

Abstract
Background
Metformin has received considerable attention as a potential anti-cancer agent. Animal and in-vitro prostate cancer (PCa) models have demonstrated decreased tumor growth with metformin, however the precise mechanisms are unknown. We examine the effects of metformin on PCa biochemical recurrence (BCR) in a large clinical database followed by evaluating metabolic signaling changes in a cohort of men undergoing prostate needle biopsy (PNB).
Methods
Men treated for localized PCa were identified in a comprehensive clinical database between 2001 and 2010. Cox regression was performed to determine association with BCR relative to metformin use. We next identified a separate case-control cohort of men undergoing prostate needle biopsy (PNB) stratified by metformin use. Differences in mean IHC scores were compared with linear regression for phosphorylated IR, IGF-IR, AKT, and AMPK.
Results
1,734 men were evaluated for BCR with mean follow up of 41 months (range 1-121 months). ‘Ever’ metformin use was not associated with BCR (HR 1.12, 0.77-1.65), however men reporting both pre/post-treatment metformin use had a 45% reduction in BCR (HR=0.55 (0.31-0.96)). For the tissue-based study, 48 metformin users and 42 controls underwent PNB. Significantly greater staining in phosphorylated nuclear (p-IR, p-AKT) and cytoplasmic (p-IR, p-IGF-1R) insulin signaling proteins were seen in patients with PCa detected compared to those with negative PNB (p-values all < 0.006). When stratified by metformin use, IGF-1R remained significantly elevated (p=0.01) in men with PCa detected whereas p-AMPK (p=0.05) was elevated only in those without PCa.
Conclusion
Metformin use is associated with reduced BCR after treatment of localized PCa when considering pre-diagnostic and cumulative dosing. In men with cancer detected on PNB, insulin signaling markers were significantly elevated compared to negative PNB patients. The finding of IGF-1R elevation in positive PNBs versus p-AMPK elevation in negative PNBs suggests altered metabolic pathway activation precipitated by metformin use.
Keywords: Prostate cancer, Metformin, AMPK, Insulin signaling, Veterans Affairs
Introduction
Metformin use is associated with a decrease in cancer-specific mortality.(1) There are now growing data for the role of metformin as an anti-tumor agent in prostate cancer (PCa) specifically with epidemiologic evidence showing a decreased incidence of PCa in men taking metformin(2, 3) and animal and in vitro models demonstrating activity against PCa cell lines.(4, 5) Clinical studies of metformin in PCa recurrence have been mixed (6-13) with several post-radical prostatectomy (RP) studies showing no beneficial effect with metformin use.(6-9) However, most of these studies compared ‘ever’ vs. ‘never use’ of metformin, preventing cumulative dose or threshold analyses, and in studies which evaluated dosing duration, there have been conflicting results.(6, 10, 11, 13) In addition, studies using actual human prostate tissue have been limited(14) but there are several trials both active and in recruitment (www.clinicaltrials.gov; NCT01864096 (active surveillance), NCT02176161 (adjuvant therapy), NCT01243385 (advanced disease)) suggesting metformin's potential importance in PCa research.
Anti-tumorigenic effects of metformin are proposed to act through several pathways. These include increased AMP-activated protein kinase (AMPK) activation, decreased hepatic gluconeogenesis (with secondary decrease in hyperinsulinemia), improved insulin receptor function with better glucose transport, and overall improved insulin sensitivity.(15, 16) AMPK is activated in response to cellular stress and/or starvation(17) resulting in a reduction of mTOR activation, protein synthesis and cellular proliferation.(18, 19) Metformin stimulates AMPK activation by blocking complex 1 of the mitochondrial electron transport chain resulting in an ATP/AMP imbalance, increased activity of liver kinase B1 (LKB1, a tumor suppressor and upstream regulator of AMPK), and subsequent phosphorylation of AMPK.(15, 16) In pre-clinical models using PCa cell lines, metformin has been shown to activate AMPK(4, 5) with corresponding decreases in proliferation suggesting direct cellular effects.
Hyperglycemia and hyperinsulinemia have been associated with multiple malignancies(20-22) and specifically, adverse outcomes in PCa.(23-26) Signaling through the insulin receptor (IR-isoform A)(27) and the closely related IGF-1 receptor leads to mitogenic and anti-apoptotic activity predominantly through mTOR activation.(16, 22) Metformin dosing results in decreased serum insulin levels(15, 16, 28) and indirectly lowers insulin like growth factor-1 (IGF-1) serum levels through alterations in IGF-binding proteins.(22) This may result in decreased downstream activation of these mitogenic pathways and potentially, a decrease in PCa proliferation (see Figure 1).
Figure 1. Proposed model of insulin signaling relative to cellular proliferation.
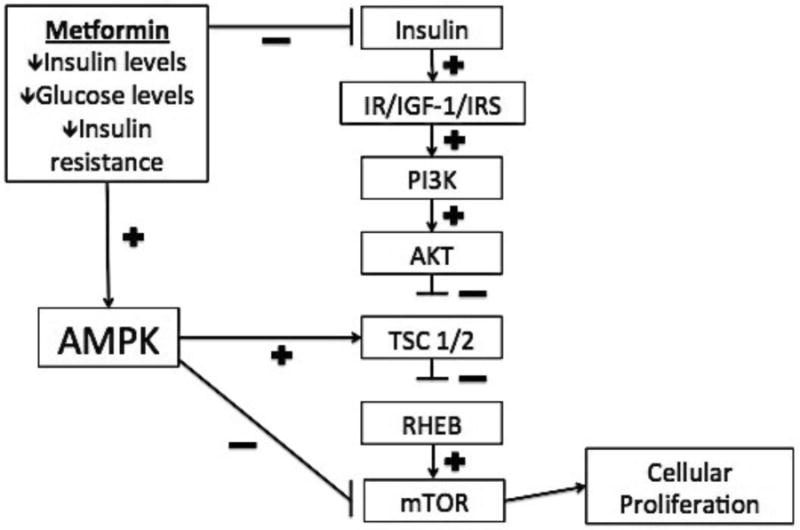
Simplified model of proposed effects of metformin and AMPK relative to insulin signaling and cellular proliferation are shown.(15, 16, 22, 45) AMPK: AMP activated protein kinase K; IR: insulin receptor; IGF-1: insulin-like growth factor 1; PI3K: phosphoinositide 3-kinase; AKT: protein kinase B; TSC1/2: tuberous sclerosis complex 1/2; RHEB: ras homologue enriched in brain; mTOR: mammalian target of rapamycin
To better define the role of metformin in PCa, we used a comprehensive computerized medical records system to determine the relationship between ‘ever’ use of metformin and BCR following treatment for PCa. We also studied whether duration and cumulative use of metformin impacts BCR. Next, to study the mechanisms underlying metformin's potential activity in PCa, we conducted a case control study in men undergoing prostate needle biopsy (PNB) to determine whether pharmacologic dosages of metformin can lead to measurable changes of metabolic signaling markers. We hypothesize that changes such as increased AMPK phosphorylation or down regulation of other metabolic markers (IR, IGF-IR and AKT) will be seen at the tissue level relative to cancer detection and metformin use.
Materials and Methods
Metformin use and PCa recurrence
Eligible men were identified from the Northwest Veterans Integrated Services Network (VISN 20) electronic medical record encompassing veterans in Washington, Oregon, Alaska, Idaho, western Montana and northern California (7 primary medical centers and 27 community-based outpatient clinics). Data were extracted from the VISN20 regional data warehouse, which obtains nearly 100% of the electronic medical record (including clinic appointments, inpatient stays, pharmacy records, laboratory values, pathology reports, imaging tests, and vital measures).(29, 30) VA IRB approval (#00399) was obtained. Men with an established primary care provider in VISN20 prior to diagnosis and had a previously assessed PSA value measured within one year of study were included to ensure capture of follow up care within the VA system. Men on active surveillance/watchful waiting and men with distant metastases at diagnosis were excluded. Men treated with primary androgen deprivation therapy (ADT) or long-term continuous ADT as part of their primary treatment were also excluded owing to the effects of ADT on glucose homeostasis.(31) Between 2001 and 2010, 1,734 men who met inclusion criteria were treated with either radical prostatectomy (RP) or radiation therapy (RT) (brachytherapy or external beam radiation therapy) for localized PCa.
Use of metformin was determined from the electronic pharmacy records. Data collected included dosage, quantity, and date prescription was filled. Additionally, the diagnosis of type 2 diabetes mellitus (DM) was collected from ICD9 diagnosis codes. Use of metformin was evaluated several different ways: ever vs. never use; cumulative dose; and cumulative duration of metformin use with men placed into quartiles based on these cumulative usages. In order to differentiate the potential effect of DM on BCR from metformin usage, we categorized the remaining men into (1) those who did not have the diagnosis of DM and were not taking metformin, and (2) those who had DM but were not taking metformin.
Serum glucose levels were also available on all patients with the glucose level closest to the date of diagnosis recorded. A glucose level was considered normal if < 100 mg/dL, according to the American Diabetes Association (ADA) classification. Demographic, clinical and tumor characteristics were also collected, including age, race, year of diagnosis, body mass index (BMI), Charlson Comorbidity Index(32),(33) Gleason score, clinical stage (all patients) and pathologic stage (available for RP patients, based on TNM classification from American Joint Committee on Cancer (AJCC, Chicago, Illinois), 7th edition.).
Biochemical recurrence (BCR) following RP was defined as any prostatic specific antigen (PSA) ≥ 0.2 ng/mL at 6 months or more after surgery in men with an undetectable PSA post-RP. For men treated with RT, the Phoenix criterion was used to define recurrence (nadir PSA + 2.0 ng/mL). Salvage therapy, in the form or RT, RP or ADT, was also considered evidence of PCa recurrence. Salvage RT was defined as pelvic RT received > 1 year after primary treatment date. Patients were considered lost to follow-up and censored at December 31st in the year of their last visit if no VA utilization in the subsequent calendar year occurred.
Multivariate Cox proportional hazards regression was performed in each group adjusting for all covariates (age, race, BMI, Charlson comorbidity index, DM, glucose level, year of diagnosis, treatment type, Gleason score, PSA and tumor stage). The proportional hazards assumption was assessed with Schoenfeld residuals. In order to account for the potential of immortal time bias (34) influencing the results, we performed additional analyses limiting the cohort to those who either never took metformin or those who were taking metformin at the time of treatment and beyond. All statistical analyses were conducted using STATA software, Version 13 (Stata, Inc., College Station, TX).
Tissue based study
The study population comes from a clinical and tissue database from the Seattle-Puget Sound Veteran's Affairs Medical Center within the VISN20 network (PI, Daniel W Lin, VA IRB # 01423) of men undergoing prostate needle biopsy for the suspicion of PCa. (IRB # 7595). We retrospectively identified men from this database who were continuously taking metformin (≥ 500 mg daily) for at least 6 months prior to the biopsy. Controls were then identified from the same database of men undergoing biopsy and not taking metformin. Exclusion criteria for potential controls included previous metformin use, current insulin use or a serum creatinine of > 1.5 mg/dL (due to relative contraindication of metformin in renal insufficiency). Cases and controls were matched 1:1 based on age (10-year age groups), race (Caucasian vs. other), body mass index (BMI, normal, overweight, obese) and whether PCa was present in the biopsy. A computerized, random selection algorithm performed matching of cases to controls.
Tissue blocks, previously fixed in formalin and embedded in paraffin, were cut and sections mounted on charged slides. Sections were rehydrated and incubated with 3% H2O2, blocked with avidin/biotin blocking solution (Vector Laboratories Inc.) and then 5% goat serum. The sections were incubated with either rabbit anti-IGF-1R (1:100; Santa Cruz Biotechnology), rabbit anti-phospho-IGF-1R (1:50; Abcam), mouse anti-insulin receptor (1:100; Santa Cruz Biotechnology), rabbit anti-phospho-insulin receptor (1:50; Abcam), rabbit anti-AMPK (1:50; Abcam), rabbit anti-phospho-AMPK (1:50; Abcam), rabbit anti-AKT (1:50; Cell Signaling), rabbit anti-phospho-AKT (1:50; Cell Signaling) or rabbit or mouse control IgG at the same concentration. The tissue was then washed and incubated with biotinylated secondary antibody (1:100; Santa Cruz Biotechnology), developed using the Vectastain ABC kit (Vector Laboratories Inc.) and stable DAB (Vector Laboratories), counterstained with hematoxylin, dehydrated, and mounted with Permount (Fisher). A clinical and research pathologist (XZ) blinded to case vs. control status scored the staining using the following method: 0 = no staining, 1 = faint/present staining, 2 = strong/intense staining. The percentage of staining cells was then estimated and a composite score calculated.
Mean IHC scores were compared between groups based on metformin exposure using linear regression adjusted for the matched variables age, race, BMI and fasting glucose. Adjustment for fasting glucose was performed due to significant differences identified between cases and controls. Analyses were performed separately for men with and without PCa detected. All statistical analyses were conducted using STATA software, Version 13 (Stata, Inc., College Station, TX).
Results
Metformin use and BCR
Demographic and clinical characteristics of the PCa treatment cohort are shown in Supplementary Table 1. Mean follow up for cohort was 41 months (range 1-121 months). The majority of men were <65 years old (56%) and Caucasian (85%). 42% of men were considered obese with a body mass index (BMI) of >30 kg/m2. Prostate specific antigen (PSA) was most commonly in the 4-9.9 ng/mL range (63%) with the majority of patients having intermediate risk disease prior to treatment. Table 1 describes metformin use relative to risk of BCR. 21% (n=366) of men used metformin while 6% (n=103) of men had DM but did not take metformin. Of those using metformin, median duration of use was > 3.5 years with the highest quartile of users reporting > 6 years of use. BCR was seen in 281/1734 (16%) overall with 64/366 (17%) in patients with ‘ever-use’ of metformin and 217/1,368 (16%) in those who never took metformin. Among those taking metformin both before and after treatment (n=143), only 11% experienced BCR. On multivariate analysis, ‘ever-use’ of metformin was not associated with the risk of PCa recurrence (HR 0.91, 95% CI 0.65-1.28). Metformin use after diagnosis was also not associated with benefit, however, in those patients taking metformin before and after diagnosis, there was a decreased risk of PCa recurrence (HR 0.55, 95% CI 0.31-0.96). Further, when the total metformin dosage or cumulative duration of dosing was considered, those in the highest quartile had a statistically significant 58% reduction in the risk of BCR compared to those who had never taken metformin (Table 1).
Table 1. Metformin Use and Adjusted Risk of Prostate Cancer (PCa) Recurrence After Primary Therapy.
| N=1734 | % | HR, 95% CI | |
|---|---|---|---|
| Any Metformin Use | |||
| No Diabetes mellitus and no metformin use | 1265 | 73 | 1.0, referent |
| Diabetes mellitus but never took metformin | 103 | 6 | 0.75, 0.42-1.32 |
| Metformin use | 366 | 21 | 0.91, 0.65-1.28 |
| Timing of Metformin Use relative to PCa treatment | |||
| No Diabetes mellitus and no metformin use | 1265 | 73 | 1.0, referent |
| Metformin user before and < 1 year after treatment | 34 | 2 | 0.74, 0.26-2.08 |
| Metformin user only after treatment | 181 | 11 | 1.12, 0.77-1.65 |
| Metformin user before and after treatment | 143 | 8 | 0.55, 0.31-0.96* |
| Days of metformin use | |||
| No Diabetes mellitus and no metformin use | 1265 | 73 | 1.0, referent |
| Days of metformin | |||
| 0-539 | 92 | 5 | 1.02, 0.59-1.74 |
| 540-1349 | 91 | 5 | 1.38, 0.85-2.24 |
| 1350-2249 | 91 | 5 | 0.94, 0.54-1.64 |
| • • • | 92 | 5 | 0.42, 0.21-0.83* |
| Cumulative dosages of metformin taken | |||
| No Diabetes mellitus and no metformin use | 1265 | 73 | 1.0, referent |
| Total Sum metformin taken (mg) | |||
| 0-315,000 | 92 | 5 | 1.16, 0.70-1.95 |
| 315,000-855,000 | 90 | 5 | 1.34, 0.82-2.20 |
| 855,000 – 1,560,000 | 93 | 5 | 0.85, 0.48-1.51 |
Tissue based study
Given the potential benefits of metformin use on BCR, we identified a small, separate cohort of patients undergoing PNB to look for measurable changes at the tissue level relative to metformin use. 48 cases and 42 matched controls with clinical and pathologic data are shown in Supplementary Table 2. The majority of the men were Caucasian (82%), obese (59%) and over the age of 65 (58%). PCa was detected in 44% overall (n=40). Median fasting glucose levels were higher in those taking metformin (138 mg/dL; IQR 107-158) compared to those not taking metformin ((102 mg/dL; IQR 94-114), p < 0.001 by Wilcoxon rank-sum test).
Table 2 reveals the differences in staining score between those with and without PCa detected on PNB with insulin signaling pathway markers highlighted in grey. Significantly greater staining in phosphorylated nuclear (p-IR, p-AKT) and cytoplasmic (p-IR, p-IGF-1R) insulin signaling proteins were seen in patients with PCa detected compared to those with negative PNB (p-values all < 0.006). Total AMPK was significantly increased in PCa patients; however, the active form (p-AMPK) was not significantly elevated in either nuclear or cytoplasmic staining.
Table 2. Mean Immunohistochemistry (IHC) Scores for Men with PCa Detected on Prostate Needle Biopsy Compared to Those Without PCa Nuclear IHC Staining.
| No Cancer | Prostate Cancer | ||||
|---|---|---|---|---|---|
| Stain+ | Mean | SD | Mean | SD | P-value* |
| IGF-1a-R | |||||
| p-IGF-IR | |||||
| IR | 4.5 | 29.8 | 9.4 | 28.8 | 0.46 |
| p-IR | 142.2 | 63.2 | 176.8 | 42.7 | 0.006 |
| AKT | 65.4 | 42.4 | 126.1 | 62.2 | < 0.001 |
| p-AKT | 79.4 | 62.8 | 134.2 | 58.8 | 0.001 |
|
| |||||
| AMPK | 171.0 | 32.4 | 189.1 | 17.6 | 0.003 |
| p-AMPK | 146.0 | 48.6 | 141.6 | 57.8 | 0.72 |
|
| |||||
| Cytoplasmic IHC Staining | |||||
|
| |||||
| No Cancer | Prostate Cancer | ||||
| Stain+ | Mean | SD | Mean | SD | P-value* |
|
| |||||
| IGF-1a-R | 140.7 | 54.2 | 174.5 | 43.1 | 0.002 |
| p-IGF-IR | 69.3 | 63.2 | 126.1 | 56.8 | < 0.001 |
| IR | 55.7 | 50.7 | 140.9 | 43.2 | < 0.001 |
| p-IR | 56.2 | 57.6 | 122.1 | 53.9 | < 0.001 |
| AKT | 163.8 | 39.9 | 197.0 | 10.4 | < 0.001 |
| p-AKT | 33.9 | 44.9 | 47.6 | 56.9 | 0.23 |
|
| |||||
| AMPK | 79.7 | 54.6 | 124.7 | 58.9 | < 0.001 |
| p-AMPK | 191.7 | 25.2 | 200.0 | 0.0 | 0.07 |
Shaded regions indicate markers of the insulin signaling cascade.
On staining, p-[marker] indicates phosphorylated form.
P-values calculated from linear regression model adjusted for age, race, BMI, and fasting glucose, significance set at 0.05. IGF-1 is only found in the cytoplasm and thus not evaluated in nuclear stains. IHC: Immunohistochemistry; PCa: prostate cancer; IR: insulin receptor; IGF-1: insulin-like growth factor 1; AKT: protein kinase B; AMPK: AMP activated protein kinase K.
Table 3 shows the mean scores, standard deviations, and adjusted p-values for each stain in men with positive PNB relative to metformin use. Cytoplasmic IGF-1R was found to be greater in those taking metformin compared to controls (p=0.01). In Table 4, the mean scores and standard deviations for each stain are provided for men with negative PNB. Higher mean scores for nuclear p-AMPK were seen in those taking metformin (p=0.05). Representative staining images are shown in Figure 2.
Table 3. Mean IHC Scores Relative to Metformin Use in Men with PCa Nuclear IHC Staining.
| No Metformin | Metformin | ||||
|---|---|---|---|---|---|
| Stain+ | Mean | SD | Mean | SD | P-value* |
| IGF-1a-R | |||||
| p-IGF-IR | |||||
| IR | 7.5 | 25.2 | 11.1 | 32.3 | 0.85 |
| p-IR | 168.5 | 52.5 | 183.5↑ | 32.9 | 0.60 |
| AKT | 129.2 | 66.4 | 124.3 | 61.4 | 0.75 |
| p-AKT | 125.3 | 70.2 | 141.4↑ | 48.4 | 0.79 |
|
| |||||
| AMPK | 185.4 | 23.1 | 192.1 | 11.2 | 0.16 |
| p-AMPK | 141.2 | 48.6 | 141.8↔ | 64.6 | 0.65 |
|
| |||||
| Cytoplasmic IHC Staining | |||||
|
| |||||
| No Metformin | Metformin | ||||
| Stain+ | Mean | SD | Mean | SD | P-value* |
|
| |||||
| IGF-1a-R | 147.3 | 56.9 | 192.2 | 15.2 | 0.01 |
| p-IGF-IR | 113.7 | 72.4 | 135.0↑ | 42.2 | 0.45 |
| IR | 142.8 | 47.8 | 139.2 | 40.0 | 0.83 |
| p-IR | 107.5 | 54.3 | 133.8↑ | 52.0 | 0.49 |
| AKT | 197.9 | 7.2 | 196.4 | 12.0 | 0.61 |
| p-AKT | 46.9 | 54.4 | 48.2↑ | 59.9 | 0.63 |
|
| |||||
| AMPK | 133.8 | 69.7 | 117.5 | 49.4 | 0.26 |
| p-AMPK | 200.0 | 0.0 | 200.0↔ | 0.0 | 0.38 |
Shaded regions indicate markers of the insulin signaling cascade. Arrows (↑↓↔) indicate direction of mean change.
On staining, p-[marker] indicates phosphorylated form.
P-values calculated from linear regression model adjusted for age, race, BMI, and fasting glucose, significance set at 0.05. IGF-1 is only found in the cytoplasm and thus not evaluated in nuclear stains. IHC: Immunohistochemistry; IR: insulin receptor; IGF-1: insulin-like growth factor 1; AKT: protein kinase B; AMPK: AMP activated protein kinase K.
Table 4. Mean IHC Scores Relative to Metformin Use in Men without PCa Nuclear IHC Staining.
| No Metformin | Metformin | ||||
|---|---|---|---|---|---|
| Stain+ | Mean | SD | Mean | SD | P-value* |
| IGF-1a-R | |||||
| p-IGF-IR | |||||
| IR | 8.8 | 41.7 | 0.0 | 0.0 | 0.32 |
| p-IR | 136.5 | 63.9 | 146.5↑ | 62.0 | 0.65 |
| AKT | 66.2 | 45.5 | 68.3 | 41.4 | 0.69 |
| p-AKT | 86.5 | 70.2 | 69.0↓ | 56.1 | 0.37 |
|
| |||||
| AMPK | 176.5 | 22.2 | 165.4 | 38.2 | 0.47 |
| p-AMPK | 138.0 | 44.2 | 149.8↑ | 53.8 | 0.05 |
|
| |||||
| Cytoplasmic IHC Staining | |||||
|
| |||||
| No Metformin | Metformin | ||||
| Stain+ | Mean | SD | Mean | SD | P-value* |
|
| |||||
| IGF-1a-R | 136.1 | 54.6 | 148.5 | 52.8 | 0.43 |
| p-IGF-IR | 82.1 | 73.1 | 75.6↓ | 65.6 | 0.51 |
| IR | 67.0 | 56.2 | 50.6 | 51.3 | 0.25 |
| p-IR | 61.1 | 55.2 | 58.7↓ | 58.5 | 0.90 |
| AKT | 165.2 | 39.2 | 166.3 | 40.2 | 0.79 |
| p-AKT | 45.0 | 49.7 | 25.6↓ | 41.6 | 0.56 |
|
| |||||
| AMPK | 77.3 | 52.8 | 83.3 | 55.1 | 0.73 |
| p-AMPK | 188.2 | 31.3 | 192.1↑ | 24.5 | 0.26 |
Shaded regions indicate markers of the insulin signaling cascade. Arrows (↑↓) indicate direction of mean change.
On staining, p-[marker] indicates phosphorylated form.
P-values calculated from linear regression model adjusted for age, race, BMI, and fasting glucose, significance set at 0.05. IGF-1 is only found in the cytoplasm and thus not evaluated in nuclear stains. IHC: Immunohistochemistry; IR: insulin receptor; IGF-1: insulin-like growth factor 1; AKT: protein kinase B; AMPK: AMP activated protein kinase K.
Figure 2. IHC staining of prostate needle biopsy specimens.
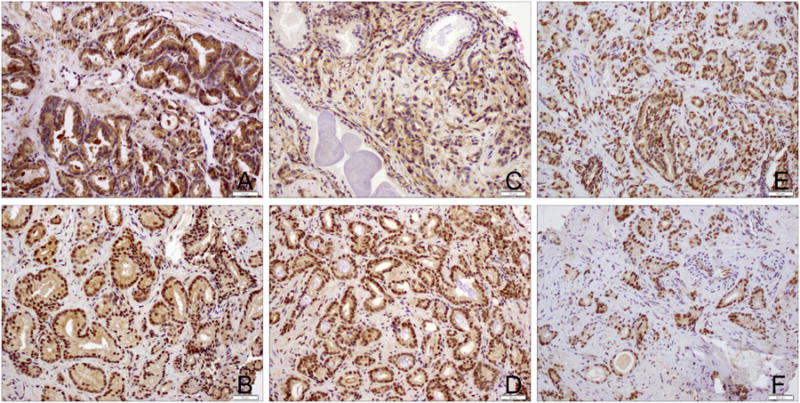
Representative IHC staining from prostate needle biopsy specimens for (A) Insulin Receptor; (B) phosphorylated Insulin Receptor; (C) IGF-1 Receptor; (D) phosphorylated IGF-1 Receptor; (E) AMPK; and (F) phosphorylated AMPK. IGF-1: insulin-like growth factor 1; AMPK: AMP activated protein kinase K.
Discussion
The role of metformin as an effective anti-cancer agent in PCa remains unclear, however, there is growing evidence to support this claim. In this study, we looked at the risk of BCR in PCa followed by examining metabolic signaling at the tissue level relative to metformin use. In our cohort of patients, ‘ever’ use of metformin did not influence the risk of BCR. However, when looking at pre- and post-treatment use of metformin (adjusted HR 0.55, 95% CI 0.31-0.96), cumulative metformin use in the highest quartile (adjusted HR 0.42, 95%CI 0.21-0.83), and cumulative dose in the highest quartile (adjusted HR 0.42, 95% CI 0.21-0.83), revealed a significant decrease in the risk of BCR. In patients undergoing PNB, individuals with PCa had significant increases in p-IR, p-IGF-1R, and p-AKT compared to those without cancer. When stratified by metformin use, IGF-1R remained elevated (p=0.01) in men with PCa detected whereas p-AMPK (p=0.05) was elevated only in those without PCa. These tissue level changes suggest metformin may alter signaling in the prostate with potential influence on PCa development.
The beneficial anti-tumor effects of metformin are proposed to occur through several pathways including increased AMPK activation, decreased circulating insulin, and improved insulin sensitivity.(15, 16) While the definitive role of AMPK in cancer remains unclear, there is evidence supporting its role given LKB1 regulation (a tumor suppressor). Histologic evaluation of breast cancer has shown reduced p-AMPK staining in primary breast cancer compared to normal epithelium.(18, 35) However, in thyroid cancer, p-AMPK staining was greater in cancer cells compared to matched normal tissue.(36) Thus, although an increased p-AMPK could decrease mTOR signaling and resultant proliferation, it may also promote some cancer cell survival by molecular crosstalk and inhibition of other cellular energy pathways.(37) As described, there are considerable data linking hyperglycemia and hyperinsulinemia with multiple malignancies(20-22) and specifically adverse outcomes in PCa.(23-26)
Previous studies have found mixed results of metformin's benefit with several post-RP studies and one brachytherapy study showing no association.(6-9, 12) A retrospective cohort study of external beam radiation therapy (EBRT) patients by Spratt et al.(10) found decreased BCR risk and PCa-specific mortality (PCSM) in those with ‘ever’ use of metformin. Interestingly, when they analyzed metformin based on duration of use (median dosing 58 months (range 38-88)), there was no effect of metformin on PCSM with Cox proportional hazards modeling (p=0.31). Conversely, Margel et al.(11) in a population-based, retrospective cohort of predominantly watchful waiting and patients treated with ADT, found a dose dependent decrease in PCSM with metformin use (median dosing 19 months (range 6.3-40), HR 0.80, 0.77-0.85). Another interesting study by Zannella et al., focusing on tissue oxygenation and metformin, compared a clinical cohort of RT patients taking metformin (31% with neoadjuvant and concurrent ADT) and found both ‘ever’ use and duration to be associated with decreased BCR.(13) Paradoxically, Allot et al. evaluated post-RP patients and showed no significant associations with metformin duration and BCR, however found that high dose metformin therapy (>2000mg/day) may play a role in increasing the risk of PCSM.(6) Finally, in an attempt to summarize the existing epidemiologic literature, a meta-analysis was recently published by Yu et al.(38) with principle findings suggesting an overall decrease in BCR risk relative to metformin use (combined HR 0.81, 0.68-0.98) with no difference in all-cause mortality (combined HR 0.86, 0.64-1.14). To our knowledge, this is the first study to look at total metformin dosing and the risk of BCR.
The dose dependent effect is important in interpreting the existing literature and may account for some of the conflicting data on metformin use.(6, 10, 11, 13) As an analogy, HMG-COA reductase inhibitors (statins) have suggested benefits in PCa.(39-42) A recent study by Yu et al. found a decreased risk of PCa-specific mortality (PCSM) and all-cause mortality in patients with post-diagnostic use. However, on further analysis, the authors found these benefits were more pronounced in those using statins prior to diagnosis, indicating a possible pre-treatment effect on tumorigenesis.(41) Our findings were similar in that the beneficial effect of metformin on BCR was only seen in those taking metformin both pre and post-primary treatment. Therefore, the timing of metformin use in future studies should be a primary focus to help clarify this relationship.
At the tissue level, previous authors have shown increased activity of insulin signaling markers in PCa.(43) Recently, Joshua et al.(14) attempted neoadjuvant metformin treatment prior to RP. With a median duration of treatment at 41 days (range 18-81), the authors found significantly reduced proliferation (via Ki-67 analysis) and decreased p-4EBP1 (a downstream effector of MTOR) in pathologic specimens with no significant findings on p-AMPK in cancer vs. controls. Our data reveal similar significant increases in multiple insulin signaling markers (p-IGF-1R, p-IR, p-AKT) in patients with PCa detected on PNB. Total AMPK was significantly elevated in the nucleus and cytoplasm of patients with PCa on PNB (p<0.003), yet p-AMPK was not significantly changed in the nucleus or cytoplasm. This suggests a preferential activation of insulin signaling in PCa, which may result in cellular proliferation with the overall role of AMPK remaining unclear.
Interestingly, when these results were limited to those with PCa detected on PNB stratified by metformin use, all insulin signaling markers were elevated in PCa patients regardless of metformin use, whereas negative PNB patients were more likely to have a decrease in insulin signaling markers relative to metformin use (see trend arrows (↑↓↔), Tables 3 and 4). We also found significant increases in IGF-1a-R (p=0.01) in patients with cancer detected, while p-AMPK (p=0.05) was elevated only in those without cancer. These findings suggest preferential activation of insulin signaling and proliferation in patients with PCa, potentially overpowering any protective effect of metformin and/or AMPK activation. Conversely, in metformin users without cancer detected, the down-regulation of insulin signaling markers combined with an increase in AMPK activation, suggests a potential therapeutic benefit. While these IGF-1a-R and p-AMPK values did reach statistical significance, they should be interpreted with caution. For instance, the mean change between IGF-1a-R IHC scores relative to metformin use was small (no metformin: 147.3 ± 56.9 vs. metformin: 192.2 ± 15.2, Table 3). Further, there was no difference in p-IGF-1a-R relative to metformin, suggesting this is not necessarily consistent with pathway activation. The difference between p-AMPK relative to metformin was even smaller (no metformin: 138.0 ± 44.2 vs. metformin 149.8 ± 53.8, Table 4) which may be negligible in the context of IHC scoring.
Taken together, metformin may have a dose dependent, beneficial effect in preventing PCa recurrence after primary therapy and may lead to measurable changes in prostate signaling with a potential (but unclear) role for AMPK in PCa tumorigenesis.
There are limitations to this study. In the database analysis, limits include the retrospective nature and the number of patients who were diabetic and not taking metformin (6%). Conversely, the comprehensive computer database of the VISN20 network with excellent pharmacy records adds strength of the analysis. We corrected for all covariates in our BCR model, however, stratified analyses for PSA, Gleason grade, stage, etc. may have added additional insight. Statin use was also not queried and thus any further protective effects of statins individually or combined with metformin use were not detected in this analysis. In order to account for the potential of immortal time bias (34) influencing the results, we performed additional analyses relative to metformin use (see methods). While this showed no significant change of BCR results relative to metformin use, this is a possible limit to the BCR analysis. Limits for the tissue-based study include that metformin was taken for indication and findings may be due to clinical and pathologic factors other than metformin use. Limits to IHC interpretation have been discussed previously. Further, there is heterogeneity in metformin absorption, bioavailability, and tissue penetration (44), which may lead to differential efficacy at the tissue level. Finally, the tissue study had a small sample size, was non-randomized, and retrospective analysis cannot show causation. Despite these limitations, these data support further the study of metformin use in PCa outcomes and metabolic signaling in the prostate.
Conclusion
We found total metformin use led to a reduction in the risk of BCR in a dose dependent fashion. We also demonstrate several metabolic pathway effectors (IR, IGF-1R, AMPK) present in higher levels in men with PCa detected on PNB compared to negative PNB. When stratified by metformin use, we found trends towards preferential activation of insulin signaling in those with PCa compared to those without, with the overall protective role of p-AMPK remaining unclear. Further prospective and interventional studies to define the precise role of metformin in BCR and metabolic signaling are required.
Supplementary Material
Acknowledgments
This material is the result of work supported by resources from the VA Puget Sound Health Care System, Seattle, Washington.
Funding: NIH Grant: P50CA097186 from the National Cancer Institute; with additional support from the Fred Hutchinson Cancer Research Center.
Footnotes
Conflicts of interest: None.
References
- 1.Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. Bmj. 2005;330(7503):1304–5. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wright JL, Stanford JL. Metformin use and prostate cancer in Caucasian men: results from a population-based case-control study. Cancer Causes Control. 2009;20(9):1617–22. doi: 10.1007/s10552-009-9407-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murtola TJ, Tammela TL, Lahtela J, Auvinen A. Antidiabetic medication and prostate cancer risk: a population-based case-control study. Am J Epidemiol. 2008;168(8):925–31. doi: 10.1093/aje/kwn190. [DOI] [PubMed] [Google Scholar]
- 4.Zakikhani M, Dowling RJ, Sonenberg N, Pollak MN. The effects of adiponectin and metformin on prostate and colon neoplasia involve activation of AMP-activated protein kinase. Cancer Prev Res (Phila Pa) 2008;1(5):369–75. doi: 10.1158/1940-6207.CAPR-08-0081. [DOI] [PubMed] [Google Scholar]
- 5.Ben Sahra I, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P, et al. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene. 2008;27(25):3576–86. doi: 10.1038/sj.onc.1211024. [DOI] [PubMed] [Google Scholar]
- 6.Allott EH, Abern MR, Gerber L, Keto CJ, Aronson WJ, Terris MK, et al. Metformin does not affect risk of biochemical recurrence following radical prostatectomy: results from the SEARCH database. Prostate cancer and prostatic diseases. 2013;16(4):391–7. doi: 10.1038/pcan.2013.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patel T, Hruby G, Badani K, Abate-Shen C, McKiernan JM. Clinical Outcomes After Radical Prostatectomy in Diabetic Patients Treated With Metformin. Urology. 2010 doi: 10.1016/j.urology.2010.03.059. [DOI] [PubMed] [Google Scholar]
- 8.Rieken M, Kluth LA, Xylinas E, Fajkovic H, Becker A, Karakiewicz PI, et al. Association of diabetes mellitus and metformin use with biochemical recurrence in patients treated with radical prostatectomy for prostate cancer. World J Urol. 2013 doi: 10.1007/s00345-013-1171-7. [DOI] [PubMed] [Google Scholar]
- 9.Kaushik D, Karnes RJ, Eisenberg MS, Rangel LJ, Carlson RE, Bergstralh EJ. Effect of metformin on prostate cancer outcomes after radical prostatectomy. Urol Oncol. 2014;32(1):43 e1–7. doi: 10.1016/j.urolonc.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spratt DE, Zhang C, Zumsteg ZS, Pei X, Zhang Z, Zelefsky MJ. Metformin and prostate cancer: reduced development of castration-resistant disease and prostate cancer mortality. Eur Urol. 2013;63(4):709–16. doi: 10.1016/j.eururo.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Margel D, Urbach DR, Lipscombe LL, Bell CM, Kulkarni G, Austin PC, et al. Metformin use and all-cause and prostate cancer-specific mortality among men with diabetes. J Clin Oncol. 2013;31(25):3069–75. doi: 10.1200/JCO.2012.46.7043. [DOI] [PubMed] [Google Scholar]
- 12.Taira AV, Merrick GS, Galbreath RW, Morris M, Butler WM, Adamovich E. Metformin is not associated with improved biochemical free survival or cause-specific survival in men with prostate cancer treated with permanent interstitial brachytherapy. J Contemp Brachytherapy. 2014;6(3):254–61. doi: 10.5114/jcb.2014.45757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zannella VE, Dal Pra A, Muaddi H, McKee TD, Stapleton S, Sykes J, et al. Reprogramming metabolism with metformin improves tumor oxygenation and radiotherapy response. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19(24):6741–50. doi: 10.1158/1078-0432.CCR-13-1787. [DOI] [PubMed] [Google Scholar]
- 14.Joshua AM, Zannella VE, Downes MR, Bowes B, Hersey K, Koritzinsky M, et al. A pilot ‘window of opportunity’ neoadjuvant study of metformin in localised prostate cancer. Prostate cancer and prostatic diseases. 2014;17(3):252–8. doi: 10.1038/pcan.2014.20. [DOI] [PubMed] [Google Scholar]
- 15.Anwar MA, Kheir WA, Eid S, Fares J, Liu X, Eid AH, et al. Colorectal and Prostate Cancer Risk in Diabetes: Metformin, an Actor behind the Scene. J Cancer. 2014;5(9):736–44. doi: 10.7150/jca.9726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pernicova I, Korbonits M. Metformin--mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol. 2014;10(3):143–56. doi: 10.1038/nrendo.2013.256. [DOI] [PubMed] [Google Scholar]
- 17.Hardie DG. Minireview: the AMP-activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology. 2003;144(12):5179–83. doi: 10.1210/en.2003-0982. [DOI] [PubMed] [Google Scholar]
- 18.Hadad SM, Fleming S, Thompson AM. Targeting AMPK: a new therapeutic opportunity in breast cancer. Crit Rev Oncol Hematol. 2008;67(1):1–7. doi: 10.1016/j.critrevonc.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 19.Dowling RJ, Zakikhani M, Fantus IG, Pollak M, Sonenberg N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 2007;67(22):10804–12. doi: 10.1158/0008-5472.CAN-07-2310. [DOI] [PubMed] [Google Scholar]
- 20.Pollak MN. Investigating metformin for cancer prevention and treatment: the end of the beginning. Cancer Discov. 2012;2(9):778–90. doi: 10.1158/2159-8290.CD-12-0263. [DOI] [PubMed] [Google Scholar]
- 21.Belfiore A. The role of insulin receptor isoforms and hybrid insulin/IGF-I receptors in human cancer. Curr Pharm Des. 2007;13(7):671–86. doi: 10.2174/138161207780249173. [DOI] [PubMed] [Google Scholar]
- 22.Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer. 2008;8(12):915–28. doi: 10.1038/nrc2536. [DOI] [PubMed] [Google Scholar]
- 23.Wright JL, Plymate SR, Porter MP, Gore JL, Lin DW, Hu E, et al. Hyperglycemia and prostate cancer recurrence in men treated for localized prostate cancer. Prostate Cancer Prostatic Dis. 2013;16(2):204–8. doi: 10.1038/pcan.2013.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goodwin PJ, Ennis M, Pritchard KI, Trudeau ME, Koo J, Madarnas Y, et al. Fasting insulin and outcome in early-stage breast cancer: results of a prospective cohort study. J Clin Oncol. 2002;20(1):42–51. doi: 10.1200/JCO.2002.20.1.42. [DOI] [PubMed] [Google Scholar]
- 25.Hammarsten J, Hogstedt B. Hyperinsulinaemia: a prospective risk factor for lethal clinical prostate cancer. Eur J Cancer. 2005;41(18):2887–95. doi: 10.1016/j.ejca.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 26.Ma J, Li H, Giovannucci E, Mucci L, Qiu W, Nguyen PL, et al. Prediagnostic body-mass index, plasma C-peptide concentration, and prostate cancer-specific mortality in men with prostate cancer: a long-term survival analysis. Lancet Oncol. 2008;9(11):1039–47. doi: 10.1016/S1470-2045(08)70235-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heni M, Hennenlotter J, Scharpf M, Lutz SZ, Schwentner C, Todenhofer T, et al. Insulin receptor isoforms A and B as well as insulin receptor substrates-1 and -2 are differentially expressed in prostate cancer. PloS one. 2012;7(12):e50953. doi: 10.1371/journal.pone.0050953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bailey CJ, Turner RC. Metformin. N Engl J Med. 1996;334(9):574–9. doi: 10.1056/NEJM199602293340906. [DOI] [PubMed] [Google Scholar]
- 29.Maynard C, Chapko MK. Data resources in the Department of Veterans Affairs. Diabetes Care. 2004;27(Suppl 2):B22–6. doi: 10.2337/diacare.27.suppl_2.b22. [DOI] [PubMed] [Google Scholar]
- 30.Zeliadt SB, Sekaran NK, Hu EY, Slatore CC, Au DH, Backhus L, et al. Comparison of demographic characteristics, surgical resection patterns, and survival outcomes for veterans and nonveterans with non-small cell lung cancer in the Pacific Northwest. Journal of thoracic oncology: official publication of the International Association for the Study of Lung Cancer. 2011;6(10):1726–32. doi: 10.1097/JTO.0b013e31822ada77. [DOI] [PubMed] [Google Scholar]
- 31.Basaria S, Muller DC, Carducci MA, Egan J, Dobs AS. Hyperglycemia and insulin resistance in men with prostate carcinoma who receive androgen-deprivation therapy. Cancer. 2006;106(3):581–8. doi: 10.1002/cncr.21642. [DOI] [PubMed] [Google Scholar]
- 32.Charlson ME, Pompei P, Ales KL, MacKenzie CR. A new method of classifying prognostic comorbidity in longitudinal studies: development and validation. Journal of chronic diseases. 1987;40(5):373–83. doi: 10.1016/0021-9681(87)90171-8. [DOI] [PubMed] [Google Scholar]
- 33.Klabunde CN, Legler JM, Warren JL, Baldwin LM, Schrag D. A refined comorbidity measurement algorithm for claims-based studies of breast, prostate, colorectal, and lung cancer patients. Annals of epidemiology. 2007;17(8):584–90. doi: 10.1016/j.annepidem.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 34.Levesque LE, Hanley JA, Kezouh A, Suissa S. Problem of immortal time bias in cohort studies: example using statins for preventing progression of diabetes. Bmj. 2010;340:b5087. doi: 10.1136/bmj.b5087. [DOI] [PubMed] [Google Scholar]
- 35.Hadad SM, Baker L, Quinlan PR, Robertson KE, Bray SE, Thomson G, et al. Histological evaluation of AMPK signalling in primary breast cancer. BMC Cancer. 2009;9:307. doi: 10.1186/1471-2407-9-307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vidal AP, Andrade BM, Vaisman F, Cazarin J, Pinto LF, Breitenbach MM, et al. AMP-activated protein kinase signaling is upregulated in papillary thyroid cancer. Eur J Endocrinol. 2013;169(4):521–8. doi: 10.1530/EJE-13-0284. [DOI] [PubMed] [Google Scholar]
- 37.Jeon SM, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. 2012;485(7400):661–5. doi: 10.1038/nature11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu H, Yin L, Jiang X, Sun X, Wu J, Tian H, et al. Effect of metformin on cancer risk and treatment outcome of prostate cancer: a meta-analysis of epidemiological observational studies. PloS one. 2014;9(12):e116327. doi: 10.1371/journal.pone.0116327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bansal D, Undela K, D'Cruz S, Schifano F. Statin use and risk of prostate cancer: a meta-analysis of observational studies. PloS one. 2012;7(10):e46691. doi: 10.1371/journal.pone.0046691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Geybels MS, Wright JL, Holt SK, Kolb S, Feng Z, Stanford JL. Statin use in relation to prostate cancer outcomes in a population-based patient cohort study. Prostate. 2013;73(11):1214–22. doi: 10.1002/pros.22671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu O, Eberg M, Benayoun S, Aprikian A, Batist G, Suissa S, et al. Use of statins and the risk of death in patients with prostate cancer. J Clin Oncol. 2014;32(1):5–11. doi: 10.1200/JCO.2013.49.4757. [DOI] [PubMed] [Google Scholar]
- 42.Mucci LA, Stampfer MJ. Mounting evidence for prediagnostic use of statins in reducing risk of lethal prostate cancer. J Clin Oncol. 2014;32(1):1–2. doi: 10.1200/JCO.2013.53.2770. [DOI] [PubMed] [Google Scholar]
- 43.Cox ME, Gleave ME, Zakikhani M, Bell RH, Piura E, Vickers E, et al. Insulin receptor expression by human prostate cancers. Prostate. 2009;69(1):33–40. doi: 10.1002/pros.20852. [DOI] [PubMed] [Google Scholar]
- 44.Graham GG, Punt J, Arora M, Day RO, Doogue MP, Duong JK, et al. Clinical pharmacokinetics of metformin. Clin Pharmacokinet. 2011;50(2):81–98. doi: 10.2165/11534750-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 45.Goodwin PJ, Ligibel JA, Stambolic V. Metformin in breast cancer: time for action. J Clin Oncol. 2009;27(20):3271–3. doi: 10.1200/JCO.2009.22.1630. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.