

Spinal neuropeptide signaling and inflammatory mediator expression supports nociceptive sensitization in a fracture model of complex regional pain syndrome.
Keywords: Fracture, Complex regional pain syndrome, Cytokines, Nerve growth factor, Substance P, Calcitonin gene-related peptide, Pain
Abstract
Tibia fracture induces exaggerated substance P (SP) and calcitonin gene–related peptide (CGRP) signaling and neuropeptide-dependent nociceptive and inflammatory changes in the hind limbs of rats similar to those seen in complex regional pain syndrome. Inflammatory changes in the spinal cord contribute to nociceptive sensitization in a variety of animal pain models. This study tested the hypothesis that fracture-induced exaggerated neuropeptide signaling upregulates spinal inflammatory mediator expression, leading to postfracture hind limb nociceptive sensitization. At 4 weeks after performing tibia fracture and casting in rats, we measured hind limb allodynia, unweighting, warmth, edema, and spinal cord neuropeptide and inflammatory mediator content. The antinociceptive effects of intrathecally injected neuropeptide and inflammatory mediator receptor antagonists were evaluated in fracture rats. Transgenic fracture mice lacking SP or the CGRP RAMP1 receptor were used to determine the effects of neuropeptide signaling on postfracture pain behavior and spinal inflammatory mediator expression. Hind limb allodynia, unweighting, warmth, edema, increased spinal SP and CGRP, and increased spinal inflammatory mediator expression (TNF, IL-1, IL-6, CCL2, and nerve growth factor) were observed at 4 weeks after fracture in rats. Fracture-induced increases in spinal inflammatory mediators were not observed in fracture mice lacking SP or the CGRP receptor, and these mice had attenuated postfracture nociceptive sensitization. Intrathecal injection of selective receptor antagonists for SP, CGRP, TNF, IL-1, IL-6, CCL2, or nerve growth factor each reduced pain behaviors in the fracture rats. Collectively, these data support the hypothesis that facilitated spinal neuropeptide signaling upregulates the expression of spinal inflammatory mediators contributing to nociceptive sensitization in a rodent fracture model of complex regional pain syndrome.
1. Introduction
Population-based studies indicate that distal limb fracture is the most common cause of complex regional pain syndrome (CRPS),11,36 and we have developed a tibia fracture model in the rat and mouse that closely resembles CRPS. Distal tibia–fractured rats treated with 4-week cast immobilization exhibited paw allodynia, unweighting, increased spinal Fos-immunoreactivity, increased hind paw skin temperature, edema, increased spontaneous protein extravasation, facilitated neuropeptide signaling, regional periarticular bone loss, keratinocyte proliferation and epidermal thickening, and increased keratinocyte expression of tumor necrosis factor α (TNF), interleukin 1 (IL-1), interleukin 6 (IL-6), and nerve growth factor (NGF) inflammatory mediators in the affected skin.13,14,25,27,34,35,46,47
Using the rat fracture model of CRPS, we have shown that upregulated expression of neuropeptides such as substance P (SP) and calcitonin gene–related peptide (CGRP) and their cognate receptors46 leads to inflammation and pain sensitization by the activation of neuropeptide receptors on the surface of cutaneous keratinocytes, mast cells, and vascular endothelial cells, causing mast cell degranulation,26 vasodilatation and protein extravasation,46 keratinocyte proliferation and activation, and keratinocyte expression of high levels of inflammatory cytokines (TNF, IL-1, IL-6) and NGF.15,25,27,34,35,47 Experimental maneuvers blocking these inflammatory mediators can inhibit the nociceptive and vascular changes that develop over the first 4 weeks in the CRPS fracture model.26,27,34,35,47 Similarly, clinical studies in patients with CRPS have reported therapeutic responses with treatments that inhibit innate immune mechanisms, but these treatments are not effective for every patient.8,17,21,40 Using bilateral skin punch biopsies from 55 patients with CRPS, we recently demonstrated unilateral mast cell and keratinocyte proliferation, epidermal thickening, and keratinocyte inflammatory cytokine expression (TNF, IL-6) in early (<3 month duration) CRPS-affected skin.4 This study also found that increased keratinocyte and mast cell proliferation gradually resolved over time, despite persistent pain symptoms. These results are consistent with studies on skin blister fluid, which indicate a gradual resolution of cutaneous inflammatory mediator (IL-6, TNF) expression over time in CRPS-affected skin.49 Collectively, these data support the hypothesis that cutaneous keratinocyte and mast cell–mediated innate immune responses contribute to the development of posttraumatic CRPS, but cannot completely account for the inflammation and pain observed in early and, particularly, in chronic CRPS.
It has been hypothesized that neuropeptides released from the central terminals of spinal primary sensory afferent neurons can initiate glial activation and inflammatory mediator expression contributing to chronic pain.42 Interestingly, several studies have reported elevated cerebrospinal fluid levels of IL-1 and IL-6 in patients with chronic CRPS,2,3 but other investigators have been unable to confirm these findings.30 Elevated cerebrospinal fluid levels of cytokines and neurotrophic factors have also been observed in other chronic pain states.6 We postulated that facilitated spinal neuropeptide signaling upregulates the expression of spinal inflammatory mediators contributing to the development of CRPS, and this study tests this hypothesis in the rodent fracture model of CRPS.
2. Materials and methods
All experiments were approved by our Institutional Animal Care and Use Committee (IACUC) and followed the animal subject guidelines of the International Association for the Study of Pain. Adult (9-month-old) male Sprague-Dawley rats (Simonsen Laboratories, Gilroy, CA) and adult (3-month-old) male C57BL/6 wild-type (WT) mice, SP-deficient mice (TAC1−/−; Jackson Laboratories, Sacramento, CA), and CGRP RAMP1 receptor–deficient mice (RAMP1−/−, generated by Dr Kazutake Tsujikawa at the Department of Immunology, Osaka University) were used in these experiments. Rats were housed individually in isolator cages with solid floors covered with 3 cm of soft bedding, whereas mice were housed for 4 to a cage. Food and water were provided ad libitum.
2.1. Surgery
The tibia was fractured in the rat under isoflurane anesthesia as we have previously described.13 The right distal tibia was fractured using pliers with an adjustable stop (Vise grip; Newell Rubbermaid, Atlanta, GA) that had been modified with a 3-point jaw. The right hind limb was then wrapped in casting tape so the hip, knee, and ankle were flexed. The cast extended from the metatarsals of the hind paw up to a spica formed around the abdomen. To prevent the rats from chewing at their casts, the cast material was wrapped in galvanized wire mesh. The animals were given subcutaneous saline and buprenorphine (0.03 mg/kg) immediately after procedure and on the first day after fracture for postoperative hydration and analgesia. At 4 weeks after fracture, the rats were anesthetized with isoflurane and the cast removed with a vibrating cast saw. The next day, the rats underwent behavioral testing for hind paw von Frey allodynia, unweighting, edema, and warmth. The tibia fracture protocol in mice was similar to the procedure describe in rats, but a 6-inch straight hemostat was used for making the fracture.15 In mice, the cast was removed at 3 weeks after fracture and behavioral testing was performed the next day. All animals used in this study had union at the fracture site at the time of cast removal.
2.2. Drug administration
All drugs were injected intrathecally in 10 μL of saline under isoflurane analgesia. The following drugs were used: LY303870 (20 μg), an SP neurokinin 1 (NK1) receptor antagonist (Eli Lily Co, Indianapolis, IN); CGRP8-37 (20 μg), a CGRP receptor antagonist (Sigma, St Louis, MO); etanercept (100 μg), a TNF inhibitor (Pfizer, New York, NY); anakinra (100 μg), an IL-1 receptor antagonist (Amgen, San Francisco, CA); TB-2081 (20 μg), an IL-6 receptor antagonist (a generous gift from Dr Kenner Rice, National Institute on Drug Abuse); RS504393 (3 μg), a C-C chemokine receptor type 2 (CCR2) antagonist (Sigma); muMab 911 (12.4 μg), an anti-NGF antibody (Pfizer); and PD98059 (20 μg), a selective inhibitor of extracellular signal-regulated kinase 1/2 (ERK1/2) mitogen-activated protein kinase (MAPK) phosphorylation (Sigma).
2.3. Hind paw allodynia and unweighting measurements
To measure mechanical allodynia in the rats and mice, an up–down von Frey testing paradigm was used as we previously described.15 Test animals were placed in a clear plastic cylinder (20 cm in diameter for rats and 10 cm in diameter for mice) with a wire mesh bottom and allowed to acclimate for 15 minutes. The paw was tested with von Frey fibers of sequentially increasing stiffness applied against the hind paw plantar skin at the same site and pressed upward to cause a slight bend in the fiber and left in place for 6 seconds. Withdrawal of or licking the hind paw after fiber application was scored as a response. When no response was obtained, the next stiffest fiber in the series was applied to the same paw; if a response was obtained, a less stiff fiber was applied. Testing proceeded in this manner until 4 fibers had been applied after “negative + positive or positive + negative” response. Estimation of the mechanical withdrawal threshold by data fitting algorithm permitted the use of parametric statistics for analysis.32 Baseline von Frey withdrawal thresholds were identical in WT, Tac1−/−, and RAMP1−/− mice. An incapacitance device (IITC Inc; Life Science, Woodland Hills, CA) was used to measure hind paw unweighting. The rats and mice were manually held in a vertical position over the apparatus with the hind paws resting on separate metal scale plates and the entire weight of the animal supported on the hind paws. The duration of each measurement was 6 seconds, and 10 consecutive measurements for rats were taken at 60-second intervals. Mice were tested 6 consecutive times. Eight readings (excluding the highest and lowest ones) for rats and 6 for mice were averaged to calculate the bilateral hind paw weight-bearing values.
2.4. Hind paw volume
A laser sensor technique was used to determine the dorsal–ventral thickness of the hind paw, as we have previously described.13 For laser measurements, each animal was briefly anesthetized with isoflurane and then held vertically so the hind paw rested on a table top below the laser. The paw was gently held flat on the table with a small metal rod applied to the top of the ankle joint. Using optical triangulation, a laser with a distance-measuring sensor was used to determine the distance to the table top and to the top of the hind paw dorsal skin over the midpoint of the third metatarsal, and the difference was used to calculate the dorsal–ventral paw thickness. The measurement sensor device used in these experiments (Limab) has a measurement range of 200 mm with a 0.01-mm resolution.
2.5. Hind paw temperature
The temperature of the hind paw was measured using a fine wire thermocouple (Omega Engineering, Stamford, CT) applied to the paw skin, as previously described.13 The investigator held the wire using an insulating Styrofoam block. Three sites were tested over the dorsum of the hind paw: the space between the first and second metatarsals (medial), the second and third metatarsals (central), and the fourth and fifth metatarsals (lateral). After a site was tested in one hind paw, the same site was immediately tested in the contralateral hind paw. The testing protocol was medial dorsum right then left, central dorsum right then left, lateral dorsum right then left, medial dorsum left then right, central dorsum left then right, and lateral dorsum left then right. The 6 measurements for each hind paw were averaged for the mean temperature.
2.6. Quantitative real-time polymerase chain reaction in spinal cord tissues
At 4 weeks after fracture, the rats were killed by CO2 inhalation and the ipsilateral spinal cord (L4,5 lumbar enlargement) was collected after behavioral testing and frozen immediately on dry ice. In mice, the spinal cord was collected at 3 weeks after fracture. Total RNA was extracted using the RNeasy Mini Kit (Qiagen, Venlo, the Netherlands), and the purity and concentration were determined spectrophotometrically. Next, cDNA was synthesized from 1 μg RNA using an iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Hercules, CA). Real-time polymerase chain reactions (PCRs) were conducted using the SYBR Green PCR master mix (Applied Biosystems, Waltham, MA) and performed on an ABI 7900HT sequencing detection system (Applied Biosystems). To validate the primer sets used, we performed dissociation curves to document single-product formation, and agarose gel analysis was conducted to confirm the size (Table 1). The data from real-time PCR experiments were analyzed as described in the manual for the ABI prism 7000 real-time systems. All results were confirmed by repeating the experiment 3 times.
Table 1.
Primers used for real-time polymerase chain reaction.
2.7. Enzyme immunoassay for substance P, calcitonin gene–related peptide, TNF, IL-1, IL-6, and nerve growth factor levels in the spinal cord
At 4 weeks after fracture, the rats were killed by CO2 inhalation and the ipsilateral spinal cord (L4,5 lumbar enlargement) was collected after behavioral testing and frozen immediately on dry ice. The spinal cord tissue was cut into fine pieces in ice-cold phosphate-buffered saline (PBS), pH 7.4, containing protease inhibitors (Sigma) followed by homogenization using a Dounce tissue grinder. Homogenates were centrifuged for 15 minutes at 12,000g, 4°C. Supernatants were transferred to precooled Eppendorf tubes. Triton X-100 was added at a final concentration 0.01%. The samples were centrifuged again for 5 minutes at 12,000g at 4°C. The supernatants were aliquoted and stored at −80°C. TNF-α was detected using a TNF-α enzyme immunoassay (EIA) kit (BD Biosciences, Franklin Lakes, NJ). IL-1β, IL-6, and chemokine (C-C motif) ligand 2 (CCL2) protein levels were measured using EIA kits from R&D Systems (Minneapolis, MN). The NGF concentrations were determined using the NGF Emax ImmunoAssay System kit (Promega, Madison, WI) according to the manufacturer's instructions. The OD of the reaction product was read on a microplate reader at 450 nm, and values were normalized per mg of protein assayed. The concentrations of TNF-α, IL-1β, IL-6, NGF, and CCL2 proteins were calculated from the standard curve for each assay. Each protein concentration was expressed as pg/mg total protein. Total protein content was determined using a Coomassie Blue Protein Assay Kit (Pierce, Life Technologies, Waltham, MA).
Substance P and CGRP content in the rat spinal cord was measured as we have previously described.46 Briefly, samples were minced in 1 mL of 3:1 ethanol/0.7 M HCl and homogenized for 20 seconds. The homogenates were shaken for 2 hours at 4°C and centrifuged at 3000g for 20 minutes at 4°C. The supernatants were lyophilized and stored at −80°C. The lyophilized products were reconstituted with EIA buffer before assay. After rehydration, the extracts were assayed in duplicate using SP and CGRP ELISA (Cayman Chemical, Ann Arbor, MI) according to the manufacturer's instructions.
2.8. Western blotting of the spinal cord
At 4 weeks after fracture, the rats were killed by CO2 inhalation and the ipsilateral spinal cord (L4,5 lumbar enlargement) was collected after behavioral testing and frozen immediately on dry ice. Spinal cord samples were later homogenized in modified RIPA buffer (50 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1% Igepal CA-630, 0.1% SDS, 50 mM NaF, and 1 mM NaVO3) containing protease inhibitors (Sigma). The homogenate was centrifuged at 10,000g for 30 minutes at 4°C. Total protein concentration of the homogenate was measured using a Coomassie Blue Protein Assay (Bio-Rad, Hercules, CA) and bovine serum albumin protein standard (Pierce). Equal amounts of protein (50 μg) were subjected to SDS-PAGE (Bio-Rad) and electrotransferred onto a polyvinylidene difluorided membrane (Millipore, Billerica, MA). The blots were blocked overnight with 5% nonfat dry milk or normal serum in Tris-buffered saline with 0.5% Tween-20 (TBST), then incubated with primary antibodies against β-actin, NK1, total ERK1/2, and phosphorylated ERK1/2 (Santa Cruz Biotechnology, Dallas, TX), and total p38 and phosphorylated p38 (Cell Signaling Technology, Beverely, MA) for 1 hour on a rocking platform at room temperature. After washing in TBST, the blots were incubated with horseradish peroxidase–conjugated secondary antibody (Santa Cruz Biotechnology) for 1 hour at room temperature. Then, the blots were washed in TBST again, and the proteins were detected using ECL chemiluminescence reagent (GE Healthcare, Little Chalfont, United Kingdom) and scanned by PhosphoImager (Typhoon; GE Healthcare). The band intensity was analyzed using ImageQuant 5.2 software (Molecular Dynamics, Sunnyvale, CA) and normalized with the corresponding internal loading control band. The specific protein expression is expressed as protein/actin band intensity ratio to demonstrate the change of the specific protein after treatments.
2.9. Tissue processing and immunofluorescence confocal microscopy
At 4 weeks after fracture, the rats were perfused with 4% paraformaldehyde in PBS, pH 7.4, through the ascending aorta; the lumbar spinal cord was removed and postfixed in 4% paraformaldehyde overnight, then the tissues were treated with 30% sucrose in PBS at 4°C before embedding in OCT (Sakura Finetek, Torrance, CA). After embedding, 20-μm thick slices were made using a cryostat, mounted onto Superfrost microscope slides (Fisher Scientific, Waltham, MA), and stored at −80°C.
Frozen sections were permeabilized and blocked with PBS containing 10% donkey serum and 0.3% Triton X-100, followed by exposure to the primary antibodies overnight at 4°C in PBS containing 2% serum. On detection of the first antigen, primary antibody from a different species against the second antigen was applied to the sections and visualized using an alternative fluorophore-conjugated secondary antibody. Sections were then rinsed in PBS and incubated with fluorophore-conjugated secondary antibodies against the immunoglobulin of the species from which the primary antibody was generated. After 3 washes, the sections were mounted with antifade mounting medium (Invitrogen, Carlsbad, CA). Images were obtained using confocal microscopy (Zeiss LSM/710; Carl Zeiss, Jena, Germany) and stored on digital media. The sources of primary antibodies were as follows: monoclonal mouse antiphospho-ERK, 1:100 (clone E4; Santa Cruz Biotechnology), monoclonal mouse anti-GFAP, 1:1000 (Clone GA5; EMD Millipore, Billerica, MA), polyclonal rabbit anti-Iba1, 1:1000 (Wako, Richmond, VA). The sources of secondary antibodies were as follows: donkey anti-mouse IgG (1:400) conjugated with cyanine dye 3, or donkey anti-rabbit IgG (1:400) conjugated with fluorescein secondary antibodies (Jackson ImmunoResearch Laboratories, Westgrove, PA), incubated with respective primary antibodies. Control experiments included incubation of slices in primary and secondary antibody-free solutions both of which led to low intensity nonspecific staining patterns in preliminary experiments (data not shown).
2.10. Statistical analysis
Statistical analysis was performed using Prism 4.02 (GraphPad Software, La Jolla, CA). Sample sizes were been based on a power analysis of preliminary and previously published data generated from using each of the proposed assays in fracture animals. Based on this analysis we calculated that the proposed experiments would require 8 animals per cohort to provide 80% power to detect 25% differences between groups. Animals were randomized to experimental groups using computer generated random numbers and all testing was performed in a blinded fashion when possible. No animals were excluded after enrollment into the experimental cohorts. The normal distribution of the data was confirmed using the D'Agostino-Pearson omnibus normality test. All data were evaluated using an analysis of variance followed by Bonferroni post hoc multiple comparison testing. For simple comparisons of 2 means, 2-tailed t tests were performed. Data are presented as the mean ± SEM, and a value of P < 0.05 was considered statistically significant.
The hind paw von Frey mechanical nociceptive threshold, temperature, and thickness were analyzed as the difference between the fracture side (right, R) and the contralateral untreated side (left, L). Right hind paw weight-bearing data were analyzed as a ratio between twice the right hind paw weight bearing and the sum of the right (R) and left (L) hind paw weight-bearing values ((2R/(R + L)) × 100%).
3. Results
3.1. Exaggerated neuropeptide signaling in the lumbar spinal cord after fracture
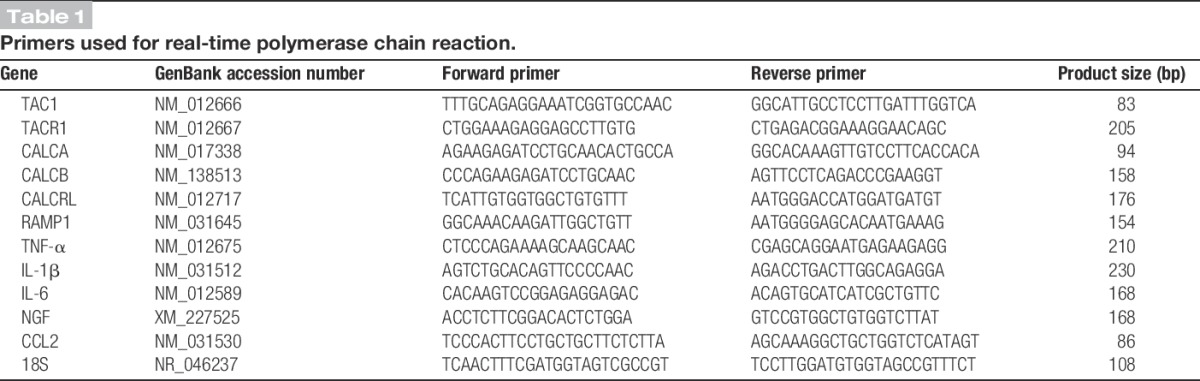
At 4 weeks after tibia fracture in rats, there was increased expression of TAC1 (SP gene), TACR1 (SP neurokinin 1 receptor gene), CALCA (alpha-CGRP gene) and CALCB (beta-CGRP gene) mRNA in the L4 and L5 spinal cord segments ipsilateral to the fracture (Fig. 1A). No changes were observed in mRNA expression for CALCRL (calcitonin receptor-like receptor, CGRP receptor gene) and RAMP1 (receptor activity modifying protein 1, CGRP receptor chaperone gene). Substance P and CGRP protein levels, as measured by EIA, also increased in the L4,5 spinal cord segments at 4 weeks after fracture (Fig. 1B, C). Detected by western immunoblot assay, Figure 1D illustrates that protein levels of the SP neurokinin 1 (NK1) receptor increased 2-fold in the spinal cord after fracture. These data demonstrate the upregulated expression of SP and CGRP signaling components in the lumbar spinal cord at 4 weeks after fracture.
Figure 1.
Distal tibia fracture–facilitated spinal neuropeptide signaling in the ipsilateral L4,5 spinal cord segments at 4 weeks after fracture (FX, n = 8), compared with control rats (controls, n = 8). (A) Tibia fracture caused increased spinal expression of the substance P (SP) gene (TAC1), the SP neurokinin 1 (NK1) receptor gene (TACR1), the α-calcitonin gene–related peptide (α-CGRP) gene (CALCA), and the β-CGRP gene (CALCB). Fracture had no effect on the spinal expression of the CGRP calcitonin receptor-like receptor (CRLR) gene (CALCRL) or on the CGRP receptor activity-modifying protein (RAMP1) gene (RAMP1). Substance P (B) and CGRP (C) spinal protein levels, as measured by enzyme immunoassay, also increased at 4 weeks after fracture. A western immunoblot assay was used to demonstrate a 2-fold increase in SP NK1 receptor protein levels after fracture (D). Values are mean ± SE. One-way analysis of variance (P < 0.001) with Bonferroni post hoc testing (A) and t-tests (B-D) *P < 0.05, **P < 0.01, and ***P < 0.001 for FX vs control.
3.2. Inhibition of spinal substance P and calcitonin gene–related peptide signaling reverses allodynia and unweighting
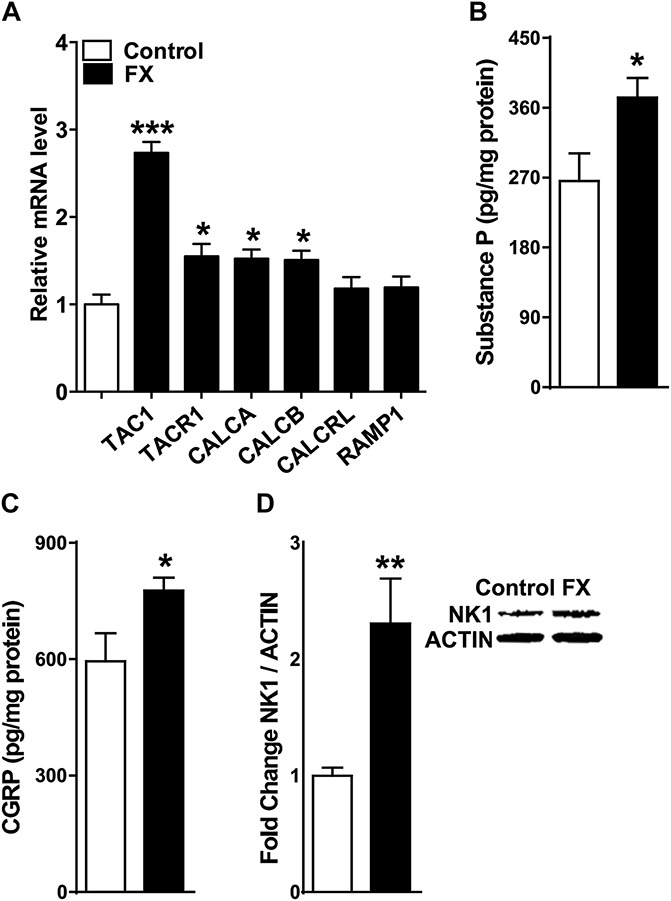
The anti-nociceptive effects of a single intrathecal injection of the SP NK1 receptor antagonist LY303870 and of the CGRP receptor antagonist CGRP8-37 were evaluated in fracture rats. At 4 weeks after fracture hind paw von Frey nociceptive thresholds were reduced, but intrathecal LY303870 and CGRP8-37 were both capable of reversing this mechanical allodynia (Fig. 2A). Similarly, intrathecal injection of LY303870 or CGRP8-37 after fracture partially restored after fracture hind paw weight bearing (Fig. 2B). These data indicate that spinal neuropeptide signaling contributes to nociceptive sensitization at 4 weeks after fracture.
Figure 2.
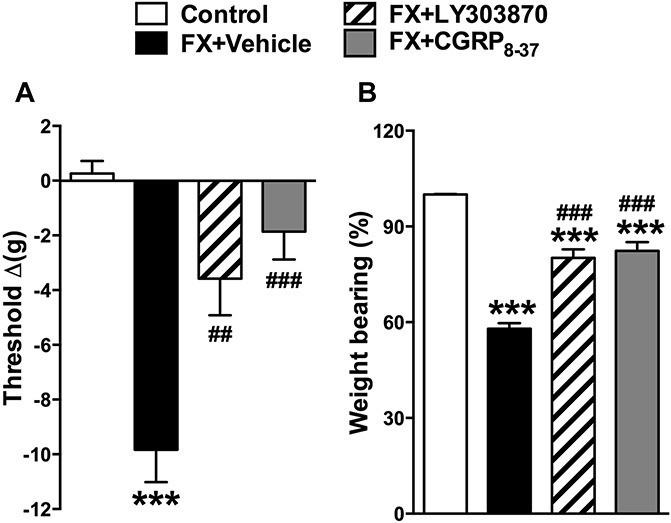
Spinally administered neuropeptide receptor antagonists reduced allodynia and unweighting at 4 weeks after fracture. Fracture (FX) rats were intrathecally injected with 20 μg of the substance P NK1 receptor antagonist LY303870 or 20 μg of the CGRP CRLR receptor antagonist CGRP8-37. Both LY303870 and CGRP8-37 reduced hind paw mechanical allodynia (A) and unweighting (B) at 30 minutes after injection, compared with FX + vehicle. Values are mean ± SE. One-way analysis of variance (P < 0.001) with Bonferroni post hoc testing **P < 0.01 and ***P < 0.001 for FX + vehicle (n = 8), FX + LY303870 (n = 8), or FX + CGRP8-37 (n = 8) vs control (n = 8), ###P < 0.001 for FX + LY303870 (n = 8), or FX + CGRP8-37 (n = 8) vs FX + vehicle (n = 8).
3.3. Cytokine induction in the lumbar spinal cord after fracture
The TNF, IL-1, IL-6, NGF, and CCL2 mRNA and protein levels in the spinal cord were measured by real-time PCR and EIA in fracture rats. Figure 3 demonstrates that the expression of these inflammatory mediators is upregulated in the lumbar spinal cord at 4 weeks after fracture.
Figure 3.
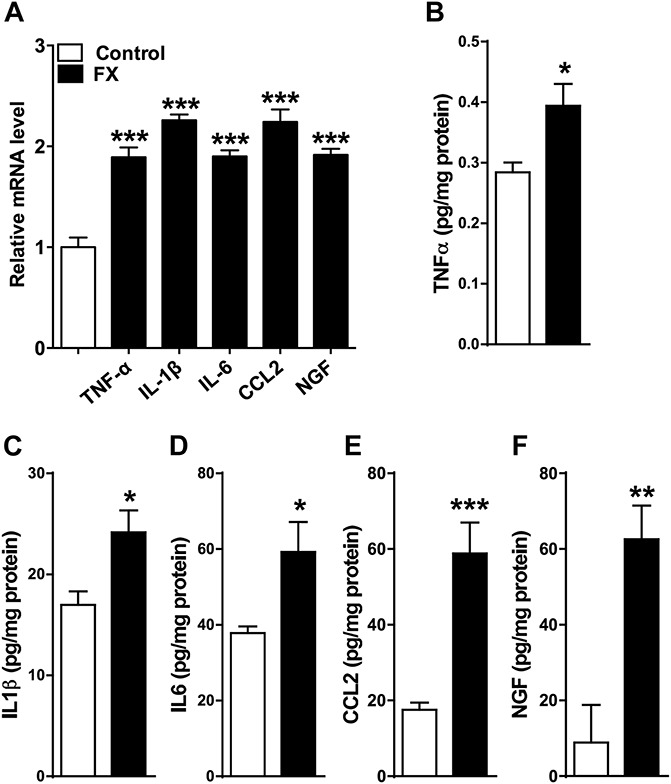
Fracture caused an increase in spinal cord inflammatory mediator expression. Tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), interleukin-6 (IL-6), chemokine (C-C motif) ligand 2 (CCL2), and nerve growth factor gene expression measured by real-time polymerase chain reaction (A) and protein levels determined by enzyme immunoassay (B-F) increased in the rat spinal cord after fracture. Values are mean ± SE. One-way analysis of variance (P < 0.001) with Bonferroni post hoc testing (A) and t tests (B-F) *P < 0.05, **P < 0.01, and ***P < 0.001 for FX (n = 8) vs control (n = 8).
3.4. Inhibition of inflammatory mediator signaling reverses allodynia and unweighting
To test the hypothesis that postfracture upregulated inflammatory mediator expression in the lumbar spinal cord (Fig. 3) contributes to hind paw nociceptive sensitization, we intrathecally injected 4 weeks after fracture rats with selective cytokine receptor antagonists (TNF-α receptor antagonist etanercept, IL-1 receptor inhibitor anakinra, IL-6 antagonist TB2081, or CCL2 receptor antagonist RS504393) or with the anti-NGF antibody muMab 911. As shown in Figure 4, all these inhibitors/antagonists partially reversed hind paw allodynia and unweighting in the fracture rats. These data indicate that upregulated cytokine and NGF expression in the spinal cord can contribute to nociceptive sensitization at 4 weeks after fracture.
Figure 4.
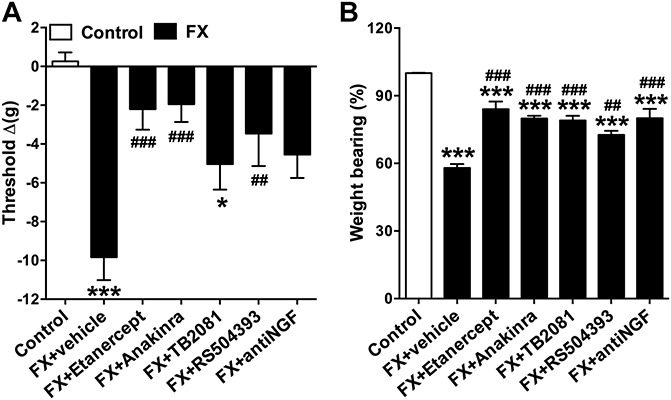
Spinally administered inflammatory mediator receptor antagonists/inhibitors reduced allodynia and unweighting at 4 weeks after fracture. Fracture (FX) rats were intrathecally injected with either: (1) vehicle, (2) the TNF inhibitor etanercept (100 μg), (3) the IL-1 receptor antagonist anakinra (100 μg), (4) the IL-6 receptor antagonist TB-2081 (20 μg), (5) the chemokine (C-C motif) receptor type 2 (CCR2) antagonist RS504393 (3 μg), and (6) the anti-nerve growth factor (NGF) antibody muMab 911 (12.4 μg). Behavioral testing was performed at various time intervals after intrathecal injection based on preliminary pilot time course studies identifying the peak analgesic effects for each drug (etanercept 3 hours, anakinra 30 minutes, TB2081 15 minutes, and RS504393 and muMab 911 1 hour). All receptor antagonists/inhibitors reduced postfracture hind paw allodynia (A) and unweighting (B). There were no effects on hind paw temperature or thickness. Values are mean ± SE, n = 8 per cohort. One-way analysis of variance (P < 0.001) with Bonferroni post hoc testing *P < 0.05. **P < 0.01 and ***P < 0.001 for FX + vehicle, FX + etanercept, FX + anakinra, FX + TB2081, FX + anti-NGF antibody or FX + RS504394 vs control, ##P < 0.01 and ###P < 0.001 for FX + etanercept, FX + anakinra, FX + TB2081, FX + anti-NGF antibody, or FX + RS504394 vs FX + vehicle.
3.5. Neuropeptide signaling contributes to postfracture vascular and nociceptive changes
Transgenic mice were used to test the hypothesis that neuropeptide signaling mediates the postfracture development of hind paw nociceptive sensitization and vascular changes. Figure 5 demonstrates that at 3 week, postfracture WT mice developed hind paw von Frey allodynia, unweighting, warmth, and edema, but SP-deficient (SP KO, TAC1−/−) mice and CGRP RAMP1 receptor–deficient (RAMP1 KO, RAMP1−/−) mice had significantly less allodynia, unweighting, and warmth than did WT fracture mice. SP-deficient mice also failed to develop hind paw edema after facture, but RAMP1-deficient fracture mice had a similar degree of hind paw edema as did WT fracture mice (Fig. 5D). These results confirm that neuropeptide signaling is required for the development of postfracture hind paw nociceptive sensitization, warmth, and edema.
Figure 5.
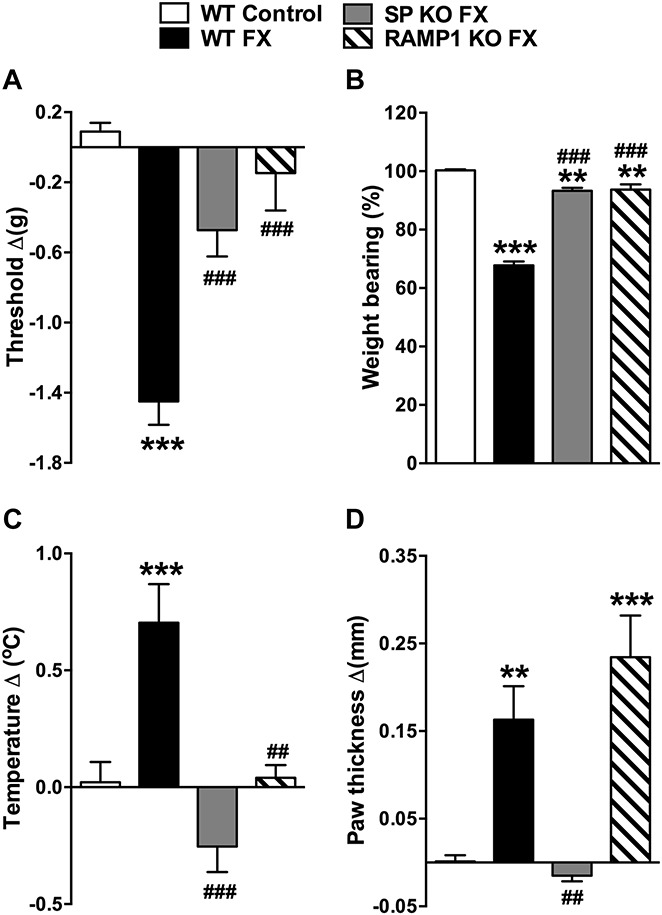
Substance P–deficient (SP KO) and CGRP RAMP1 receptor–deficient (RAMP1 KO) fracture (FX) mice had reduced hind paw allodynia, unweighting, warmth, and edema, compared with wild-type (WT) fracture mice. WT fracture mice exhibited hind paw von Frey allodynia (A), unweighting (B), warmth (C), and edema (D) at 3 weeks after fracture. The SP-deficient fracture mice had attenuated hind paw allodynia (A) and unweighting (B), and failed to develop hind paw warmth (C) or edema (D). Similarly, the RAMP1 receptor–deficient fracture mice had no von Frey allodynia (A), attenuated unweighting (B), and no warmth (C), but the development of postfracture hind paw edema was unaffected by the loss of calcitonin gene–related peptide signaling (D), compared with WT FX mice. Values are mean ± SE, n = 8 per cohort. One-way analysis of variance (P < 0.001) with Bonferroni post hoc testing **P < 0.01 and ***P < 0.001 for WT FX, SP KO FX, or RAMP1 KO FX vs WT control, ##P < 0.01 and ###P < 0.001 for SP KO FX, or RAMP1 KO FX vs WT FX.
3.6. Neuropeptide signaling contributes to postfracture inflammatory mediator expression in the lumbar spinal cord
Figure 6 illustrates the postfracture lumbar spinal cord expression of TAC1, TACR1, CALCA, CALCB, CALCRL, RAMP1, TNF, IL-1, IL-6, CCL2, and NGF mRNA in wild-type, SP-deficient, and the CGRP RAMP1 receptor–deficient mice. Compared with no fracture WT controls, the 3-week postfracture WT mice exhibited a 2- to 5-fold increase in the spinal cord expression for all these genes. The SP-deficient fracture mice had upregulated TACR1, CALCA, TNF, and NGF expression, compared with no fracture WT controls, but this increase was less than the increase observed in the fracture WT mice. The increase in IL-6 expression in fracture SP-deficient mice was same as that observed in the fracture WT mice. As expected, there was no TAC1 mRNA in the SP-deficient mice, and there was no postfracture increase in spinal CALCB, CALCRL, RAMP1, IL-1, or CCL2. At 3 weeks after fracture, there was increased expression of TAC1, CALCA, TNF, and CCL2 in the RAMP1-deficient mice, compared with control WT mice, but this increase was less than the increase observed in the fracture WT mice. TACR1, CALCB, CALCRL, IL-1, IL-6, and NGF expression was unchanged in the fracture RAMP1-deficient mice, compared with no fracture WT controls. As expected, there was no RAMP1 mRNA in the RAMP1-deficient mice. These data indicate that neuropeptide signaling contributes to the postfracture amplification of neuropeptide signaling and upregulated inflammatory mediator expression in the lumbar spinal cord.
Figure 6.
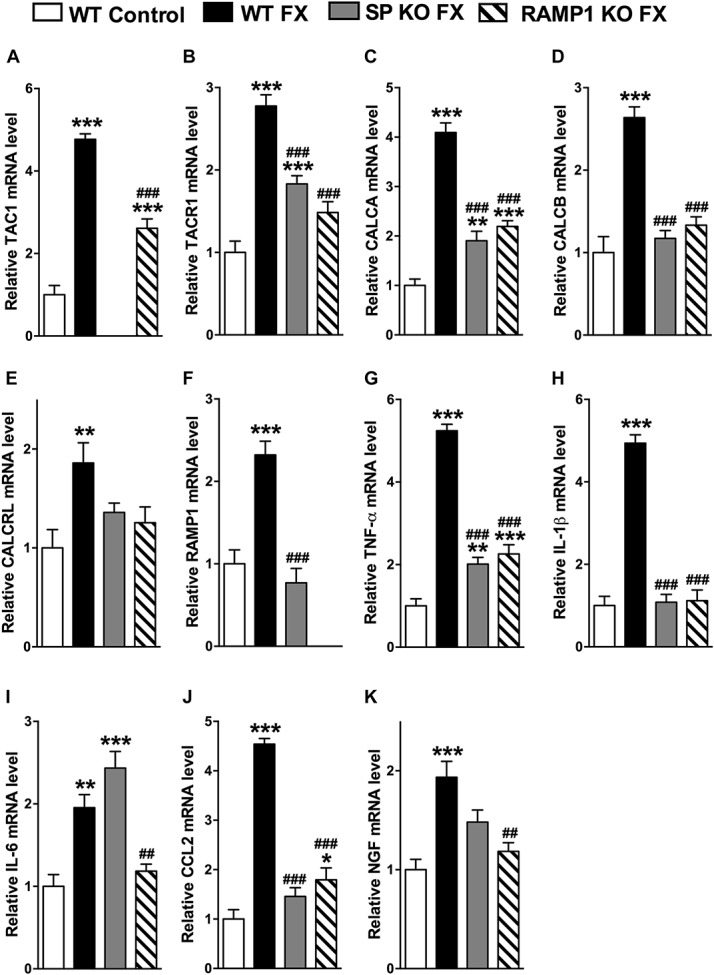
Substance P–deficient (SP KO) and CGRP RAMP1 receptor–deficient (RAMP1 KO) fracture (FX) mice failed to develop exaggerated spinal neuropeptide signaling and inflammatory mediator expression at 3 weeks after fracture, measured by real-time polymerase chain reaction. Similar to the changes observed in 4-week postfracture rats (Figs. 1 and 3), in wild-type (WT) fracture (FX) mice, there was increased gene expression of (A) SP (TAC1), (B) SP NK1 receptor (TACR1), (C, D) CGRP (CALCA and CALCB), (E) CGRP CRLR receptor (CALCRL), (F) CGRP RAMP1 receptor (RAMP1), (G) TNF-α, (H) IL-1β, (I) IL-6, (J) CCL2, and (K) nerve growth factor (NGF), compared with WT mice with no fracture (WT control). Substance P–deficient fracture (SP KO FX) mice had attenuated postfracture spinal cord changes compared with WT FX mice, and there was no increase in the expression of (D) CALCB, (E) CALCRL, (F) RAMP1, (H) IL1β, and (K) CCL2 in the SP KO FX mice, compared with WT control mice. CGRP RAMP1 receptor–deficient fracture (RAMP1 KO FX) mice also exhibited attenuated postfracture spinal cord changes, compared with WT FX mice, and there was no increase in (B) TACR1, (D) CALCB, (E) CALCRL, (H) IL-1β, (I) IL-6, and (J) NGF mRNA levels, compared with WT controls. (A) TAC1 mRNA was not detected in the TAC1-deficient mice, and (F) RAMP1 mRNA was not measurable in the RAMP1 receptor–deficient mice. Values are mean ± SE, n = 8 per cohort. One-way analysis of variance (P < 0.01 for graph E, P < 0.001 for all other graphs), with Bonferroni post hoc testing *P < 0.05, **P < 0.01, and ***P < 0.001 for WT FX, SP KO FX, or RAMP1 KO FX vs WT control, #P < 0.05, ##P < 0.01, and ###P < 0.001 for SP KO FX, or RAMP1 KO FX vs WT FX.
3.7. Spinal ERK1/2 activation contributes to postfracture nociceptive sensitization and upregulated TNF expression in the spinal cord
Western immunoblot was used to evaluate phosphorylation of the mitogen-activated protein kinases (MAPKs) extracellular signal-regulated kinase 1/2 (ERK) and p38 in the L4,5 spinal cord segments ipsilateral to fracture. At 4 weeks after fracture in rats, there was a 50% increase in phosphorylated ERK, compared with controls (Fig. 7G), but no change was observed in p38 phosphorylation (data not shown). To elucidate the involvement of ERK in postfracture behavioral changes and spinal inflammatory mediator expression, we intrathecally injected 4-week postfracture rats with PD98059, a selective ERK phosphorylation inhibitor. PD98059 treatment reduced fracture-induced hind paw allodynia and unweighting (Fig. 7A, B), partially blocked the postfracture increase in TNF mRNA expression, but had no effects on IL-1, IL-6, and NGF expression at 4 weeks after fracture (Fig. 7C–F). Intrathecal injection of the SP NK1 receptor antagonist LY303870 only partially inhibited fracture-induced phosphorylation of ERK, but the CGRP receptor antagonist CGRP8-37 completely reversed ERK phosphorylation (Fig. 7G). To identify the cell types in which ERK phosphorylation occurred, sections of the lumbar cord were immunostained for glial cell markers and phosphorylated ERK. Representative confocal images in Figure 8 demonstrate that ERK activation was frequently observed in Iba1-positive microglia and occasionally in GFAP-positive astrocytes in the rat spinal cord dorsal horn at 4 weeks after fracture. These results suggest that CGRP induced phosphorylation of ERK in spinal microglia and astrocytes can stimulate spinal TNF expression and induce nociceptive sensitization after fracture.
Figure 7.
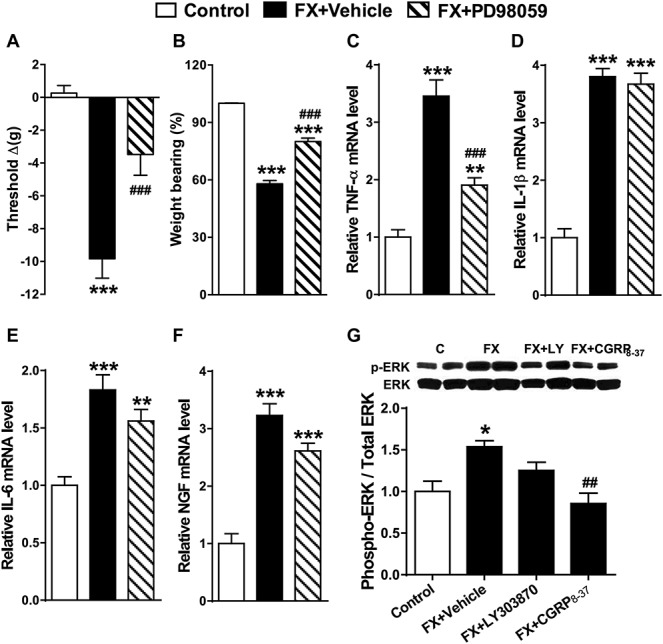
Spinal extracellular signal-regulated kinase 1/2 (ERK1/2) mitogen-activated protein kinase (MAPK) phosphorylation increased after fracture and contributed to upregulated spinal TNF-α expression and hind paw nociceptive sensitization. At 4 weeks, postfracture rats underwent intrathecal injection with vehicle (FX + Vehicle) or the ERK1/2 MAPK phosphorylation inhibitor PD98059 (20 μg). Peak analgesic effects for hind paw von Frey allodynia (A) and unweighting (B) were observed at 1 hour after injection. PD98059 intrathecal injection also partially inhibited postfracture increases in spinal TNF-α mRNA levels (C), but had no effect on postfracture increases in IL-1β (D), IL-6 (E), and nerve growth factor (F), compared with the FX + vehicle group. At 4 weeks after fracture, there was an increased ratio of phosphorylated ERK (pERK) to total ERK1/2 (ERK) in the lumbar spinal cord, indicating ERK1/2 activation (G). Intrathecal injection of the substance P NK1 antagonist LY303870 (20 μg) in fracture rats (FX + LY) did not significantly inhibit ERK1/2 phosphorylation, but injection of the calcitonin gene–related peptide antagonist CGRP8-37 (20 μg) (FX + CGRP8-37) completely blocked postfracture-induced phosphorylation of ERK1/2. Values are mean ± SE, n = 8 per cohort. One-way analysis of variance (P < 0.001) with Bonferroni post hoc testing *P < 0.05, **P < 0.01, and ***P < 0.001 for FX + vehicle, or FX + PD98059 vs control, ###P < 0.001 for FX + PD98059, or FX + CGRP8-37 vs wild-type FX.
Figure 8.
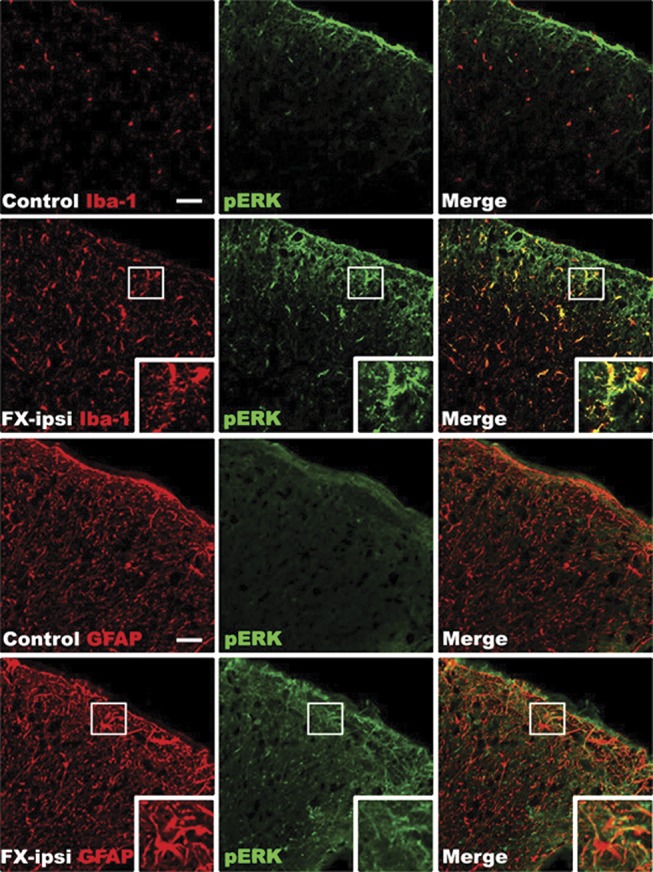
Representative confocal images of coimmunostaining for glial cell markers and phosphorylated ERK1/2 (pERK) in the lumbar spinal cord at 4 weeks after fracture. Top and third row panels are from a normal control rat, second and fourth row panels are from a fracture rat. Minimal pERK (green) immunostaining was seen in the spinal cord from normal control. In contrast, a substantial increase in pERK immunostaining was observed in the cord at 4 weeks after fracture. Double labeling demonstrates that ERK is activated in the Iba1-positive microglia (red) and GFAP-positive astrocytes (red) in the spinal cord dorsal horn after fracture (n = 4 per cohort). Scale bar = 50 μm. FX, fracture; ipsi, ipsilateral.
4. Discussion
Neurogenic inflammation is mediated by sensory afferent release of SP and CGRP, neurotransmitters that activate their receptors in the dermal vasculature to induce extravasation and vasodilatation. Electrically evoked extravasation and vasodilatation responses are enhanced in patients with CRPS, and when SP is microdialyzed in the skin, there is an exaggerated extravasation response.24,44 Furthermore, serum levels of SP and CGRP are elevated in patients with CRPS.5,7,37 Similarly, after fracture in rats, we have observed increased SP and CGRP expression in the sciatic nerve and serum, upregulated SP NK1 receptor expression in the endothelial cells and keratinocytes of the paw skin, and enhanced SP-evoked extravasation and edema responses in the fracture limb.46 Treating fracture rats with an NK1 receptor antagonist reduced hind paw allodynia, warmth, and edema, providing evidence that exaggerated SP signaling contributes to the development of CRPS-like changes.13 We also observed that fracture mice developed hind paw allodynia, unweighting, warmth, and edema at 3 weeks after fracture, but that in SP-deficient fracture mice allodynia and unweighting were attenuated, and there was no warmth and edema.15 Fracture mice lacking the CGRP RAMP1 receptor had a similar presentation. Collectively, these data support the hypothesis that neuropeptide signaling is amplified in the skin and vasculature of patients with CRPS and in the fracture rats and mice, contributing to CRPS-like changes.
At 4 weeks after fracture, the expression of SP and its NK1 receptor were upregulated in the ipsilateral lumbar spinal cord (Fig. 1). There was also a postfracture increase in the spinal expression of CGRP, but no change was observed in the expression of the CGRP receptor dimers CALCRL and RAMP1 (Fig. 1). Although the primary sources of dorsal horn SP and CGRP are the small sensory fiber terminals, other potential sources for upregulated intrinsic spinal neuropeptide mRNA expression are spinal interneurons expressing SP9 and ventral motor neurons expressing CGRP.12 Previously, we observed a postfracture increase in SP and CGRP mRNA and protein expression in the ipsilateral L4,5 dorsal root ganglia and sciatic nerve, respectively.46 Collectively, these data suggest that primary afferent and intrinsic neuropeptide signaling in the spinal cord is enhanced at 4 weeks after fracture. Similarly, several previous studies using inflammatory and neuropathic pain models have also observed exaggerated spinal neuropeptide signaling.1,22,28 Intrathecal injection of either an SP or CGRP receptor antagonist partially reversed hind paw von Frey allodynia and unweighting in the 4-week fracture rats (Fig. 2), but had no effect on the contralateral von Frey thresholds, providing evidence that spinal neuropeptide signaling contributes to postfracture nociceptive sensitization but not to basal pain thresholds.
When SP or CGRP is dialyzed through normal48 or CRPS-affected skin,24 there is no immediate painful response, leading us to postulate that SP and CGRP act as intermediate mediators in the development of posttraumatic pain. Increased levels of TNF and IL-6 are observed in experimental blister fluid or skin biopsies from the affected limbs, but not the contralateral limbs of patients with CRPS,16,18,23 and we recently demonstrated activated keratinocytes expressing TNF and IL-6 in CRPS-affected skin.4 TNF, IL-1, IL-6, and NGF levels are increased in the fracture hind paw skin, and fracture rats treated systemically with the TNF inhibitor etanercept, the IL-1 receptor antagonist anakinra, or the NGF antibody tanezumab had reduced allodynia and hind limb unweighting, indicating the contribution of cytokine and growth factor signaling in the development of trauma-induced chronic pain.27,34,35
The TNF, IL-1, and NGF inhibitors we tested in the fracture rats are large molecules that do not cross the blood-brain barrier, suggesting that the pronociceptive effects of these inflammatory mediators occur at the nociceptor level in hind paw skin. Proinflammatory cytokines and NGF can immediately evoke spontaneous firing and sensitization in sensory afferents.10,20,31 Furthermore, we have observed that intraplantar injection of these mediators into normal hind paw skin rapidly induces nociceptive sensitization and have identified proliferating keratinocytes as the primary cellular source of these mediators in the fracture hind paw.25 These convergent data support the premise that postfracture keratinocyte proliferation and inflammatory mediator expression can induce nociceptive sensitization.
Previously, we observed that SP and CGRP applied to keratinocytes in vitro stimulated keratinocyte expression of TNF, IL-1, IL-6, and NGF through cell surface receptor activation.38 After plantar injection of SP in normal rats, we observed a gradual increase in keratinocyte expression of TNF, IL-1, IL-6, and NGF that resolved after 48 hours, temporally correlating with the development and resolution of allodynia.45 In another study, we observed that WT fracture mice expressed elevated levels of TNF, IL-1, IL-6, and NGF in the hind paw skin but that in SP-deficient fracture mice and CGRP RAMP1 receptor–deficient fracture mice, only IL-6 levels increase in the hind paw skin.15 These results demonstrate that neuropeptide signaling directly induces keratinocyte expression of inflammatory mediators in vitro and in vivo.
In this study, at 4 weeks after fracture, TNF, IL-1, IL-6, CCL2, and NGF expression was elevated in the lumbar spinal cord (Fig.3). These inflammatory mediators are upregulated in spinal cord tissue in a variety of neuropathic and inflammatory pain models.19,33,42 Intrathecal injections of specific antagonists or inhibitors for each of these inflammatory mediators partially reduced allodynia and unweighting in the fracture hind paw (Fig. 4), confirming their association with nociceptive sensitization in the fracture model.
Transgenic fracture mice deficient for SP or the RAMP1 receptor were used to examine the relationship between neuropeptide signaling, nociceptive sensitization, and expression of spinal inflammatory mediators. As previously noted, the SP- and RAMP1 receptor–deficient fracture mice had attenuated hind paw allodynia and unweighting at 3 weeks after fracture, compared with WT fracture mice (Fig. 5).15 Postfracture increases in the spinal expression of SP and CGRP and of their cognate receptors were reduced in the SP- and CGRP receptor–deficient mice (Fig. 6A–F). These results suggest that exaggerated postfracture neuropeptide signaling in the spinal cord can upregulate intrinsic neuropeptide and neuropeptide receptor expression, further amplifying neuropeptide signaling in the spinal cord. Furthermore, SP- and RAMP1-deficient mice had reduced postfracture increases in spinal TNF, IL-1, IL-6, CCL2, and NGF, indicating a crucial role for neuropeptide signaling in postfracture spinal cord inflammatory mediator expression (Fig. 6G–K). Additional support for this hypothesis comes from the observation that exaggerated spinal SP signaling contributes to allodynia and elevated spinal TNF levels in rats exhibiting opiate withdrawal allodynia.41
Spinal ERK phosphorylation was increased in fracture rats, and this increase was inhibited by intrathecal administration of the CGRP receptor antagonist CGRP8-37 (Fig. 7G). Intrathecal injection of an ERK phosphorylation inhibitor (PD98058) partially reversed postfracture pain behavior (Fig. 7A, B) and partially blocked the increase in TNF, but not IL-1, IL-6, or NGF expression (Fig. 7C–F). Immunostaining for phosphorylated ERK and the microglial marker Iba-1 or the astrocyte marker GFAP in the dorsal horn of the lumbar cord demonstrated extensive ERK phosphorylation in microglia and occasionally in astrocytes (Fig. 8). Macrophages and microglia are both immune cells derived from monocytes, and it has been reported that the in vitro activation of macrophage NK1 receptors induces phosphorylation ERK and stimulates production of proinflammatory mediators such as CCL2.39
It has been proposed that intense nociceptive afferent activation triggers neurotransmitter release in the dorsal horn, thus inducing spinal glia to express and release inflammatory mediators.42,43 Interestingly, the intensity of pain in the first week after wrist fracture is the best predictor of the development of CRPS in the ensuing 4 months.29 The results of this study support the hypothesis that intense postfracture nociceptive signaling induces neuropeptide release in the spinal cord, with resultant amplification of spinal neuropeptide signaling and inflammatory mediator expression. We postulate that inflammatory mediator expression in the spinal cord, acting in concert with the inflammatory mediators released by the cutaneous keratinocytes and mast cells in the injured limb, can initiate the development of regional nociceptive sensitization in the fracture model of CRPS. The spinal cord inflammatory changes observed after fracture may also play a critical role in the maintenance of nociceptive sensitization in chronic CRPS. Over time, keratinocyte and mast cell proliferation and inflammatory mediator expression return to normal levels in most patients with CRPS,4,49 but in some patients, the pain and nociceptive sensitization persist, perhaps because of the residual overexpression of spinal inflammatory mediators.2,3 Future studies on the fracture model looking at changes over time in the expression of inflammatory mediators in the skin and spinal cord may further our understanding of the postfracture temporal evolution of neurokeratinocyte and neuroglial inflammatory signaling and its nociceptive sequelae.
To our knowledge, this is the first study to examine spinal cord neuropeptide signaling and inflammatory mediator expression in a CRPS model. This study has several limitations, including those inherent to using animal models of pain and the translational value of the data acquired. Clinical investigations are required to determine whether upregulated spinal neuropeptide signaling and inflammatory mediator expression contribute to CRPS. The results of this study do provide preclinical support for CRPS treatments that inhibit spinal nociceptive processing, such as NMDA antagonists, intrathecal drugs, and spinal cord stimulation.
Conflict of interest statement
This study was supported by National Institutes of Health grants NS072168 and NS072143, and Department of Veterans Affairs, Rehabilitation Research and Development Merit grants F7137R.
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
References
- [1].Abbadie C, Brown JL, Mantyh PW, Basbaum AI. Spinal cord substance P receptor immunoreactivity increases in both inflammatory and nerve injury models of persistent pain. Neuroscience 1996;70:201–9. [DOI] [PubMed] [Google Scholar]
- [2].Alexander GM, Perreault MJ, Reichenberger ER, Schwartzman RJ. Changes in immune and glial markers in the CSF of patients with complex regional pain syndrome. Brain Behav Immun 2007;21:668–76. [DOI] [PubMed] [Google Scholar]
- [3].Alexander GM, van Rijn MA, van Hilten JJ, Perreault MJ, Schwartzman RJ. Changes in cerebrospinal fluid levels of pro-inflammatory cytokines in CRPS. PAIN 2005;116:213–9. [DOI] [PubMed] [Google Scholar]
- [4].Birklein F, Drummond PD, Li W, Schlereth T, Albrecht N, Finch PM, Dawson LF, Clark JD, Kingery WS. Activation of cutaneous immune responses in complex regional pain syndrome. J Pain 2014;15:485–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Birklein F, Schmelz M, Schifter S, Weber M. The important role of neuropeptides in complex regional pain syndrome. Neurology 2001;57:2179–84. [DOI] [PubMed] [Google Scholar]
- [6].Bjurstrom MF, Giron SE, Griffis CA. Cerebrospinal fluid cytokines and neurotrophic factors in human chronic pain populations: a comprehensive review. Pain Pract 2014. [DOI] [PubMed] [Google Scholar]
- [7].Blair SJ, Chinthagada M, Hoppenstehdt D, Kijowski R, Fareed J. Role of neuropeptides in pathogenesis of reflex sympathetic dystrophy. Acta Orthop Belg 1998;64:448–51. [PubMed] [Google Scholar]
- [8].Brunner F, Schmid A, Kissling R, Held U, Bachmann LM. Biphosphonates for the therapy of complex regional pain syndrome I–systematic review. Eur J Pain 2009;13:17–21. [DOI] [PubMed] [Google Scholar]
- [9].Cassam AK, Llewellyn-Smith IJ, Weaver LC. Catecholamine enzymes and neuropeptides are expressed in fibres and somata in the intermediate gray matter in chronic spinal rats. Neuroscience 1997;78:829–41. [DOI] [PubMed] [Google Scholar]
- [10].De Jongh RF, Vissers KC, Meert TF, Booij LH, De Deyne CS, Heylen RJ. The role of interleukin-6 in nociception and pain. Anesth Anal 2003;96:1096–103. [DOI] [PubMed] [Google Scholar]
- [11].de Mos M, de Bruijn AG, Huygen FJ, Dieleman JP, Stricker BH, Sturkenboom MC. The incidence of complex regional pain syndrome: a population-based study. PAIN 2007;129:12–20. [DOI] [PubMed] [Google Scholar]
- [12].Gibson SJ, Polak JM, Giaid A, Hamid QA, Kar S, Jones PM, Denny P, Legon S, Amara SG, Craig RK, Bloom SR, Penketh RJA, Rodek C, Ibrahim NBN, Dawson A. Calcitonin gene-related peptide messenger RNA is expressed in sensory neurones of the dorsal root ganglia and also in spinal motoneurones in man and rat. Neurosci Lett 1988;91:283–8. [DOI] [PubMed] [Google Scholar]
- [13].Guo TZ, Offley SC, Boyd EA, Jacobs CR, Kingery WS. Substance P signaling contributes to the vascular and nociceptive abnormalities observed in a tibial fracture rat model of complex regional pain syndrome type I. PAIN 2004;108:95–107. [DOI] [PubMed] [Google Scholar]
- [14].Guo TZ, Wei T, Kingery WS. Glucocorticoid inhibition of vascular abnormalities in a tibia fracture rat model of complex regional pain syndrome type I. PAIN 2006;121:158–67. [DOI] [PubMed] [Google Scholar]
- [15].Guo TZ, Wei T, Shi X, Li WW, Hou S, Wang L, Tsujikawa K, Rice KC, Cheng K, Clark DJ, Kingery WS. Neuropeptide deficient mice have attenuated nociceptive, vascular, and inflammatory changes in a tibia fracture model of complex regional pain syndrome. Mol Pain 2012;8:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Huygen FJ, De Bruijn AG, De Bruin MT, Groeneweg JG, Klein J, Zijistra FJ. Evidence for local inflammation in complex regional pain syndrome type 1. Mediators Inflamm 2002;11:47–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Huygen FJ, Niehof S, Zijlstra FJ, van Hagen PM, van Daele PL. Successful treatment of CRPS 1 with anti-TNF. J Pain Symptom Manage 2004;27:101–3. [DOI] [PubMed] [Google Scholar]
- [18].Huygen FJ, Ramdhani N, van Toorenenbergen A, Klein J, Zijlstra FJ. Mast cells are involved in inflammatory reactions during complex regional pain syndrome type 1. Immunol Lett 2004;91:147–54. [DOI] [PubMed] [Google Scholar]
- [19].Ji RR, Berta T, Nedergaard M. Glia and pain: is chronic pain a gliopathy? PAIN 2013;154(suppl 1):S10–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Jin X, Gereau RW. Acute p38-mediated modulation of tetrodotoxin-resistant sodium channels in mouse sensory neurons by tumor necrosis factor-alpha. J Neurosci 2006;26:246–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kalita J, Vajpayee A, Misra UK. Comparison of prednisolone with piroxicam in complex regional pain syndrome following stroke: a randomized controlled trial. QJM 2006;99:89–95. [DOI] [PubMed] [Google Scholar]
- [22].Kar S, Rees RG, Quirion R. Altered calcitonin gene-related peptide, substance P and enkephalin immunoreactivities and receptor binding sites in the dorsal spinal cord of the polyarthritic rat. Eur J Neurosci 1994;6:345–54. [DOI] [PubMed] [Google Scholar]
- [23].Kramer HH, Eberle T, Uceyler N, Wagner I, Klonschinsky T, Muller LP, Sommer C, Birklein F. TNF-alpha in CRPS and “normal” trauma–significant differences between tissue and serum. PAIN 2011;152:285–90. [DOI] [PubMed] [Google Scholar]
- [24].Leis S, Weber M, Isselmann A, Schmelz M, Birklein F. Substance-P-induced protein extravasation is bilaterally increased in complex regional pain syndrome. Exp Neurol 2003;183:197–204. [DOI] [PubMed] [Google Scholar]
- [25].Li WW, Guo TZ, Li XQ, Kingery WS, Clark JD. Fracture induces keratinocyte activation, proliferation, and expression of pro-nociceptive inflammatory mediators. PAIN 2010;151:843–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Li WW, Guo TZ, Liang DY, Sun Y, Kingery WS, Clark JD. Substance P signaling controls mast cell activation, degranulation, and nociceptive sensitization in a rat fracture model of complex regional pain syndrome. Anesthesiology 2012;116:882–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Li WW, Sabsovich I, Guo TZ, Zhao R, Kingery WS, Clark JD. The role of enhanced cutaneous IL-1beta signaling in a rat tibia fracture model of complex regional pain syndrome. PAIN 2009;144:303–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Marlier L, Poulat P, Rajaofetra N, Privat A. Modifications of serotonin-, substance P- and calcitonin gene-related peptide-like immunoreactivities in the dorsal horn of the spinal cord of arthritic rats: a quantitative immunocytochemical study. Exp Brain Res 1991;85:482–90. [DOI] [PubMed] [Google Scholar]
- [29].Moseley GL, Herbert RD, Parsons T, Lucas S, Van Hilten JJ, Marinus J. Intense pain soon after wrist fracture strongly predicts who will develop complex regional pain syndrome: prospective cohort study. J Pain 2014;15:16–23. [DOI] [PubMed] [Google Scholar]
- [30].Munts AG, Zijlstra FJ, Nibbering PH, Daha MR, Marinus J, Dahan A, van Hilten JJ. Analysis of cerebrospinal fluid inflammatory mediators in chronic complex regional pain syndrome related dystonia. Clin J Pain 2008;24:30–4. [DOI] [PubMed] [Google Scholar]
- [31].Pezet S, McMahon SB. Neurotrophins: mediators and modulators of pain. Annu Rev Neurosci 2006;29:507–38. [DOI] [PubMed] [Google Scholar]
- [32].Poree LR, Guo TZ, Kingery WS, Maze M. The analgesic potency of dexmedetomidine is enhanced after nerve injury: a possible role for peripheral alpha2-adrenoceptors. Anesth Analg 1998;87:941–48. [DOI] [PubMed] [Google Scholar]
- [33].Ren K, Dubner R. Interactions between the immune and nervous systems in pain. Nat Med 2010;16:1267–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sabsovich I, Guo TZ, Wei T, Zhao R, Li X, Clark DJ, Geis C, Sommer C, Kingery WS. TNF signaling contributes to the development of nociceptive sensitization in a tibia fracture model of complex regional pain syndrome type I. PAIN 2008;137:507–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Sabsovich I, Wei T, Guo TZ, Zhao R, Shi X, Li X, Yeomans DC, Klyukinov M, Kingery WS, Clark JD. Effect of anti-NGF antibodies in a rat tibia fracture model of complex regional pain syndrome type I. PAIN 2008;138:47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Sandroni P, Benrud-Larson LM, McClelland RL, Low PA. Complex regional pain syndrome type I: incidence and prevalence in Olmsted county, a population-based study. PAIN 2003;103:199–207. [DOI] [PubMed] [Google Scholar]
- [37].Schinkel C, Gaertner A, Zaspel J, Zedler S, Faist E, Schuermann M. Inflammatory mediators are altered in the acute phase of posttraumatic complex regional pain syndrome. Clin J Pain 2006;22:235–9. [DOI] [PubMed] [Google Scholar]
- [38].Shi X, Wang L, Clark JD, Kingery WS. Keratinocytes express cytokines and nerve growth factor in response to neuropeptide activation of the ERK1/2 and JNK MAPK transcription pathways. Regul Pept 2013;186:92–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sun J, Ramnath RD, Zhi L, Tamizhselvi R, Bhatia M. Substance P enhances NF-kappaB transactivation and chemokine response in murine macrophages via ERK1/2 and p38 MAPK signaling pathways. Am J Physiol Cell Physiol 2008;294:C1586–96. [DOI] [PubMed] [Google Scholar]
- [40].Tran de QH, Duong S, Bertini P, Finlayson RJ. Treatment of complex regional pain syndrome: a review of the evidence. Can J Anaesth 2010;57:149–66. [DOI] [PubMed] [Google Scholar]
- [41].Tumati S, Largent-Milnes TM, Keresztes AI, Yamamoto T, Vanderah TW, Roeske WR, Hruby VJ, Varga EV. Tachykinin NK(1) receptor antagonist co-administration attenuates opioid withdrawal-mediated spinal microglia and astrocyte activation. Eur J Pharmacol 2012;684:64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Watkins LR, Milligan ED, Maier SF. Glial activation: a driving force for pathological pain. Trends Neurosci 2001;24:450–5. [DOI] [PubMed] [Google Scholar]
- [43].Watkins LR, Milligan ED, Maier SF. Glial proinflammatory cytokines mediate exaggerated pain states: implications for clinical pain. Adv Exp Med Biol 2003;521:1–21. [PubMed] [Google Scholar]
- [44].Weber M, Birklein F, Neundorfer B, Schmelz M. Facilitated neurogenic inflammation in complex regional pain syndrome. PAIN 2001;91:251–7. [DOI] [PubMed] [Google Scholar]
- [45].Wei T, Guo TZ, Li WW, Hou S, Kingery W, Clark JD. Keratinocyte expression of inflammatory mediators plays a crucial role in substance P-induced acute and chronic pain. J Neuroinflamm 2012;9:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wei T, Li WW, Guo TZ, Zhao R, Wang L, Clark DJ, Oaklander AL, Schmelz M, Kingery WS. Post-junctional facilitation of substance P signaling in a tibia fracture rat model of complex regional pain syndrome type I. PAIN 2009;144:278–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Wei T, Sabsovich I, Guo TZ, Shi X, Zhao R, Li W, Geis C, Sommer C, Kingery WS, Clark DJ. Pentoxifylline attenuates nociceptive sensitization and cytokine expression in a tibia fracture rat model of complex regional pain syndrome. Eur J Pain 2009;13:253–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Weidner C, Klede M, Rukwied R, Lischetzki G, Neisius U, Skov PS. Acute effects of substance P and calcitonin gene-related peptide in human skin-a microdialysis study. J Invest Dermatol 2000;115:1015–20. [DOI] [PubMed] [Google Scholar]
- [49].Wesseldijk F, Huygen FJ, Heijmans-Antonissen C, Niehof SP, Zijlstra FJ. Six years follow-up of the levels of TNF-alpha and IL-6 in patients with complex regional pain syndrome type 1. Mediators Inflamm 2008;2008:469439. [DOI] [PMC free article] [PubMed] [Google Scholar]