

Abstract
Chagas disease, caused by the protozoan parasite Trypanosoma cruzi (T. cruzi), is a life threatening global health problem with only two drugs available for treatment (benznidazole and nifurtimox), both having variable efficacy in the chronic stage of the disease and high rates of adverse drug reactions. Inhibitors of sterol 14α-demethylase (CYP51) have proven effective against T. cruzi in vitro and in vivo in animal models of Chagas disease. Consequently two azole inhibitors of CYP51 (posaconazole and ravuconazole) have recently entered clinical development by the Drugs for Neglected Diseases initiative. Further new drug treatments for this disease are however still urgently required, particularly having a different mode of action to CYP51 in order to balance the overall risk in the drug discovery portfolio. This need has now been further strengthened by the very recent reports of treatment failure in the clinic for both posaconazole and ravuconazole. To this end and to prevent enrichment of drug candidates against a single target, there is a clear need for a robust high throughput assay for CYP51 inhibition in order to evaluate compounds active against T. cruzi arising from phenotypic screens. A high throughput fluorescence based functional assay using recombinantly expressed T. cruzi CYP51 (Tulahuen strain) is presented here that meets this requirement. This assay has proved valuable in prioritising medicinal chemistry resource on only those T. cruzi active series arising from a phenotypic screening campaign where it is clear that the predominant mode of action is likely not via inhibition of CYP51.
Author Summary
Chagas disease, caused by the parasite Trypanosoma cruzi (T. cruzi), is endemic in Latin America and emerging in North America and Europe through human migration. It is a severe global health problem with 8–10 million people infected and an estimated 12,000 deaths annually. Current treatment options are poorly efficacious and have severe side effects. New drugs are therefore urgently required. Two of these potential new drugs, posaconazole and ravuconazole, both targeting an enzyme in T. cruzi called CYP51, have recently failed in clinical development. Therefore, in light of these recent clinical failures and in order to better balance the overall risk in the drug discovery portfolio for Chagas disease, it has become prudent to assess whether new chemical start points for drug discovery programmes have a mode of action predominantly driven by T. cruzi CYP51 inhibition. In this paper we report a fluorescence based assay to determine whether compounds inhibit T. cruzi CYP51. This provides a high throughput screen to help prioritise medicinal chemistry resource on those T. cruzi active new chemical series that do not have a mode of action predominantly driven by CYP51 inhibition.
Introduction
Chagas disease is a tropical parasitic disease caused by the flagellate eukaryotic (protozoan) parasite Trypanosoma cruzi (T. cruzi), endemic in Latin America and now emerging in North America and Europe through human migration. It is becoming a severe global health problem with approximately 8–10 million people infected, an estimated 12,000 deaths per year, and placing 100 million people at risk. Transmission to humans and other mammals is predominantly by an insect vector, the blood-sucking "kissing bugs" of the subfamily Triatominae (family Reduviidae) [1]. Transmission has also been reported to occur through contaminated food, blood transfusions and from mother to child.
Clinical Chagas disease can be classified into two distinct phases, acute and chronic. In the acute phase, lasting a few weeks, parasites begin to multiply in the organs and tissues. Symptoms are usually mild and non-specific with patients rarely being diagnosed. However, life-threatening myocarditis or meningoencephalitis can occur during the acute phase with a death rate for people in this phase of about ten percent. Ten to fifty percent of infected survivors develop chronic Chagas disease. People in the chronic phase can be asymptomatic for many years, with parasites generally undetectable in the blood. However, the disease causes organ and tissue damage, particularly potentially lethal cardiopathy and megacolon or megaoesophagus, caused by the sequential induction of inflammatory response to the parasite. Nitroheterocyclic compounds, benznidazole and nifurtimox, developed in the 1960’s [2], are currently the only two drugs used for the treatment of Chagas disease. Both have low efficacy in the chronic stage and, with prolonged dosing regimens, both drugs have significant side effects including skin irritation, neurotoxicity, and digestive system disorders [3]. Newer, safer and more efficacious treatments are therefore in desperate need.
Inhibition of sterol 14α-demethylase (CYP51) has been considered a viable target against T. cruzi for over 30 years [2,4,5,6,7,8]. Found in a broad variety of organisms including animals, plants, fungi and protozoa, this enzyme plays an essential role in the sterol biosynthetic pathway, catalysing the oxidative removal of the 14α-methyl group from sterol precursors such as lanosterol or eburicol [9]. The products of the pathway, cholesterol in humans or ergosterol in fungi, are required for the integrity of the eukaryotic cell membrane. These sterols are required for membrane function in T. cruzi. Inhibition of CYP51 activity is lethal as the T. cruzi parasites are unable to scavenge and utilise host cholesterol [10]. The CYP51 gene is known to be expressed in all stages of the parasite life cycle and indeed it has also been shown to be up-regulated in multiplying forms [9]. As with other members of the Cytochrome family, CYP51 is a haem containing protein located on the membrane of the endoplasmic reticulum that relies upon electron transfer by NADPH reductase for activation [11].
Azole inhibitors, which interfere with sterol biosynthesis, essential in eukaryotic cells, have already been used with success in humans in the treatment of fungal infections. Several of these drugs have been considered as possible treatments for Chagas disease [12]. Ketoconazole, fluconazole, itraconazole, ravuconazole and posaconazole are known to inhibit CYP51 in vitro, competitively binding to the haem within CYP51 and occupying the active site preventing any substrate from binding. Although ketoconazole and itraconazole have not demonstrated significant curative activity in humans with chronic Chagas disease [6], other azoles, with greater potency and improved pharmacokinetic properties, which have been shown to have potent activity against T. cruzi, including posaconazole [13,14] and ravuconazole (Fig 1), are in clinical development with the Drugs for Neglected Diseases initiative (DNDi).
Fig 1. Clinical candidates for Chagas disease.

To prevent enrichment of candidates against a single target, and thus reduce risk in the overall drug discovery portfolio for Chagas disease, it has therefore become necessary to evaluate and prioritise medicinal chemistry resource on new chemical series active against T. cruzi but with such activity not likely driven via T. cruzi CYP51 inhibition. Recent findings from clinical trials with posaconazole [15] and ravuconazole [16] has indicated re-emergence of parasitaemia in two thirds of patients once dosing has been completed, thus reinforcing the need to strengthen the overall drug discovery portfolio for Chagas disease with new chemical lead series not working via this mechanism of action.
Evaluating compounds as potential inhibitors of T. cruzi CYP51 has previously been demonstrated measuring the apparent dissociation constants (Kd) by spectral titration [4,17,18] utilising the shift of the haem iron soret band in response to binding [18]. One of the drawbacks to this methodology is that micromolar protein concentrations are required for screening causing potential interference with the optical properties and/or solubility of test compounds [4]. There are many potential reasons why affinity estimates measured by binding may not correlate with functional inhibition. These include allosteric sites, non or uncompetitive modes of inhibition or slow kinetics [19].
Inhibition of endogenous substrate lanosterol, eburicol and obtusifoliol has also been used as an in vitro tool using recombinant expressed human CYP51 enzyme [20]. In particular, measuring effect on CYP51 driven metabolism of lanosterol to follicular fluid meiosis activating sterol (FF-MAS) in the presence of test substances [21] is well established (Fig 2). However, FF-MAS detection requires mass spectrometry limiting the number of compounds that can be tested and consequently limiting the value of such an assay for triaging large numbers of phenotypic screening T. cruzi hits toward identifying modes of action away from CYP51.
Fig 2.

a: Traditional endogenous substrate (Lanosterol) and CYP51 specific metabolite (FF-MAS); b: Fluorogenic probe substrate BOMCC is metabolised to CHC by CYP51.
Metabolism of fluorogenic probe substrate to a product, detectable by fluorescence is well established with recombinantly expressed cytochrome P450 enzymes (CYP’s) for the purpose of assessing possible drug-drug interactions [22]. Measuring CYP inhibition by this method provides a high throughput screening approach, avoiding time consuming analysis by mass spectrometry and minimising use of expensive substrates. The O-dealkylation of Vivid substrate benzyloxymethylocyanocoumarin (BOMCC) to fluorescent product cyanohydroxycoumarin (CHC) is commonly used to evaluate CYP3A4 activity in recombinantly expressed membrane preparations (Fig 2). Valuably, O-dealkylation activity in the presence of recombinantly expressed T. cruzi CYP51 was observed. This has enabled the creation of a fast, high-throughput, 96 and 384 well microtitre method to assess the inhibitory potential of compounds against T. cruzi CYP51, which is described in this paper.
Materials and Methods
Chemicals and reagents
Benzyloxymethyloxycyanocoumarin (BOMCC) was obtained from ThermoFisher Scientific. Fluconazole, ketoconazole, itraconazole, NADP, NADPH, glucose-6-phosphate, glucose-6-phosphate dehydrogenase, Cytochrome C from horse heart and sodium bicarbonate were obtained from Sigma Aldrich. 50 mM potassium phosphate (pH 7.4) buffer was prepared from dibasic and monobasic forms of potassium phosphate obtained from Sigma Aldrich.
Cloning and expression of T. cruzi CYP51 in E. coli
E. coli membrane fractions containing the T. cruzi CYP51 (Tulahuen strain) (Bactosomes) were provided by Cypex Ltd.
The cDNAs coding for T. cruzi CYP51 and NADPH P450 reductase were synthesized and supplied in pUC vectors by Genescript. The CYP51 cDNA was cloned into the expression vector pCW with an ompA N terminal leader and the reductase was cloned into a pACYC184 derived expression vector with a pelB N terminal leader. E. coli JM109 was used for the co-expression of the recombinant proteins.
Data analysis
For each concentration of test compound the rate of fluorescence units per minute was calculated as a percentage of the average rate of the solvent only control wells.
The percentage of solvent control values was then plotted against the concentration range. Using the following 4 parameter fit equation, an IC50 value can be determined.
Accession numbers
Accession number for T. cruzi cDNA is AY283022; T. cruzi reductase is DQ857724 and mosquito reductase is AY183375.
Results and Discussion
T. cruzi CYP51 reductase partner
Despite the presence of the N terminal pelB leader sequence, the recombinant T. cruzi NADPH P450 reductase proved to be a cytosolic protein (as might be expected from previous data [10]) and was not present in the E.coli membrane fraction with the T. cruzi CYP51. Reconstitution of the cytosolic fraction containing the T. cruzi reductase with the membrane fraction containing the T. cruzi CYP51 did not result in active CYP51 so, in order to generate an active T. cruzi CYP51 system, the T. cruzi CYP51 was co-expressed with human (expression construct supplied by Cypex Ltd) or mosquito (expression construct supplied by the Liverpool School of Tropical Medicine) NADPH P450 reductase. Screening of CYP51 activity showed that the T. cruzi CYP51 co-expressed with the mosquito reductase gave the highest rate of BOMCC turnover. The mosquito reductase expression cassette was cloned from the pACYC vector into the pCW based CYP51 expression vector to allow both proteins to be expressed from the same plasmid resulting in a relatively higher level of mosquito reductase in the membrane fraction with a concomitant increase in BOMCC turnover. Supplementation of the T. cruzi CYP51 / mosquito NADPH P450 reductase bactosomes with partially purified mosquito cytochrome b 5 at 10 fold excess over the CYP51 resulted in a further increase in BOMCC turnover. This bactosome preparation was then scaled up for all further work.
Determination of kinetic parameters
To determine kinetic parameters, 100 μM BOMCC O-dealkylase activity was assessed in incubations containing 25, 50, 75, 100, 150, 200, 250, 300, 400, 500 and 600 pmoles/mL T. cruzi CYP51 enzyme. Protein concentrations were normalised using control protein containing no CYP activity. Incubations were pre-warmed at 37°C before addition of NADPH regenerating system. Production of CHC was measured at 1 minute intervals at 37°C (Exc 410 nm, Em 460 nm) for 10 minutes (Fig 3a).
Fig 3.

a: Linearity of CHC production (expressed as average rate of fluorescence units (AFU)) with increasing T. cruzi CYP51 enzyme concentration (96 well format; average points (n = 3) with error bars); b: Michaelis-Menten Kinetic determination (96 well format; average points (n = 3) with error bars).
Km and Vmax parameters for BOMCC were determined by incubating 37 pmoles/mL T. cruzi CYP51 enzyme (0.24 mg/mL bactosomes) with 25, 50, 75, 100, 150, 200 and 250 μM BOMCC. Incubations were pre-warmed at 37°C before addition of NADPH regenerating system. Production of CHC was measured at 1 minute intervals at 37°C (Exc 410 nm, Em 460 nm) for 10 minutes (Fig 3b). Rates of BOMCC metabolism appeared linear up to 100 pmoles/mL with a Km value determined as 191± 55 μM. Assay conditions of 37 pmoles/mL of T. cruzi CYP51 in bactosomes and 100 μM BOMCC provided a good dynamic range of metabolism.
Metabolic activation of 100 μM BOMCC by T. cruzi CYP51 (37 pmoles/mL) was evaluated in the presence of 2% v/v DMSO, methanol or acetonitrile. Incubations were pre-warmed at 37°C before addition of NADPH regenerating system. Production of CHC was measured at 1 minute intervals at 37°C (Exc 410 nm, Em 460 nm) for 10 minutes. The average rate of fluorescence units (AFU) per minute was compared to a solvent only control (Fig 4a). There did not appear to be a significant decrease in metabolic activity in the presence of 2% v/v DMSO or 2% v/v methanol.
Fig 4.

a: The effect of solvent on T. cruzi CYP51 activity; b: Cytochrome C reductase activity of recombinant preparations.
Implementation of microtitre-based assay for compound screening
With kinetic parameters established, a microtitre-based assay to measure inhibition of recombinantly expressed T. cruzi CYP51 by test compounds was then implemented. Using a selection of ‘azole’ inhibitors, including those previously shown to competitively bind to CYP51, and known xenobiotic CYP450 inhibitors (Table 1), the assay was validated to ensure robustness. The incubation times and protein concentrations employed were within the linear range for each assay. Incubations containing 37 pmoles/mL of T. cruzi CYP51 in bactosomes, 100 μM BOMCC, 10, 3.3, 1.0, 0.33, 0.1, 0.033 or 0.01 μM test compound (2% v/v solvent) in 50 mM potassium phosphate buffer (pH 7.4), were pre-incubated at 37°C for 5 minutes. Upon addition of NADPH regenerating system (7.8 mg [28 μM] glucose-6-phosphate, 1.7 mg [2.2 μM] NADP, 6 units/mL glucose-6-phosphate dehydrogenase in 2% w/v sodium bicarbonate buffer) production of fluorescent metabolite CHC was measured (Exc 410 nm, Em 460 nm) at 1 minute intervals over a 10 minute period. Rates of metabolite production per minute were compared to uninhibited controls and plotted against test compound concentration to obtain an IC50. The assay was then miniaturized to 20 μL and moved from kinetic to single endpoint stopped readout (60 minute reaction before quenching with posaconazole to generate a long stable signal) to increase throughput.
Table 1. IC50 Determination of a test set of compounds.
| Test Compound | Structure | IC50 (μM), n = 3 | Reference, Kd (μM) |
|---|---|---|---|
| Ketoconazole | 0.014 a | 0.2 [18] | |
| Miconazole | 0.057 a | ||
| Itraconazole | 0.029 a | ||
| Posaconazole | 0.048 a | 0.06 [24], 0.018 [20] | |
| Fluconazole | 0.880 a | 0.23 [18], 0.43 [24], 0.15 [25] | |
| Methimazole | >8 | ||
| Ticlopidine | >8 | ||
| Sulphaphenazole | >8 | ||
| Sulphamethoxazole | >8 | ||
| Quinidine | >8 | ||
| Furafylline | >8 |
a Coefficient of variation for all IC50’s were lower than 1%
Ketoconazole, itraconazole, posaconazole and miconazole all showed potent inhibition of T. cruzi CYP51 activation of substrate BOMCC with IC50 values of 0.014, 0.029, 0.048 and 0.057 μM, respectively. Indeed, it is likely that these compounds are even more potent than they appear to be as the T. cruzi CYP51 enzyme concentration used in the assay defines a minimum IC50 of approximately 0.02 μM. Fluconazole also inhibited activity (IC50 0.88 μM) although this was at least 20 fold less inhibitory than observed with other azole type compounds. Methimazole did not appear to inhibit activity (IC50 > 8 μM), perhaps as a result of an inability to adequately fill the active site and prevent substrate binding.
Compounds which did not appear to inhibit T. cruzi CYP51 in this assay included known CYP450 inhibitors ticlopidine (CYP2C19), sulphaphenazole (CYP2C9), sulphamethoxazole (CYP2C8/9), quinidine (CYP2D6) and furafylline (CYP1A2). This was expected as, unlike other drug metabolising CYP450s, CYP51 has a very narrow substrate specificity, being limited to endogenous sterols including eburicol and lanosterol [23].
Assessment of cytochrome c reductase activity in presence of test compounds
As previously discussed, addition of an equivalent reductase enzyme (mosquito, A. gambiae) was required to deliver metabolic activity of T. cruzi CYP51. Due to the artificial nature of this pairing it was therefore necessary to confirm that any potent inhibition of CHC production was not the result of indirect inhibition of the electron transfer by NADPH Reductase. Thus, cytochrome c reductase activity of the bactosome preparation was monitored in the presence of all test inhibitor compounds individually. Incubations containing 0.82 pmoles/mL of T. cruzi CYP51 in bactosomes, 50 μM cytochrome c, 10, 3.3, 1.0, 0.33, 0.1, 0.033 or 0.01 μM test compound (2% solvent) in 50mM potassium phosphate buffer (pH 7.4), were pre-incubated at 37°C for 5 minutes. Absorbance at 550 nm was then measured for 3 minutes to ascertain a background level. After addition of NADPH (final concentration 80 μg/mL), absorbance was further measured over 5 minutes. The rate of reduction of cytochrome c at each test compound concentration was compared to an uninhibited control (Fig 4b). The assay was then miniaturized to 50 μL and moved from kinetic to single endpoint readout (60 minutes reaction) to increase throughput. No decrease in activity of cytochrome c reductase was observed for any of the test inhibitors. Furthermore, subsequent evaluation of a much larger set of T. cruzi CYP51 inhibitors has since been carried out and none have yet decreased cytochrome c reductase activity confirming that inhibition of BOMCC O-dealkylation observed is by direct inhibition of T. cruzi CYP51 and not by its effect on the cytochrome c reductase nor the regeneration system. This strongly suggests that the artificial nature of the pairing of T. cruzi CYP51 with A. gambiae NADPH reductase in the bactosomes used for this assay will not deliver false positives.
Implementation in Chagas disease drug discovery screening cascade
Identifying the fluorogenic probe BOMCC has enabled development of a fluorescence based high throughput functional assay to determine T. cruzi CYP51 inhibition, suitable for compound triaging of T. cruzi phenotypic screening hits. This assay is now embedded in our Chagas disease screening cascade to prioritise compound series for progression into hit to lead and lead optimisation.
Following a phenotypic screen of approximately 200,000 compounds against the T. cruzi parasite, inhibition of T. cruzi CYP51 was measured for compounds from 22 trypanocidal series and 16 singleton compounds. Compounds were derived from a variety of sources including diversity and target-centric libraries from the Dundee Drug Discovery Unit and GSK corporate collections. At least three compounds from each chemical series displaying as wide a range of T. cruzi activity as possible were tested (a total of 129 compounds; Fig 5a).
Fig 5.
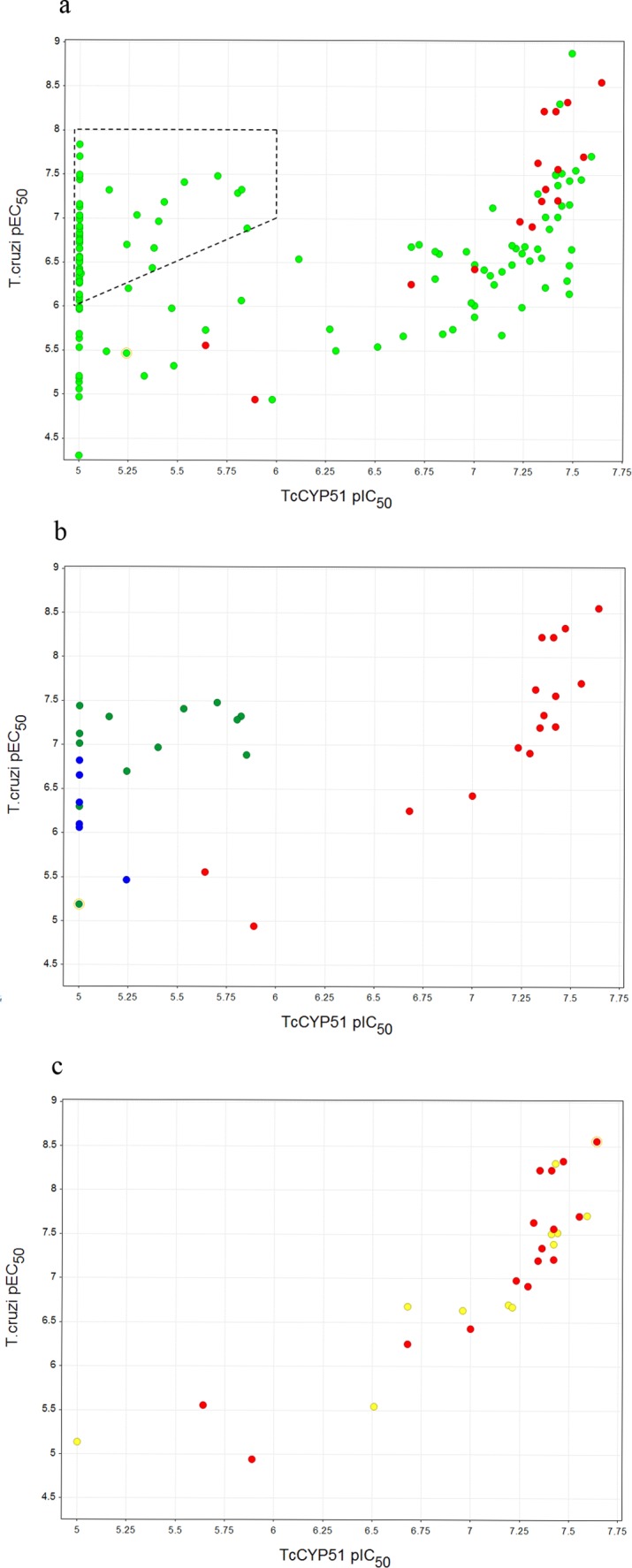
a: T. cruzi CYP51 inhibition plotted against T. cruzi activity for compounds from the different chemical series (coloured green), in addition to 16 known inhibitors of human CYP51 used as a reference set to define the correlation curve (coloured red). T. cruzi active series within the dotted box are prioritised for further medicinal chemistry; b: T. cruzi CYP51 inhibition plotted against T. cruzi activity for series one (green), and series two (blue) compared with the human CYP51 training set (red); c: T. cruzi CYP51 inhibition plotted against T. cruzi activity for a third series (yellow) compared with the human CYP51 training set (red). pEC50 or pIC50 is the −log of EC50 or IC50 respectively in M.
With the assumption that those series which show a good correlation between T. cruzi activity and T. cruzi CYP51 enzyme inhibition have a T. cruzi CYP51 mediated mode of action, a remarkable enrichment for the T. cruzi CYP51 mode of action was observed. Correlation between T. cruzi activity and T. cruzi CYP51 inhibition was evident for 11 out of the 22 hit series, with an additional two series showing sporadic association with T. cruzi CYP51 inhibition. Similarly high rates were observed with the singletons, with 11 out of 16 showing antiparasitic activity in line with T. cruzi CYP51 inhibition. 13/22 series and 11/16 singletons were therefore de-prioritised from further medicinal chemistry resourcing and focus given to those hits demonstrating at least one log unit divergence between T. cruzi activity and T. cruzi CYP51 activity, providing confidence that the predominant mode of action was not T. cruzi CYP51 driven. It is clearly important to look at the overall correlation between inhibition of T. cruzi CYP51 and antiparasitic activity for a given series to ensure that they are not tracking.
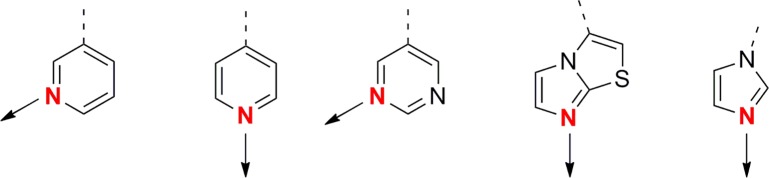
As it transpires, the majority of the chemical series removed from progression bear aza-heterocyclic groups which are already well known to exert inhibition of cytochromes through co-ordination to the haem iron (Fig 6) [9] and could have perhaps been eliminated “by- eye”. However, the great benefit of this assay to our drug discovery effort is that it has allowed identification of T. cruzi active series which do contain potential cytochrome binding motifs, but which do not inhibit T. cruzi CYP51. For instance, two particularly promising series (Fig 5b) having potent T. cruzi activity were found not to have an obvious correlation with T. cruzi CYP51 inhibition in spite of containing heterocycles with known potential for haem-binding in cytochromes. This was in stark contrast to a third series (Fig 5c) where T. cruzi activity tracked with T. cruzi CYP51 inhibition, akin to the known human CYP51 training set. The work described here has therefore enabled us to advance the first two series into hit-to-lead studies with a greater degree of confidence and oral activity with compounds from both series has subsequently been demonstrated in mouse models of Chagas disease.
Fig 6. Chemical moieties associated with CYP inhibition.
The 96 well plate based assay used in this analysis has now been further miniaturized and adapted to automated liquid dispensers and microplate readers for high-throughput screening in 384 well endpoint format. The T. cruzi CYP51 FLINT assay (Fig 7a) can be run in 20 μL and the cytochrome c reductase absorbance assay (Fig 7b) in 50 μL final volumes. This allows a throughput increase to up to more than 10,000 wells in a single experiment with Z’ > 0.7 and with a significant reduction in screening cost and time. Sensitivity to described inhibitors was maintained during assay miniaturization.
Fig 7. Trends of control 1 (closed circles, solvent only control, 0% inhibition), control 2 (open circles, reaction in absence of bactosomes, 100% inhibition) and Z’ (grey squares) of representative high-throughput screening runs of T. cruzi CYP51 FLINT (a) and cytochrome c reductase absorbance (b) assays in 384 well format.

Error bars are the standard deviations of 16 replicates for each control and plate.
Acknowledgments
We thank Mark Paine at the Liverpool school of tropical medicine for providing us with mosquito reductase and José Julio Martín Plaza and Fernando Ramón for their contributions, helpful advice, and discussions.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
Funding under Wellcome Trust strategic award WT092340 and WT100476 supported this work for JR, SB and KR. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Rassi A Jr., Rassi A, Marin-Neto JA (2010) Chagas disease. Lancet 375: 1388–1402. 10.1016/S0140-6736(10)60061-X [DOI] [PubMed] [Google Scholar]
- 2. Buckner FS, Urbina JA (2012) Recent developments in sterol 14-demethylase inhibitors for Chagas disease. International Journal for Parasitology: Drugs and Drug Resistance 2: 236–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Castro JA, de Mecca MM, Bartel LC (2006) Toxic side effects of drugs used to treat Chagas' disease (American trypanosomiasis). Hum Exp Toxicol 25: 471–479. [DOI] [PubMed] [Google Scholar]
- 4. Gunatilleke SS, Calvet CM, Johnston JB, Chen CK, Erenburg G, et al. (2012) Diverse inhibitor chemotypes targeting Trypanosoma cruzi CYP51. PLoS Negl Trop Dis 6: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Apt W (2010) Current and developing therapeutic agents in the treatment of Chagas disease. Drug Des Devel Ther 4: 243–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Buckner FS (2008) Sterol 14-demethylase inhibitors for Trypanosoma cruzi infections. Adv Exp Med Biol 625: 61–80. 10.1007/978-0-387-77570-8_6 [DOI] [PubMed] [Google Scholar]
- 7. Docampo R, Moreno SNJ, Turrens JF, Katzin AM, Gonzalez-Cappa SM, et al. (1981) Biochemical and ultrastructural alterations produced by miconazole and econazole in Trypanosoma cruzi. Molecular and Biochemical Parasitology 3: 169–180. [DOI] [PubMed] [Google Scholar]
- 8. Choi JY, Podust LM, Roush WR (2014) Drug Strategies Targeting CYP51 in Neglected Tropical Diseases. Chemical Reviews. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lepesheva GI, Villalta F, Waterman MR (2011) Targeting Trypanosoma cruzi sterol 14alpha-demethylase (CYP51). Adv Parasitol 75: 65–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lepesheva GI, Hargrove TY, Ott RD, Nes WD, Waterman MR (2006) Biodiversity of CYP51 in trypanosomes. Biochem Soc Trans 34: 1161–1164. [DOI] [PubMed] [Google Scholar]
- 11. Portal P, Fernandez Villamil S, Alonso GD, De Vas MG, Flawia MM, et al. (2008) Multiple NADPH-cytochrome P450 reductases from Trypanosoma cruzi suggested role on drug resistance. Mol Biochem Parasitol 160: 42–51. [DOI] [PubMed] [Google Scholar]
- 12. Urbina JA (2002) Chemotherapy of Chagas disease. Curr Pharm Des 8: 287–295. [DOI] [PubMed] [Google Scholar]
- 13. Pinazo MJ, Espinosa G, Gallego M, Lopez-Chejade PL, Urbina JA, et al. (2010) Successful treatment with posaconazole of a patient with chronic Chagas disease and systemic lupus erythematosus. Am J Trop Med Hyg 82: 583–587. 10.4269/ajtmh.2010.09-0620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lívia de Diniz Figueiredo, * Urbina Julio A.,2 de Andrade Isabel Mayer,1 Mazzeti Ana Lia,1 Martins Tassiane Assíria F,1 Caldas Ivo Santana,1 Talvani André,1 Ribeiro Isabela,3 and Bahia Maria Terezinha1 (2013) Benznidazole and Posaconazole in Experimental Chagas Disease: Positive Interaction in Concomitant and Sequential Treatments. PLoS Negl Trop Dis 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Molina I, Gómez i Prat J, Salvador F, Treviño B, Sulleiro E, et al. (2014) Randomized Trial of Posaconazole and Benznidazole for Chronic Chagas' Disease. New England Journal of Medicine 370: 1899–1908. 10.1056/NEJMoa1313122 [DOI] [PubMed] [Google Scholar]
- 16.DNDi (2013) Drug Trial for Leading Parasitic Killer of the Americas Shows Mixed Results but Provides New Evidence for Improved Therapy. First placebo-controlled study in adults with Chagas disease highlights urgent need to scale up treatment for millions of patients at risk. DNDi Press Release.
- 17. Andriani G, Amata E, Beatty J, Clements Z, Coffey BJ, et al. (2013) Antitrypanosomal Lead Discovery: Identification of a Ligand-Efficient Inhibitor of Trypanosoma cruzi CYP51 and Parasite Growth. Journal of Medicinal Chemistry 56: 2556–2567. 10.1021/jm400012e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lepesheva GI, Ott RD, Hargrove TY, Kleshchenko YY, Schuster I, et al. (2007) Sterol 14α-Demethylase as a Potential Target for Antitrypanosomal Therapy: Enzyme Inhibition and Parasite Cell Growth. Chemistry & Biology 14: 1283–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Segel IH (1975) Enzyme kinetics: behavior and analysis of rapid equilibrium and steady state enzyme systems. New York: Wiley. [Google Scholar]
- 20. Hargrove TY, Wawrzak Z, Alexander PW, Chaplin JH, Keenan M, et al. (2013) Complexes of Trypanosoma cruzi sterol 14alpha-demethylase (CYP51) with two pyridine-based drug candidates for Chagas disease: structural basis for pathogen selectivity. J Biol Chem 288: 31602–31615. 10.1074/jbc.M113.497990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Trosken ER, Adamska M, Arand M, Zarn JA, Patten C, et al. (2006) Comparison of lanosterol-14 alpha-demethylase (CYP51) of human and Candida albicans for inhibition by different antifungal azoles. Toxicology 228: 24–32. [DOI] [PubMed] [Google Scholar]
- 22. Bell L, Bickford S, Nguyen PH, Wang J, He T, et al. (2008) Evaluation of fluorescence- and mass spectrometry-based CYP inhibition assays for use in drug discovery. J Biomol Screen 13: 343–353. 10.1177/1087057108317480 [DOI] [PubMed] [Google Scholar]
- 23. Lepesheva GI, Waterman MR (2007) Sterol 14alpha-demethylase cytochrome P450 (CYP51), a P450 in all biological kingdoms. Biochim Biophys Acta 3: 467–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Villalta F, Dobish MC, Nde PN, Kleshchenko YY, Hargrove TY, et al. (2013) VNI cures acute and chronic experimental Chagas disease. J Infect Dis 208: 504–511. 10.1093/infdis/jit042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen CK, Doyle PS, Yermalitskaya LV, Mackey ZB, Ang KK, et al. (2009) Trypanosoma cruzi CYP51 inhibitor derived from a Mycobacterium tuberculosis screen hit. PLoS Negl Trop Dis 3: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.