplasma. 2013;250:639–650. doi: 10.1007/s00709-012-0448-9. [DOI] [PubMed] [Google Scholar]
- 10.Filichkin SA, Priest HD, Givan SA, Shen R, Bryant DW, Fox SE, et al. Genome-wide mapping of alternative splicing in Arabidopsis thaliana. Genome Res. 2010;20:45–58. doi: 10.1101/gr.093302.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang PG, Huang SZ, Pin AL, Adams KL. Extensive divergence in alternative splicing patterns after gene and genome duplication during the evolutionary history of Arabidopsis. Mol Biol Evol. 2010;27:1686–1697. doi: 10.1093/molbev/msq054. [DOI] [PubMed] [Google Scholar]
- 12.Marquez Y, Brown JW, Simpson C, Barta A, Kalyna M. Transcriptome survey reveals increased complexity of the alternative splicing landscape in Arabidopsis. Genome Res. 2012;22:1184–1195. doi: 10.1101/gr.134106.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Syed NH, Kalyna M, Marquez Y, Barta A, Brown JW. Alternative splicing in plants - coming of age. Trends Plant Sci. 2012;17:616–623. doi: 10.1016/j.tplants.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang B, Brendel V. Genome wide comparative analysis of alternative splicing in plants. Proc Natl Acad Sci U S A. 2006;103:7175–7180. doi: 10.1073/pnas.0602039103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.VanBuren R, Walters B, Ming R, Min XJ. Analysis of expressed sequence tags and alternative splicing genes in sacred lotus (Nelumbo nucifera Gaertn.) Plant Omics J. 2013;6:311–317. [Google Scholar]
- 16.Vitulo N, Forcato C, Carpinelli EC, Telatin A, Campagna D, D'Angelo M, et al. A deep survey of alternative splicing in grape reveals changes in the splicing machinery related to tissue, stress condition and genotype. BMC Plant Biol. 2014;14:99. doi: 10.1186/1471-2229-14-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walters B, Lum G, Sablok G, Min XJ. Genome-wide landscape of alternative splicing events in Brachypodium distachyon. DNA Res. 2013;20:163–171. doi: 10.1093/dnares/dss041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stamm S, Ben-Ari S, Rafalska I, Tang Y, Zhang Z, Toiber D, et al. Function of alternative splicing. Gene. 2005;344:1–20. doi: 10.1016/j.gene.2004.10.022. [DOI] [PubMed] [Google Scholar]
- 19.Chang CY, Lin WD, Tu SL. Genome-wide analysis of heat-sensitive alternative splicing in Physcomitrella patens. Plant Physiol. 2014;165:826–840. doi: 10.1104/pp.113.230540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang X, Zhang H, Li L. Alternative mRNA processing increases the complexity of microRNA-based gene regulation in Arabidopsis. Plant J. 2012;70:421–431. doi: 10.1111/j.1365-313X.2011.04882.x. [DOI] [PubMed] [Google Scholar]
- 21.Mao H, Sun S, Yao J, Wang C, Yu S, Xu C, et al. Linking differential domain functions of the GS3 protein to natural variation of grain size in rice. Proc Natl Acad Sci U S A. 2010;107:19579–19584. doi: 10.1073/pnas.1014419107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Staiger D, Brown JW. Alternative splicing at the intersection of biological timing, development, and stress responses. Plant Cell. 2013;25:3640–3656. doi: 10.1105/tpc.113.113803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Campbell MA, Haas BJ, Hamilton JP, Mount SM, Buell CR. Comprehensive analysis of alternative splicing in rice and comparative analyses with Arabidopsis. BMC Genomics. 2006;7:327. doi: 10.1186/1471-2164-7-327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Panahi B, Abbaszadeh B, Taghizadeghan M, Ebrahimie E. Genome-wide survey of alternative splicing in Sorghum bicolor. Physiol Mol Biol Plants. 2014;20:323–329. doi: 10.1007/s12298-014-0245-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thatcher SR, Zhou W, Leonard A, Wang BB, Beatty M, Zastrow-Hayes G, et al. Genome-wide analysis of alternative splicing in Zea mays: landscape and genetic regulation. Plant Cell. 2014;26:3472–3487. doi: 10.1105/tpc.114.130773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ner-Gaon H, Leviatan N, Rubin E, Fluhr R. Comparative cross-species alternative splicing in plants. Plant Physiol. 2007;144:1632–1641. doi: 10.1104/pp.107.098640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang X, Madan A. CAP3: A DNA sequence assembly program. Genome Res. 1999;9:868–877. doi: 10.1101/gr.9.9.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu TD, Watanabe CK. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics. 2005;21:1859–1875. doi: 10.1093/bioinformatics/bti310. [DOI] [PubMed] [Google Scholar]
- 29.Goodstein DM, Shu S, Howson R, Neupane R, Hayes RD, Fazo J, et al. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 2012;40:D1178–1186. doi: 10.1093/nar/gkr944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ouyang S, Zhu W, Hamilton J, Lin H, Campbell M, Childs K, et al. The TIGR rice genome annotation resource: improvements and new features. Nucleic Acids Res. 2007;35:D883–887. doi: 10.1093/nar/gkl976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, et al. The B73 maize genome: complexity, diversity, and dynamics. Science. 2009;326:1112–1125. doi: 10.1126/science.1178534. [DOI] [PubMed] [Google Scholar]
- 32.Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood J, Gundlach H, et al. The Sorghum bicolor genome and the diversification of grasses. Nature. 2009;457:551–556. doi: 10.1038/nature07723. [DOI] [PubMed] [Google Scholar]
- 33.Yu J, Hu S, Wang J, Wong GK, Li S, Liu B, et al. A draft sequence of the rice genome (Oryza sativa L. ssp indica) Science. 2002;296:79–92. doi: 10.1126/science.1068037. [DOI] [PubMed] [Google Scholar]
- 34.Min XJ. ASFinder: a tool for genome-wide identification of alternatively spliced transcripts from EST-derived sequences. Int J Bioinformatics Res Appl. 2013;9:221–226. doi: 10.1504/IJBRA.2013.053603. [DOI] [PubMed] [Google Scholar]
- 35.Florea L, Hartzell G, Zhang Z, Rubin GM, Miller W. A computer program for aligning a cDNA sequence with a genomic DNA sequence. Genome Res. 1998;8:967–974. doi: 10.1101/gr.8.9.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Foissac S, Sammeth M. ASTALAVISTA: dynamic and flexible analysis of alternative splicing events in custom gene datasets. Nucleic Acids Res. 2007;35:W297–299. doi: 10.1093/nar/gkm311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Min XJ, Butler G, Storms R, Tsang A. OrfPredictor: predicting protein-coding regions in EST-derived sequences. Nucleic Acids Res. 2005;33:W677–680. doi: 10.1093/nar/gki394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Min XJ, Butler G, Storms R, Tsang A. TargetIdentifier: a web server for identifying full-length cDNAs from EST sequences. Nucleic Acids Res. 2005;33:W669–W672. doi: 10.1093/nar/gki436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McCarthy FM, Wang N, Magee GB, Williams WP, Luthe DS, Burgess SC. AgBase: a functional genomics resource for agriculture. BMC Genomics. 2006;7:229. doi: 10.1186/1471-2164-7-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mascarenhas D, Mettler IJ, Pierce DA, Lowe HW. Intron-mediated enhancement of heterologous gene expression in maize. Plant Mol Biol. 1990;15:913–920. doi: 10.1007/BF00039430. [DOI] [PubMed] [Google Scholar]
- 41.Baek JM, Han P, Iandolino A, Cook DR. Characterization and comparison of intron structure and alternative splicing between Medicago truncatula, Populus trichocarpa. Arabidopsis Rice Plant Mol Biol. 2008;67:499–510. doi: 10.1007/s11103-008-9334-4. [DOI] [PubMed] [Google Scholar]
- 42.Labadorf A, Link A, Rogers MF, Thomas J, Reddy ASN, Ben-Hur A. Genome-wide analysis of alternative splicing in Chlamydomonas reinhardtii. BMC Genomics. 2010;111:14. doi: 10.1186/1471-2164-11-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hickey SF, Sridhar M, Westermann AJ, Qin Q, Vijayendra P, Liou G, et al. Transgene regulation in plants by alternative splicing of a suicide exon. Nucleic Acids Res. 2012;40:4701–4710. doi: 10.1093/nar/gks032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu L, Zhang Y, Zhang W, Yang S, Chen JQ, Tian D. Patterns of exon-intron architecture variation of genes in eukaryotic genomes. BMC Genomics. 2009;10:47. doi: 10.1186/1471-2164-10-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Niu D-K, Yang Y-F. Why eukaryotic cells use introns to enhance gene expression: Splicing reduces transcription-associated mutagenesis by inhibiting topoisomerase I cutting activity. Biol Direct. 2011;6:24. doi: 10.1186/1745-6150-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang H. In plants, expression breadth and expression level distinctly and non-linearly correlate with gene structure. Biol Direct. 2009;4:45. doi: 10.1186/1745-6150-4-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rose AB, Beliakoff JA. Intron-mediated enhancement of gene expression independent of unique intron sequences and splicing. Plant Physiol. 2000;122:535–542. doi: 10.1104/pp.122.2.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maas C, Laufs J, Grant S, Korfhage C, Werr W. The combination of a novel stimulatory element in the first exon of the maize Shrunken-1 gene with the following intron 1 enhances reporter gene expression up to 1000-fold. Plant Mol Biol. 1991;16:199–207. doi: 10.1007/BF00020552. [DOI] [PubMed] [Google Scholar]
- 49.Sinibaldi RM, Mettler IJ. Intron splicing and intron-mediated enhanced expression in monocots. In: Cohn WE, Moldave K, editors. Progress in Nucleic Acid Research and Molecular Biology. New York: Academic Press; 1992. pp. 229–257. [DOI] [PubMed] [Google Scholar]
- 50.Donath M, Mendel R, Cerff R, Martin W. Intron-dependent transient expression of the maize GapA1 gene. Plant Mol Biol. 1995;28:667–676. doi: 10.1007/BF00021192. [DOI] [PubMed] [Google Scholar]
- 51.Rethmeier N, Seurinck J, Van Montagu M, Cornelissen M. Intron-mediated enhancement of transgene expression in maize is a nuclear, gene-dependent process. Plant J. 1997;12:895–899. doi: 10.1046/j.1365-313X.1997.12040895.x. [DOI] [PubMed] [Google Scholar]
- 52.McElroy D, Zhang W, Cao J, Wu R. Isolation of an efficient actin promoter for use in rice transformation. Plant Cell. 1990;2:163–171. doi: 10.1105/tpc.2.2.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu Y, Yu H, Hall TC. Rice triosephosphate isomerase gene 5′ sequence directs β-glucuronidase activity in transgenic tobacco but requires an intron for expression in rice. Plant Physiol. 1994;106:459–467. doi: 10.1104/pp.106.2.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kelemen O, Convertini P, Zhang Z. Function of alternative splicing. Gene. 2013;514:1–30. doi: 10.1016/j.gene.2012.07.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu KL. The WRKY family of transcription factors in rice and Arabidopsis and their origins. DNA Res. 2005;12:9–26. doi: 10.1093/dnares/12.1.9. [DOI] [PubMed] [Google Scholar]
- 56.Xie Z. Annotations and functional analyses of the rice WRKY gene superfamily reveal positive and negative regulators of abscisic acid signaling in aleurone cells. Plant Physiol. 2005;137:176–189. doi: 10.1104/pp.104.054312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang S, Tang F, Zhu H. Alternative splicing in plant immunity. Int J Mol Sci. 2014;15:10424–10445. doi: 10.3390/ijms150610424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Feng B, Yang S, Du H, Hou X, Zhang J, Liu H, et al. Molecular characterization and functional analysis of plant WRKY genes. African J Biotechnol. 2012;11:13606–13613. [Google Scholar]
- 59.Peng Y. OsWRKY62 is a negative regulator of basal and Xa21- mediated defense against Xanthomonas oryzae pv. Oryzae in rice Mol Plant. 2008;1:446–458. doi: 10.1093/mp/ssn024. [DOI] [PubMed] [Google Scholar]
- 60.Li J, Li X, Guo L, Lu F, Feng X, He K, et al. A subgroup of MYB transcription factor genes undergoes highly conserved alternative splicing in Arabidopsis and rice. J Exp Bot. 2006;57:1263–1273. doi: 10.1093/jxb/erj094. [DOI] [PubMed] [Google Scholar]
- 61.Zhao C, Beers E. Alternative splicing of Myb-related genes MYR1 and MYR2 may modulate activities through changes in dimerization, localization, or protein folding. Plant Signal Behav. 2013;11:e27325. doi: 10.4161/psb.27325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Morello L, Breviario D. Plant spliceosomal introns: not only cut and paste. Curr Genomics. 2008;9:227–238. doi: 10.2174/138920208784533629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kalyna M, Simpson CG, Syed NH, Lewandowska D, Marquez Y, Kusenda B, et al. Alternative splicing and nonsense-mediated decay modulate expression of important regulatory genes in Arabidopsis. Nucleic Acids Res. 2012;40:2454–69. doi: 10.1093/nar/gkr932. [DOI] [PMC free article] [PubMed] [Google Scholar]