

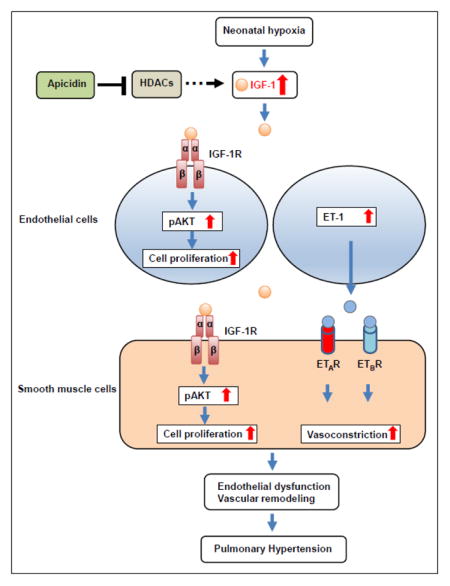
Abstract
Pulmonary hypertension is a fatal disease characterized by a progressive increase in pulmonary artery pressure accompanied by pulmonary vascular remodeling and increased vasomotor tone. Although some biological pathways have been identified in neonatal hypoxia-induced pulmonary hypertension (PH), little is known regarding the role of growth factors in the pathogenesis of PH in neonates. In this study, using a model of hypoxia-induced PH in neonatal mice, we demonstrate that the growth factor insulin-like growth factor-1 (IGF-1), a potent activator of the AKT signaling pathway, is involved in neonatal PH. After exposure to hypoxia, IGF-1 signaling is activated in pulmonary endothelial and smooth muscle cells in vitro, and the IGF-1 downstream signal pAKTS473 is upregulated in lungs of neonatal mice. We found that IGF-1 regulates ET-1 expression in pulmonary endothelial cells and that IGF-1 expression is regulated by histone deacetylases (HDACs). In addition, there is a differential cytosine methylation site in the IGF-1 promoter region in response to neonatal hypoxia. Moreover, inhibition of HDACs with apicidin decreases neonatal hypoxia-induced global DNA methylation levels in lungs and specific cytosine methylation levels around the pulmonary IGF-1 promoter region. Finally, HDAC inhibition with apicidin reduces chronic hypoxia-induced activation of IGF-1/pAKT signaling in lungs and attenuates right ventricular hypertrophy and pulmonary vascular remodeling. Taken together, we conclude that IGF-1, which is epigenetically regulated, is involved in the pathogenesis of pulmonary hypertension in neonatal mice. This study implicates a novel HDAC/IGF-1 epigenetic pathway in the regulation of hypoxia-induced PH and warrants further study of the role of IGF-1 in neonatal pulmonary hypertensive disease.
Keywords: IGF-1, neonatal hypoxia, pulmonary hypertension, histone deacetylase, endothelin-1, DNA methylation
Graphical abstract
1. Introduction
Unlike in the adult, the perinatal and neonatal pulmonary vascular response to chronic hypoxic exposure is much more rapid and severe which can result in the failure of the fetal or neonatal circulation to adapt to postnatal life. This in turn contributes to the pathogenesis of persistent pulmonary hypertension of the newborn. There is increasing evidence in humans and experimental animals that perinatal and neonatal factors may be linked with the development of adult diseases such as chronic obstructive pulmonary disease and PH in later life [1; 2]. As such, furthering our understanding of the mechanisms and pathology of neonatal PH is important to help us understand adult disease and to support more effective therapeutic target identification for neonates and adults.
The insulin-like growth factor (IGF)/insulin family of growth factors is an evolutionally conserved system which plays a crucial role in the growth and development of many tissues and in the regulation of overall growth and metabolism [3; 4]. Within the family, IGF-1 is a single-chain polypeptide with a high sequence homology to pro-insulin. IGF-1 interacts with the IGF-1 receptor (IGF-1R) whose signaling has pleiotropic effects on cell behaviors that control cell proliferation, differentiation, and cell migration and also regulate the apoptotic machinery [5; 6; 7]. High levels of circulating IGF-1 are associated with an increased risk of several cancers. However, the role of IGF-1 signaling in neonatal hypoxia-induced PH is largely unknown.
Epigenetic programming, dynamically regulated by histone acetylation, is a key mechanism impacting cell proliferation, survival, and apoptosis [8; 9]. Previous studies have shown that lungs from patients with idiopathic pulmonary arterial hypertension exhibit increased expression of HDACs compared with control lung tissue, and inhibition of HDAC activity provides a beneficial effect in hypoxia-induced PH in adult mice [10; 11]. Although there are several factors and pathways found to be involved in perinatal and neonatal hypoxia-induced PH [12; 13; 14; 15; 16; 17; 18; 19], little is known regarding the role of growth factors and epigenetic signaling in neonatal hypoxia-induced PH.
In this study, we test the hypothesis that IGF-1 is epigenetically regulated in neonatal hypoxia-induced PH. We use a neonatal mouse model to investigate the effects of HDAC inhibition on hypoxia-induced PH. The cellular and molecular mechanisms underlying abnormal expression of IGF-1 in response to hypoxia are also examined.
2. Materials and methods
2.1. Animal model of hypoxia-induced PH
All experimental protocols described in mice were reviewed and approved by the Animal Care and Use Committee at the University of Illinois. Mice were cared for in accordance with the University of Illinois at Chicago Guide for the Care and Use of Laboratory Animals.
Newborn C57BL/6 mouse pups along with their dams (Charles River Laboratories, Wilmington, MA) were exposed to normobaric hypoxia (11% oxygen) shortly after birth for two weeks in a Plexiglas chamber flushed with nitrogen gas to maintain hypoxic conditions using a ProOx-110 oxygen controller (BioSpherix, NY). Carbon dioxide from the chamber was removed by absorption with Amsorb Plus pellets (Bell Medical, St. Louis, MO). Control pups were left in room air (normoxia) for two weeks.
To inhibit HDAC activity, apicidin (dissolved in 50% DMSO in physiological saline) was administered (2 mg/kg, i.p.) starting on postnatal day 5 (P5) three times per week to pups maintained in room air and hypoxia. Increasing the frequency of drug treatment was found to increase mortality, hence this particular frequency was chosen. Mouse pups given 50% DMSO in physiological saline (vehicle) and maintained in room air or hypoxia served as controls. Mice were sacrificed immediately following the completion of normoxia or hypoxia at postnatal day 14 (P14). Some mice were recovered in room air for a month after exposure to hypoxia and sacrificed at day 45 (P45).
2.2. Cell culture and reagents
Human pulmonary artery smooth muscle cells (HPASMCs) and human pulmonary artery endothelial cells (HPAECs) were cultured according to the manufacture’s recommendations. All other biochemical reagents were obtained from Sigma (St. Louis, MO, USA) unless specified.
2.3. In vitro IGF-1 and inhibitor treatments
HPASMCs were seeded at 50–60% confluence in complete media and made quiescent in serum-free media for 7 h. The cells were then pretreated with inhibitors of IGF-1R (OSI-906, 150 nM), AKT (MK-2206, 130 nM), and PI3K (PF-04691502, 32 nM) for 1 h before incubation with hIGF-1 (100 ng/ml, EMD Millipore, Billerica, MA, USA) for 14 h. The inhibitors OSI-906, MK-2206, and PF-04691502 were obtained from Selleck Chemicals (Houston, TX, USA). Cells were washed with PBS, and total RNA and protein were extracted with TRIzol (Invitrogen, Carlsbad, CA, USA) and RIPA buffer, respectively, for q-PCR and Western blot analysis.
2.4. In vitro hypoxia
HPAECs were seeded at 50–60% confluence in complete media and made quiescent in serum-free media overnight. The cells were exposed to air (21% oxygen) or hypoxia (1% oxygen) in the presence or absence of the class I HDAC inhibitor apicidin (2.5 μg/ml) for 24 h. The cells were washed with PBS, and total RNA and protein were extracted with TRIzol and RIPA buffer, respectively, for q-PCR and Western blot analysis.
2.5. siRNA transfection
HPAECs were transfected with siRNA for HDAC1, HDAC2, HDAC3, IGF-1 (50 nM) or a nontargeting negative control (NC) using Lipofectamine 2000 (Invitrogen, CA) according to the manufacturer’s protocol adjusted for the concentration of siRNA (see Table 1).
Table 1.
Primer and target sequencs for Q-PCR and siRNA
| Gene | F/R | primers or target sequences | Assay | species | Accession Number |
|---|---|---|---|---|---|
| IGF-1 | Forward | TGCAACCTGGGTCACACAA | q-PCR | mouse | NM_010512 |
| IGF-1 | Reverse | GCACCGAGAGGTGGAGTGAT | q-PCR | mouse | NM_010512 |
| 18S | Forward | CGGACCAGAGCGAAAGCA | q-PCR | mouse | NR_003278 |
| 18S | Reverse | CCTCCGACTTTCGTTCTTGATT | q-PCR | mouse | NR_003278 |
| EDN-1 | Forward | TTGCTGAAATGTTTTTCCTTGTGT | q-PCR | human | J05008 |
| EDN-1 | Reverse | TTCTTATGATTATTCCAGTCTTTCTCCAT | q-PCR | human | J05008 |
| IGF-1 | Forward | TGCTTCCGGAGCTGTGATCT | q-PCR | human | X00173 |
| IGF-1 | Reverse | CGGACAGAGCGAGCTGACTT | q-PCR | human | X00173 |
| IGF-1 r | Forward | CTTAACTGACATGGGCCTTTA | q-PCR | human | NM_000875 |
| IGF-1 r | Reverse | AGGAGAGAGAAAGGGAGAAC | q-PCR | human | NM_000875 |
| EDNRA | Forward | GAGTCGTGTTCAGGGAATTG | q-PCR | human | L06622 |
| EDNRA | Reverse | CAGGAATGGCCAGGATAAAG | q-PCR | human | L06622 |
| EDNRB | Forward | GCTGGTGCCTTTCATACA | q-PCR | human | L06623 |
| EDNRB | Reverse | ACAGCTCGATATCTGTCAATAC | q-PCR | human | L06623 |
| IGF-1r | Forward | GTCACTGGGAGATACTGTTTG | q-PCR | human | NM_010513 |
| IGF-1r | Reverse | AGGTATGTGCGAGAGGATAG | q-PCR | human | NM_010513 |
| Ednra | Forward | CACCGAGTGTTGGTACTTTAT | q-PCR | human | NM_010332 |
| Ednra | Reverse | CCTCTTCCTCTCCTCTTTCT | q-PCR | human | NM_010332 |
| ednrb | Forward | GGCTGTTCAGTTTCTACTTCT | PCR | human | NM_007904 |
| ednrb | Reverse | CTGCATACCGCTCTTCTTC | q-PCR | human | NM_007904 |
| IGF-1 | Forward | ACACTGACGACATGGTTCTACATTTTGGGAAAGTATATTTGGAGA | NGS | mouse | |
| IGF-1 | Reverse | TACGGTAGCAGAGACTTGGTCTTCATTCAAATCCCTCAACTAAA | NGS | mouse | |
| HDAC-1 | target | CACCCGGAGGAAAGTCTGTTA | siRNA | human | |
| HDAC-2 | target | TCCCAATGAGTTGCCATATAA | siRNA | human | |
| HDAC-3 | target | GACCATGACAATGACAAGGAA | siRNA | human | |
| IGF-1 | target | ACGGTAATACGTGAAAGCAAA | siRNA | human |
2.6. Isolation of mouse pulmonary arterial smooth muscle cells
Mouse pulmonary arterial smooth muscle cels (mPASMCs) were isolated using a modification of previously described methods [20; 21]. Mouse lungs were flushed with PBS in-situ through the right ventricle to remove the blood. A warm solution of M199 containing HEPES (25 mM), penicillin and streptomycin (P/S, 1%), agarose (0.5% w/v), and iron particles (0.5% w/v) was then injected into the pulmonary vasculature via the right ventricle. Because the iron particles are too large to pass through the capillaries, they become trapped in the arteries only. The lungs were then inflated via the trachea with a warm solution of M199 containing HEPES (25 mM), P/S (1%), and agarose (10% w/v) without iron. The heart and lungs were removed en bloc and placed in PBS on ice for the agarose to harden. The lung lobes were then dissected free and finely minced in a Petri dish, then resuspended in M199 containing collagenase (80 U/ml) and incubated at 37°C for 50–60 min. To remove extravascular tissue, the fragments were drawn through an 18-guage needle and then washed in M199 containing 20% FBS five times with the use of a magnet to retain the pieces containing iron. The resulting iron-containing fragments were resuspended in M199 containing 20% FBS and P/S and plated in a T-25 flask. The flasks were incubated at 37°C with CO2 (5%) in air for 1 day. After 1 day, the iron-containing fragments were removed, washed with growth medium, and replated in a new T-25 flask. The adherent myocytes were maintained using standard cell culture techniques and used for experiments at passage 2. Cells were confirmed to be pulmonary artery myocytes by fluorescent staining for smooth muscle alpha-actin.
2.7. In vivo hemodynamic assessments
C57BL/6 mice (P45) were anesthetized with isoflurane and intubated with an 18-gauge angiocath sleeve. Surgical anesthesia was maintained using isoflurane delivered through a vaporizer with compressed oxygen gas connected in series to a rodent ventilator. The heart was exposed through a bilateral incision in the diaphragm, and a Scisense pressure transducer (Transonic Systems, Ithaca, NY) was introduced apically into the right ventricle following a small stab wound with a 28-gauge needle. The transducer was advanced to just outside the pulmonary outflow tract, and the mouse was given 10 min to acclimate before right ventricular systolic pressures (RVSP) were taken. Immediately following this procedure, the mouse was euthanized by explantation of the heart.
2.8. Measurement of ventricular weights
C57BL/6 mice were weighed and then sacrificed. The right ventricle (RV) was dissected from the left ventricle and interventricular septum (LV + S), and the ratios of their weights [RV / (LV + S)] (Fulton’s index) and [RV / body weight] were calculated as indices of right heart hypertrophy. Values are represented as mean ± SEM.
2.9. Lung morphometry
The chest cavity was opened and lungs were inflation fixed via the trachea with 10% formalin at 20 cm H2O pressure, and the right ventricle was perfused sequentially with PBS and 10% formalin at the same pressure. Fixed lungs were paraffin-embedded, cut into 5-μm sections, stained with hematoxylin and eosin, and scanned using Aperio ScanScope (Leica Biosystems). Pulmonary arteries adjacent to bronchioles and less than 80 μm in diameter were scored for wall thickness represented by the difference between the area of the entire vessel and area of the lumen divided by the area of the entire vessel. Values are represented as mean ± SEM.
2.10. cDNA synthesis and SYBR green real-time PCR
RNA was isolated using TRIzol reagent. Reverse transcription was performed using Superscript III (Invitrogen) and 50 μM oligo(dT)12-18 at 50°C for 50 min. SYBR Green real-time PCR (q-PCR) reactions were set up containing 1X Power SYBR Green Master Mix (Applied Biosystems, Foster City, CA), 250 nM forward and reverse primers in a 20 μl reaction. All assays were carried out in either 96-well or 384-well format. Real-time fluorescent detection of PCR products was performed with ViiA7 Real-Time PCR System (Applied Biosystems) using the following thermocycling conditions: 1 cycle of 95°C for 10 min; 40 cycles of 95°C for 30 s and 60°C for 1 min. The sequences of the primers were designed using the Primer Express software (Applied Biosystems) and are shown in Table 1. 18S ribosome RNA was used as an endogenous control for gene expression. For data analysis, the comparative method (ΔΔCt) was used to calculate relative quantities of a nucleic acid sequence.
2.11. Western blot analysis
Total proteins from cells and lung tissue were extracted by lysing or homogenizing (for tissue samples) in RIPA buffer containing protease and phosphatase inhibitor cocktails, and the protein concentrations were determined using the Bradford protein assay kit (Bio-Rad, Hercules, CA). Equal amounts of total protein (10–25 μg) from samples were subjected to SDS-PAGE. Proteins were transferred to nitrocellulose membrane for 90 min at 100 V. Membranes were blocked for 1 h at room temperature in Tris-buffered saline (TBS) containing 5% nonfat powdered milk and probed with primary antibody in TBS at concentrations from 1:250 to 1:5,000 overnight according to the manufacturer’s instructions for each antibody. Antibodies against pAKTS473, pAKTT308, and pan AKT were obtained from Cell Signaling Technology (Beverly, MA, USA). Antibodies against β-actin were obtained from Santa Cruz Biotechnology (Dallas, TX, USA). Me3K9H3 antibody was obtained from Abcam (Cambridge, MA). In all cases, a secondary antibody labeled with horseradish peroxidase (Bio-Rad) was used at concentrations from 1:5,000 to 1:10,000 for 1 h at room temperature, and immuno-reactive bands were detected by using SuperSignal West Pico Chemi-luminescent Substrate (Thermo Fisher Scientific, Waltham, MA) and recorded on photosensitive film. The relative intensities of immuno-reactive bands detected by Western blot analysis were quantified by densitometry using NIH ImageJ software (NIH) and normalized with pan AKT and β-actin levels.
2.12. DNA methylation analysis
Amplicon sequencing on next-generation sequencing (NGS) platform was used to determine the DNA methylation status of the mIGF-1 promoter region. Genomic DNA samples from four mouse lungs in each experimental group were treated with sodium bisulfite as previously described [8], and PCR amplifications were performed using the primers listed in Table 1. PCR products were sequenced on Ion Torrent PGM and raw sequence data were imported into the software package CLC genomics workbench (CLC bio, Qiagen). Data were quality trimmed (0.01 or Q20) and primer and linker sequences were removed using the trimming algorithm implemented within CLC genomics. Subsequently, trimmed data were mapped against the reference converted sequence, assuming no methylation. Variant calling using the quality variant algorithm was performed using a minimum threshold of 0.1%. A table of variants relative to the reference was generated, and known CpG sites were identified. Percent methylation was determined through variant abundance value for each site.
Global DNA methylation analysis was performed on the lungs of animals exposed to normoxia or hypoxia and treated with vehicle control or apicidin, and total cytosine methylation levels were measured by liquid chromatography-mass spectrometry (LC/MS) as described previously [22]. Briefly, DNA was hydrolyzed to nucleosides by adding 5U nuclease P1 (Sigma) at 37°C for 2 h, 0.002 units of venom phosphodiesterase I (Sigma) at 37°C for 2 h, 0.5 units of alkaline phosphatase at 37°C for 1 h. Stock solutions of 2′-deoxycytidine (Sigma) and 5-methyl-2′-deoxycytidine (ChemGenes, Wilmington, MA) were prepared in water. An eight-point stock mixture of a standard was carefully prepared to give an exact known concentration ratio of 2′-deoxycytidine and 5-methyl-2′-deoxycytidine. The concentration of 2′-deoxycytidine and 5-methyl-2′-deoxycytidine in each sample was calculated from the standard curve. Each DNA sample was injected into the LC and run through an Atlantis DC18 silica column (Waters Corporation, Milford, MA). Identification of 2′-deoxycytidine and 5-methyl-2′-deoxycytidine was obtained by mass spectrometry of chromatographic peaks. Values were expressed as percentage of values in lungs from the vehicle-treated normoxic group.
2.13. Statistical analysis
Statistical analysis of the data was performed using a single factor analysis of variance (ANOVA) and standard two-sample Student’s t-test assuming unequal variances of the two data sets. Statistical significance was determined using a two-tailed distribution assumption and was set at a 5% level (p < 0.05).
3. Results
3.1. IGF-1 is upregulated in neonatal hypoxia-induced pulmonary hypertension
Figure 1A is a recapitulation of the experimental protocol with newborn mice. After chronic hypoxia exposure for 2 weeks, neonatal mice at P14 have developed RV hypertrophy (Figure 1B, C). Although the RV hypertrophy is relatively decreased at P45 compared to P14, sustained RV hypertrophy is observed in the hypoxia group (Figures 1D, E). In addition, RVSP is increased at P45 in mice exposed to neonatal hypoxia (Figure 1F).
Figure 1. Neonatal hypoxia induces RV hypertrophy and increased RVSP and upregulates IGF-1 expression in mice.

(A) Schematic of the study design. N=Normoxia, H=Hypoxia. (B) Weight ratio of RV to (LV + septum) is increased in P14 mice after exposure to hypoxia compared with room air controls. (C) The ratio of RV weight to body weight (BW) is increased in hypoxic mice compared to normoxic mice at P14. (D) The RV to (LV + septum) weight ratio is increased in mice exposed to neonatal hypoxia at P45 compared with normoxic controls. (E) There is a sustained increase in the ratio of RV weight to body weight in mice exposed to neonatal hypoxia compared to control mice at P45. (F) RVSP is increased in hypoxic mice at P45 as compared to normoxic mice. (G) IGF-1 mRNA expression is upregulated in hypoxic lungs at P14 compared to normoxic lungs. * p<0.05 compared to normoxia controls.
Next, we performed q-PCR analysis to determine the expression levels of IGF-1. As shown in Figure 1G, IGF-1 expression at P14 is significantly upregulated in mouse lungs exposed to hypoxia when compared with normoxic lungs.
3.2. IGF-1 activates pAKTS473 in mPASMCs
We investigated whether IGF-1 signaling is active in PASMCs. As shown in Figures 2A and B, IGF-1 is able to increase the levels of phosphorylated AKT at residue position S473 in mPASMCs. Next, we determined whether IGF-1 signaling can be inhibited using inhibitors of IGF-1R, PI3K, and AKT. Since two residues (S473, T308) on AKT can be phosphorylated, antibodies against residues at S473 and T308 were used to characterize the IGF-1-induced AKT phosphorylation. As shown in Figures 2C and D, IGF-1 augments pAKTS473 but not pAKTT308 in HPASMC. Furthermore, inhibition of IGF-1R (OSI-906), PI3K (PF-04691502) and AKT (MK-2206) decreases IGF-1-induced elevation of pAKTS473 levels.
Figure 2. IGF-1 activates pAKT in PASMC.

(A) IGF-1 increases pAKTS473 in mouse PASMC. PASMC were serum-starved overnight and then incubated with IGF-1 (100 ng/ml) for 24 h. Cell lysates were prepared and subjected to Western blot analysis using an antibody against pAKTS473. Pan AKT and β-actin were used as loading controls. (B) Densitometry quantification of pAKTS473 levels in the mPASMC in the presence or absence of IGF-1 for Figure 1A. * p<0.05 compared with the untreated control. (C) Inhibitors of IGF-1R (OSI-906), AKT (MK-2206), and PI3K (PF-04691502) decrease IGF-1-induced pAKT473 in HPASMC. However, inhibitors of AKT (MK-2206) and PI3K (PF-04691502) are capable of decreasing the levels of pAKTT308, while the IGF-1R inhibitor (OSI-906) is not. (D) Densitometry quantification of pAKTS473 levels for Figure 2C. * p<0.05 compared with the untreated control. # p<0.05 compared to IGF-1-treated control (IGF-1 +, no inhibitor).
3.3. Regulation of Endothelin-1 expression by IGF-1
Endothelin-1 (ET-1) plays an important role in ventricular hypertrophy and vascular constriction following hypoxia. To determine the molecular link between IGF-1 and ET-1, ET-1 expression was measured in HPAECs in the presence or absence of IGF-1. As shown in Figure 3A, IGF-1 augments ET-1 expression in HPAECs. Next, we reduced IGF-1 expression using an siRNA knockdown approach. As shown in Figure 3B, IGF-1 siRNA decreases IGF-1 expression, however, ET-1 expression is not notably altered in IGF-1-diminished HPAECs, suggesting that IGF-1 augments ET-1 expression through some paracrine effect.
Figure 3. IGF-1 regulates ET-1 expression.

(A) PAECs were serum-starved overnight and exposed to 100 ng/ml of IGF-1 for 24 h. RNA was extracted and cDNA was synthesized for q-PCR analysis to measure ET-1 expression. * p<0.05 compared to untreated control cells. (B) IGF-1 siRNA (50 nM) was introduced into PAEC for 48 h. RNA was extracted and subjected to cDNA synthesis and q-PCR analysis to measure IGF-1 and ET-1 expression. * p<0.05 compared to negative control (NC). HPASMCs were serum-starved overnight and exposed to 100 ng/ml of IGF-1 for 24 h. RNA was extracted and cDNA was synthesized for q-PCR analysis to measure mRNA expression for the following genes: (C) EDNRA; (D) EDNRB; (E) IGF-1R.
To determine whether IGF-1 regulates the expression of IGF-1 receptors or ET-1 receptors in HPASMC, HPASMC were treated with IGF-1 for 24h, and total RNA was isolated and subjected to cDNA synthesis and quantitative PCR. As shown in Figures 3C, D and E, IGF-1 treatment does not affect the mRNA expression of EDNRA, EDNRB or IGF-1R in HPASMCs.
3.4. IGF-1 expression and signaling are regulated by HDACs
To determine whether the hypoxia-induced increase in IGF-1 is modulated by HDACs, HPAECs were exposed to hypoxia for two days in the presence or absence of the class I HDAC inhibitor apicidin. As shown in Figure 4A, hypoxia increases IGF-1 expression. Apicidin treatment decreases the elevated levels of IGF-1 expression induced by hypoxia. In addition, apicidin treatment decreases the basal levels of IGF-1 expression in normoxia.
Figure 4. HDAC inhibition with apicidin regulates IGF-1 expression and IGF-1 signaling.

(A) Human PAECs were cultured in normoxia or hypoxia (1% oxygen) for 2 days in the presence or absence of apicidin. Total RNA was isolated and cDNA was synthesized. q-PCR was performed to measure the expression levels of IGF-1. * p<0.05 compared to vehicle controls. # p<0.05 compared to normoxia controls. (B) Apicidin administration attenuates hypoxia-induced increased expression of IGF-1 expression in mouse lungs. (C) Apicidin administration attenuates hypoxia-induced activation of pAKT in mouse lungs. (D) The mRNA expression levels of IGF-1R was examined by real-time PCR in lung tissues from mice exposed to normoxia or hypoxia. E) The mRNA expression levels of EDNRA was examined by real-time PCR in lung tissues from mice exposed to normoxia or hypoxia. (F) The mRNA expression levels of EDNRB was examined by real-time PCR in lung tissues from mice exposed to normoxia or hypoxia.
To determine whether pulmonary IGF-1 signaling is involved in neonatal hypoxia-induced PH, the IGF-1 mRNA levels were measured in mouse lungs from three experimental groups. As shown in Figure 4B, apicidin treatment reduces the hypoxia-induced increase in IGF-1 expression in lungs. Moreover, apicidin administration attenuates hypoxia-induced elevated levels of pAKTS473 in lungs (Figure C).
To determine the involvement of IGF1R, EDNRA, and EDNRB in hypoxia-induced pulmonary hypertension, expression levels of these genes were measured by q-PCR in the lungs. As shown in Figures 4D, 4E and 4F, neonatal hypoxia decreases the expression levels of EDNRB, but not IGF1R and EDNRA. In addition, apicidin administration does not alter the expression levels of EDNRA, ENNRB, and IGF1R under hypoxic conditions.
To determine which HDACs may be involved in IGF-1 regulation, siRNA knockdown experiments were performed. As shown in Figures 5A and 5B, reduction of HDAC1, HDAC2, and HDAC3 levels by siRNA knockdown individually elicited no change in IGF-1 expression. However, a combination knockdown of HDACs decreases IGF-1 expression in HPAEC.
Figure 5. HDACs regulate IGF-1 expression in PASMCs.

(A) PASMCs were transfected individually with HDAC1, HDAC2, and HDAC3 siRNA (50 nM), or in combination for 2 days. q-PCR was performed to measure the mRNA expression of HDAC1, HDAC2 and HDAC3. * p<0.05 compared to negative control. (B) q-PCR was performed to determine the expression level of IGF-1 in HDAC1-, HDAC2- and HDAC3- siRNA-treated PAECs. Combination, but not individual, knock-down of HDAC1, HDAC2, and HDAC3 decreases expression of IGF-1. * p<0.05 compared to negative control.
3.5. Inhibition of HDACs decreases chronic hypoxia-induced right heart hypertrophy and pulmonary vascular remodeling
To investigate whether inhibition of HDACs prevents chronic hypoxia-induced PH in neonates, we used a neonatal mouse model of hypoxia-induced PH (Figure 6A). Apicidin attenuates chronic hypoxia-induced RV hypertrophy (Figure 6B). To evaluate pulmonary vascular remodeling, the medial thickness of the pulmonary arterial walls was measured. As shown in Figures 6C and 6D, two weeks of neonatal hypoxia causes significant increases in the thickness of the pulmonary vascular walls as compared to normoxia (p<0.05). However, administration of apicidin attenuates neonatal hypoxia-induced pulmonary wall thickening (p<0.05). Our data suggests that inhibition of HDACs with apicidin provides a beneficial effect on the pathogenesis of PH induced by hypoxia.
Figure 6. Inhibition of HDACs with apicidin suppresses neonatal hypoxia-induced pulmonary hypertension.

(A) Schematic of the study design. (B) C57BL/6 mouse pups were exposed to normoxia or 11% hypoxia from birth for two weeks and administered either apicidin (2 mg/kg, i.p.) or vehicle control three days per week from P5. RV hypertrophy was assessed at P14. (C) Ratio of right ventricular weight to body weight was measured at P14. (D) Representative photomicrographs of pulmonary arteries at P14 in mouse pups exposed to normoxia or hypoxia and given either vehicle or apicidin. Scale bar = 50 μm. NV=normoxia vehicle HV=hypoxia vehicle HT=hypoxia apicidin-treated (E) Quantitative analysis of wall thickness of pulmonary arteries at P14 of mice in normoxia or hypoxia and given either vehicle or apicidin. * p<0.05 compared with normoxia vehicle. # p<0.05 compared with hypoxia vehicle group.
3.6. Neonatal hypoxia modulates lung DNA methylation in the IGF-1 promoter region
To determine whether neonatal hypoxia exposure modulates DNA methylation of the mIGF-1 promoter, we employed the state-of–the-art technology, targeted NGS, and characterized the novel differential cytosine methylation sites in the IGF-1 promoter region in the normoxia and hypoxia groups. Figure 7A shows the location of the examined CpG sites around the mIGF-1 promoter region. As shown in Figure 7B, four out of five CpG sites (mouse Chr 10, 87858009, 87858096, 87858038, 87858170) around the mIGF-1 promoter region show no differences in cytosine methylation levels between the normoxia and hypoxia groups. However, cytosine methylation at position 87857993 (mouse Chr10, CpG1 site) is significantly increased in the lungs of mice exposed to hypoxia as compared to normoxia controls. Interestingly, apicidin treatment normalizes the increased levels of cytosine methylation due to hypoxia at CpG1 site (Figure 7C).
Figure 7. Chronic neonatal hypoxia modulates lung DNA methylation in IGF-1 promoter region.

(A) Location of CpG sites along proximal mouse IGF-1 promoter region. (B) Genomic DNA isolated from normoxic and hypoxic lungs was analyzed for cytosine methylation at multiple sites along the mIGF-1 promoter region in the lungs. * p<0.05 compared to normoxia control. (C) HDAC inhibition with apicidin normalizes hypoxia-induced elevation of cytosine methylation at CpG1 site (position 87857993) along pulmonary IGF-1 promoter region (Chr10). * p<0.05 compared to normoxia controls. # p<0.05 compared to hypoxia vehicle controls.
3.7. HDAC inhibition with apicidin alters global DNA methylation levels in mouse lung
We tested whether neonatal hypoxia alters global DNA methylation in mouse lungs. LC/MS data showed that neonatal hypoxia increases global cytosine methylation, which is associated with increased cytosine methylation levels in the mIGF-1 promoter region (Figure 8). Moreover, apicidin treatment in-vivo decreases hypoxia-induced elevated levels of global DNA methylation.
Figure 8. Inhibition of HDACs with apicidin attenuates hypoxia-induced global DNA methylation in vivo.

Global DNA methylation was determined by LC/MS. Each genomic DNA sample was analyzed in triplicate. * p<0.05 compared to normoxia controls. # p<0.05 compared to hypoxia vehicle controls.
4. Discussion
There is increasing evidence in humans and experimental animals that adverse stimuli, especially hypoxia, can impair lung growth and function during a critical period of lung development in a variety of animal species [1; 23]. In addition, early exposure to hypoxia alters the pulmonary vasculature and increases the susceptibility to development of pulmonary hypertension later in life. In neonatal rats, brief perinatal hypoxia followed by reexposure to hypoxia increases the severity of PH, including increased RVSP and RV hypertrophy [24]. Although a number of signaling pathways have been reported to be involved in chronic hypoxia-induced PH in neonatal animals, the role of IGF-1 in neonatal chronic hypoxia-induced PH is largely unknown. It has been reported that IGF-1 signaling is activated in many types of cancer cells [25; 26; 27]. In this study, we demonstrate that the pAKT pathway is activated in response to IGF-1 exposure in both PAECs and PASMCs. pAKTS473, but not pAKTT308, was remarkably increased in the presence of IGF-1. Inhibitors of IGF-1R, AKT, and PI3K strongly suppress pAKTT473 activation in response to IGF-1 stimulation, suggesting that IGF-1 induces elevated activation of pAKT473 through IGF-1R and PI3K in PASMCs. Our in vivo data demonstrate that expression of IGF-1 is upregulated in the lungs of neonatal mice after hypoxia exposure which is paralleled with elevated activation of pAKT473. In addition, the expression levels of IGF1R are not altered under hypoxia condition as compared to normoxia condition. These data suggested that IGF-1 as a ligand triggers the IGF-1 signaling without altering the expression of IGF-1 receptor under hypoxia condition.
A number of recent studies demonstrate the importance of the epigenetic code in normal human development as well as the burden of disease that occurs when the epigenetic code or machinery malfunctions. One of the interesting findings in this study is that IGF-1 expression is modulated by histone modifications, and inhibition of HDACs attenuates the chronic hypoxia-induced RVSP, RV hypertrophy, and vascular remodeling in neonatal mice. Importantly, apicidin administration also attenuates hypoxia-induced IGF-1 expression and elevated activation of pAKT473. The downregulation of IGF-1 in neonatal lungs by apicidin is consistent with our in vitro data showing that apicidin treatment decreases IGF-1 expression in HPASMCs. HDACs have emerged as key targets to reverse aberrant epigenetic alterations associated with many diseases including PH [10; 11]. HDACs may modulate intracellular signaling cascades through their interaction with protein phosphates.
Our study suggests that class I HDACs play a role in IGF-1/AKT signaling pathway activation. Our study is in line with others that show growth factor-mediated signaling pathways are epigenetically regulated [28]. Zinkhan et al. have demonstrated that maternal hyperglycemia disrupts histone 3 lysine 36 trimethylation of the IGF-1 gene. Maternal food restriction results in intrauterine growth-restricted newborns with significantly lower liver weight in association with altered histone codes along the rat hepatic IGF-1 gene [29; 30]. Beside IGF-1, other growth factor-related pathways have been reported to be regulated epigenetically. Nordin et al. have reported that the IGF-2/H19 gene-cluster is epigenetically regulated leading to parent-specific expression [31]. In addition, the expression of vascular endothelial growth factor receptor-3 has been linked to epigenetic regulation. TSA (an HDAC inhibitor) treatment leads to the accumulation of acetylated histones H3/H4 at the VEGFR3 gene promoter, upregulation of its mRNA levels, and transactivation of promoter reporter constructs in endothelial cells. Moreover, methylation inhibition by 5-Aza-dC triggers upregulation of VEGF3 mRNA levels and increased promoter activity [32]. The evidence in our study along with others show that increased IGF-1 signaling is positively correlated with increased HDAC activity in response to hypoxia. Our study further demonstrates that inhibition of HDACs with apicidin decreases IGF-1 expression both in vitro and in vivo, suggesting that IGF-1 may be a target of HDACs in our experimental mouse model and that increased HDAC activity contributes to the vascular pathology of PH. We performed siRNA experiments to ascertain which HDAC members play an important role in regulating IGF-1 expression. Individual knockdown exhibits no change of IGF-1 expression. This is likely due to dose or compensation effects, which is consistent with our finding that a combination knockdown showed a decrease in IGF-1 expression. However, it remains to be further investigated how IGF-1 is involved in the pathogenesis of hypoxic PH.
The observed benefits of apicidin treatment in this study on RVSP, RV hypertrophy, and vascular remodeling in response to neonatal hypoxia are partial, which can probably be attributed to incomplete drug efficacy and multiple signaling pathways implicated in the pathogenesis of hypoxic PH. PASMCs proliferate in response to hypoxia, which involves other growth factors such as PDGF, FGF, and EGF, suggesting that multiple growth factors that activate their receptors and downstream signaling are involved [33; 34; 35]. Although reduced signaling through IGF-1R with apicidin treatment in hypoxia is observed in this study, the other pathways triggered by PDGF, FGF, and EGF were not examined and need further investigation.
ET-1 has been described to have an important role in vascular hypertrophy and proliferation leading to hypertension [36; 37]. ET-1 is a potent vasoconstrictor peptide that also exhibits mitogenic and hypertrophic properties. In addition, ET-1 has been shown to stimulate vascular smooth muscle cell proliferation, migration, contraction, extracellular matrix remodeling and deposition, and the secretion of growth factors [38]. EDNRA encodes the ET-1 receptor termed ETA receptor, which is a member of the endothelin receptor group of G-protein-coupled receptors that also includes ETB receptor. ET-1 binds to ETA-R and ETB-R on PASMC, which leads to vasoconstriction, proliferation and migration of the PASMCs [39; 40; 41]. However, the mechanism underlying increased ET-1 expression in hypoxia is largely unknown. Olave et al. have previously demonstrated that inhibition of TGF-β signaling reduces ET-1 in vivo, which is confirmed in vitro in mouse pulmonary endothelial cells. Reduction of ET-1 via inhibition of TGF-β signaling indicates that TGF-β is upstream of ET-1 during hypoxia-induced signaling in the newborn lung [23]. In this study, we demonstrate that neonatal hypoxia decreases the expression of EDNRB but not EDNRA. However apicidin administration did not alter the expression of either EDNRA or EDNRB. Moreover, we demonstrate a novel finding that IGF-1 regulates ET-1, which was exhibited in IGF-1-treated PAECs. However, using an siRNA knockdown experiment, ET-1 expression is not altered in IGF-1-diminished PAECs. Therefore, further investigation is required to elucidate the mechanisms underlying the regulation of ET-1 by IGF-1.
Generation of an epigenetic signature by chronic hypoxia has been reported in various types of cells and tissues, including prostate [42] and lung [43]. In this study, we demonstrate that neonatal hypoxia increases gene-specific cytosine methylation levels along the IGF-1 promoter region, which is associated with increased global DNA methylation levels in mouse lungs after exposure to hypoxia. In addition, we demonstrate a global hypermethylation in the lungs that is reversible after administration of apicidin in neonatal hypoxic lungs. Although we have characterized differentially methylated hotspots induced by hypoxia in our experimental model, the distal region and regulatory elements around the IGF-1 promoter need to be extensively investigated. Furthermore, it should be emphasized that these studies were performed using a preclinical animal model, and characterization of differential epigenetic signaling in patients with PH will provide more clinically relevant data to increase our understanding of the role of epigenetics in the pathogenesis of PH. Our studies suggest that epigenetic changes may be used as a potential signature of response to early insults such as neonatal hypoxia.
5. Conclusion
In conclusion, our findings indicate that IGF-1 signaling is involved in chronic hypoxia-induced pathogenesis of PH in neonatal mice. IGF-1 signaling is activated in the lungs of neonatal mice exposed to hypoxia, and inhibition of HDACs with apicidin is capable of decreasing chronic hypoxia-induced IGF-1 expression and activation of AKT in vivo. We show that the expression of the potent vasoconstrictor ET-1 is regulated by IGF-1 in HPAECs, suggesting that IGF-1 may modulate pulmonary arterial vascular tone. Furthermore, neonatal hypoxia alters specific cytosine methylation around the IGF-1 promoter region, and inhibition of HDACs normalizes not only global DNA methylation, but also a specific methylation site in the mIGF-1 promoter region. Our study may provide new insights for the prevention and treatment of hypoxia-related PH.
Acknowledgments
We are grateful to Greg Weypa, Northwestern Memorial Hospital, IL for assistance with mPASMC isolation protocol. This work was supported in part by National Institutes of Health grants HL075187, HL059435 and HL110829 (to J.U.R.)
Footnotes
Author Disclosure: None of the authors have a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jones RD, Morice AH, Emery CJ. Effects of perinatal exposure to hypoxia upon the pulmonary circulation of the adult rat. Physiological research / Academia Scientiarum Bohemoslovaca. 2004;53:11–7. [PubMed] [Google Scholar]
- 2.Yang Q, Sun M. Perinatal and neonatal hypoxia in pulmonary vascular dysfunction. Cardiol pharmacol. 2013;3:e123. [Google Scholar]
- 3.Yee D. Insulin-like growth factor receptor inhibitors: baby or the bathwater? Journal of the National Cancer Institute. 2012;104:975–81. doi: 10.1093/jnci/djs258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arcaro A. Targeting the insulin-like growth factor-1 receptor in human cancer. Frontiers in pharmacology. 2013;4:30. doi: 10.3389/fphar.2013.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vincent EE, Elder DJ, Curwen J, Kilgour E, Hers I, Tavare JM. Targeting non-small cell lung cancer cells by dual inhibition of the insulin receptor and the insulin-like growth factor-1 receptor. PloS one. 2013;8:e66963. doi: 10.1371/journal.pone.0066963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang Y, Yee D. Targeting insulin and insulin-like growth factor signaling in breast cancer. Journal of mammary gland biology and neoplasia. 2012;17:251–61. doi: 10.1007/s10911-012-9268-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Limesand KH, Chibly AM, Fribley A. Impact of targeting insulin-like growth factor signaling in head and neck cancers. Growth hormone & IGF research: official journal of the Growth Hormone Research Society and the International IGF Research Society. 2013;23:135–40. doi: 10.1016/j.ghir.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang QW, Liu S, Tian Y, Salwen HR, Chlenski A, Weinstein J, Cohn SL. Methylation-associated silencing of the thrombospondin-1 gene in human neuroblastoma. Cancer research. 2003;63:6299–310. [PubMed] [Google Scholar]
- 9.Yang Q, Tian Y, Liu S, Zeine R, Chlenski A, Salwen HR, Henkin J, Cohn SL. Thrombospondin-1 peptide ABT-510 combined with valproic acid is an effective antiangiogenesis strategy in neuroblastoma. Cancer research. 2007;67:1716–24. doi: 10.1158/0008-5472.CAN-06-2595. [DOI] [PubMed] [Google Scholar]
- 10.Cavasin MA, Demos-Davies K, Horn TR, Walker LA, Lemon DD, Birdsey N, Weiser-Evans MC, Harral J, Irwin DC, Anwar A, Yeager ME, Li M, Watson PA, Nemenoff RA, Buttrick PM, Stenmark KR, McKinsey TA. Selective class I histone deacetylase inhibition suppresses hypoxia-induced cardiopulmonary remodeling through an antiproliferative mechanism. Circulation research. 2012;110:739–48. doi: 10.1161/CIRCRESAHA.111.258426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao L, Chen CN, Hajji N, Oliver E, Cotroneo E, Wharton J, Wang D, Li M, McKinsey TA, Stenmark KR, Wilkins MR. Histone deacetylation inhibition in pulmonary hypertension: therapeutic potential of valproic acid and suberoylanilide hydroxamic acid. Circulation. 2012;126:455–67. doi: 10.1161/CIRCULATIONAHA.112.103176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ambalavanan N, Nicola T, Hagood J, Bulger A, Serra R, Murphy-Ullrich J, Oparil S, Chen YF. Transforming growth factor-beta signaling mediates hypoxia-induced pulmonary arterial remodeling and inhibition of alveolar development in newborn mouse lung. American journal of physiology. Lung cellular and molecular physiology. 2008;295:L86–95. doi: 10.1152/ajplung.00534.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nicola T, Ambalavanan N, Zhang W, James ML, Rehan V, Halloran B, Olave N, Bulger A, Oparil S, Chen YF. Hypoxia-induced inhibition of lung development is attenuated by the peroxisome proliferator-activated receptor-gamma agonist rosiglitazone. American journal of physiology. Lung cellular and molecular physiology. 2011;301:L125–34. doi: 10.1152/ajplung.00074.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bierer R, Nitta CH, Friedman J, Codianni S, de Frutos S, Dominguez-Bautista JA, Howard TA, Resta TC, Bosc LV. NFATc3 is required for chronic hypoxia-induced pulmonary hypertension in adult and neonatal mice. American journal of physiology. Lung cellular and molecular physiology. 2011;301:L872–80. doi: 10.1152/ajplung.00405.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Young KC, Torres E, Hatzistergos KE, Hehre D, Suguihara C, Hare JM. Inhibition of the SDF-1/CXCR4 axis attenuates neonatal hypoxia-induced pulmonary hypertension. Circulation research. 2009;104:1293–301. doi: 10.1161/CIRCRESAHA.109.197533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sartina E, Suguihara C, Ramchandran S, Nwajei P, Rodriguez M, Torres E, Hehre D, Devia C, Walters MJ, Penfold ME, Young KC. Antagonism of CXCR7 attenuates chronic hypoxia-induced pulmonary hypertension. Pediatr Res. 2012;71:682–8. doi: 10.1038/pr.2012.30. [DOI] [PubMed] [Google Scholar]
- 17.Blood AB, Terry MH, Merritt TA, Papamatheakis DG, Blood Q, Ross JM, Power GG, Longo LD, Wilson SM. Effect of chronic perinatal hypoxia on the role of rho-kinase in pulmonary artery contraction in newborn lambs. American journal of physiology. Regulatory, integrative and comparative physiology. 2013;304:R136–46. doi: 10.1152/ajpregu.00126.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dumitrascu R, Weissmann N, Ghofrani HA, Dony E, Beuerlein K, Schmidt H, Stasch JP, Gnoth MJ, Seeger W, Grimminger F, Schermuly RT. Activation of soluble guanylate cyclase reverses experimental pulmonary hypertension and vascular remodeling. Circulation. 2006;113:286–95. doi: 10.1161/CIRCULATIONAHA.105.581405. [DOI] [PubMed] [Google Scholar]
- 19.Peyter AC, Muehlethaler V, Liaudet L, Marino M, Di Bernardo S, Diaceri G, Tolsa JF. Muscarinic receptor M1 and phosphodiesterase 1 are key determinants in pulmonary vascular dysfunction following perinatal hypoxia in mice. American journal of physiology. Lung cellular and molecular physiology. 2008;295:L201–13. doi: 10.1152/ajplung.00264.2007. [DOI] [PubMed] [Google Scholar]
- 20.Chi AY, Waypa GB, Mungai PT, Schumacker PT. Prolonged hypoxia increases ROS signaling and RhoA activation in pulmonary artery smooth muscle and endothelial cells. Antioxidants & redox signaling. 2010;12:603–10. doi: 10.1089/ars.2009.2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson BA, Lowenstein CJ, Schwarz MA, Nakayama DK, Pitt BR, Davies P. Culture of pulmonary microvascular smooth muscle cells from intraacinar arteries of the rat: characterization and inducible production of nitric oxide. American journal of respiratory cell and molecular biology. 1994;10:604–12. doi: 10.1165/ajrcmb.10.6.7516171. [DOI] [PubMed] [Google Scholar]
- 22.Shah MY, Vasanthakumar A, Barnes NY, Figueroa ME, Kamp A, Hendrick C, Ostler KR, Davis EM, Lin S, Anastasi J, Le Beau MM, Moskowitz IP, Melnick A, Pytel P, Godley LA. DNMT3B7, a truncated DNMT3B isoform expressed in human tumors, disrupts embryonic development and accelerates lymphomagenesis. Cancer research. 2010;70:5840–50. doi: 10.1158/0008-5472.CAN-10-0847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olave N, Nicola T, Zhang W, Bulger A, James M, Oparil S, Chen YF, Ambalavanan N. Transforming growth factor-beta regulates endothelin-1 signaling in the newborn mouse lung during hypoxia exposure. American journal of physiology. Lung cellular and molecular physiology. 2012;302:L857–65. doi: 10.1152/ajplung.00258.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang JR, Le Cras TD, Morris KG, Jr, Abman SH. Brief perinatal hypoxia increases severity of pulmonary hypertension after reexposure to hypoxia in infant rats. American journal of physiology. Lung cellular and molecular physiology. 2000;278:L356–64. doi: 10.1152/ajplung.2000.278.2.L356. [DOI] [PubMed] [Google Scholar]
- 25.Mulvihill MJ, Cooke A, Rosenfeld-Franklin M, Buck E, Foreman K, Landfair D, O’Connor M, Pirritt C, Sun Y, Yao Y, Arnold LD, Gibson NW, Ji QS. Discovery of OSI-906: a selective and orally efficacious dual inhibitor of the IGF-1 receptor and insulin receptor. Future medicinal chemistry. 2009;1:1153–71. doi: 10.4155/fmc.09.89. [DOI] [PubMed] [Google Scholar]
- 26.Kim JS, Kim ES, Liu D, Lee JJ, Solis L, Behrens C, Lippman SM, Hong WK, Wistuba, Lee HY. Prognostic implications of tumoral expression of insulin like growth factors 1 and 2 in patients with non-small-cell lung cancer. Clinical lung cancer. 2014;15:213–21. doi: 10.1016/j.cllc.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hirakawa T, Yashiro M, Murata A, Hirata K, Kimura K, Amano R, Yamada N, Nakata B, Hirakawa K. IGF-1 receptor and IGF binding protein-3 might predict prognosis of patients with resectable pancreatic cancer. BMC cancer. 2013;13:392. doi: 10.1186/1471-2407-13-392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zinkhan EK, Fu Q, Wang Y, Yu X, Callaway CW, Segar JL, Scholz TD, McKnight RA, Joss-Moore L, Lane RH. Maternal Hyperglycemia Disrupts Histone 3 Lysine 36 Trimethylation of the IGF-1 Gene. Journal of nutrition and metabolism. 2012;2012:930364. doi: 10.1155/2012/930364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tosh DN, Fu Q, Callaway CW, McKnight RA, McMillen IC, Ross MG, Lane RH, Desai M. Epigenetics of programmed obesity: alteration in IUGR rat hepatic IGF1 mRNA expression and histone structure in rapid vs. delayed postnatal catch-up growth. American journal of physiology. Gastrointestinal and liver physiology. 2010;299:G1023–9. doi: 10.1152/ajpgi.00052.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fu Q, Yu X, Callaway CW, Lane RH, McKnight RA. Epigenetics: intrauterine growth retardation (IUGR) modifies the histone code along the rat hepatic IGF-1 gene. The FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2009;23:2438–49. doi: 10.1096/fj.08-124768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nordin M, Bergman D, Halje M, Engstrom W, Ward A. Epigenetic regulation of the Igf2/H19 gene cluster. Cell proliferation. 2014;47:189–99. doi: 10.1111/cpr.12106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hertel J, Hirche C, Wissmann C, Ebert MP, Hocker M. Transcription of the vascular endothelial growth factor receptor-3 (VEGFR3) gene is regulated by the zinc finger proteins Sp1 and Sp3 and is under epigenetic control: transcription of vascular endothelial growth factor receptor 3. Cell Oncol (Dordr) 2014;37:131–45. doi: 10.1007/s13402-014-0169-5. [DOI] [PubMed] [Google Scholar]
- 33.Dahal BK, Cornitescu T, Tretyn A, Pullamsetti SS, Kosanovic D, Dumitrascu R, Ghofrani HA, Weissmann N, Voswinckel R, Banat GA, Seeger W, Grimminger F, Schermuly RT. Role of epidermal growth factor inhibition in experimental pulmonary hypertension. American journal of respiratory and critical care medicine. 2010;181:158–67. doi: 10.1164/rccm.200811-1682OC. [DOI] [PubMed] [Google Scholar]
- 34.Chanakira A, Dutta R, Charboneau R, Barke R, Santilli SM, Roy S. Hypoxia differentially regulates arterial and venous smooth muscle cell proliferation via PDGFR-beta and VEGFR-2 expression. American journal of physiology. Heart and circulatory physiology. 2012;302:H1173–84. doi: 10.1152/ajpheart.00411.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grimminger F, Schermuly RT. PDGF receptor and its antagonists: role in treatment of PAH. Advances in experimental medicine and biology. 2010;661:435–46. doi: 10.1007/978-1-60761-500-2_28. [DOI] [PubMed] [Google Scholar]
- 36.Schiffrin EL. Endothelin: potential role in hypertension and vascular hypertrophy. Hypertension. 1995;25:1135–43. doi: 10.1161/01.hyp.25.6.1135. [DOI] [PubMed] [Google Scholar]
- 37.Ambalavanan N, Bulger A, Murphy-Ullrich J, Oparil S, Chen YF. Endothelin-A receptor blockade prevents and partially reverses neonatal hypoxic pulmonary vascular remodeling. Pediatr Res. 2005;57:631–6. doi: 10.1203/01.PDR.0000159512.55862.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kapakos G, Bouallegue A, Daou GB, Srivastava AK. Modulatory Role of Nitric Oxide/cGMP System in Endothelin-1-Induced Signaling Responses in Vascular Smooth Muscle Cells. Current cardiology reviews. 2010;6:247–54. doi: 10.2174/157340310793566055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.MacLean MR, McCulloch KM, Baird M. Endothelin ETA- and ETB-receptor-mediated vasoconstriction in rat pulmonary arteries and arterioles. Journal of cardiovascular pharmacology. 1994;23:838–45. doi: 10.1097/00005344-199405000-00022. [DOI] [PubMed] [Google Scholar]
- 40.Ivy D, McMurtry IF, Yanagisawa M, Gariepy CE, Le Cras TD, Gebb SA, Morris KG, Wiseman RC, Abman SH. Endothelin B receptor deficiency potentiates ET-1 and hypoxic pulmonary vasoconstriction. American journal of physiology. Lung cellular and molecular physiology. 2001;280:L1040–8. doi: 10.1152/ajplung.2001.280.5.L1040. [DOI] [PubMed] [Google Scholar]
- 41.Sun M, Yang Q. Neonatal hyperoxia and pulmonary hypertension. Pediatri neonatal nurs open J. 2014;1:1–5. [Google Scholar]
- 42.Watson JA, Watson CJ, McCrohan AM, Woodfine K, Tosetto M, McDaid J, Gallagher E, Betts D, Baugh J, O’Sullivan J, Murrell A, Watson RW, McCann A. Generation of an epigenetic signature by chronic hypoxia in prostate cells. Human molecular genetics. 2009;18:3594–604. doi: 10.1093/hmg/ddp307. [DOI] [PubMed] [Google Scholar]
- 43.Yang Q, Lu Z, Ramchandran R, Longo LD, Raj JU. Pulmonary artery smooth muscle cell proliferation and migration in fetal lambs acclimatized to high-altitude long-term hypoxia: role of histone acetylation. American journal of physiology. Lung cellular and molecular physiology. 2012;303:L1001–10. doi: 10.1152/ajplung.00092.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]