

Abstract
The interaction of primary autoxidation products of cholesterol, 25- and 20ξ-hydroperoxides, with the four principal cholesterol-metabolizing cytochrome P450 enzymes is reported. Addition of cholesterol 25-hydroperoxide to CYP27A1 and CYP11A1 induced well-defined spectral changes while generating 25-hydroxycholesterol as major product along with small amounts of triols. The 20ξ-hydroperoxides induced spectral shifts in CYP27A1 and CYP11A1, yet glycol metabolites were detected only with CYP11A1. CYP7A1 and CYP46A1 failed to give metabolites with any of the hydroperoxides. A P450 hydroperoxide-shunt reaction is proposed, where the hydroperoxides serve both as donor for reduced oxygen and as substrate. For the first time, CYP27A1 is shown to mediate the reduction of cholesterol 25-hydroperoxide to 25-hydroxycholesterol, a role of potential significance for cholesterol-rich tissues with high oxidative stress. CYP27A1 may participate in these tissues in removal of harmful autoxidation products, while providing a complementary source for 25-hydroxycholesterol, a modulator of immune cell function and mediator of viral cell entry.
Keywords: Cholesterol 25-hydroperoxide, cytochrome P450, autoxidation, biocatalysis, 25-hydroxycholesterol
Cholesterol plays a major role in many biological processes that are considered central to the well-being of most living organisms. Apart from being the precursor of steroid hormones and bile acids, cholesterol plays a structural role and is an important component of cellular membranes. Cholesterol excess in many tissues is prevented in part through the action of different cytochrome P450 enzymes that hydroxylate cholesterol at specific positions to facilitate its elimination.[1] These enzymes include the 7-, 24- and 27-hydroxylases (CYP7A1, CYP46A1 and CYP27A1, respectively) as well as the cholesterol side-chain cleavage enzyme CYP11A1.[1,2] CYP7A1 and CYP46A1 are microsomal enzymes, whereas CYP27A1 and CYP11A1 are mitochondrial monooxygenases. CYP7A1 is expressed only in the liver, CYP46A1 is mainly found in the neurons, CYP27A1 is ubiquitous, and CYP11A1 is the main cholesterol hydroxylase in steroidogenic tissues. For cholesterol to be metabolized by these P450s, they need NADPH as a source of reducing equivalents, and protein(s) that transfer electrons from NADPH to the P450; cytochrome P450 oxidoreductase for microsomal CYP7A1 and CYP46A1, and ferredoxin oxidoreductase/ferredoxin for mitochondrial CYP27A1 and CYP11A1.[3] The principal intermediate in P450-mediated hydroxylations has been identified as Compound I (Cpd I), the highly reactive hydroperoxi-Fe(IV)=O+• species.[4] In addition to the P450 hydroxylases, various tissues express cholesterol 25-hydroxylase (CH25H), an endoplasmic reticulum-bound protein and a member of non-haem lipid hydroxylases using diiron-oxygen as a cofactor.[5] The past few years have seen important advances in understanding the function of 25-hydroxycholesterol (4) as a key modulator of immune cell function and inhibitor of viral entry.[6] This oxysterol may also be produced in cholesterol-rich tissues via autoxidation,[7] a possibility that has received far less attention.
Many oxysterols found in living organisms are also present in air-aged cholesterol, which led to the speculation that some of them may have fundamental roles in mammalian cells.[8] Although secondary autoxidation products have been detected in cholesterol-rich human tissues, 24-hydroxycholesterol in human brain[9] and 27(26)-hydroxycholesterol in human atherosclerotic plaques[10] are mainly of enzymatic origin. Most oxysterols found in nature are derived from singlet oxygen attack at the cholesterol Δ5 double bond to give the 5- and 7-hydroperoxide derivatives and their decomposition products.[11] In contrast ground-state, bi-radical triplet oxygen attacks at tertiary side-chain carbons to give cholesterol 25-hydroperoxide (1) as a major product, together with small amounts of the epimeric 20ξ-hydroperoxides 2 and 3 (Scheme 1).[7] Minute amounts of cholesterol 26(27)- and 24-hydroperoxides have also been isolated from air-aged cholesterol.[12] Decomposition of these hydroperoxides accounts for the formation of a spate of secondary rearrangement products, including the reduced hydroxy analogs,[13] as well as a complex mixture of volatile products.[14] Thermal decomposition of the isomeric 20α- and 20β-hydroperoxides 2 and 3 yields pregnenolone as a major product, while mitochondrial CYP11A1 converts 2 and 3 to 20α,22R-dihydroxycholesterol (7) and 20β,21-dihydroxy-20-iso-cholesterol (8), respectively.[15] Even though 7 is a natural intermediate in the CYP11A1 mediated cholesterol side-chain cleaving reaction, the 20α-hydroperoxide 2 is not. Instead, the enzymatic convertion of cholesterol to pregnenolone is believed to proceed via three consecutative oxidation steps, to give 22R-hydroxycholesterol and 20α,22R-dihydroxycholesterol as enzyme-bound intermediates,[16] prior to cleaving the C20-22 bond to release pregnenolone from the enzyme.[17]
Scheme 1.

Structures of cholesterol 25-hydroperoxide (1), cholesterol 20α-hydroperoxide (2) and 20-iso-cholesterol 20β-hydroperoxide (3) and their metabolites. Incubation of (1) with either CYP27A1 or CYP11A1 gave 25-hydroxycholesterol (4) as major product. Secondary products include the 25,26-dihydroxycholesterol (5) from CYP27A1 and 25,22-dihydroxycholesterol (6) from CYP11A1. Incubation of 2 with CYP11A1 gave 20α,22R-dihydroxycholesterol (7), whereas the epimeric 20β-hydroperoxide 3 gave 20β,21-dihydroxy-20-iso-cholesterol (8) as single product.
Spectral changes in CYP27A1 upon addition of 25-hydroperoxide 1 gave a difference spectrum with a through shifting within 10 min from 422 to 416 nm (Figure 1A). Thin layer chromatography (TLC) of the MeCl2 extract showed that most of 1 had been reduced to the corresponding 25-hydroxycholesterol (4) as confirmed by identical gas chromatography-mass spectrometry (GC-MS) properties of the TMS derivative as compared to authentic 4. A small polar spot on the TLC suggested the presence of a triol product, that correlated to a new peak in the GC-MS profile of the TMS derivative (<30% yield rel. to 2), to which we assign the 25,27-dihydroxycholesterol (5) structure (Scheme 1) based on its characteristic GC-MS fragmentation pattern. Addition of 20α-hydroperoxide 2 to CYP27A1 induced difference spectrum with a maximum at 404 nm and a minimum at 425 nm (Figure 1B), a spectral response that does not seem to correspond to any of the reported types of P450 responses.[18] TLC analysis showed a small brown spot in the triol region and a small peak (<10%) in the GC-MS analysis with a MS fragmentation pattern consistent with 20α,27-dihydroxycholesterol. Addition of the 20β-hydroperoxide 3 to CYP27A1 also induced a difference spectrum similar to that elicited by the 20α-hydroperoxide 2 with a maximum at 408 nm and a minimum at 425 nm (Figure 1C). However, no metabolites were detected in the MeCl2 extracts as indicated by TLC and GC-MS analyses. Likewise, addition of hydroperoxides 1, 2 and 3 to CYP7A1 or CYP46A1 did not produce major reaction products as observed by TLC and GC-MS analysis. Spectral changes in the difference spectra of CYP7A1 suggested significant light scattering as indicated by a lack of well-defined peaks or troughs. Yet there was no visible protein precipitation in the cuvette, which could be a reason for increased solution light scattering. Light scattering was the smallest upon the addition of 25-hydroperoxide 1 and produced difference spectrum with a maximum at 384 nm and a minimum at 426 nm, parameters close to those of type 1 spectral response (maximum at 380–390 nm and minimum at 415–420 nm).[19]
Figure 1.

Difference spectra developed within 1–10 min after addition of cholesterol 25-hydroperoxide (1), cholesterol 20α-hydroperoxide (2) or 20-iso-cholesterol 20β-hydroperoxide (3) to CYP27A1 and CYP11A1. A) CYP27A1 and 1, B) CYP27A1 and 2, C) CYP27A1 and 3, D) CYP11A1 and 1, E) CYP11A1 and 2, F) CYP11A1 and 3.
Spectral changes in CYP11A1 upon addition of the 25-hydroperoxide 1 developed within 3 min to give a stable spectrum with a maximum at 388 nm and a minimum at 419 nm (Figure 1D). This type of a response is indicative of water displacement from the coordination sphere of the haem iron and is usually elicited by P450 substrates.[20] Analysis of the MeCl2 extract showed that most of 1 had been reduced to the 25-hydroxy analog 4, which was in all respects identical to authentic 4 (Scheme 1). TLC analysis of the MeCl2 extract and GC-MS analysis of the TMS derivatives revealed the presence of a minor product to which we assign the 22,25-dihydroxycholesterol (6) structure based on its characteristic mass spectral fragmentation pattern (Scheme 1). The positon of the second hydroxy in 5 and 6 corresponds to the natural cholesterol hydroxylation sites of CYP27A1 and CYP11A1, i.e. C27 and C22, respectively, suggesting specific binding of the 25-hydroperoxide 1 in the P450 haem pockets. Addition of the 20α-hydroperoxide 2 to CYP11A1 induced within 1 min a classical type 1 difference spectrum with a maximum at 388 nm and a minimum at 419 nm (Figure 1E). The isomeric 20β-hydroperoxide 3 gave a similar type of response; except the maximum was shifted to 403 nm and minimum to 423 nm and it took a longer time (~10 min) for the spectrum to develop (Figure 1F). Extraction of the incubation mixtures with MeCl2 followed by TLC analysis showed that each isomer 2 and 3 was transformed to a single glycol product identified as the 20α,22R-glycol 7 and the 20β,21-glycol 8, respectively (Scheme 1). Assigned structures were confirmed by comparison of GC-MS fragmentation pattern to those of authentic samples.[12c]
The stereospecific 20ξ-hydroperoxide-glycol reactions can be envisaged by substituting 22R-hydroxycholesterol with 20α-hydroperoxide 2 or 20β-hydroperoxide 3 in the active site of the previously determined 22R-hydroxycholesterol-CYP11A1 co-complex[2b] (Figure 2). The model shows the hydroperoxide oxygen of 2 at a distances of 2.7 Å from the Fe(III) haem iron allowing formation of the transient haem-Fe(IV)=O+• species (Cpd I) that subsequently can oxidize the adjacent 22R-position of 2 (4 Å from the haem iron) to yield the 20α,22R-glycol 7. Changing the configuration about C20 to the 20β-isomer 3 places the hydroperoxide oxygen at a distance of 3.1 Å and the 21-methyl at 3.5 Å from the haem iron, favouring the formation of the 20β,21-glycol 8 (Figure 2). The involvement of a reactive transient intermediate Cpd I in the hydroperoxide-glycol conversion was previously proposed using adrenal bovine mitochondrial CYP11A1 and supported by intermediate product analysis,[21] stop-flow UV/Vis spectral data, and electron paramagnetic resonance measurements.[22]
Figure 2.
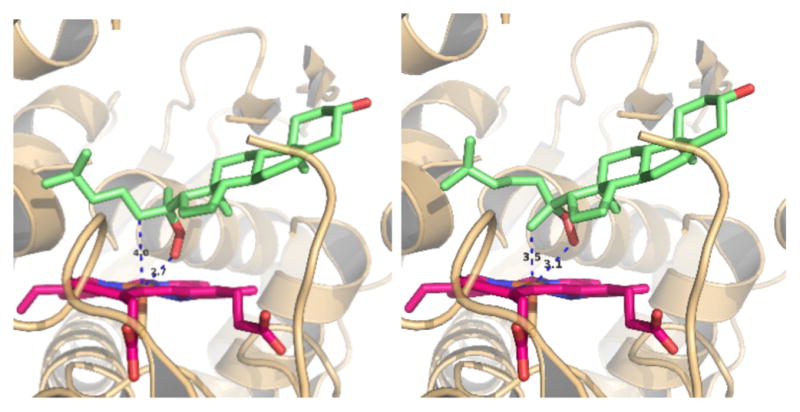
Putative positions of cholesterol 20α-hydroperoxide (2) (left panel) and 20-iso-cholesterol 20β-hydroperoxide (3) (right panel) in the active site of CYP11A1. Steroids were placed instead of 22R-hydroxycholesterol in the crystal structure of the previously determined 22R-hydroxycholesterol-CYP11A1 co-complex.[2b] The model shows the 20α-hydroperoxide oxygen of 2 at a distances of 2.7 Å from the Fe(III) haem iron permitting formation of the transient haem-Fe(IV)=O+• species (Cmp I) that subsequently hydroxylates the adjacent 22R-position of 2 (4 Å from the haem iron) to yield the 20α,22R-glycol 7. In the case of the 20β-isomer 3 the hydroperoxide oxygen is at a distance of 3.1 Å and the 21-methyl at 3.5 Å the Fe(III) haem iron resulting in the formation of the 20β,21-glycol 8 (Scheme 1).
Our data conform a reaction mechanism (Scheme 2) whereby the cholesterol hydroperoxide substrates 1-3 bind in the CYP27A1 or CYP11A1 haem pocket providing a reduced oxygen atom to support a hydroperoxide-shunt reaction to yield the intermediate haem-Fe(IV)=O+• species (Cpd I). This shunt does not require CYP27A1 redox partners nor endogenous NADPH, a source of reducing equivalents. Depending on the proximity of the reactive oxygen of Cpd I to the cholesterol side-chain, the latter may be hydroxylated to give dihydroxycholesterols, or dissipate while the reduced hydroxy compound is released. This shunt is well established for xenobiotic metabolizing P450[3] but here for the first time shown to be operative for CYP27A1 and CYP11A1. The metabolism of 25-hydroperoxide 1 by CYP27A1, a mitochondrial enzyme, may complement the 25-hydroxycholesterol (4) production from cholesterol by C25H, a microsomal enzyme, and thus provide a source of a biologically actie oxysterol to different cellular compartments. Metabolism of 25-hydroperoxide (1) by CYP27A1 may also serve a purpose of elimination of a highly reactive hydroperoxide that may further oxidize other lipids and thereby damage the cells where it is produced.
Scheme 2.

Hydroperoxide-shunt in the catalysis of cholesterol 25-hydroperoxide (1) by CYP27A1 showing the proposed intermediates (adapted from ref. 3). The 25-hydroperoxide 1 binds to the oxidized (FeIII) haem-iron of CYP27A1 (A) with the release of a hydrogen ion (H+) to give intermediate (B). Heterolysis of the O-O hydroperoxide bond and H+ addition gives Cpd I (C) and 25-hydroxycholesterol (4), which is either released with the dissipation of Cpd I (broken arrow), or the latter reacts with the nearby C27 carbon of 4 to give complex D from which the 25,27-dihydroxycholesterol (5) is released. In the case of the reaction of 1 and CYP11A1 the secondary triol product is 22,25-dihydroxycholesterol (6). Both CYP27A1 and CYP11A1 give 25-hydroxycholesterol (4) is the principal product. In the case of the interaction of the isomeric 20ξ-hydroperoxides 2 and 3 and CYP11A1, specific binding in the haem pocket (Figure 2) results in formation of triols 7 and 8, respectively, without release of the intermediate 20-hydroxy analogs.
In summary, the isolation of cholesterol 25-hydroperoxide (1) and the epimeric 20-hydroperoxides 2 and 3 from air-aged cholesterol, as well as the availability of all four purified recombinant, cholesterol-metabolizing cytochrome P450 enzymes provided us with the unique opportunity to evaluate a potential biological role of their interactions using spectroscopy and metabolite analysis. Here we have shown for the first time that both CYP27A1 and CYP11A1 reduce cholesterol 25-hydroperoxide (1) to 25-hydroxycholesterol (4) while generating small amounts of dihydroxycholesterols. The epimeric 20ξ-hydroperoxides 2 and 3 reacted with CYP11A1 only to give characteristic glycol products. A reaction mechanism involving a P450 hydroperoxide-shunt reaction is proposed, whereby the sterol hydroperoxides provide a reduced oxygen atom to generate Cpd I and the corresponding reduced hydroxysterol, while simultaneously serving as substrate for further hydroxylation. Our results provide for the first time in vitro evidence for a role of CYP27A1 in cholesterol-rich tissues with high oxidative stress to function as catalyst for the reduction of cholesterol 25-hydroperoxide to 25-hydroxycholesterol, a modulator of immune cell function and mediator of viral cell entry. Elimination of highly reactive cholesterol hydroperoxides by CYP21A1 may prevent peroxidative chain reactions that can inflict cellular damage.
Experimental Section
The cholesterol 25-hydroperoxide (1), cholesterol 20α-hydroperoxide (2) and the 20-iso-cholesterol 20β-hydroperoxide (3), were isolated from a >15 year old sample of 500 g cholesterol (Scheme 1).[7,12c] The 20-hydroxycholesterols, 22R-hydroxycholesterol, 25-hydroxycholesterol (2), 27-hydroxycholesterol, and pregnenolone were from Steraloid Inc. (Pawling, N.Y.). The 20α,22R-dihydroxycholesterol (7) was prepared by established methods.[23] The 20α,21-dihydroxycholesterol and epimeric 20β,21-dihydroxy-20-iso-cholesterol (8) were prepared from 21-hydroxypregnenolone and addition of the side chain via a Grignard reaction.[12a,d] The assigned configurations were confirmed by X-ray crystallography.
Bovine recombnant CYP11A1 and human recombinant CYP27A1, CYP46A1 and CYP7A1 were expressed and purified as described.[2b,24] P450 difference spectra were recorded as described,[25] except two double, not single, cuvettes were used to compensate for a residual hydroperoxide absorption in the visible region. The concentrations of P450 and cholesterol hydroperoxide were 5 μM and 10 μM, respectively. Incubations were carried out at 24 °C. The incubation buffers were 50 mM potassium phosphate (KPi), pH 7.2, containing 1 mM EDTA (for CYP11A1 and CYP7A1); 50 mM KPi, pH 7.2, containing 0.5 M NaCl, 10% glycerol, and 1 mM EDTA (for CYP27A1); and 50 mM KPi, pH 7.2, containing 0.1 M NaCl (for CYP46A1). For product analyses, P450s were incubated with cholesterol hydroperoxides for 20 min followed by sterol extraction with MeCl2 and analysis by TLC as previously described.[15e] The remaining sample was derivatized with bis-(trimethylsilyl) trifluoroacetamide/trimethylchlorosilane (TMS) and analyzed by GC-MS as described.[26]
Supplementary Material
Acknowledgments
Supported in part by the Jeanne and J.-Louis Lévesque Chair in radiobiology of the Université de Sherbrooke (to J.E.v.L.) and United States Public Health Service Grant GM62882 (to I.A.P). J.E.v.L. is a member of the Fonds de la Recherche en Santé du Québec-supported CRCHUS, Sherbrooke, QC, Canada. I.A.P. is a recipient of the Jules and Doris Stein Professorship from the Research to Prevent Blindness. The authors thank Hasrat Ali, Guillaume Langlois and Yves Dory for technical support.
Contributor Information
Johan E. van Lier, Email: Johan.E.vanlier@USherbrooke.ca.
Irina A. Pikuleva, Email: iap8@case.edu.
References
- 1.Pikuleva IA. Drug Metab Dispos. 2006;34:513–520. doi: 10.1124/dmd.105.008789. [DOI] [PubMed] [Google Scholar]
- 2.a) Mast N, White MA, Bjorkhem I, Johnson EF, Stout CD, Pikuleva IA. Proc Natl Acad Sci USA. 2008;105:9546–9551. doi: 10.1073/pnas.0803717105. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mast N, Annalora AJ, Lodowski DT, Palczewski K, Stout CD, Pikuleva IA. J Biol Chem. 2011;286:5607–5613. doi: 10.1074/jbc.M110.188433. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Strushkevich N, MacKenzie F, Cherkesova T, Grabovec I, Usanov S, Park HW. Proc Natl Acad Sci USA. 2011;108:10139–10143. doi: 10.1073/pnas.1019441108. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Tempel W, Grabovec I, MacKenzie F, Dichenko YV, Usanov SA, Gilep AA, Park HW, Strushkevich N. J Lipid Res. 2014;55:1925–1932. doi: 10.1194/jlr.M050765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luthra A, Denisov IG, Sligar SG. Arch Biochem Biophys. 2011;507:26–35. doi: 10.1016/j.abb.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Rittle J, Green M. Science. 2010;330:933–937. doi: 10.1126/science.1193478. [DOI] [PubMed] [Google Scholar]; McQuarters AB, Wolf MW, Hunt AP, Lehnert N. Angew Chem Int Edit. 2014;53:4750–4752. doi: 10.1002/anie.201402404. [DOI] [PubMed] [Google Scholar]
- 5.a) Russell DW. Annu Rev Biochem. 2003;72:137–174. doi: 10.1146/annurev.biochem.72.121801.161712. [DOI] [PubMed] [Google Scholar]; b) Lund EG, Kerr TA, Sakai J, Li WP, Russell DW. J Biol Chem. 1998;273:34316–34327. doi: 10.1074/jbc.273.51.34316. [DOI] [PubMed] [Google Scholar]
- 6.a) Cyster JG, Dang EV, Reboldi A, Yi T. Nature. 2014;14:731–742. doi: 10.1038/nri3755. [DOI] [PubMed] [Google Scholar]; b) Reboldi A, Dang EV, McDonald JG, Liang G, Russell DW, Cyster JG. Science. 2014;345:679–684. doi: 10.1126/science.1254790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Lier JE, Smith LL. J Org Chem. 1970;35:2627–2632. doi: 10.1021/jo00833a031. [DOI] [PubMed] [Google Scholar]
- 8.a) Smith LL. Cholesterol Autoxidation. New York: Plenum Press; 1981. [Google Scholar]; b) Smith LL. Lipids. 1996;31:453–487. doi: 10.1007/BF02522641. [DOI] [PubMed] [Google Scholar]; c) Yavitt NB. Steroids. 2008;73:149–157. doi: 10.1016/j.steroids.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 9.Schubert K, Rose G, Buerger M. H-S Z Physiol Chem. 1961;326:235–241. doi: 10.1515/bchm2.1961.326.1.235. [DOI] [PubMed] [Google Scholar]
- 10.van Lier JE, Smith LL. Biochemistry. 1967;6:3269–3278. doi: 10.1021/bi00862a037. [DOI] [PubMed] [Google Scholar]
- 11.Teng JI, Kulig MJ, Smith LL, Kan G, van Lier JE. J Org Chem. 1973;38:119–123. doi: 10.1021/jo00941a024. [DOI] [PubMed] [Google Scholar]
- 12.a) van Lier JE, Smith LL. J Org Chem. 1971;36:1007–1009. doi: 10.1021/jo00806a038. [DOI] [PubMed] [Google Scholar]; b) van Lier JE, Kan G. J Org Chem. 1972;37:145–147. [PubMed] [Google Scholar]
- 13.van Lier JE, Smith LL. Steroids. 1970;15:483–503. doi: 10.1016/s0039-128x(70)80078-2. [DOI] [PubMed] [Google Scholar]
- 14.van Lier JE, DaCosta AL, Smith LL. Chem Phys Lipids. 1975;14:327–335. doi: 10.1016/0009-3084(75)90069-9. [DOI] [PubMed] [Google Scholar]
- 15.a) van Lier JE, Smith LL. Biochem Biophys Acta. 1970;210:153–163. doi: 10.1016/0005-2760(70)90070-6. [DOI] [PubMed] [Google Scholar]; b) van Lier JE, Smith LL. Biochim Biophys Acta. 1970;218:320–326. doi: 10.1016/0005-2760(70)90070-6. [DOI] [PubMed] [Google Scholar]; c) van Lier JE, Smith LL. Biochem Biophys Res Comm. 1970;40:510–516. doi: 10.1016/0006-291x(70)90931-9. [DOI] [PubMed] [Google Scholar]; d) van Lier JE, Rousseau J. FEBS Letters. 1976;70:23–27. doi: 10.1016/0014-5793(76)80718-1. [DOI] [PubMed] [Google Scholar]; e) Larroque C, van Lier JE. J Biol Chem. 1986;261:1083–1087. [PubMed] [Google Scholar]
- 16.Larroque C, Rousseau J, van Lier JE. Biochemistry. 1981;20:925–929. doi: 10.1021/bi00507a043. [DOI] [PubMed] [Google Scholar]
- 17.a) Burstein S, Gut M. Steroids. 1976;28:315–311. doi: 10.1016/0039-128x(76)90131-8. [DOI] [PubMed] [Google Scholar]; b) Duque C, Morisaki M, Ikekawa N, Shikita M. Biochem Biophys Res Commun. 1978;82:179–187. doi: 10.1016/0006-291x(78)90593-4. [DOI] [PubMed] [Google Scholar]
- 18.a) Schenkman JB, Remmer H, Estabrook RW. Mol Pharmacol. 1967;3:113–123. [PubMed] [Google Scholar]; b) Dawson JH, Andersson LA, Sono M. 1982;257:3606–3617. [PubMed] [Google Scholar]
- 19.Mailman RB, Kulkarni AP, Baker RC, Hodgson E. Drug metabolism and disposition: the biological fate of chemicals. 1974;2:301–308. [PubMed] [Google Scholar]
- 20.Poulos TL, Finzel BC, Howard AJ. J Mol Biol. 1987;195:687–700. doi: 10.1016/0022-2836(87)90190-2. [DOI] [PubMed] [Google Scholar]
- 21.a) Larroque C, van Lier JE. J Biol Chem. 1986;261:1083–1087. [PubMed] [Google Scholar]; b) van Lier JE, Rousseau J, Langlois R, Fisher GJ. Biochem Biophys Acta. 1977;487:395–399. doi: 10.1016/0005-2760(77)90016-9. [DOI] [PubMed] [Google Scholar]
- 22.a) Larroque C, van Lier JE. Biochem Biophys Res Commun. 1983;112:655–662. doi: 10.1016/0006-291x(83)91513-9. [DOI] [PubMed] [Google Scholar]; b) Larroque C, Lange R, Morin L, Bienvenue A, van Lier JE. Arch Biochem Biophys. 1990;282:198–201. doi: 10.1016/0003-9861(90)90104-7. [DOI] [PubMed] [Google Scholar]
- 23.Morisaki M, Sato S, Ikekawa N. Chem Pharm Bull. 1977;25:2576–2583. [Google Scholar]
- 24.a) Mast N, Murtazina D, Liu H, Graham SE, Bjorkhem I, Halpert JR, Peterson J, Pikuleva IA. Biochemistry. 2006;45:4396–4404. doi: 10.1021/bi052654w. [DOI] [PubMed] [Google Scholar]; b) White MA, Mast N, Bjorkhem I, Johnson EF, Stout CD, Pikuleva IA. Acta crystallographica. 2008;64:487–495. doi: 10.1107/S0907444908004046. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Nakayama K, Puchkaev A, Pikuleva IA. J Biol Chem. 2001;276:31459–31465. doi: 10.1074/jbc.M103943200. [DOI] [PubMed] [Google Scholar]
- 25.Mast N, Zheng W, Stout CD, Pikuleva IA. J Biol Chem. 2013;288:4613–4624. doi: 10.1074/jbc.M112.438754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mast N, Reem R, Bederman I, Huang S, DiPatre PL, Bjorkhem I, Pikuleva IA. Invest Ophthalmol Vis Sci. 2011;52:594–603. doi: 10.1167/iovs.10-6021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.