

Summary
Encounters between immune cells and invading bacteria ultimately determine the course of infection. These interactions are usually measured in populations of cells, masking cell-to-cell variation that may be important for infection outcome. To characterize gene expression variation that underlies distinct infection outcomes, we developed an experimental system that combines single-cell RNA-seq with fluorescent markers, monitoring infection phenotypes. Probing the responses of individual macrophages to invading Salmonella, we find that variation between individual infected host cells is determined by the heterogeneous activity of bacterial factors in individual infecting bacteria. We illustrate how variable PhoPQ activity in the population of invading bacteria drives variable host Type I IFN responses by modifying LPS in a subset of bacteria. This work demonstrates a causative link between host and bacterial variability, with cell-to-cell variation between different bacteria being sufficient to drive radically different host immune responses. This co-variation has implications for host-pathogen dynamics in vivo.
Keywords: Single-cell RNA-seq, host-pathogen interaction, Type I interferon response, Salmonella typhimurium, macrophage
Introduction
Interactions between a pathogen and its host involve both a complex virulence program executed by pathogens and activation of an orchestrated defense response by the host (Schwan et al., 2000). Genomic approaches – profiling either the host, the pathogen or both – have been employed in recent years to uncover substantial molecular details about host and bacterial factors that underlie infection outcomes (Eriksson et al., 2003). However, to date, these genomic studies have been typically based on averaging cellular behaviors across populations (Helaine et al., 2010), whereas the heterogeneous, stochastic, and dynamic nature of both host and pathogens suggests that descriptions of average behavior may fail to accurately characterize their interactions (Jaitin et al., 2014). For example, studies using flow cytometry and microscopy indeed indicate that disparate Salmonella-macrophage encounters give rise to diverse subpopulations with dramatically different individual outcomes (Claudi et al., 2014). Recent advances in single-cell expression analysis provide an attractive approach to probe subpopulations and cell-to-cell variability (Shalek et al., 2014).
One of the best-studied cellular models of the host-pathogen interaction is infection of macrophages with the enteric pathogen Salmonella enterica serovar typhimurium (S. typhimurium). S. typhimurium is a facultative, intracellular, Enterobacteriaceae that causes a range of enteric diseases in mammalian hosts. It has evolved to evade host defenses by sensing the transition from extracellular to intravacuolar environments, triggering a global modulation of gene expression that activates diverse virulence strategies, including alterations of pathogen-associated molecular patterns (PAMPs) and secretion of compounds to alter macrophage response (Galan and Collmer, 1999). In a single population, both in vitro and in vivo, S. typhimurium has been shown to display significant cell-to-cell variation in attributes such as growth rate, expression of virulence factors, and sensitivity to antibiotics (Claudi et al., 2014). Using receptors that recognize PAMPs (e.g., lipopolysaccharides (LPS) by Toll-like receptor 4 (Tlr4)), macrophage detection of invading bacteria results in a transcriptional response that leads to the production of inflammatory cytokines and a variety of effector defense mechanisms (Rosenberger et al., 2000). Like S. typhimurium, macrophages and other innate immune cells have been observed to display extensive cell-to-cell variation upon exposure to even homogeneous ligands (Shalek et al., 2014). Recent studies using single-cell RNA-seq have found subsets of dendritic cells with differential responses to LPS stimulation both in vitro (Shalek et al., 2014) and in vivo (Jaitin et al., 2014).
The heterogeneous, stochastic, and dynamic nature of both macrophage and Salmonella populations suggests that their interaction is likely to result in a variety of subpopulations with different, complex phenotypes (Helaine et al., 2010). Indeed, infection of macrophages with Salmonella generates well-documented diverse outcomes: some macrophages engulf the bacteria, while others remain uninfected (McIntrye et al., 1967); some macrophages lyse the ingested bacteria, while others are permissive to intracellular bacterial survival (McIntrye et al., 1967); some macrophages will undergo cell death with bacterial release (Monack et al., 1996), while others survive and allow bacteria to multiply or persist intracellularly (Helaine et al., 2010). Despite longstanding observations of these diverse outcomes however, we currently lack an understanding of the underlying molecular mechanisms in either the host or pathogen.
How macrophages integrate signals from bacterial PAMPs to determine cell fate, and how bacteria regulate different virulence strategies to optimize pathogenicity in the host environment are fundamental to understanding infection biology and finding novel treatment options for infectious disease. Understanding the basis and significance of heterogeneity could inform strategies that result in a more beneficial outcome to the host. The discovery that distinct subpopulations of immune cells vary in their transcriptional responses to uniform PAMPs (Shalek et al., 2014) suggests that there may be some variability in the intrinsic state of the host cells that accounts for their differential response. Adding complexity, infection with live bacteria, which have diverse regulatory states themselves, might result in an even wider range of transcriptional interactions with implications for infection outcome.
Here, we set out to test whether and how distinct infection outcomes are reflected in the transcriptional status of individual host cells, to decipher the mechanistic underpinnings of this variation in both the host and bacteria, and to examine the relationship of this variation to infection outcomes in vivo.
Results
Heterogeneous outcomes of Salmonella-macrophage encounters
To quantitatively characterize outcomes of individual S. typhimurium-macrophage interactions, we developed a fluorescent system using GFP-expressing bacteria stained with the red dye pHrodo (Experimental Procedures), which binds to the cell wall of bacteria and increases in fluorescence in the low pH environment of macrophage lysosomes. In the early stages after S. typhimurium challenge, there are three possible outcomes (Figure 1A and S1A): (1) no infection, (2) infection with intracellular survival of a bacterium, and (3) infection resulting in an intracellular dead bacterium. While live bacteria display both red and green fluorescence, dead bacteria fluoresce only red due to degradation of GFP. Exposed but uninfected macrophages do not fluoresce (Figure 1A). Importantly, using the GFP and pHrodo reporters we could distinguish cells that had been initially infected but cleared the infecting bacterium (pHrodo+, GFP–) from those that had never been infected (pHrodo–, GFP–). We used this system to follow mouse bone marrow-derived macrophages (BMMs) exposed to pHrodo-stained, GFP-expressing S. typhimurium at a multiplicity of infection (MOI) of 1:1 for 24 hours. Importantly, we used a low MOI to ensure that infected macrophages are generally infected with only one bacterium.
Figure 1. Heterogeneous outcomes of BMM-Salmonella encounters are captured by single-cell expression analysis.

(A) Schematic representation of the experimental model, using BMMs infected with pHrodo-labeled, GFP-expressing S. typhimurium. (B) Representative images of mouse BMMs exposed to S. typhimurium reveals heterogeneity in infection phenotype including uninfected macrophages, and infected macrophages containing live (yellow) or dead (red) bacteria at early (4 hours; top) and late (24 hours; bottom) time points. (C) FACS analysis of fluorescently labeled populations (unexposed-left, exposed for 4 hours-right). (D) CFU enumerated from individual fluorescently labeled macrophages. Unexposed, uninfected and pHrodo+,GFP– cells had no or minimal surviving bacteria. GFP+ cells contain different numbers of cells over time (left y-axis). The red line indicates the percentage of pHrodo-only infected cells demonstrating the increase in the number of dead bacteria over time (right Y axis). (E) Single macrophages have distinct transcriptional responses depending on infection phenotype. 96 single cells from (C) were analyzed by RNA-seq and principle component analysis. Shown are the first two principal components (PC1 and PC2, 5 and 3 percent of the total variation respectively, Table S1B).
Microscopy and FACS revealed diverse phenotypes, including uninfected cells and cells infected with single or multiple, live (yellow) or dead (red) bacteria, as has been previously described (McIntrye et al., 1967) (Figures 1B, 1C). This variability is neither simply a transient phenomenon nor a mere outcome of the specific MOI chosen, since it is sustained throughout the 24 hour time course (Figure S1B) and with increasing MOI (Figure S1C). To better quantify bacterial burden in single cells, we sorted macrophages according to fluorescence phenotype and enumerated the number of intracellular bacteria by plating for colony forming units (CFU) (Experimental Procedures). As expected, no viable bacteria were recovered from uninfected or pHrodo+, GFP– (dead bacteria) infected cells. GFP+, pHrodo+ cells contained a range of bacteria (Figure 1D), which correlated with GFP intensity (Figures S1D, S1E), and showed reduction in bacterial burden during the twenty-four hours time course, similar to other studies using primary cells infected at low MOIs (Monack et al., 1996; Schwan et al., 2000). Thus, individual host cells may vary widely in their ability to phagocytose bacteria and/or restrict bacterial growth after uptake.
Single-cell RNA-seq of exposed macrophages accurately distinguishes transcriptional changes associated with extracellular and intracellular bacterial detection
To determine if cell-to-cell transcriptional variation in host cells may underlie some of these different outcomes, we used FACS to sort single macrophages based on fluorescence and generated single-cell RNA-seq libraries from individually sorted cells, as well as a time course of sorted populations of 150 cells (using SMART-Seq (Trombetta et al., 2014); Experimental Procedures). To ensure that cell sorting and library construction methods did not significantly alter the measured cellular response, we also analyzed a time course of bulk RNA-seq libraries from entire exposed populations (5×105 cells) using Illumina's Tru-seq library construction method (Experimental Procedures). We found good agreement in expression patterns (Figure S1F, G) and differentially expressed genes (Figure S1H, I) among all three data sets, despite lower sensitivity from single cells particularly for low abundance transcripts, as previously reported (Shalek et al., 2014).
Single-cell profiles clearly distinguished cells with different phenotypic states. We used a list of 535 genes that are upregulated in exposed macrophages in our single-cell libraries (DEseq p<0.05 and fold change > 2 fold, Experimental Procedures, Table S1B) to perform Principal Component Analysis (PCA) on the single-cell expression data. PCA clearly distinguished both between exposed/unexposed macrophages (mostly on PC1) and between infected and uninfected macrophages (mostly on PC2) (Figures 1E, S1J). Taken together, the ability to distinguish these different phenotypes suggests that some pathways respond primarily to extracellular cues of bacterial presence, while others respond to intracellular cues.
To better understand these distinct responses, we calculated a metric we term the “intracellular to extracellular response ratio” reflecting the magnitude of induction accounted for by bacterial infection verses extracellular bacterial exposure (Figure 2A, Experimental Procedures). We then classified genes based on their mode of response: Cluster I contains genes that respond primarily to extracellular cues of bacterial presence, and Cluster II contains genes that respond primarily to intracellular cues. Supporting our classification, many Cluster I genes (responding to bacterial exposure - i.e., PC1 above) are known to be associated with the classic LPS response (e.g., Tnf and NFκB) and many Cluster II genes (responding to intracellular bacteria - i.e., PC2 above) are known to be associated with antibacterial defense (e.g., Nos2 and IL12b). While Cluster I was relatively stable across different time points, many genes in Cluster II were found to be induced also in uninfected cells at later time points (Figure S2A). At these early time points (8 hours) we did not detect differences between pHrodo+,GFP+ and pHrodo+, GFP– infected cells, so these groups were merged for further analysis. While it will be interesting to also study possible differences between these infected populations at later time points (12-24 hours), in this work we focused on analyzing the variation between single infected cells, as discussed below for a third cluster (Cluster III).
Figure 2. Single-cell expression profiling reveals macrophage subpopulations in infected cells.

(A) Expression levels of genes (rows) in single BMMs (columns) were measured using single-cell RNA-seq after infection with S. typhimurium and grouped by their infection phenotype (unexposed (white, n=23), uninfected (grey, n=24), infected (green, n=42)). Genes are categorized into two cluseters as described. The number of genes in each cluster is denoted next to the heat map. Genes are arranged by the extracellular or intracellular ratio (IC/EC ratio, left bars indicate distribution of scores for each cluster, Table S2A). (B) Analysis of gene correlations across single cells revealed a cluster of bimodally expressed genes in infected cells (Cluster III). Cells in (A) and (B) are sorted according to average expression of Cluster III. (C) Highly variable genes in infected cells are enriched for immune response pathways (Table S2C). Localized regression was used to estimate the mean/variance relationship for genes in infected macrophages. Genes were assigned a variance score based on distance from the fitted relationship (solid line). (D) Shown are box plots of variance score for either exposure (Cluster I) or infection response genes (Cluster II), at three time points following infection. Infection response genes have reproducibly higher variance then exposure response genes (p<0.01 by Wilcoxon rank-sum test, Table S2D). (E) Representative examples of single-cell gene expression distributions in infected cells from Cluster I, II and III.
Bimodal induction of Type I IFN response genes in infected macrophages
It has been previously suggested that immune networks may be structured to produce subpopulations of cells with distinct physiologies (Jin et al., 2014). Thus, we searched for additional clusters of genes that co-vary across multiple time points using weighted gene correlation network analysis (Experimental Procedures). We identified three gene clusters (Clusters III, IV, and V) that met these criteria (Figures 2B, S2B, S2C). Cluster III was particularly interesting as it was significantly enriched for the Type I IFN response (Table S2B), which has previously been shown to play a role in non-canonical inflammasome activation in response to infection with S. typhimurium (Rathinam et al., 2012). Cluster III is induced in approximately one third of infected macrophages beginning at 4 hours post exposure and continues to show bimodal expression at 8 hours, suggesting that this induction is not a transient phenomenon (Figure S2C). Notably, this cluster is induced also in uninfected cells at 8 hours. This may not be surprising, given that interferon is a secreted soluble factor that may result in a non-cell autonomous induction of this cluster later in uninfected cells (Honda and Taniguchi, 2006). Cluster IV is enriched for cell-cycle genes, is bimodal in unexposed cells, and decreases in expression upon exposure. It does not differentiate between uninfected, pHrodo+,GFP− or pHrodo+,GFP+ cells at any time point (Mann-Whitney test, p>0.05). Cluster V is highly expressed in all unexposed cells and has reduced expression in some cells upon exposure (becomes bimodal).
We verified representative expression patterns in Cluster II and III genes using single-molecule RNA fluorescence in situ hybridization (FISH). Using pHrodo to identify infected cells, we confirmed both the induction of Cluster II in all infected cells (e.g., Il1b, Il12b, and Nos2) and the bimodal induction of Cluster III (e.g., Irf7 and Ifit2) in infected cells (Figure S2D). This method also allowed us to directly verify that the expression of Cluster III was not correlated with GFP fluorescence, indicating that the heterogeneity we observe is not merely due to differences in bacterial burden (Figure S2E). It is important to note that single cell analysis was required to identify the induction of Cluster III between infected and uninfected cells. Analyzing sorted populations (Figure S2F) failed to identify these genes as they are not highly induced when averaged over all cells.
Infected macrophages display high cell-to-cell variation in genes from immune response pathways
Motivated by the high variation of the Type I IFN response between infected cells, we next examined whether immune responsive pathways in general show high variation between infected cells. We developed a scoring system based on localized linear regression to estimate each gene's variance in a manner that is largely independent of mean expression (Experimental Procedures, Figure 2C). We then tested each pathway (as annotated in MSigDB) for its enrichment in variable genes using GSEA (Experimental Procedures), and identified those pathways that are consistently variable across multiple time points post exposure in infected macrophages. As expected from previous reports (Shalek et al., 2013), many pathways associated with housekeeping functions, such as ribosome function and oxidative phosphorylation, show consistently low variation. On the other hand, many pathways involved in the immune response including Toll-like receptor signaling, cyctokine-cytokine receptor interactions and Rig-I receptor signaling show consistently high variance up to at least 8 hours after bacterial infection (Table S2C). Furthermore, at all time points evaluated, genes induced primarily by the intracellular bacterial signals of infection (Cluster II) were more variable than those induced by extracellular exposure cues in infected macrophages (Cluster I; Figures 2D, 2E). This difference suggests that within a seemingly homogenous population of infected cells there exists extensive cell-to-cell variation in the response to infection. This variation is characteristic of responses to intracellular cues of infection more than those to extracellular cues, possibly due to variability in intracellular bacterial state, bacterial burden, or bacterial clearance.
Intracellular Tlr4 signaling through Trif and Irf3 determines the activation of the Type I IFN response in infected cells
It has been previously suggested that LPS accounts for all the transcriptional responses to infection, including intracellular bacterial detection (Rosenberger et al., 2000). LPS is detected by Tlr4, which signals through two different adaptor proteins Myd88 or Trif, depending on whether LPS is sensed at the cell membrane or at a phagosome, respectively (Kagan et al., 2008). Specifically, induction of the Type I IFN response was shown to be mediated by Trif through the interferon regulatory factors Irf3 and Irf7 (Fitzgerald et al., 2003). We hypothesized that the differential activation of Cluster III in infected cells may depend on key components of Tlr4/LPS signaling. Thus, we measured the transcriptional response of wild-type (WT), Tlr4−/−, Trif−/−, and Myd88−/− immortalized BMMs (iBMMs) (Experimental Procedures) to infection with S. typhimurium at the single-cell level by monitoring an expression signature of 96 genes representative of Clusters I, II, and III using qRT-PCR (Figure S3A, Table S2D, Experimental Procedures). Compared to WT cells, we found ablated activation of all three clusters in Tlr4−/− cells (Figure 3A), suggesting that LPS and Tlr4 sensing dominate the transcriptional responses to infection, as previously suggested (Rosenberger et al., 2000). Next, to analyze the transcriptional response to infection of Myd88−/− and Trif−/− cells, we defined a “Trif-Myd88 ratio” to assess the dependence of each gene's expression on Trif versus Myd88 (Figure 3A, Experimental Procedures). We found that in Cluster I and Cluster II regulation is partitioned, with some genes being regulated by Myd88 and some by Trif. Cluster III on the other hand, is regulated almost entirely through Trif (Figure 3A). Interestingly, Myd88 knockout upregulated this cluster in both infected and uninfected cells, which may indicate Myd88-dependent negative feedback inhibition of Cluster III induction.
Figure 3. Analysis of macrophage pathways regulating the bimodal induction of the Type I IFN response.

(A) Induction of Cluster III is solely dependent on Trif signaling. iBMMs from WT, Tlr4−/−, Myd88−/− or Trif−/− mice were infected with S. typhimurium and expression of single cells was analyzed. Genes are arranged by a score summarizing their Myd88 or Trif dependence (MTR, left bars indicate distribution of scores for each cluster, Table S3A). (B) BMMs from Irf3−/− and Irf7−/− mice were infected with pHrodo-stained GFP-labeled S. typhimurium. Decreased induction of representative genes from Cluster III was evident in Irf3−/− cells, compared to increased induction levels in Irf7−/− cells. (Table S3B) (C) BMMs were infected with pHrodo-labeled GFP-labeled S. typhimurium, in the presence of BI2536 and BX795. While BI2536 inhibited mostly Cluster III genes but also genes from Cluster I and II, BX795 specifically inhibits only the induction of only Cluster III genes. (Table S3C) (D) Schematic representation of the gene regulatory networks that control the response of macrophages to S. typhimurium infection. The induction of the Type I IFN response is due to activation of Tbk1 and Irf3 in only a subset of infected cells. (E) Plots summarize the expression of each gene cluster in BMMs infected with live bacteria (top panel) or with LPS coated beads (bottom panel) using a weighted average of scaled expression values (x-axis) verses the frequency of single cells (y-axis). In contrast to the bimodal activation of the Type I IFN response in cells infected with live bacteria, there was a much higher proportion of cells that activated Cluster III among the cells that had taken up LPS coated beads.
Next, we infected BMMs from WT, Irf3−/− and Irf7−/− mice and found that knockout of Irf3 exclusively ablates the activity of Cluster III in infected cells, while Irf7 knockout enhances its activation (Figure 3B). This suggests that while Trif has a role in the induction of all clusters, its activation of Irf3 is specific to Cluster III and occurs in only a subset of infected cells. Based on these results, we tested two known inhibitors of the Type I IFN response, BX795 (a TBK1 inhibitor, (Lee et al., 2013)) and BI2536 (a PLK inhibitor, (Chevrier et al., 2011)) and found that while BI2536 inhibited genes from all three clusters, BX795 specifically inhibited only Cluster III genes (Figure 3C).
Overall, these data are consistent with a model in which single-cell transcriptional responses of macrophages to S. typhimurium exposure include a homogenous inflammatory response to bacterial exposure (Cluster I) and a more variable antibacterial response to intracellular invasion (Cluster II). Both responses are mediated by a combination of Myd88 and Trif activity. A third response also occurs in a fraction of infected cells, involving intracellular LPS detection by Tlr4 which signals through Trif and Irf3 and results in a bimodal Type I IFN response (Figure 3D).
Live bacteria but not LPS-coated beads elicit a variable Type I IFN response in infected cells
To study the molecular mechanisms that lead to activation of the Type I IFN response in only a subset of infected macrophages, we explored this variation over time and in different infection models. A recent study showed that in dendritic cells exposed to LPS, the Type I IFN response is initially bimodally expressed and then uniformly induced over the entire population by four hours due to paracrine signaling (Shalek et al., 2014). In contrast, we have observed that the Type I IFN response in macrophages infected with bacteria has sustained bimodal expression during the entire time course (8 hours). While we also observe additional non-cell autonomous effects of Type I IFN activation at late time points in uninfected cells (reminiscent of the induction pattern seen in dendritic cells exposed to LPS), these additional effects do not eliminate the bimodal response in cells infected with live bacteria. This discrepancy between a transient and sustained bimodal Type I IFN response might be due to a difference between stimulation with soluble LPS versus LPS associated with an intact, infecting bacterium, other additional components of an intracellular bacterium, or a difference between host cell types. To examine these possibilities, we compared transcriptional responses between macrophages exposed to live S. typhimurium and macrophages exposed to fluorescently labeled latex beads coated with LPS extracted from S. typhimurium (Experimental Procedures). Macrophages exposed to LPS-coated beads indeed activated Clusters I, II and III (Figure S3B). To compare between different subpopulations after treatment with LPS-coated beads or live bacteria, we summarized the expression of each cluster with a single “eigen-gene” and calculated the density of these values across single cells (Experimental Procedures). Interestingly, compared to cells infected with bacteria, a much higher proportion of cells activated Cluster III among the cells that had taken up LPS-coated beads (Figures 3E, S3C). This difference in activation was not the result of different levels of LPS exposure, since there was a uniform but reduced induction of Clusters I and II in cells exposed to LPS coated beads compared to live bacteria (Figure 3E). This result suggests that there may be a bacterial factor that varies (e.g., displays bimodal behavior) among individual invading bacteria that accounts for the heterogeneous expression of the Type I IFN response upon bacterial uptake. However, on isolated LPS-coated beads this factor's heterogeneity is less pronounced. We also observe a stronger non-cell autonomous effect in uninfected cells exposed to LPS-coated beads that may be due to the release of more interferon from infected cells.
The variation in the Type I IFN response is driven by bimodal activity of the bacterial PhoPQ two-component system in infecting bacteria
Based on the hypothesis that the bimodal induction of the Type I IFN response may be due to heterogeneity in the infecting bacteria, we sought to identify bacterial factors that may influence Type I IFN expression. In the nucleus, Irf3 binds to the IFN-stimulated response element (ISRE, (Honda and Taniguchi, 2006)), a process that can be monitored at the single-cell level using a fluorescent reporter and FACS. We used iBMMs stably transduced with an ISRE fused to GFP as a reporter of the Type I IFN response in individual cells (iBMM-ISRE, Figure 4A, Experimental Procedures). We infected iBMM-ISRE with RFP-expressing bacteria, sorted ISRE-positive and ISRE-negative infected populations, and used RNA-seq to simultaneously profile host and bacterial transcripts in each population (Experimental Procedures). We confirmed that indeed, the Type I IFN response is more strongly induced in sorted ISRE-positive cells compared to sorted ISRE-negative cells (induction>1.5 fold, pFWER<0.05 GSEA, Figure 4B). Comparing the expression of bacterial pathways in these two populations, we found that targets of the bacterial transcription factor PhoP were significantly upregulated in ISRE-positive cells compared to ISRE-negative cells (pFWER<0.05, GSEA analysis, Figures 4B, S4A). In fact, both phoP and the associated phoQ gene were in the top 50 differentially expressed genes between these two populations, while hilA, a gene known to be repressed by PhoP, was among the most downregulated (Figure 4B). PhoP is the response regulator of a two-component system (with its cognate sensor kinase PhoQ) that is activated after a Salmonella bacterium is taken up by macrophages and induces the expression of genes important for intramacrophage survival (Groisman, 2001). We therefore hypothesized that variation in PhoP activity may underlie the variation in the Type I IFN response.
Figure 4. Heterogeneity of the invading bacterial populations shapes a heterogeneous host Type I IFN response.

(A) Schematic of iBMMs with a transcriptional reporter (6XISRE-GFP) of the activity of the Type I IFN gene cluster. (B). Shown is an MA plot of the induction levels of host and bacterial transcripts in ISRE-positive over ISRE-negative cells (y-axis) versus average absolute read counts (x-axis). Infected ISRE- positive cells expressing high levels of Cluster III genes (green dots) are infected with bacteria expressing higher levels of PhoP regulated genes (red dots) compared with ISRE-negative cells. Inset indicates the enrichment of PhoPQ regulated genes and Cluster III (GSEA analysis, p=0.007 and p<0.001 respectively). (C) Schematic of S. typhimurium with the transcriptional reporter of PhoP activity (phoP-GFP, top). PhoP displayed bimodal activity in infected macrophages, as analyzed by FACS (bottom, infected cells were identified by pHrodo). (D) Cells infected with bacteria expressing high phoP-GFP show higher expression of Cluster III genes compared to cells infected with low phoP-GFP. (E) Plots summarize the expression of the Type I IFN response in BMMs infected with WT, PhoP–, or PhoPc strains of S. typhimurium with a weighted average based score (x-axis) and display it versus the frequency of single cells (y-axis). Infection with PhoPc results in induction of the Type I IFN response in almost all infected cells, compared to cells infected with WT or PhoP– strains. (Table S4)
To test this hypothesis, we first assessed the variation in PhoP activity among intracellular bacteria using an engineered reporter with a PhoP-sensitive promoter upstream of GFP (phoP-GFP). We infected BMMs with pHrodo-labeled S. typhimurium carrying the phoP-GFP reporter. Consistent with our hypothesis, we found that PhoP indeed has bimodal activity in the population of infected cells (Figure 4C). We then sorted GFP-high and GFP-low macrophage populations and confirmed the difference in the expression levels of phoP between GFP-high and GFP-low infected cells by real-time qPCR (Figure S4B). We found increased expression of the Type I IFN response in the PhoP -high compared to PhoP-low infected cells (over 5-fold increase at 4 hours and over 3-fold increase at 8 hours, p<0.05 at both time points by a bootstrap analysis, Figures 4D, S4C). Importantly, this difference in Type I IFN expression was not observed when using a constitutive GFP reporter (Fig S4D) implying that the host cell is not responding primarily to differences in bacterial burden but to unique properties of PhoP-low and PhoP-high bacteria. No significant difference was observed in the expression of Cluster I or Cluster II between PhoP-high and PhoP-low infected cells (Figure S4E). Together, these results demonstrate a correlation between PhoPQ activity and the host Type I IFN response.
To establish whether PhoP activity functionally determines Type I IFN expression in the host cell, we infected macrophages with a phoP null mutant (PhoP–) and a strain with a single mutation in the phoQ gene that renders it constitutively active (PhoPc) (Miller and Mekalanos, 1990). Analyzing sorted infected populations, we found that cells infected with PhoPc bacteria induce the Type I IFN response more strongly than WT infected cells, while cells infected with PhoP– bacteria induce a weaker response (Figure S4F). Interestingly, at the single cell level, we found that infection with PhoPc, like stimulation with LPS-coated beads, increased the fraction of cells inducing the Type I IFN response (Figure 4E). PhoPc exposure also elicited a Type I IFN response in more uninfected cells than WT or PhoP– exposure, again implicating non-cell autonomous effects. Similar proportions of PhoP– and WT infected cells induced the Type I IFN response. Notably, no differences in the induction of Clusters I or II were observed between the phoP mutants (Figure S4F, S4G). These results indicate that the Type I IFN response is both correlated with and functionally the result of the activity of PhoPQ.
Intracellular recognition of PhoPQ-mediated LPS modifications results in induction of the Type I IFN response
PhoPQ is a global regulator of S. typhimurium virulence, involved in numerous cellular processes including activation of Type III secretion and cell wall alterations (Groisman, 2001). To test which of these processes might impact host Type I IFN expression, we treated BMMs with supernatants or heat-killed bacteria from PhoPc and PhoP– cultures. Culture supernatants failed to elicit a differential Type I IFN response excluding the involvement of factors secreted by PhoP-regulated Type III secretion systems (Figure S5A, bottom). Treatment with heat-killed cultures elicited a differential Type I IFN response, corresponding to infection with live mutants (Figure S5A, top). This result would be consistent with cell wall alterations playing a role in Type I IFN induction.
These results, together with reports implicating PhoPQ as regulator of LPS modification (Guo et al., 1997), led us to hypothesize that PhoPQ may exert its influence on the Type I IFN response through LPS modification. To test this hypothesis, we extracted LPS from WT, PhoP– and PhoPc strains and used it to stimulate BMMs. We used a standard limulus amebocyte lysate (LAL) test to normalize LPS concentrations from the different extractions (Experimental Procedures). Similar to infection with live bacteria, LPS from PhoPc induced higher levels of Type I IFN responsive genes compared to WT (over 9-fold higher at 2 hours post exposure, p<0.05 by bootstrap analysis), while LPS from PhoP– induced lower levels (over 4-fold lower at 2 hours post exposure and over 40-fold lower at 4 hours post exposure, p<0.05 at each time point by bootstrap analysis, Figures 5A, S5B). Notably, stimulation of cells with commercially available LPS from S. typhimurium resulted in induction levels similar to LPS from WT (Figure S5C), validating our extraction method and quantifications of LPS. These results demonstrate that PhoPQ's modification of LPS is responsible for the induction of the Type I IFN response.
Figure 5. Cell-to-cell variation in LPS modifications mediated by PhoPQ determines the bimodal induction of the Type I IFN response.

(A) Cells stimulated with LPS from the PhoPc strain induce higher levels of Type I IFN responsive genes compared to cells stimulated with LPS from the WT strain. Cells stimulated with LPS from the PhoP– strain showed less induction of this cluster. (Table S5A) (B) BMMs were stimulated with a mixture of red and green fluorescent beads coated with LPS extracted from PhoPc and PhoP– respectively. Induction of the Type I IFN response is evident in a larger proportion of cells taking up beads coated with PhoPc LPS (blue) than in cells taking up beads coated with LPS from the PhoP– strain (red). 74% of PhoPc compared to 26% of PhoP– induce more than the highest unexposed cells (white), p=0.003 using a two-population proportion z-test. (Table S5B) (C+D) Schematic representation of the differences in the responses of BMMs to infection with live bacteria and to stimulation with LPS coated beads. Live bacteria are more heterogeneous than LPS coated beads.
We next sought to test whether variations in LPS on the surface of individual bacteria are sufficient to drive a bimodal Type I IFN response. As it is not currently technically possible to query LPS modifications at the single-cell level, we simulated a heterogeneous population of “bacteria” by coating red fluorescent beads with LPS from the PhoP– strain and green fluorescent beads with LPS from the PhoPc strain. We then treated macrophages with an equal mixture of red and green LPS coated beads, sorted macrophages according to the color of beads they had taken up, and examined induction of genes at the single-cell level. We used a low MOI treatment to preclude the uptake of more then one bead in a given cell. We observed no difference in induction of Cluster I and Cluster II between cells that took up beads with LPS from PhoP– or PhoPc strains (Figure 5B, inset). In contrast, there was a clear shift in Cluster III induction, with induction of this cluster in a larger proportion of cells taking up beads coated with PhoPc LPS (74%) than in cells taking up beads coated with PhoP– LPS (26%) (Figure 5B; p=0.003 using a two-population proportion z-test). Similar to exposure to live bacterial strains, a non-cell autonomous effect was also evident in uninfected cells exposed to beads coated with PhoPc LPS (Figure S5D). As controls, induction levels of green or red beads coated with LPS extracted from WT S. typhimurium were similar, no induction was observed using beads not coated with LPS, and comparable results were obtained in bead-color swap experiments (Figure S5D). Additionally, no significant expression changes were noted between our brightest and dimmest cells infected with beads coated with WT LPS (demonstrating that differences in LPS burden cannot explain our results, Figure S5E). These results indicate that the bimodal Type I IFN response within a population of infected cells can be recapitulated by infecting with LPS coated beads from PhoP– and PhoPc mutant strains. Thus, differences in the induction of the Type I IFN response are determined not only by the internal state of the host cells or non-cell autonomous effects between host cells, but also as a direct result of the state of the infecting bacterium. Specifically, the extent of PhoPQ-regulated LPS modification of the invading bacterium accounts for the differences between individual intracellular “bacteria” that drive different host responses (Figures 5C, 5D).
LPS modifications mediated by PhoPQ impact the in vivo Type I IFN response and infection outcome
To confirm bimodal induction of the Type I IFN response of infected macrophages that had been naturally differentiated in vivo, we collected all cells from the peritoneal cavity of mice (Experimental Procedures) and immediately infected them with GFP-labeled S. typhimurium. After two hours we sorted infected resident macrophages (Figure S6A) and analyzed the induction of Clusters I, II, and III. We found similar expression patterns for all three clusters to those we observed in infected BMMs. Importantly, we found bimodal induction of the Type I IFN response (Figure 6A), indicating that this pattern of response to infection is generalizable to macrophages from different tissues.
Figure 6. LPS modifications mediated by PhoPQ impact in vivo infection outcomes.
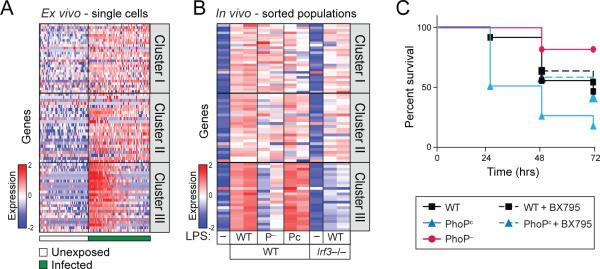
(A) Peritoneal macrophages, when infected ex vivo with GFP-labeled S. typhimurium show bimodal induction of Cluster III, like BMMs. (Table S6A) (B) Activation of the Type I IFN response in vivo was enhanced after stimulation with LPS extracted from PhoPc and reduced after stimulation with LPS extracted from PhoP– strain, compared to LPS extracted from WT S. typhimurium. As a control, no induction of the Type I IFN response was measured in Irf3−/− mice. (Table S6B) (C) Mice challenged with LPS extracted from PhoPc (blue, n=12) showed reduced survival compared to mice challenged with WT LPS (black, n=11). Inhibition of the Type I IFN response by co-administration of BX795 improved survival from PhoPc challenge, restoring it to WT levels (dotted blue, n=12). Mice challenged with LPS extracted from PhoP– (red, n=11) showed enhanced survival compared to WT.
To determine the physiological importance of the relationship between bacterial PhoPQ, LPS variation, and the host Type I IFN response, we next sought to demonstrate the same correlation in mice using LPS stimulation. We injected mice intraperitoneally (i.p.) with sub-lethal, normalized doses of LPS extracted from WT, PhoP– and PhoPc strains (Experimental Procedures). After two hours we isolated peritoneal macrophages from treated mice (Figure S6A), and analyzed the in vivo induction of Clusters I, II, and III. Notably, LPS from the PhoPc strain induced higher levels of Cluster III, while LPS from the PhoP– strain had the opposite effect, thereby mirroring the in vitro infection results (Figure 6B). Minimal differences in Clusters I and II were observed in vivo, similar to what was observed in vitro. Additionally, we confirmed the Irf3 dependence of Cluster III by performing this same experiment in Irf3−/− mice (Figure 6B). These results demonstrate a relationship between modified LPS and Type I IFN expression in mice and suggest that PhoPQ is an important regulator of the Type I IFN response in vivo.
Ifnar1−/− mice were previously shown to have prolonged survival after S. typhimurium challenge, demonstrating an important role for the Type I IFN response in determining infection outcome (Robinson et al., 2012). We thus sought to test whether bacterial PhoPQ activity, through its activation of the Type I IFN response, has an impact on infection outcome similar to ablating signaling downstream of Ifnar. Because PhoP– and PhoPc strains are both avirulent in mice (Miller and Mekalanos, 1990), we turned to a mouse model of LPS-induced septic shock. Septic shock, a systemic response to severe bacterial infection, is considered an important determinant of infection outcome, as it is often associated with high mortality (Morrison and Ryan, 1987). We induced septic shock in mice using high doses of LPS extracted from WT, PhoP– or PhoPc Salmonella strains and monitored survival. Mice injected with normalized amounts of PhoPc-LPS had significantly higher mortality rates than mice injected with WT-LPS (Figure 6C, p=0.003, log-rank test). Meanwhile, mice challenged with PhoP–-LPS had higher survival rates compared to WT-LPS challenged mice (p=0.003, log-rank test). We then co-administered LPS extracted from PhoPc with the small molecule BX795, which we had previously shown to be a specific inhibitor of the Type I IFN response (Figure 3C), and found significantly improved survival rates of the PhoPc-LPS challenged mice (p=0.031, log-rank test). We further verified that these effects were mirrored in the transcriptional responses of peritoneal macrophages. The co-administration of the BX795 inhibitor together with LPS extracted from PhoPc strain abrogated the induction of Type I IFN response, reducing it to levels similar to mice challenged with WT LPS (Figure S6B). These results demonstrate that the extent of LPS modification by PhoPQ and its interaction with the cognate host Type I IFN response are important determinants of infection outcome in vivo.
Discussion
A general approach to characterize the transcriptional underpinnings of phenotypic heterogeneity in host-pathogen encounters
Heterogeneity between individual cells is a common feature of dynamic cellular processes, including signaling, transcription, and cell fate (Elowitz et al., 2002). Phenotypic heterogeneity has similarly long been observed as an important feature of infection resulting from individual cellular encounters that involve highly dynamic, adaptable cells and bacteria. However, to date, tools for probing the variation in host-pathogen interactions have been limited and studies of host-pathogen interactions have relied on bulk, population-level measurements. Thus, the specific mechanisms underlying this heterogeneity remain largely unknown and demonstrations of its effects in vivo still incomplete. Here, we present a generalizable approach to identify and characterize transcriptional heterogeneity in subpopulations that may underlie phenotypic variation of infection by directly probing individual macrophage-bacteria encounters. We use microscopy to map infection phenotypes to transcriptional states as determined by single-cell RNA-seq, resulting in a high resolution view of host-pathogen interactions.
Heterogeneity of pathogen populations as a mechanism to shape the host immune response
We revealed specific genetic pathways that show unexpectedly large amounts of variation between what otherwise appear to be identically infected cells. One such pathway is the Type I IFN response, which was only fully induced in a fraction of infected macrophages. Upon further investigation, we found that the level of Type I IFN induction in infected macrophages is determined by the level of PhoPQ activity in the invading bacterium (Figure 4D, E).
Heterogeneity of transcriptional responses has been reported and traditionally ascribed to stochastic variation or intrinsic state of the cell. For example, a recent publication suggests that the induction of the antiviral response in dendritic cells in response to bacterial LPS stimulation is dependent on the existence of a relatively small fraction of “precocious” cells that initiate the response that eventually spreads through the population via paracrine responses (Shalek et al., 2014). Our work highlights the fact that immune activation also depends on the state of the invading pathogen. This demonstrates an alternative source of host heterogeneity, whereby intrinsic variation in bacterial populations shapes the host immune response. The in vivo experiments indicate functional consequences during infection of the variable factors identified, and point to heterogeneity as a feature of pathogen populations that impacts infection.
Studies of immune responses in the context of heterogeneous bacterial ligands
Different types of LPS have been shown to produce dramatically different host responses, with diversity in LPS structures having been described between bacterial populations exposed to different environments (Paciello et al., 2013), different bacterial mutants (Guo et al., 1997), and different LPS variants resulting from different isolation procedures (Gutschow et al., 2013). We now show that heterogeneity also exists within a single population of wild-type bacteria. While this alone may not be altogether surprising, we demonstrate that this variability has functional consequence.
There is accumulating evidence that cell-to-cell variation exists in the expression of numerous bacterial factors in addition to LPS, including other PAMPs and virulence factors. For example, bacteria in the same culture can be in either a motile (flagella positive) or a non-motile (flagella negative) state (Cummings et al., 2006), or contain very different levels of effector proteins (Schlumberger et al., 2005). Importantly, immunological studies of such molecules have often implicitly neglected pathogen variability by relying on measurements of host cell response to what is assumed to be a homogenous ligand, ignoring the reality that such ligands actually result from a heterogeneous, diverse population. In this study, we show that coating beads with LPS isolated from a pooled, heterogeneous population of bacteria artificially limits heterogeneity by mixing modified and unmodified LPS stemming from different individual bacteria onto the same bead. This system thus fails to recapitulate the diversity of actual pathogens and the diversity of the cognate host response. The heterogeneity of the host response can be restored by reinstating the heterogeneity in the chemical stimulus (coating two sets of beads with LPS isolated from two different bacterial mutants (phoP– and phoPc), followed by mixing of the two sets of beads (Figure 5D)).
Importantly, although we show that bacterial heterogeneity in PhoPQ mediated LPS modification has a significant effect in mediating the host Type I IFN response, this is by no means the only determining factor, nor is it solely responsible for the heterogeneity we observe. It is well known that the Type I IFN response can be induced by non-cell autonomous effects such as paracrine signaling, given that interferon is a soluble secreted molecule (Honda and Taniguchi, 2006). Indeed, we also observe induction of this cluster in uninfected cells at later time points during infection (Figure S2C). We also observe this paracrine signaling in a larger fraction of uninfected cells treated with PhoPc LPS, probably due to the fact that a larger fraction of infected cells are inducing the Type I response.
We also observe the induction of the Type I IFN response in a small population of cells infected with the PhoP– strain (Figure 4E). This demonstrates that the infection with PhoP mutant strains does not perfectly mirror the naturally occurring low and high PhoP populations that we observe during WT infection as genetically altering the strains cannot provide the fine-tuned regulation and variation that occurs in WT bacteria. It is a relatively common phenomenon that genetic knockout does not abolish an activity for a protein that is revealed by overexpression (Kitano, 2004); in fact it has been demonstrated before that the PhoP– strain does not show the opposite phenotype of the phoPc strain (Strandberg et al., 2012). This is generally indicative of redundant pathways and suggests that PhoPQ does not fully account for the variability observed in the host response. Other complementary bacterial pathways are also known to control LPS modifications and it is likely that some of these also play a similar role in modulating Type I IFN response. For example, one such possible candidate is the bacterial PmrAB two component system (Perez and Groisman, 2007)) and understanding the role of such additional regulators merits further investigation.
Possible advantages of bimodal expression of bacterial factors within a population in the course of in vivo infection
It has been previously reported that while virulence factors allow growth and survival of the pathogen within the host, their activity elicits changes that seem both beneficial and detrimental to the bacteria (Ackermann et al., 2008). For example, while PhoPQ activation plays a key role in permitting intracellular survival by making Salmonella more resistant to environmental stressors, it is also associated with decreased transcytosis by epithelial cells and decreased replication rates (Groisman, 2001). This suggests that the utility of these factors may be highly dependent on environmental context. In changing environments, bistability or diversification of bacterial populations has been shown to be beneficial (Kussell and Leibler, 2005).
Recently, it has been shown that cooperation between virulent and avirulent subpopulations is essential for S. enterica pathogenicity (Diard et al., 2013). The effects of this cooperation were demonstrated using co-infection with genetically distinct mutant strains. Our work suggests that this strategy need not be restricted to mixed genetic subpopulations, but could occur between isogenic subpopulations during WT infection. For example, one could imagine a beneficial cooperation in which a population with high PhoPQ activity could induce a more robust immune response, as has previously shown to be helpful in overcoming the commensal microflora (Lupp et al., 2007), paying a metabolic cost that benefits a population with low PhoPQ activity. In support of this, it is interesting that both the PhoP– and PhoPc mutants, that are unable to diversify PhoP activity, are attenuated (Miller and Mekalanos, 1990). Thus, in order to succeed in the complex host environments encountered throughout infection, Salmonella could tune the variation of factors such as PhoPQ to create distinct subpopulations that ensure that some pathogen subsets prevail in infection.
To conclude, this work establishes a mechanism by which transcriptional heterogeneity can have functional consequences for host-pathogen interactions, in this case through differences in pathogen detection. The ability of immune cells to respond to differences between individual pathogens implies that pathogen heterogeneity is a key feature of pathogen populations that impacts host response. This work suggests that further investigation of the role of bacterial heterogeneity as a mechanism to drive different host responses and the extent to which this strategy is employed by diverse pathogens is warranted to fully uncover its role in bacterial pathogenesis and ultimately, in determining infection outcome.
Experimental Procedures
Mice, cell lines and bacterial strains
C57BL/6 WT mice were obtained from Jackson Laboratory (Bar Harbor, Maine). All animals were housed and maintained in a conventional pathogen-free facility at the Massachusetts General Hospital in Boston, Massachusetts. All experiments were performed in accordance to the guidelines outlined by the MGH Committee on Animal Care (Boston, MA). WT, Tlr4−/−, Trif−/− and Myd88−/− iBMMs were obtained from BEI resources (Manassas, Virginia). Irf3−/− and Irf7−/− BMMs were a generous gift from Dr. Nir Hacohen (Broad Institute, Cambridge, Massachusetts).
All S. Typhimurium strains used in this study were derived from wild-type strain ATCC14028s or SL1344. 14028 mutant strains PhoPc with pho-24 and PhoP– with phoP::Tn10d-Cam (Miller and Mekalanos, 1990) were a generous gift from Dr. Sam Miller (University of Washington, Seattle, Washington).
Cultures of S.typhimurium labeled with GFP (pFPV25.1; Addgene, Cambridge, MA) were grown in Luria-Bertani (LB) medium at 37 °C, washed in PBS and incubated for 1 hour with pHrodo dye (Life Technologies, Carlsbad, CA). BMMs were infected at an MOI of 1:1. Thirty minutes later cells were washed with media containing 15 μg/ml gentamicin to remove S.typhimurium that were not internalized.
Single cell sorting and transcript quantification
At the indicated time points, single cells were sorted by FACS and processed using the SMARter whole transcriptome amplification protocol (Clontech, Mountain View, CA). cDNA products were then converted to Illumina sequencing libraries using Nextera XT (Illumina, San Diego, CA). Samples were sequenced on an Illumina Hiseq-2500.
A mouse transcriptome was generated using Ensembl gene annotations and the Dec. 2011 (GRCm38/mm10) build of the mouse genome. Alignment was done using RSEM v1.2.3. Transcript abundance was estimated using Transcripts per Million (TPM).
Bacterial reads were aligned to a composite mouse-salmonella transcriptome built by combining the mouse transcriptome above with the NCBI build of the SL1344 genome ((NC_017718.1). Alignment of bacterial reads was done using BWA 0.7.10-r789 and an in-house script was used for transcript enumeration.
Differential expression analysis was done using DEseq version 1.10.1, treating each cell in a given condition as replicate. Genes were considered differentially expressed only if they had an FDR of less than or equal to 0.05 and an average fold change of at least 2 fold.
Heatmaps and density plots
To generate heatmaps, gene TPM values were transformed into log space (log2(expression +1)) and scaled (by gene) to mean 0 and unit standard deviation prior to plotting. Rows and columns were ordered as described in each figure legend. For density plots summarizing the behavior of a gene cluster, PCA (biomark) or a weighted average approach taking to account overall library quality (RNA-seq) was used to generate a single estimate per cell. Density estimates of these summary values were plotted and given the same maximum height for easy visualization.
Mouse LPS stimulation
In vivo experiments were performed in C57BL/6J mice injected intraperitoneally with a sub-lethal (20 μg per mouse) or lethal (700 ug per mouse) dose of LPS extracted from WT, PhoP− or PhoPc Salmonella strains and survival of mice was followed for 5 days.
Additional computational analyses and experiments performed using RNAtag-seq for simultaneous detection of host and intracellular bacterial transcripts, RNA-flowFISH (Panomics), knockout mice and bacterial mutants as described in the Extended Experimental Procedures.
Supplementary Material
Acknowledgments
We thank Alan Grossman, Dan Kahne, Jon Kagan, Nir Hacohen, Itai Yanai and Amy Barczak for discussions and comments. We thank Eduardo Villablanca for advise with in vivo experiments. We thank Ken Livak for help with Biomark experiments. We thank Leslie Gafney for help with graphical illustrations and figures. This work was supported by NHGRI grant HG002295 to N.H.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributions:
R.A and D.T.H conceived and designed the study. R.A., N.H., D.B., C.P., H.B.J. performed experiments. N.H. performed computational analysis. R.A., N.H. and D.T.H. wrote the manuscript with extensive input from all authors.
Datasets
Gene Expression data are accessible through GEO (http://ncbi.nlm.nih.gov/geo) under accession numbers GSE66528, GSE65529, GSE65530, and GSE65531
References
- Ackermann M, Stecher B, Freed NE, Songhet P, Hardt WD, Doebeli M. Self-destructive cooperation mediated by phenotypic noise. Nature. 2008;454:987–990. doi: 10.1038/nature07067. [DOI] [PubMed] [Google Scholar]
- Chevrier N, Mertins P, Artyomov MN, Shalek AK, Iannacone M, Ciaccio MF, Gat-Viks I, Tonti E, DeGrace MM, Clauser KR, et al. Systematic discovery of TLR signaling components delineates viral-sensing circuits. Cell. 2011;147:853–867. doi: 10.1016/j.cell.2011.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claudi B, Sprote P, Chirkova A, Personnic N, Zankl J, Schurmann N, Schmidt A, Bumann D. Phenotypic variation of Salmonella in host tissues delays eradication by antimicrobial chemotherapy. Cell. 2014;158:722–733. doi: 10.1016/j.cell.2014.06.045. [DOI] [PubMed] [Google Scholar]
- Cummings LA, Wilkerson WD, Bergsbaken T, Cookson BT. In vivo, fliC expression by Salmonella enterica serovar Typhimurium is heterogeneous, regulated by ClpX, and anatomically restricted. Mol Microbiol. 2006;61:795–809. doi: 10.1111/j.1365-2958.2006.05271.x. [DOI] [PubMed] [Google Scholar]
- Diard M, Garcia V, Maier L, Remus-Emsermann MN, Regoes RR, Ackermann M, Hardt WD. Stabilization of cooperative virulence by the expression of an avirulent phenotype. Nature. 2013;494:353–356. doi: 10.1038/nature11913. [DOI] [PubMed] [Google Scholar]
- Elowitz MB, Levine AJ, Siggia ED, Swain PS. Stochastic gene expression in a single cell. Science. 2002;297:1183–1186. doi: 10.1126/science.1070919. [DOI] [PubMed] [Google Scholar]
- Eriksson S, Lucchini S, Thompson A, Rhen M, Hinton JC. Unravelling the biology of macrophage infection by gene expression profiling of intracellular Salmonella enterica. Mol Microbiol. 2003;47:103–118. doi: 10.1046/j.1365-2958.2003.03313.x. [DOI] [PubMed] [Google Scholar]
- Fitzgerald KA, Rowe DC, Barnes BJ, Caffrey DR, Visintin A, Latz E, Monks B, Pitha PM, Golenbock DT. LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the toll adapters TRAM and TRIF. J Exp Med. 2003;198:1043–1055. doi: 10.1084/jem.20031023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galan JE, Collmer A. Type III secretion machines: bacterial devices for protein delivery into host cells. Science. 1999;284:1322–1328. doi: 10.1126/science.284.5418.1322. [DOI] [PubMed] [Google Scholar]
- Groisman EA. The pleiotropic two-component regulatory system PhoP-PhoQ. J Bacteriol. 2001;183:1835–1842. doi: 10.1128/JB.183.6.1835-1842.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Lim KB, Gunn JS, Bainbridge B, Darveau RP, Hackett M, Miller SI. Regulation of lipid A modifications by Salmonella typhimurium virulence genes phoP-phoQ. Science. 1997;276:250–253. doi: 10.1126/science.276.5310.250. [DOI] [PubMed] [Google Scholar]
- Gutschow MV, Hughey JJ, Ruggero NA, Bajar BT, Valle SD, Covert MW. Single-cell and population NF-kappaB dynamic responses depend on lipopolysaccharide preparation. PloS one. 2013;8:e53222. doi: 10.1371/journal.pone.0053222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helaine S, Thompson JA, Watson KG, Liu M, Boyle C, Holden DW. Dynamics of intracellular bacterial replication at the single cell level. Proc Natl Acad Sci U S A. 2010;107:3746–3751. doi: 10.1073/pnas.1000041107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, Taniguchi T. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol. 2006;6:644–658. doi: 10.1038/nri1900. [DOI] [PubMed] [Google Scholar]
- Jaitin DA, Kenigsberg E, Keren-Shaul H, Elefant N, Paul F, Zaretsky I, Mildner A, Cohen N, Jung S, Tanay A, et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science. 2014;343:776–779. doi: 10.1126/science.1247651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SQ, Li YY, Pan RG, Zou XF. Characterizing and controlling the inflammatory network during influenza A virus infection. Sci Rep-Uk. 2014;4 doi: 10.1038/srep03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nature immunology. 2008;9:361–368. doi: 10.1038/ni1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitano H. Biological robustness. Nature reviews Genetics. 2004;5:826–837. doi: 10.1038/nrg1471. [DOI] [PubMed] [Google Scholar]
- Kussell E, Leibler S. Phenotypic diversity, population growth, and information in fluctuating environments. Science. 2005;309:2075–2078. doi: 10.1126/science.1114383. [DOI] [PubMed] [Google Scholar]
- Lee MN, Roy M, Ong SE, Mertins P, Villani AC, Li WB, Dotiwala F, Sen J, Doench JG, Orzalli MH, et al. Identification of regulators of the innate immune response to cytosolic DNA and retroviral infection by an integrative approach. Nature immunology. 2013;14:179–185. doi: 10.1038/ni.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupp C, Robertson ML, Wickham ME, Sekirov I, Champion OL, Gaynor EC, Finlay BB. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell host & microbe. 2007;2:204. doi: 10.1016/j.chom.2007.08.002. [DOI] [PubMed] [Google Scholar]
- McIntrye J, Rowley D, Jenkin CR. The functional heterogeneity of macrophages at the single cell level. The Australian journal of experimental biology and medical science. 1967;45:675–680. doi: 10.1038/icb.1967.67. [DOI] [PubMed] [Google Scholar]
- Miller SI, Mekalanos JJ. Constitutive expression of the phoP regulon attenuates Salmonella virulence and survival within macrophages. J Bacteriol. 1990;172:2485–2490. doi: 10.1128/jb.172.5.2485-2490.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monack DM, Raupach B, Hromockyj AE, Falkow S. Salmonella typhimurium invasion induces apoptosis in infected macrophages. Proc Natl Acad Sci U S A. 1996;93:9833–9838. doi: 10.1073/pnas.93.18.9833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison DC, Ryan JL. Endotoxins and disease mechanisms. Annual review of medicine. 1987;38:417–432. doi: 10.1146/annurev.me.38.020187.002221. [DOI] [PubMed] [Google Scholar]
- Paciello I, Silipo A, Lembo-Fazio L, Curcuru L, Zumsteg A, Noel G, Ciancarella V, Sturiale L, Molinaro A, Bernardini ML. Intracellular Shigella remodels its LPS to dampen the innate immune recognition and evade inflammasome activation. Proc Natl Acad Sci U S A. 2013;110:E4345–4354. doi: 10.1073/pnas.1303641110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez JC, Groisman EA. Acid pH activation of the PmrA/PmrB two-component regulatory system of Salmonella enterica. Mol Microbiol. 2007;63:283–293. doi: 10.1111/j.1365-2958.2006.05512.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathinam VA, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, Leong JM, Fitzgerald KA. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell. 2012;150:606–619. doi: 10.1016/j.cell.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson N, McComb S, Mulligan R, Dudani R, Krishnan L, Sad S. Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nature immunology. 2012;13:954–962. doi: 10.1038/ni.2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberger CM, Scott MG, Gold MR, Hancock RE, Finlay BB. Salmonella typhimurium infection and lipopolysaccharide stimulation induce similar changes in macrophage gene expression. J Immunol. 2000;164:5894–5904. doi: 10.4049/jimmunol.164.11.5894. [DOI] [PubMed] [Google Scholar]
- Schlumberger MC, Muller AJ, Ehrbar K, Winnen B, Duss I, Stecher B, Hardt WD. Real-time imaging of type III secretion: Salmonella SipA injection into host cells. Proc Natl Acad Sci U S A. 2005;102:12548–12553. doi: 10.1073/pnas.0503407102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwan WR, Huang XZ, Hu L, Kopecko DJ. Differential bacterial survival, replication, and apoptosis-inducing ability of Salmonella serovars within human and murine macrophages. Infection and immunity. 2000;68:1005–1013. doi: 10.1128/iai.68.3.1005-1013.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalek AK, Satija R, Adiconis X, Gertner RS, Gaublomme JT, Raychowdhury R, Schwartz S, Yosef N, Malboeuf C, Lu DN, et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature. 2013;498:236–240. doi: 10.1038/nature12172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalek AK, Satija R, Shuga J, Trombetta JJ, Gennert D, Lu D, Chen P, Gertner RS, Gaublomme JT, Yosef N, et al. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature. 2014;510:363–369. doi: 10.1038/nature13437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strandberg KL, Richards SM, Gunn JS. Cathelicidin antimicrobial peptide expression is not induced or required for bacterial clearance during salmonella enterica infection of human monocyte-derived macrophages. Infection and immunity. 2012;80:3930–3938. doi: 10.1128/IAI.00672-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trombetta JJ, Gennert D, Lu D, Satija R, Shalek AK, Regev A. Preparation of Single-Cell RNA-Seq Libraries for Next Generation Sequencing. Current protocols in molecular biology / edited by Frederick M Ausubel [et al] 2014;107:4 22 21–24 22 17. doi: 10.1002/0471142727.mb0422s107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.