

Abstract
Thyroid hormone action is predominantly mediated by thyroid hormone receptors (THRs), which are encoded by the thyroid hormone receptor α (THRA) and thyroid hormone receptor β (THRB) genes. Patients with mutations in THRB present with resistance to thyroid hormone β (RTHβ), which is a disorder characterized by elevated levels of thyroid hormone, normal or elevated levels of TSH and goitre. Mechanistic insights about the contributions of THRβ to various processes, including colour vision, development of the cochlea and the cerebellum, and normal functioning of the adult liver and heart, have been obtained by either introducing human THRB mutations into mice or by deletion of the mouse Thrb gene. The introduction of the same mutations that mimic human THRβ alterations into the mouse Thra and Thrb genes resulted in distinct phenotypes, which suggests that THRA and THRB might have non-overlapping functions in human physiology. These studies also suggested that THRA mutations might not be lethal. Seven patients with mutations in THRα have since been described. These patients have RTHα and presented with major abnormalities in growth and gastrointestinal function. The hypothalamic–pituitary–thyroid axis in these individuals is minimally affected, which suggests that the central T3 feedback loop is not impaired in patients with RTHα, in stark contrast to patients with RTHβ.
Introduction
The narrow range of serum concentrations of the thyroid hormones, T4 and T3, in mammals is critical for regulation of intrauterine and postnatal development, as well as for maintenance of metabolic pathways after development.1–3 Such tight control is mediated by a negative feedback loop that decreases the release of both pituitary TSH and hypothalamic thyrotropin-releasing hormone (TRH) (Box 1).3,4 After conversion of the inactive pro-hormone, T4, to the active form, T3, negative feed-back and other important functions of active thyroid hormone are mediated by specific nuclear hormone receptors that function as modulators of gene expression.5,6 Two thyroid hormone receptor (THR) proteins have been described, THRα and THRβ; these receptors are encoded by the THRA and THRB genes, respectively. The promoter regions of genes that are regulated by T3contain thyroid-hormone responsive elements (TREs), which provide binding sites for THRs (Figure 1).1,7 This Review focuses on the functions and mechanisms of action of THRs in the context of transcriptional regulation. Clinical findings in patients with mutations in THRA and THRB are reviewed, and emerging results from preclinical models that have provided insights into THR functions are also discussed.
Box 1. The hypothalamic–pituitary–thyroid axis.
Circulating thyroid hormone levels are maintained within a narrow range by a negative feedback loop involving thyrotropin-releasing hormone (TRH), which is secreted by the hypothalamus; TSH secreted by thyrotroph cells in the anterior pituitary gland; and the thyroid gland.3 TRH is a tripeptide that is secreted by parvocellular neurons of the anterior hypothalamus that project to the median eminence. TRH then travels via the portal circulation to the anterior pituitary where it simulates thyrotroph cells to produce and secrete glycosylated TSH.3
TSH is composed of two subunits; a specific β-subunit and an α-subunit that is shared with the other glycoprotein hormones: luteinizing hormone, follicle-stimulating hormone and human chorionic gonadotropin.3 TSH enters the systemic circulation and stimulates follicular cells of the thyroid gland to synthesize and osecrete thyroid hormones (T3 and T4).
Thyroid hormones in turn feedback, at both the level of the pituitary and the hypothalamus, to reduce TSH and TRH synthesis and secretion, respectively.4 Studies suggest this process is mediated by DNA-bound THRβ.11,55
Figure 1.

Overview of thyroid hormone action. T3 enters the cell via thyroid hormone transporters, or is generated locally by cytoplasmic deiodinases (not shown). In the nucleus T3 binds to THR-containing dimers, which bind to genomic TREs to regulate gene transcription. Abbreviations: RXR, retinoic acid receptor; THR, thyroid hormone receptor; TRE, thyroid-hormone responsive element.
THRs have been extensively studied since they were first isolated and described in 1986.8,9 In the past two decades, several naturally occurring mutations in THRB have been identified in patients who presented with the classic form of resistance to thyroid hormone (RTHβ). This disorder is characterized by elevated thyroid hormone levels and concentrations of TSH that are either within the normal range or slightly elevated.2,10 The majority of patients with RTHβ are heterozygous for dominant negative THRB mutations, which result in the production of a mutant receptor that inhibits the function of wild-type THR.1 This inhibition of wild-type receptors can lead to elevated thyroid hormone signalling through THRα receptors, a situation that has hampered efforts to determine the specific functions of THRβ in different tissues and organs. Furthermore, different mutations in THRB have varying effects on THRβ function and THRβ expression varies among organs, which makes understanding the relationship between genotype and phenotype in patients with RTHβ challenging. Given the limitations of investigating the mechanisms of THRβ function in humans, use of gene manipulation techniques in mice has helped to clarify the role of this receptor in developmental processes and adult mammals. Patients with homozygous mutations in THRB and homozygous mutant mice exhibit profound features of RTHβ that include large goitre, hearing impairment and severe deregulation of the hypothalamic–pituitary–thyroid (HPT) axis.11–14
Previously, the failure to identify patients with THRA mutations raised the question of whether these mutations were lethal or if the phenotypes of such patients are extremely difficult to identify in a clinical setting. Generation of a mouse model in which a THRB mutation detected in patients with RTHβ was introduced into the mouse Thra gene suggested that mutations in THRA might not be lethal and supported the hypothesis that there could be a human phenotype associated with such mutations.15
Key points.
The main isoforms of thyroid hormone receptors, THRα1, THRβ1 and THRβ2, are predominantly responsible for mediating thyroid hormone action, which is critical for normal development, growth and metabolism
Patients with mutations in either THRA or THRB have been described and have strikingly different clinical phenotypes known as resistance to thyroid hormone (RTH)α and RTHβ, respectively
Patients with RTHβ frequently present with elevated thyroid hormone levels, normal or elevated TSH levels and goitre, which suggests a critical role for THRB in negative-feedback regulation
Currently only seven patients with RTHα have been described; these individuals have near-normal levels of thyroid hormones and TSH but display hypothyroidism, delayed growth and constipation
Studies of mutations associated with RTH disorders using transgenic mouse models have provided novel insights into the divergent roles of THRA and THRB in physiology
In the past 2 years, four reports of patients with heterozygous mutations in THRA have emerged.16–19 Patients with mutations in THRA have prominent growth retardation and abnormalities in gastrointestinal function. However, unlike in patients with RTHβ, the HPT axis in individuals with RTHα is only minimally affected, which feedback loop is not impaired suggests that the central T3 in this disorder.16,20
Thyroid hormone receptors
The THRs belong to the nuclear receptor superfamily that includes the estrogen receptor, vitamin D receptor, peroxisome proliferator-activated receptors (PPARs), retinoic acid receptor (RAR) and retinoid X receptor (RXR).1,21 These receptors are formed from a single peptide that is folded into three modular functional domains: an amino-terminal domain (A–B domain), a central DNA-binding domain (DBD) and a carboxyl-terminal ligand-binding domain (LBD) (Figure 2). The function of the activation 1 (AF-1) region is poorly understood22 and this region is localized to within the A–B domain. The LBD dictates the specificity of the receptor for its ligand and modulates its capacity to form either homodimers or heterodimers with other members of the nuclear receptor superfamily.1,23
Figure 2.

The THRα and THRβ isoforms have considerable homology. The DBDs of all THR isoforms are highly homologous and each of these proteins can form heterodimers and homodimers. Alternative splicing and use of alternative transcription sites results in the generation of four THR isoforms that able to bind T3 (THRα1 and THRβ1-3). The AF-1 and AF-2 regions of the proteins have some sequence variability across the isoforms; the THRα2 and THRα3 isoforms are truncated and, as a result of having no AF-2 domains, they are unable to bind T3. Abbreviations: AF-1, activation function 1; AF-2, activation function 2; DBD, DNA-binding domain; LBD, ligand-binding domain.
Expression of thyroid hormone receptor genes
The human THR proteins are transcribed from two separate genes, THRA and THRB, each of which has a homologue in mice, known as Thra and Thrb, respectively.
THRα1 isoforms
Three mRNA species are transcribed from the THRA gene, which is located on human chromosome 17. THRα1 binds T3 and can form dimers with the truncated THRA gene products, THRα2 and THRα3, that lack the ability to bind this hormone.2 THRα2 and THRα3 differ in length and amino acid composition in the C-terminal region when compared with THRα1 (Figure 2). Although the exact physiological importance of these truncated proteins is not known, heterodimerization of these isoforms with full-length THR proteins in vitro results in antagonization of T3-mediated transcriptional activation.24,25
THRβ isoforms
The THRB gene is located on human chromosome 3, and is expressed as either of two T3-binding isoforms, THRβ1 or THRβ2; a third isoform, THRβ3 is found only in rats.26 THRβ1 or THRβ2 mainly differ in their tissue distribution and in the length of the A–B domain.21 The DBD and LBD, which bind to very similar TREs, are entirely conserved between THRβ isoforms (Figure 2).
Patterns of gene expression
Comparing the relative levels of expression of each of the THR isoforms in different tissues during development and in adults is challenging, especially at the protein level.1 The availability of specific antibodies that are well-validated is lacking, which means that investigators have predominantly studied the mRNA levels of Thra and Thrb instead. Analysis of mRNA distributions in mice revealed that Thra1 and Thrb1 are expressed in all tissues studied; however, Thra1 mRNA is predominantly expressed in the heart and brain,27 whereas Thrb1 mRNA is more highly expressed in skeletal muscle, kidney and liver than in other tissues. Expression of Thrb2 mRNA has a distinct pattern of tissue distribution that is higher in the brain, pituitary gland, retina and inner ear than in other tissues.2,28,29 By contrast, studies in rats indicate that Thrb3 mRNA is expressed only in the kidneys, liver and lungs.1 Data for the tissue distributions of expression of the THRA and THRB isoforms in human tissues are scarce; however, THRA1 and THRB1 are highly expressed in trophoblast and stromal cells in the lacenta.30
Functions of the thyroid hormone receptors
The binding of T3 to THRs can result in either a decrease or an increase of the transcription rates of target genes.9,31,32 THRs can constitutively bind to the TREs of T3 target genes. Furthermore, these receptors can act in a ligand-independent manner, such that the transcription rate of target genes can change depending on whether or not the THR is bound to T3.
The carboxyl-terminal portion of the LBD contains an activation function 2 (AF-2) region that adopts a distinct conformation when bound to T3.33–35 Simulations of molecular dynamics have shown that a conformational change alters the position of helix 12 in the AF-2 domain when T3 binds to the THR,36 which allows it to bind to other proteins (cofactors), including co-activators, such as the nuclear receptor co-activator 1 (also known as NCoA-1 or SRC-1),35 and co-repressors, such as nuclear receptor-interacting protein 1 (nuclear factor RIP140).12 The importance of helix 12 to THR function has been demonstrated by studying both naturally occurring and artificial mutations in this structure that specifically block recruitment of cofactors to the AF-2 domain, which results in inhibition of both T3-mediated activation and repression of gene transcription.12,36,37
The AF-1 domain is localized to the amino–terminal portion of the THR. In the absence of T3, the THR exists in an unliganded conformation and AF-1 interacts with cofactors, such as nuclear receptor co- repressor 1 (N-CoR) or nuclear receptor co-repressor 2 (N-CoR2).38,39 The structure of the AF-1 domain is poorly understood; however, this domain seems to be involved in determination of THR specificity for TREs.40 Each of the THR isoforms has a distinct AF-1 domain, which influences the composition of the dimers preferentially formed by the THR proteins and the specificity of the DNA targets to which they bind (Figure 2). For example, the amino-terminal portion of THRα1 preferentially forms heterodimers with RXRs and other nuclear receptors to positively regulate transcription at target TREs.40
THRs can either homodimerize or form heterodimers with other THRs or nuclear receptor superfamily proteins, such as RXRs,2 liver X receptor41 and PPARs.42 Findings from in vitro studies suggest that dimerization drives THR binding to specific TREs.40,43 Several TREs have been characterized,1 and emerging studies of genome-wide analysis of THR binding sites demonstrated that THRβ homodimers bound predominantly near transcription start sites of T3-induced genes in a liver-derived cell line.44 Genome-wide analysis of chromatin occupancy of THRα1 or THRβ1 homodimers in a neuronal cell line revealed that THRs were bound at shared and unique binding sites throughout the genome and identified the DR+4 consensus sequence as the only known binding element at these sites.45
A general model of T3 action has been developed on the basis of findings from studies that used crystallography techniques in combination with in vitro mutation analyses and transgenic animal models (Figure 3). In the absence of T3, a positively regulated target gene will have a TRE to which the THR binds and recruits a co-repressor, such as N-CoR or N-CoR2.46 The co-repressor forms a complex with histone deacetylases, which modify the chromatin structure resulting in a subsequent a decrease in gene transcription.46 In the presence of T3, the repressive complex is destabilized and the co-repressors are released, which leads to closure of helix 12, exposure of the AF-2 domain and recruitment of co-activators, such as SRC-1 (Figure 3).36,47,48 Co-activators induce remodeling of chromatin by acetylating or methylating histones or altering the DNA conformation, which changes the interactions among RNA polymerase and other transcriptional factors.49
Figure 3.
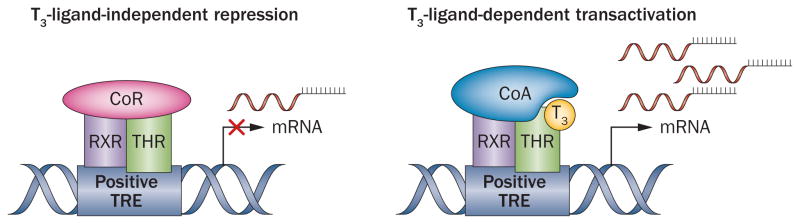
Model of gene regulation by thyroid hormones. In the absence of T3, a co-repressor is bound to the RXR–THR heterodimer at the positive TRE, thereby actively repressing target gene expression. When T3 binds to the THR dimer, the o-repressor is released and co-activators are recruited, which results in activation of gene transcription. Abbreviations: CoA, co-activator; CoR, co-repressor; RXR, retinoid X receptor; THR, thyroid hormone receptor; TRE, thyroid-hormone responsive element.
There is no widely accepted model for T3 action on negatively regulated target genes, although some features of a potential model have been established. In vitro studies have demonstrated that THRs can bind to sequences of putative negative TREs, which leads to repression of gene transcription and we and others have postulated that this repression occurs via mechanisms that are the inverse of those that regulate transcriptional activation.50–52 A mouse model with a targeted disruption in the DBD of THRβ confirmed that THR binding to DNA is needed for negative regulation of the HPT axis.11 However, the specific roles of co-repressors and co-activators remain unknown. SRC-1 is a prototypic co-activator that can function as either a positive or negative regulator of TSH.12,53,54 Furthermore, the co-repressor N-CoR has been demonstrated to drive the increase in TSH levels that results from lack of negative feedback when thyroid hormone levels are low.55 In vitro data also indicate that N-CoR targets NR1D1 (also known as Rev-Erbα) not THR at the Tshb promoter to regulate basal circadian expression patterns of this gene.56 It is also possible that a completely different set of cofactors is recruited when THR is bound to a negative TRE. For example, nuclear factor RIP140 interacts with THRα–RXR or THRβ–RXR heterodimers, and this interaction might lead to inhibition of transcription mediated by THR homodimers.12,57 Moreover, findings from a study in which the genome-wide binding sites for THRβ1 in livers of hypothyroid and hyperthyroid mice were compared suggested that negative gene regulation in this tissue might be an indirect consequence of decreased THR recruitment to positive TREs in the presence of T3.58
THR isoforms and gene regulation
The THRα and THRβ proteins are structurally alike, which suggests that they might have similar functions. However, the identification of patients with mutations in THRA and their markedly different clinical presentation to that of patients with mutations in THRB suggests that the THR isoforms have distinct and/or complementary physiological roles that extend beyond differences in tissue distribution.16,20 In patients with mutations in THRA, the HPT axis is not impaired as it is in patients with RTHβ.16 These clinical observations are supported by findings from studies in mouse models in which THRβ was identified as the receptor that modulates negative regulation of the HPT axis.25,59,60 In mouse pituitary thyrotroph cells, T3 functions to regulate Tshb transcription selectively through Thrβ;7 however, it is also possible that this Thrβ-mediated regulation is antagonized by Thrα.7,60–62 Transcriptome and cistrome analyses, which determined both the level of expression of target genes and the THR binding sites on a genome-wide scale in a neuronal cell line, suggest that both THR proteins can bind to the same TREs; however, they also bind to unique sites to regulate expression of target genes in an isoform-specific manner.45 Nota4bly, this study was performed in cells grown in culture in which either THRα1 or THRβ1 was expressed exclusively and these conditions do not precisely mimic those in normal tissues where both proteins are usually present.
Aetiology and clinical features of RTHβ
Patients with mutations in THRB present with RTHβ, which is typically characterized by elevated thyroid hormone levels, concentrations of TSH either within the normal range or mildly elevated, goitre and the absence of symptoms of thyrotoxicosis. Clinical phenotypes of RTHβ can be highly variable depending on the degree of tissue responsiveness to elevated thyroid hormone levels in a given individual; the same mutation can result in different phenotypes in different patients, even within the same family (Figure 4). In general, a single copy of the mutated gene is sufficient to cause disease, and as such, RTHβ is inherited in an autosomal dominant fashion.20 The clinical presentation, together with the fact that most patients with RTHβ are heterozygous for mutations in THRB, suggests that pathogenesis of the disease is driven by dominant-negative inhibition of wild-type THRβ.63
Figure 4.

Overview of tissues and homeostatic functions affected in RTHβ. Patients with mutations in THRB can present with different phenotypes that affect a number of tissues and functions. These effects include increased levels of circulating thyroid hormones, goitre, impaired negative feedback of the HPT axis, affected vision and hearing, heart defects and abnormal neuronal development. Abbreviations: HPT, hypothalamic–pituitary–thyroid; RTHβ, resistance to thyroid hormone β; TRH, thyrotropin-releasing hormone.
Only four patients with homozygous mutations in THRB have been identified to date, and each of these patients exhibits a more severe phenotype than patients with heterozygous mutations in this gene. One patient presented with a homozygous deletion of threonine residue 337 (Thr337del) in THRB.64,65 This mutation, which is localized to the LBD, prevents T3 binding to the receptor, although DNA-binding capacity is preserved. The affected patient presented with goitre, elevated levels of T4 and TSH, delays in growth and bone development, tachycardia, heart malformation, impaired hearing and vision, and intellectual disabilities. As these patients do not have a wild-type copy of THRB, it has been suggested that the severe phenotypes characteristic of these individuals might be due to a dominant-negative effect of mutant THRβ on wild-type THRα.64
Members of a family in which RTHβ was inherited in an autosomal-recessive mode carried a mutation that resulted in deletion of the entire coding region.66 Family members that were homozygous for this mutation presented with hearing impairment, colour blindness, stippled epiphyses and growth delay but had an IQ within the normal range.66 However, patients who retain a single functioning wild-type THRB allele do not exhibit these symptoms, which supports the hypothesis that mutant THRβ exerts dominant-negative effects that are responsible for the phenotypes seen in patients with RTHβ.66
Aetiology and clinical features of RTHα
In the past 2 years, reports have emerged of patients with mutations in THRA that lead to the development of RTHα.10,16–19 The first patient identified with RTHα carried a heterozygous nonsense mutation in THRA (Glu403X) that resulted in a frameshift and loss of the helix 12 owing to the introduction of a premature stop codon.16,33 This patient presented with delay of linear growth, tooth eruption and severe constipation, in addition to decreased muscle tone and impaired gross and fine motor skills. Defects in the cardiovascular system, including decreased heart rate and blood pressure, were consistent with THRα1 being the predominant isoform expressed in the heart. This patient was not responsive to treatment with levothyroxine, which reflected resistance to T3. Conversely, circulating levels of sex-hormone-binding globulin (a hepatic marker of thyroid hormone action) were elevated and levels of serum TSH were suppressed after treatment with levotyroxine, which is consistent with THRβ predominance in the liver and pituitary gland, respectively.
Analysis of the functional activity of the Glu403X mutant protein in cultured cells revealed very low capacity of the mutant receptor to bind T3, defective activation at canonical TREs and increased repression of basal promoter activity, which occurred via recruitment of co-repressors. Together, these findings suggest that the Glu403X mutant protein functions as a dominant-negative receptor.16
Six other patients with RTHα have also been identified, all of whom have heterozygous mutations in THRA that result in disruptions to the carboxyl-terminus of the THRα protein.17–19,67 A father and daughter with a Phe406X mutation in THRα presented with clinical signs similar to that of the index patient with RTHα, an adult woman with an Ala382X mutation in THRα; a mother and two sons with Ala263Val mutations in THRα have also been described.17–19,67 Each of these six individuals presented with severe constipation, short stature, bradycardia, low levels of IGF–1 and clinical signs of hypothyroidism, such as dry skin, elevated levels of total cholesterol and LDL cholesterol, slow reflexes and delayed intellectual development that was confirmed by a low IQ evaluation (Figure 5).18,19
Figure 5.

Overview of tissues and homeostatic functions affected in RTHα. In patients with mutations in THRA the levels of circulating thyroid hormones are mildly affected. Additionally, these patients have delayed bone development, heart defects, chronic constipation and impaired neuronal development. Abbreviation: RTHα, resistance to thyroid hormone α.
In vitro studies that assessed the functions of wild-type and Phe406X THRα proteins demonstrated that Phe406X THRα acts in a dominant-negative fashion and inhibits wild-type THRα. However, studies in cell lines indicated that the dominant-negative effects of mutant THRα on wild-type THRβ were partially reversed in the presence of high levels of thyroid hormones, which might explain why total cholesterol and LDL cholesterol levels were normalized in patients with RTHα following treatment with levothyroxine.67,109
Unlike patients with RTHβ, the HPT axis in patients carrying mutations in THRA is minimally affected. Patients with RTHα have circulating TSH levels within the normal range, slightly lowered levels of free T4 and slightly elevated levels of total T3, which results in an abnormally low free T4:T3 ratio. Furthermore, treatment with levothyroxine leads to suppression of TSH levels among patients with RTHα, a finding that supports the hypothesis that the central T3 feedback loop in these patients is not impaired as it is in patients with RTHβ.20,60,67
Mouse models and THR function
Advances in techniques for manipulation of mouse genetics have enabled detailed investigations into the functions of the THR isoforms. Early studies used mouse models in which either Thra or Thrb was completely deleted. Homozygous Thrb deletion caused auditory system defects owing to impaired cochlear development; impaired vision resulting from lack of expression of the gene encoding medium-wave-sensitive opsin 1; defects in cerebellum foliation; and disruption of T3-mediated repression of Tshb and Trh genes.11,68–72
To establish the role and importance of each isoform in specific organs and cell types, naturally occurring THRB mutations that are found in patients with RTHβ,13,64,73–76 as well as artificial mutations,11,12 have been reproduced in mouse models (Table 1). In these mouse models the mutated THRs were either overexpressed or expressed in an allelic manner using homologous recombination techniques whereby the endogenous sequence is substituted by the mutated sequence, a so-called ‘knock-in’ mouse model. The primary biochemical phenotype of RTHβ, namely impairment of the HPT axis, is recapitulated in each of these mouse models. Mice had elevated levels of circulating T4 and T3 and high levels of TSH that could not be suppressed by the administration of T3.11,12,74,77 One important similarity of the mouse model phenotypes to that of patients with RTHβ was the presence of goitre, which results from exposure to high levels of circulating TSH and is the most frequent clinical sign among patients with RTHβ.12,13
Table 1.
Mouse models of mutations in Thrβ
| Mutation | Able to bind T3 | Affected systems | Reference |
|---|---|---|---|
| Thr337del* | No | Cerebellum, ear, retina, HPT axis | Hashimoto et al. (2001)73 |
| Thr448fsX17*‡ | No | Cerebellum, ear, retina, HPT axis, heart | Kaneshige et al. (2000)13 |
| Glu124Gly§ and Gly125Ser§ | Yes | Ear, retina, HPT axis | Shibusawa et al. (2003)11 |
| Glu457Ala§ | Yes | Ear, retina, HPT axis | Ortiga-Carvalho et al. (2005)12 |
| Arg429Gln* | Yes | Ear, retina, HPT axis, heart | Machado et al. (2009)74 |
Natural mutation.
PV mutant
Artificial mutation.
Abbreviation: HPT, hypothalamic–pituitary–thyroid.
Sensory systems in mice, such as hearing11,78,79 and vision,80–82 were also adversely affected by introduction of mutations in Thrb, with disruption of Thrb2 being primarily responsible for these phenotypes. However, few studies in mice have successfully addressed the implications of these mutations in humans.83
Genomic effects of thyroid hormones
As with other nuclear receptors in the superfamily, the THR DBD contains a zinc-rich structure that binds to double-stranded DNA. Monomeric, homodimeric or heterodimeric THRs bind to TREs that contain the nucleotide sequence 5’–(A/G)GGT(C/A/G)A–3’. This sequence is arranged as a direct repeat with four spacer nucleotides (DR+4), which can be arranged either as a palindrome or an inverted palindrome.1,84 To definitively address the importance of THR binding to DNA in the context of gene regulation by T3, the Thrb’s DBD sequence was mutated to resemble the sequence of the glucocorticoid receptor’s DBD, which renders the mutant THR unable to recognize TREs.11,50 Introduction of this mutation completely abolished THRβ DNA-binding and binding to canonical TREs; however, T3 ligand-induced interactions with cofactors such as N-CoR and SRC-1 were unaffected, although impaired positive and negative regulation was noted in assays of thyroid-hormone-regulated gene promoter activity.50 A knock-in mouse model of the DBD-glucocorticoid-receptor mutation in Thrb was phenotypically similar to Thrb knockout mice, but the effects were less severe with regard to hearing, although visual impairment was similar to that observed in other mutant Thrb mouse models.11,81,82 Importantly, mice that are homozygous for this mutation in the DBD have an impairment in TSH regulation by T3 that is similar to that observed in Thrb knockout mice, which confirms that the interaction between THRβ and TREs is essential for T3-mediated inhibition of Tshb.11
When the THR binds T3, the LBD undergoes a dramatic structural change that facilitates the association of co-activators.34,36,47,85,86 The physiological importance and specific role of co-activators and co-repressors that bind AF-2 to facilitate T3-driven inhibition of transcription are not well known; however, studies using a knock-in mouse model that expressed a mutant Thrβ Glu457Ala protein have demonstrated that this region is critical for T3-mediated transcriptional inhibition.12 Interestingly, double knock-in mice that were generated by crossing Thrb-mutant mice with mice expressing a mutant copy of Ncor1 showed partial correction of the increased levels of circulating TSH, T4 and T3 and a subsequent reduction in the size of goitre.87
Cardiac functions
Excess or deficiency of thyroid hormones has profound effects on cardiac function. Thyroid hormones have both genomic and non-genomic effects in the heart and the vascular system, which are difficult to distinguish in vivo.88,89 Thra and Thrb mRNAs are expressed at a 3:1 ratio in the mouse heart, and comparison of hearts of mice in which Thra or Thrb have been deleted strongly suggests that Thrα1 is the major mediator of T3 effects on cardiac function.90
Studies using a mouse model in which the human Thr337del mutation is homozygous and expressed under control of the endogenous Thrb promoter have been instructive in elucidating the role of THRβ in the heart. The Thr337del mutation causes resistance to thyroid hormones in the HPT axis. Additionally, these mice demonstrate resistance to T3-induced cardiac hypertrophy.26,73 However, when overexpressed only in the heart, this mutation resulted in cardiac-specific hypothyroidism, which manifested as increased expression of Myh7 and decreased expression of Myh6 genes, despite the presence of normal serum levels of thyroid hormones.63 Interestingly, the T3-induced cardiac hypertrophy observed in these mice was similar to that observed in wild-type mice treated with T3.63,91 Given that the heart is a tissue in which Thrα1 is the predominantly expressed THR isoform, these studies suggest that Thrβ has a dominant-negative effect on wild-type Thrα. Additionally, these findings establish the importance of Thrα in maintenance of cardiac function and suggest that Thrβ might directly or indirectly regulate T3-dependent cardiac hypertrophy.63,91
The mouse PV mutant model is based on the human THRB PV mutation,75 which is caused by a frameshift in the LBD and results in deletion of the last nine amino acids of the carboxyl-terminus of THRβ.15 Mice that were homozygous for the PV mutation demonstrated a mild decrease in heart rate, which was further decreased when the mice were rendered euthyroid by administration of anti-thyroid drugs.88 Euthyroid homozygous Thrβ PV mice also had a decrease in cardiac contractile function, which suggests that in the presence of normal thyroid hormone levels mutant Thrβ could function as a dominant-negative protein, presumably acting on wild-type Thrα. Taken together, evidence from these mouse studies suggests that Thrβ1 has an important role in regulating specific functions in the heart.88,92
Modelling RTHβ
Clinical investigations into the pathophysiology of RTHβ have yielded important information about the role of cofactors in THR-mediated gene regulation. In 1996, families with severe RTHβ phenotypes who had no discernible mutations in THRB, THRA or in the TSHB promoter region were described.35 Assays of nuclear protein extracts isolated from affected individuals revealed the presence of additional proteins that interacted with wild-type THRβ, which suggested that a lack of co-activator function could be responsible for the RTHβ observed in these individuals.35
Studies in mouse models support this hypothesis (Table 1); mice lacking Src-1 present with mild resistance to thyroid hormone.53 However, it is also possible that other co-activators and co-repressors, such as nuclear factor RIP140, bind to the same region of the AF-2 domain of THR. Introduction of a mutation in Thrb that results in a Glu457Ala mutation in the AF-2 domain and completely abolishes co-activator recruitment in vitro was found to preserve T3 binding and limit recruitment of the co-repressor nuclear factor RIP140.12 In a homozygous mouse mutant expressing Thrβ Glu457Ala, both negative and positive gene regulation mediated by T3 were impaired. Serum levels of T3, T4 and TSH were highly elevated compared with those of wild-type mice and administration of T3 failed to suppress the elevation of TSH levels.12
The mechanism by which cofactor recruitment to the ligand-bound AF-2 domain of THR leads to inhibition of transcription is presently unknown. However, the findings from studies in Glu457Ala mice suggest that an AF-2 domain of a THR that is bound to a negatively regulated gene could potentially recruit a different complement of cofactors to a THR that is bound to a positively regulated gene.12 These results are supported by findings from studies in which the Glu457Ala mutation was introduced into Src-1 knockout mouse.37 Thyroid hormone levels in homozygous Thrβ Glu457Ala mice that also lacked Src-1 were elevated compared with those in Thrβ Glu457Ala mutant mice that retained Src-1. However, when double-mutant mice were rendered hypothyroid by the use of an anti-thyroid diet (low iodine plus 5-propyl-2-thiouracil) they had only a 50% increase in TSH levels when compared with mice lacking only Src-1.37 Taken together, these data suggest that co-activators are involved in ligand-dependent repression of negatively regulated genes.12,37
Studies using in vitro systems have shown that THR–RXR heterodimerization increases the binding of THR to TREs and results in increased transcription levels of target genes.1 Mutations in patients with RTHβ, as well as experiments with transgenic mice, have provided clues to the functional differences of homodimeriza-tion versus heterodimerization of THRs in vivo in the context of gene regulation. The majority of THRB mutations found in patients with RTHβ result in reduced T3 binding; however, some mutations do not impair the ability of THRs to bind T3 yet still cause RTH in some tissues. One of these mutations is Arg429Gln,93 which is located in helix11 of the LBD, a region that contains a hydrophobic surface suitable for promotion of protein–protein interactions.34,94 Different mutations in the same region selectively interfere with dimerization.95–97 The THRβ Arg429Gln mutant functions differently to most other THRβ proteins with RTHβ-associated mutations; T3 binding is intact but there is a deficiency in homodimer formation and co-repressor recruitment.74 In a knock-in mouse model, the Arg429Gln mutation exclusively affects negative gene regulation in the liver, heart and pituitary gland. This phenotype differs from those of most Thrb mutations, which affect both negative and positive gene regulation. The Arg429Gln mutation is the only homodimer-insufficient model studied to date and has been an extremely useful tool for elucidating the mechanism of negative gene regulation that is driven by thyroid hormones.74
Modelling RTHα
In the first mouse model in which Thra was knocked out, pups failed to survive 5 weeks after birth, which suggested that functional Thrα is essential for postnatal development.98 In this model, both the Thrα1 and Thrα2 isoforms were inactivated and the mice showed delayed maturation in the small intestine and bones.98 However, the small intestine phenotype in these mice seems to be unrelated to the severe constipation seen in patients with RTHα.25
The generation of Thra mutant mouse models that contain the same mutations found in the THRB locus in patients with RTHβ showed that it was possible that mutations in THRA were not necessarily lethal and that phenotypes associated with mutation of this gene might also be present in humans.15 Several mutant Thra mouse models have been developed and findings from experiments using these mice are conflicting. Here, we will focus on the models that produced similar phenotypes to those seen in patients with RTHα, including Thrα PV, Thrα Leu400Arg, and Thrα Arg384Cys (Table 2). In all cases, the mutations in Thra resulted in reduced or abolished T3 binding and recruitment of co-activators and caused a dominant negative effect.16,98–101 Interestingly, despite having similar biochemical properties in vitro, each of these mutations resulted in slightly different in vivo phenotypes that have provided insights into the different functions of the affected protein regions.33,102–104
Table 2.
Thrα mutant mouse models phenotypically similar to patients with RTHα
| Mutation* | Able to bind T3 | Affected systems or phenotype | Reference |
|---|---|---|---|
| Pro398His | 50% decreased | Obesity, heart | Liu et al. (2003)106 |
| Thr394fsX17‡ | No | Bone, dwar sm | O’Shea et al. (2005)100 |
| Leu400Arg | Yes | Dwar sm, cerebellum | Quignodon et al. (2007)101 |
| Arg384Cys | Partially impaired | Bone, delayed growth | Tinnikov et al. (2002)104 |
Artificial mutation.
PV mutation.
Abbreviation: RTHα, resistance to thyroid hormone α
Introduction of the PV mutation15 into mouse Thra and Thrb genes demonstrated the importance of an intact THRα LBD for correct skeletal development.99,100 Specifically, Thra PV mice but not Thrb PV mice presented with growth retardation similar to that observed in patients with RTHα.16,19,103
The Thrα Leu400Arg knock-in mouse was developed on the basis of an artificial RTH mutation (Thrβ Leu454Arg). This mutation prevents the binding of Thrα to co-activators but preserves interaction of the receptor with co-repressors.101 In contrast to the Thrα PV model, Thrα Leu400Arg binds T3 normally but retains strong dominant-negative activity. In addition to dwarfism, these mice have delayed cerebellar development, which is characterized by a delayed granule-cell differentiation pattern similar to that seen in patients with congenital hypothyroidism.101 These mice have difficulty maintaining body temperature under stress and usually do not survive beyond 3 weeks after birth.101 The Leu400Arg mutation was inserted into different cerebellar cell types using a conditional expression system; in these experiments, nonfunctional THRα primarily affected Purkinje cells and the Bergman glia.102
Lastly, the Thrα Arg384Cys knock-in mouse model was developed to mimic a human THRB mutation that results in an Arg438Cys substitution that affects the LBD and decreases T3 binding by 10-fold.103 These mice exhibited slightly lowered serum levels of T3 and T4.104 Marked signs of underdevelopment were observed during the neonatal period, although this phenotype became milder in the adult mice. Patients with mutations in THRA and some Thra mutant mouse models display growth failure that persists into adulthood; however, this effect does not seem to occur in Thrα Arg384Cys mice, which suggests that this model has limited value for the study of abnormalities in growth. Alternatively, it is possible that as more patients with RTHα are identified, not all of these individuals will have substantial growth retardation.
Conclusions
Resistance to thyroid hormone was first described as a clinical entity in 1967.105 A majority of patients with this disorder present with goitre, elevated thyroid hormone levels and normal or elevated levels of TSH. The molecular pathogenesis of this syndrome was determined over 20 years later when mutations in the region of the gene that encodes the ligand-binding domain of THRβ were identified in patients with RTHβ. Interestingly, patients with mutations in THRA were not identified until 2012. Individuals with these mutations have RTHα, which is clinically distinct from RTHβ and is characterized by nearly normal thyroid hormone and TSH levels, as well as growth retardation and constipation.
Complementary studies of patients with mutations in either THRB or THRA and findings from mouse models that have been generated to recapitulate these mutations have provided strong evidence to support distinct and specific roles for each of the THR isoforms. Both receptors have an important role in central nervous system, cerebellum and heart. However, it seems that only THRβ is important for regulating the negative-feedback loop of the HPT axis that maintains circulating thyroid hormone levels in a normal range. Furthermore, THRβ is important for proper retinal and cochlear development. By contrast, THRα has a key role in bone and intestinal development and function. Further characterization of these differences will be likely to involve the identification of the full complement of cofactors that are recruited or dismissed by the THRs in response to thyroid hormone. Such investigations have the potential to further development of specific agonists and antagonists that could provide treatment options for patients with rare disorders of THR action, such as RTHα and RTHβ, as well as patients with other disorders of metabolism, growth, and intestinal motility.
Review criteria.
A search for original articles published between 1986 and 2014 and focusing on thyroid hormone receptors was performed in MEDLINE and PubMed. The search terms used were “thyroid hormone receptor”, “thyroid hormone receptor alpha”, “thyroid hormone receptor beta” and “resistance to thyroid hormone” alone and in combination. In addition, personal libraries of references were used. All articles identified were English-language, full-text papers. We also searched the reference lists of identified articles for additional relevant papers.
Acknowledgments
T.M.O.-C.’s research is supported by Fundação Carlos Chagas Filho de Amparo a Pesquisa do Estado do Rio de Janeiro (FAPERJ, CNE 102.873/2012) and Conselho Nacional de Pesquisa e Desenvolvimento (CNPq, #303,734/2012-4) and the Bill and Melinda Gates Foundation. F.E.W.’s research is supported by National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) grant R01 DK49126 and the Johns Hopkins–University of Maryland Diabetes Research Center NIDDK grant P30DK79637.
Footnotes
Competing interests
The authors declare no competing interests.
Author contributions
All authors contributed equally to all aspects of this article.
Contributor Information
Tânia M. Ortiga-Carvalho, Instituto de Biofísica Carlos Chagas Filho, Universidade Federal do Rio de Janeiro, Av. Carlos Chagas Filho, S/N, Cidade Universitária, 21941-902, Rio de Janeiro, Brazil
Aniket R. Sidhaye, Departments of Paediatrics and Medicine, Johns Hopkins University School of Medicine, 600 N. Wolfe Street, CMSC 10-113, Baltimore, MD 21287, USA
Fredric E. Wondisford, Departments of Paediatrics and Medicine, Johns Hopkins University School of Medicine, 600 N. Wolfe Street, CMSC 10-113, Baltimore, MD 21287, USA
References
- 1.Cheng SY, Leonard JL, Davis PJ. Molecular aspects of thyroid hormone actions. Endocr Rev. 2010;31:139–170. doi: 10.1210/er.2009-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brent GA. Mechanisms of thyroid hormone action. J Clin Invest. 2012;122:3035–3043. doi: 10.1172/JCI60047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chiamolera MI, Wondisford FE. Minireview: Thyrotropin-releasing hormone and the thyroid hormone feedback mechanism. Endocrinology. 2009;150:1091–1096. doi: 10.1210/en.2008-1795. [DOI] [PubMed] [Google Scholar]
- 4.Nikrodhanond AA, et al. Dominant role of thyrotropin-releasing hormone in the hypothalamic–pituitary–thyroid axis. J Biol Chem. 2006;281:5000–5007. doi: 10.1074/jbc.M511530200. [DOI] [PubMed] [Google Scholar]
- 5.Gereben B, et al. Cellular and molecular basis of deiodinase-regulated thyroid hormone signaling. Endocr Rev. 2008;29:898–938. doi: 10.1210/er.2008-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bianco AC. Minireview: cracking the metabolic code for thyroid hormone signaling. Endocrinology. 2011;152:3306–3311. doi: 10.1210/en.2011-1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chiamolera MI, et al. Fundamentally distinct roles of thyroid hormone receptor isoforms in a thyrotroph cell line are due to differential DNA binding. Mol Endocrinol. 2012;26:926–939. doi: 10.1210/me.2011-1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sap J, et al. The c-erb-A protein is a high-affinity receptor for thyroid hormone. Nature. 1986;324:635–640. doi: 10.1038/324635a0. [DOI] [PubMed] [Google Scholar]
- 9.Oetting A, Yen PM. New insights into thyroid hormone action. Best Pract Res Clin Endocrinol Metab. 2007;21:193–208. doi: 10.1016/j.beem.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 10.Refetoff S, et al. Classification and proposed nomenclature for inherited defects of thyroid hormone action, cell transport, and metabolism. Thyroid. 2014;24:407–409. doi: 10.1089/thy.2013.3393.nomen. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shibusawa N, et al. Thyroid hormone action in the absence of thyroid hormone receptor DNA-binding in vivo. J Clin Invest. 2003;112:588–597. doi: 10.1172/JCI18377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ortiga-Carvalho TM, et al. Negative regulation by thyroid hormone receptor requires an intact coactivator-binding surface. J Clin Invest. 2005;115:2517–2523. doi: 10.1172/JCI24109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaneshige M, et al. Mice with a targeted mutation in the thyroid hormone β receptor gene exhibit impaired growth and resistance to thyroid hormone. Proc Natl Acad Sci USA. 2000;97:13209–13214. doi: 10.1073/pnas.230285997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferrara AM, et al. Homozygous thyroid hormone receptor β-gene mutations in resistance to thyroid hormone: three new cases and review of the literature. J Clin Endocrinol Metab. 2012;97:1328–1336. doi: 10.1210/jc.2011-2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaneshige M, et al. A targeted dominant negative mutation of the thyroid hormone α1 receptor causes increased mortality, infertility, and dwarfism in mice. Proc Natl Acad Sci USA. 2001;98:15095–15100. doi: 10.1073/pnas.261565798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bochukova E, et al. A mutation in the thyroid hormone receptor α gene. N Engl J Med. 2012;366:243–249. doi: 10.1056/NEJMoa1110296. [DOI] [PubMed] [Google Scholar]
- 17.Moran C, et al. Resistance to thyroid hormone caused by a mutation in thyroid hormone receptor (TR)α1 and TRα2: clinical, biochemical, and genetic analyses of three related patients. Lancet Diabetes Endocrinol. doi: 10.1016/S2213-8587(14)70111-1. http://dx.doi.org/10.1016/S2213-8587(14)70111-1. [DOI] [PMC free article] [PubMed]
- 18.Moran C, et al. An adult female with resistance to thyroid hormone mediated by defective thyroid hormone receptor α. J Clin Endocrinol Metab. 2013;98:4254–4261. doi: 10.1210/jc.2013-2215. [DOI] [PubMed] [Google Scholar]
- 19.van Mullem A, et al. Clinical phenotype and mutant TRα1. N Engl J Med. 2012;366:1451–1453. doi: 10.1056/NEJMc1113940. [DOI] [PubMed] [Google Scholar]
- 20.Refetoff S, Dumitrescu AM. Syndromes of reduced sensitivity to thyroid hormone: genetic defects in hormone receptors, cell transporters and deiodination. Best Pract Res Clin Endocrinol Metab. 2007;21:277–305. doi: 10.1016/j.beem.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 21.Phan TQ, Jow MM, Privalsky ML. DNA recognition by thyroid hormone and retinoic acid receptors: 3, 4, 5 rule modified. Mol Cell Endocrinol. 2010;319:88–98. doi: 10.1016/j.mce.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Warnmark A, Treuter E, Wright AP, Gustafsson JA. Activation functions 1 and 2 of nuclear receptors: molecular strategies for transcriptional activation. Mol Endocrinol. 2003;17:1901–1909. doi: 10.1210/me.2002-0384. [DOI] [PubMed] [Google Scholar]
- 23.Figueira AC, et al. Analysis of agonist and antagonist effects on thyroid hormone receptor conformation by hydrogen/deuterium exchange. Mol Endocrinol. 2011;25:15–31. doi: 10.1210/me.2010-0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chassande O, et al. Identification of transcripts initiated from an internal promoter in the c-erbA α locus that encode inhibitors of retinoic acid receptor-α and triiodothyronine receptor activities. Mol Endocrinol. 1997;11:1278–1290. doi: 10.1210/mend.11.9.9972. [DOI] [PubMed] [Google Scholar]
- 25.Gauthier K, et al. Different functions for the thyroid hormone receptors TRα and TRβ in the control of thyroid hormone production and post-natal development. EMBO J. 1999;18:623–631. doi: 10.1093/emboj/18.3.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Williams GR. Cloning and characterization of two novel thyroid hormone receptor β isoforms. Mol Cell Biol. 2000;20:8329–8342. doi: 10.1128/mcb.20.22.8329-8342.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wallis K, et al. The thyroid hormone receptor α1 protein is expressed in embryonic postmitotic neurons and persists in most adult neurons. Mol Endocrinol. 2010;24:1904–1916. doi: 10.1210/me.2010-0175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bradley DJ, Towle HC, Young WS. 3rd Spatial and temporal expression of α- and β-thyroid hormone receptor mRNAs, including the β2-subtype, in the developing mammalian nervous system. J Neurosci. 1992;12:2288–2302. doi: 10.1523/JNEUROSCI.12-06-02288.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bradley DJ, Towle HC, Young WS., 3rd α and β thyroid hormone receptor (TR) gene expression during auditory neurogenesis: evidence for TR isoform-specific transcriptional regulation in vivo. Proc Natl Acad Sci USA. 1994;91:439–443. doi: 10.1073/pnas.91.2.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kilby MD, et al. Circulating thyroid hormone concentrations and placental thyroid hormone receptor expression in normal human pregnancy and pregnancy complicated by intrauterine growth restriction (IUGR) J Clin Endocrinol Metab. 1998;83:2964–2971. doi: 10.1210/jcem.83.8.5002. [DOI] [PubMed] [Google Scholar]
- 31.Feng X, Jiang Y, Meltzer P, Yen PM. Thyroid hormone regulation of hepatic genes in vivo detected by complementary DNA microarray. Mol Endocrinol. 2000;14:947–955. doi: 10.1210/mend.14.7.0470. [DOI] [PubMed] [Google Scholar]
- 32.Flores-Morales A, et al. Patterns of liver gene expression governed by TRβ. Mol Endocrinol. 2002;16:1257–1268. doi: 10.1210/mend.16.6.0846. [DOI] [PubMed] [Google Scholar]
- 33.de Araujo AS, Martinez L, de Paula Nicoluci R, Skaf MS, Polikarpov I. Structural modeling of high-affinity thyroid receptor-ligand complexes. Eur Biophys J. 2010;39:1523–1536. doi: 10.1007/s00249-010-0610-2. [DOI] [PubMed] [Google Scholar]
- 34.Valadares NF, Polikarpov I, Garratt RC. Ligand induced interaction of thyroid hormone receptor β with its coregulators. J Steroid Biochem Mol Biol. 2008;112:205–212. doi: 10.1016/j.jsbmb.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 35.Weiss RE, et al. Dominant inheritance of resistance to thyroid hormone not linked to defects in the thyroid hormone receptor α or β genes may be due to a defective cofactor. J Clin Endocrinol Metab. 1996;81:4196–4203. doi: 10.1210/jcem.81.12.8954015. [DOI] [PubMed] [Google Scholar]
- 36.Souza PC, et al. Helix 12 dynamics and thyroid hormone receptor activity: experimental and molecular dynamics studies of Ile280 mutants. J Mol Biol. 2011;412:882–893. doi: 10.1016/j.jmb.2011.04.014. [DOI] [PubMed] [Google Scholar]
- 37.Alonso M, et al. In vivo interaction of steroid receptor coactivator (SRC)-1 and the activation function-2 domain of the thyroid hormone receptor (TR) β in TRβ E457A knock-in and SRC-1 knockout mice. Endocrinology. 2009;150:3927–3934. doi: 10.1210/en.2009-0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lavery DN, McEwan IJ. Structure and function of steroid receptor AF1 transactivation domains: induction of active conformations. Biochem J. 2005;391:449–464. doi: 10.1042/BJ20050872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cohen RN, et al. The specificity of interactions between nuclear hormone receptors and corepressors is mediated by distinct amino acid sequences within the interacting domains. Mol Endocrinol. 2001;15:1049–1061. doi: 10.1210/mend.15.7.0669. [DOI] [PubMed] [Google Scholar]
- 40.Hollenberg AN, Monden T, Wondisford FE. Ligand-independent and -dependent functions of thyroid hormone receptor isoforms depend upon their distinct amino termini. J Biol Chem. 1995;270:14274–14280. doi: 10.1074/jbc.270.24.14274. [DOI] [PubMed] [Google Scholar]
- 41.Hashimoto K, et al. Cross-talk between thyroid hormone receptor and liver X receptor regulatory pathways is revealed in a thyroid hormone resistance mouse model. J Biol Chem. 2006;281:295–302. doi: 10.1074/jbc.M507877200. [DOI] [PubMed] [Google Scholar]
- 42.Liu YY, et al. A mutant thyroid hormone receptor α antagonizes peroxisome proliferator-activated receptor α signaling in vivo and impairs fatty acid oxidation. Endocrinology. 2007;148:1206–1217. doi: 10.1210/en.2006-0836. [DOI] [PubMed] [Google Scholar]
- 43.Brent GA, et al. Capacity for cooperative binding of thyroid hormone (T3) receptor dimers defines wild type T3 response elements. Mol Endocrinol. 1992;6:502–514. doi: 10.1210/mend.6.4.1584220. [DOI] [PubMed] [Google Scholar]
- 44.Ayers S, et al. Genome-wide binding patterns of thyroid hormone receptor β. PLoS One. 2014;9:e81186. doi: 10.1371/journal.pone.0081186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chatonnet F, Guyot R, Benoit G, Flamant F. Genome-wide analysis of thyroid hormone receptors shared and specific functions in neural cells. Proc Natl Acad Sci USA. 2013;110:E766–E775. doi: 10.1073/pnas.1210626110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Astapova I, et al. The nuclear corepressor, NCoR, regulates thyroid hormone action in vivo. Proc Natl Acad Sci USA. 2008;105:19544–19549. doi: 10.1073/pnas.0804604105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Feng W, et al. Hormone-dependent coactivator binding to a hydrophobic cleft on nuclear receptors. Science. 1998;280:1747–1749. doi: 10.1126/science.280.5370.1747. [DOI] [PubMed] [Google Scholar]
- 48.Darimont BD, et al. Structure and specificity of nuclear receptor-coactivator interactions. Genes Dev. 1998;12:3343–3356. doi: 10.1101/gad.12.21.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McKenna NJ, et al. Nuclear receptor coactivators: multiple enzymes, multiple complexes, multiple functions. J Steroid Biochem Mol Biol. 1999;69:3–12. doi: 10.1016/s0960-0760(98)00144-7. [DOI] [PubMed] [Google Scholar]
- 50.Shibusawa N, Hollenberg AN, Wondisford FE. Thyroid hormone receptor DNA binding is required for both positive and negative gene regulation. J Biol Chem. 2003;278:732–738. doi: 10.1074/jbc.M207264200. [DOI] [PubMed] [Google Scholar]
- 51.Sasaki S, et al. Ligand-induced recruitment of a histone deacetylase in the negative-feedback regulation of the thyrotropin β gene. EMBO J. 1999;18:5389–5398. doi: 10.1093/emboj/18.19.5389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Santos GM, et al. Negative regulation of superoxide dismutase-1 promoter by thyroid hormone. Mol Pharmacol. 2006;70:793–800. doi: 10.1124/mol.106.025627. [DOI] [PubMed] [Google Scholar]
- 53.Weiss RE, et al. Mice deficient in the steroid receptor co-activator 1 (SRC-1) are resistant to thyroid hormone. EMBO J. 1999;18:1900–1904. doi: 10.1093/emboj/18.7.1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kamiya Y, et al. Modulation by steroid receptor coactivator-1 of target-tissue responsiveness in resistance to thyroid hormone. Endocrinology. 2003;144:4144–4153. doi: 10.1210/en.2003-0239. [DOI] [PubMed] [Google Scholar]
- 55.Costa-e-Sousa RH, Astapova I, Ye F, Wondisford FE, Hollenberg AN. The thyroid axis is regulated by NCoR1 via its actions in the pituitary. Endocrinology. 2012;153:5049–5057. doi: 10.1210/en.2012-1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aninye IO, Matsumoto S, Sidhaye AR, Wondisford FE. Circadian regulation of Tshb gene expression by Rev-Erbα (NR1D1) and nuclear corepressor 1 (NCOR1) J Biol Chem. 2014;289:17070–17077. doi: 10.1074/jbc.M114.569723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Treuter E, Albrektsen T, Johansson L, Leers J, Gustafsson JA. A regulatory role for RIP140 in nuclear receptor activation. Mol Endocrinol. 1998;12:864–881. doi: 10.1210/mend.12.6.0123. [DOI] [PubMed] [Google Scholar]
- 58.Ramadoss P, et al. Novel mechanism of positive versus negative regulation by thyroid hormone receptor β1 (TRβ1) identified by genome-wide profiling of binding sites in mouse liver. J Biol Chem. 2014;289:1313–1328. doi: 10.1074/jbc.M113.521450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abel ED, Ahima RS, Boers ME, Elmquist JK, Wondisford FE. Critical role for thyroid hormone receptor β2 in the regulation of paraventricular thyrotropin-releasing hormone neurons. J Clin Invest. 2001;107:1017–1023. doi: 10.1172/JCI10858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gauthier K, et al. Genetic analysis reveals different functions for the products of the thyroid hormone receptor α locus. Mol Cell Biol. 2001;21:4748–4760. doi: 10.1128/MCB.21.14.4748-4760.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Macchia PE, et al. Increased sensitivity to thyroid hormone in mice with complete deficiency of thyroid hormone receptor α. Proc Natl Acad Sci USA. 2001;98:349–354. doi: 10.1073/pnas.011306998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gothe S, et al. Mice devoid of all known thyroid hormone receptors are viable but exhibit disorders of the pituitary-thyroid axis, growth, and bone maturation. Genes Dev. 1999;13:1329–1341. doi: 10.1101/gad.13.10.1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pazos-Moura C, et al. Cardiac dysfunction caused by myocardium-specific expression of a mutant thyroid hormone receptor. Circ Res. 2000;86:700–706. doi: 10.1161/01.res.86.6.700. [DOI] [PubMed] [Google Scholar]
- 64.Ono S, et al. Homozygosity for a dominant negative thyroid hormone receptor gene responsible for generalized resistance to thyroid hormone. J Clin Endocrinol Metab. 1991;73:990–994. doi: 10.1210/jcem-73-5-990. [DOI] [PubMed] [Google Scholar]
- 65.Usala SJ, et al. A homozygous deletion in the c-erbA β thyroid hormone receptor gene in a patient with generalized thyroid hormone resistance: isolation and characterization of the mutant receptor. Mol Endocrinol. 1991;5:327–335. doi: 10.1210/mend-5-3-327. [DOI] [PubMed] [Google Scholar]
- 66.Takeda K, Sakurai A, DeGroot LJ, Refetoff S. Recessive inheritance of thyroid hormone resistance caused by complete deletion of the protein-coding region of the thyroid hormone receptor-β gene. J Clin Endocrinol Metab. 1992;74:49–55. doi: 10.1210/jcem.74.1.1727829. [DOI] [PubMed] [Google Scholar]
- 67.van Mullem AA, et al. Clinical phenotype of a new type of thyroid hormone resistance caused by a mutation of the TRα1 receptor: consequences of LT4 treatment. J Clin Endocrinol Metab. 2013;98:3029–3038. doi: 10.1210/jc.2013-1050. [DOI] [PubMed] [Google Scholar]
- 68.Forrest D, Erway LC, Ng L, Altschuler R, Curran T. Thyroid hormone receptor β is essential for development of auditory function. Nat Genet. 1996;13:354–357. doi: 10.1038/ng0796-354. [DOI] [PubMed] [Google Scholar]
- 69.Forrest D, Vennstrom B. Functions of thyroid hormone receptors in mice. Thyroid. 2000;10:41–52. doi: 10.1089/thy.2000.10.41. [DOI] [PubMed] [Google Scholar]
- 70.Abel ED, et al. Divergent roles for thyroid hormone receptor β isoforms in the endocrine axis and auditory system. J Clin Invest. 1999;104:291–300. doi: 10.1172/JCI6397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dettling J, et al. Autonomous functions of murine thyroid hormone receptor TRα and TRβ in cochlear hair cells. Mol Cell Endocrinol. 2013;382:26–37. doi: 10.1016/j.mce.2013.08.025. [DOI] [PubMed] [Google Scholar]
- 72.Portella AC, et al. Thyroid hormone receptor β mutation causes severe impairment of cerebellar development. Mol Cell Neurosci. 2010;44:68–77. doi: 10.1016/j.mcn.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 73.Hashimoto K, et al. An unliganded thyroid hormone receptor causes severe neurological dysfunction. Proc Natl Acad Sci USA. 2001;98:3998–4003. doi: 10.1073/pnas.051454698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Machado DS, et al. A thyroid hormone receptor mutation that dissociates thyroid hormone regulation of gene expression in vivo. Proc Natl Acad Sci USA. 2009;106:9441–9446. doi: 10.1073/pnas.0903227106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Parrilla R, Mixson AJ, McPherson JA, McClaskey JH, Weintraub BD. Characterization of seven novel mutations of the c-erbA β gene in unrelated kindreds with generalized thyroid hormone resistance. Evidence for two “hot spot” regions of the ligand binding domain. J Clin Invest. 1991;88:2123–2130. doi: 10.1172/JCI115542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Flynn TR, et al. A novel C-terminal domain in the thyroid hormone receptor selectively mediates thyroid hormone inhibition. J Biol Chem. 1994;269:32713–32716. [PubMed] [Google Scholar]
- 77.Faustino LC, et al. Liver glutathione S-transferase α in expression is decreased by T3 hypothyroidism but not in euthyroidism in mice. Exp Physiol. 2011;96:790–800. doi: 10.1113/expphysiol.2011.058172. [DOI] [PubMed] [Google Scholar]
- 78.Cordas EA, et al. Thyroid hormone receptors control developmental maturation of the middle ear and the size of the ossicular bones. Endocrinology. 2012;153:1548–1560. doi: 10.1210/en.2011-1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Richter CP, Munscher A, Machado DS, Wondisford FE, Ortiga-Carvalho TM. Complete activation of thyroid hormone receptor β is essential for normal cochlear function and by T3 morphology in mice. Cell Physiol Biochem. 2011;28:997–1008. doi: 10.1159/000335812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pessoa CN, et al. Thyroid hormone action is required for normal cone opsin expression during mouse retinal development. Invest Ophthalmol Vis Sci. 2008;49:2039–2045. doi: 10.1167/iovs.07-0908. [DOI] [PubMed] [Google Scholar]
- 81.Ng L, et al. A thyroid hormone receptor that is required for the development of green cone photoreceptors. Nat Genet. 2001;27:94–98. doi: 10.1038/83829. [DOI] [PubMed] [Google Scholar]
- 82.Roberts MR, Srinivas M, Forrest D, Morreale de Escobar G, Reh TA. Making the gradient: thyroid hormone regulates cone opsin expression in the developing mouse retina. Proc Natl Acad Sci USA. 2006;103:6218–6223. doi: 10.1073/pnas.0509981103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Weiss AH, Kelly JP, Bisset D, Deeb SS. Reduced L- and M- and increased S-cone functions in an infant with thyroid hormone resistance due to mutations in the THRβ2 gene. Ophthalmic Genet. 2012;33:187–195. doi: 10.3109/13816810.2012.681096. [DOI] [PubMed] [Google Scholar]
- 84.Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995;83:841–850. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- 85.Hollenberg AN, Monden T, Madura JP, Lee K, Wondisford FE. Function of nuclear co-repressor protein on thyroid hormone response elements is regulated by the receptor A/B domain. J Biol Chem. 1996;271:28516–28520. doi: 10.1074/jbc.271.45.28516. [DOI] [PubMed] [Google Scholar]
- 86.Liu Y, Xia X, Fondell JD, Yen PM. Thyroid hormone-regulated target genes have distinct patterns of coactivator recruitment and histone acetylation. Mol Endocrinol. 2006;20:483–490. doi: 10.1210/me.2005-0101. [DOI] [PubMed] [Google Scholar]
- 87.Fozzatti L, et al. Resistance to thyroid hormone is modulated in vivo by the nuclear receptor corepressor (NCOR1) Proc Natl Acad Sci USA. 2011;108:17462–17467. doi: 10.1073/pnas.1107474108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Swanson EA, et al. Cardiac expression and function of thyroid hormone receptor β and its PV mutant. Endocrinology. 2003;144:4820–4825. doi: 10.1210/en.2003-0522. [DOI] [PubMed] [Google Scholar]
- 89.Suarez J, Scott BT, Suarez-Ramirez JA, Chavira CV, Dillmann WH. Thyroid hormone inhibits ERK phosphorylation in pressure overload-induced hypertrophied mouse hearts through a receptor-mediated mechanism. Am J Physiol Cell Physiol. 2010;299:C1524–C1529. doi: 10.1152/ajpcell.00168.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gloss B, et al. Cardiac ion channel expression and contractile function in mice with deletion of thyroid hormone receptor α or β. Endocrinology. 2001;142:544–550. doi: 10.1210/endo.142.2.7935. [DOI] [PubMed] [Google Scholar]
- 91.Gloss B, et al. Altered cardiac phenotype in transgenic mice carrying the Δ337 threonine thyroid hormone receptor β mutant derived from the S family. Endocrinology. 1999;140:897–902. doi: 10.1210/endo.140.2.6527. [DOI] [PubMed] [Google Scholar]
- 92.Ortiga-Carvalho TM, et al. Thyroid hormone resistance in the heart: role of the thyroid hormone receptor β isoform. Endocrinology. 2004;145:1625–1633. doi: 10.1210/en.2003-1031. [DOI] [PubMed] [Google Scholar]
- 93.Safer JD, et al. Isoform variable action among thyroid hormone receptor mutants provides insight into pituitary resistance to thyroid hormone. Mol Endocrinol. 1997;11:16–26. doi: 10.1210/mend.11.1.9867. [DOI] [PubMed] [Google Scholar]
- 94.Wagner RL, et al. A structural role for hormone in the thyroid hormone receptor. Nature. 1995;378:690–697. doi: 10.1038/378690a0. [DOI] [PubMed] [Google Scholar]
- 95.Nagaya T, Jameson JL. Thyroid hormone receptor dimerization is required for dominant negative inhibition by mutations that cause thyroid hormone resistance. J Biol Chem. 1993;268:15766–15771. [PubMed] [Google Scholar]
- 96.Yen PM, Wilcox EC, Hayashi Y, Refetoff S, Chin WW. Studies on the repression of basal transcription (silencing) by artificial and natural human thyroid hormone receptor-β mutants. Endocrinology. 1995;136:2845–2851. doi: 10.1210/endo.136.7.7789309. [DOI] [PubMed] [Google Scholar]
- 97.Monden T, et al. Leucine at codon 428 in the ninth heptad of thyroid hormone receptor β1 is necessary for interactions with the transcriptional cofactors and functions regardless of dimer formations. Thyroid. 2003;13:427–435. doi: 10.1089/105072503322021089. [DOI] [PubMed] [Google Scholar]
- 98.Fraichard A, et al. The T3R α gene encoding a thyroid hormone receptor is essential for post-natal development and thyroid hormone production. EMBO J. 1997;16:4412–4420. doi: 10.1093/emboj/16.14.4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.O’Shea PJ, et al. A thyrotoxic skeletal phenotype of advanced bone formation in mice with resistance to thyroid hormone. Mol Endocrinol. 2003;17:1410–1424. doi: 10.1210/me.2002-0296. [DOI] [PubMed] [Google Scholar]
- 100.O’Shea PJ, et al. Contrasting skeletal phenotypes in mice with an identical mutation targeted to thyroid hormone receptor α1 or β. Mol Endocrinol. 2005;19:3045–3059. doi: 10.1210/me.2005-0224. [DOI] [PubMed] [Google Scholar]
- 101.Quignodon L, Vincent S, Winter H, Samarut J, Flamant F. A point mutation in the activation function 2 domain of thyroid hormone receptor α1 expressed after CRE-mediated recombination partially recapitulates hypothyroidism. Mol Endocrinol. 2007;21:2350–2360. doi: 10.1210/me.2007-0176. [DOI] [PubMed] [Google Scholar]
- 102.Fauquier T, et al. Purkinje cells and Bergmann glia are primary targets of the TRα1 thyroid hormone receptor during mouse cerebellum postnatal development. Development. 2014;141:166–175. doi: 10.1242/dev.103226. [DOI] [PubMed] [Google Scholar]
- 103.Adams M, et al. Genetic analysis of 29 kindreds with generalized and pituitary resistance to thyroid hormone. Identification of thirteen novel mutations in the thyroid hormone receptor β gene. J Clin Invest. 1994;94:506–515. doi: 10.1172/JCI117362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tinnikov A, et al. Retardation of post-natal development caused by a negatively acting thyroid hormone receptor α1. EMBO J. 2002;21:5079–5087. doi: 10.1093/emboj/cdf523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Refetoff S, DeWind LT, DeGroot LJ. Familial syndrome combining deaf-mutism, stuppled epiphyses, goiter and abnormally high PBI: possible target organ refractoriness to thyroid hormone. J Clin Endocrinol Metab. 1967;27:279–294. doi: 10.1210/jcem-27-2-279. [DOI] [PubMed] [Google Scholar]
- 106.Liu YY, Schultz JJ, Brent GA. A thyroid hormone receptor α gene mutation (P398H) is associated with visceral adiposity and impaired catecholamine-stimulated lipolysis in mice. J Biol Chem. 2003;278:38913–38920. doi: 10.1074/jbc.M306120200. [DOI] [PubMed] [Google Scholar]