

Abstract
Background
Serum Response Factor (SRF) is an important transcription factor in castrate-resistant prostate cancer (CRPC). Since CRPC is associated with androgen receptor (AR) hypersensitivity, we investigated the relationship between SRF and AR.
Material and methods
Transcriptional activity was assessed by luciferase assay. Cell proliferation was measured by MTT and flow cytometry. Protein expression in patients was assessed by immunohistochemistry.
Results
To investigate AR involvement in SRF response to androgen, AR expression was down-regulated using siRNA. This resulted in the abrogation of SRF induction post-DHT. Moreover, DHT stimulation failed to induce SRF transcriptional activity in AR-negative PC346 DCC cells, which was only restored following AR over-expression. Next, SRF expression was down-regulated by siRNA, resulting in AR increased transcriptional activity in castrate-resistant LNCaP Abl cells but not in the parental LNCaP. This negative feedback loop in the resistant cells was confirmed by immunohistochemistry which showed a negative correlation between AR and SRF expression in CRPC bone metastases and a positive correlation in androgen-naïve prostatectomies. Cell proliferation was next assessed following SRF inhibition, demonstrating that SRF inhibition is more effective than AR inhibition in castrate-resistant cells.
Conclusion
Our data support SRF as a promising therapeutic target in combination with current treatments.
Introduction
While early detection of prostate cancer allows for curative therapies such as surgery and radiation treatments, patients with locally advanced and metastatic prostate cancer are treated with androgen ablation therapy. However, despite initial response, the majority of men will progress to develop castrate-resistant prostate cancer (CRPC) which, despite the emergence of new treatments such as abiraterone acetate and Enzalutamide (MDV3100), is still challenging to treat. Therefore defining the mechanisms of resistance represents a key question facing clinicians and scientists.
Using a combination of transcriptomics profiling and bioinformatics analysis, we have recently identified Serum Response Factor (SRF) as an important transcription factor (TF) in an in vitro model of CRPC [1]. SRF is a widely expressed TF involved in cellular proliferation and cytoskeletal organisation as well as cellular growth, differentiation and resistance to apoptosis [2, 3]. Known SRF target genes are characterised by single or multiple copies of the serum responsive elements (SRE) which contain the consensus sequence CC [A/T]2A[A/T]3GG, generally known as CArG box. SRF has been recently associated with prostate cancer by our group and others and its inhibition has been shown to decrease cellular proliferation in LNCaP cells [1, 4]. The association of SRF protein expression with prostate cancer and its relevance to patient survival has also been shown in several studies [5, 6]. We have recently validated SRF clinical relevance to CRPC by immunohistochemical staining in patients who had failed hormone ablation therapy and had received a transurethral resection of the prostate (TURP), with 95% of these patients showing SRF nuclear positivity against only 50% of localised tumours [1]. In line with a previous study which showed an association between SRF expression in primary prostate cancer tissues and poor outcome following radical prostatectomy [6], we have recently shown a negative association between SRF nuclear positivity in bone metastases of patients who died of prostate cancer and survival from time of diagnosis and time of castration-resistance [5].
While mounting evidence both in vitro and in vivo suggest a key role for SRF in prostate cancer development and progression [1, 4–8], the mechanisms underlying SRF action in prostate cancer are still poorly understood. Recently, an SRF androgen-dependent gene signature was discovered, which indicated that a proportion of androgen-responsive genes are under SRF control [7]. While a role for RhoA has been suggested for SRF activation in response to androgens [9] the question still remains whether the androgen receptor (AR) is involved in this activation, considering that recruitment of AR by SRF on serum response elements has been previously shown in myoblasts [10]. Due to the central role played by AR during prostate cancer development and progression and to the fact that a significant proportion of CRPCs remain sensitive to ligand activation of AR [11] the aim of the present study was to investigate the relationship between SRF and AR in advanced prostate cancer.
Material and Methods
Cell culture and reagents
The LNCaP Parental cells (ATCC) were routinely cultured as published previously [1]. The LNCaP Abl cell line was generated from the LNCaP cell line as described previously [12] and cultured as previously described [1]. In order to keep conditions consistent with their LNCaP Abl subline, LNCaP Parental cells were cultured in medium supplemented with charcoal-stripped FBS (Sigma, Germany) for 48 hours before DHT stimulation. The PC346 DCC cell line was a gift from Dr. Wytske M. van Weerden, Erasmus Medical Center, Rotterdam, Netherlands. This cell line was derived from the PC346C cell line, generated from the prostate tumor of a patient with non-progressive prostate adenocarcinoma (T4N0M0), upon long-term culture in charcoal-stripped medium as previously described [13, 14]. PC346 DCC cells were cultured as previously described [13]. All cell lines were maintained at 37°C in a humidified atmosphere of 5% CO2 in air.
(5α,17β)-17-Hydroxy-androstan-3-one (DHT) was purchased from Sigma. CCG-1423 was purchased from Cayman Chemicals. MDV3100 was purchased from Selleckchem, UK.
Western Blot analysis
Western blots were carried out as previously published [15]. The following primary antibodies were used: anti-AR (1:500, Santa Cruz), anti-SRF (1:1000, Santa Cruz) and β-actin (1:5000, Sigma Aldrich). Densitometry on western blot X-Ray films was performed using Image J software.
Small-interfering RNA (siRNA) and plasmids transfections
Cells were seeded in 6 well plates at a density of 250 000 cells per well. The next day, cells were transfected with siGENOME SMART pool targeting SRF, AR or siControl siRNA (all from Dharmacon), at a final concentration of 10 nM, using Lipofectamine 2000 (Invitrogen) following the manufacturer’s instructions. The pEGFP-C1-AR plasmid (Addgene plasmid 28235) was obtained by Addgene and was generated by Stenoien and colleagues as previously described [16]. PC346 DCC cells were seeded in 6 well plates at a density of 250 000 cells per well. The next day, cells were transfected with 500 ng of the pEGFP-C1-AR plasmid or with 500 ng of the empty vector (EV) using Lipofectamine 2000 (Invitrogen).
Luciferase Reporter assay
SRF transcriptional activity was assessed using the pGL4.34 vector containing a SRF responsive element (CArG box) that drives the transcription of the luciferase reporter gene luc2P (Promega, Fitchburg, Wisconsin, US). AR transcriptional activity was assessed using a pGL3-ARE-E1B-Luc [17]. A TK-Renilla luciferase plasmid was used as a transfection efficiency control. Plasmids were co-transfected using GeneJuice® Transfection Reagent (Novagen, Darmstadt, Germany) following the manufacturer’s instructions. The luciferase and renilla activities were measured using a Dual-Luciferase® reporter assay (Promega). SRF and AR transcriptional activities were expressed as the fold change of Relative Luciferase Units (RLU) (ratio between luciferase light values divided by the renilla light values) taking the untreated controls as the baseline.
RNA isolation and Real-time reverse transcriptase PCR
Total RNA was extracted by cell pellets, using the guanidine-based TRIzol reagent (Invitrogen, Carlsbad, CA, US), according to the manufacturer’s instructions. All RNA samples were DNAse (Invitrogen, Carlsbad, CA, US) treated before cDNA synthesis. Random hexamer cDNA was synthesised from 1 μg of total RNA, using the standard Superscript II kit (Invitrogen, Carlsbad, CA, US). The cDNA was subsequently used as template for gene-specific real time quantitative reverse transcription PCR (qRT-PCR) as previously published [15]. The primers and probe for Twist-1 were supplied as a pre-optimised single tube primer/probe Gene Expression Assay (Applied Biosystems, CA, US). GAPDH was used as an endogenous control for normalisation of the target gene. Its primers and probe were supplied as a pre-developed assay reagent (Applied Biosystems, CA, US). Results were analyzed using the ΔCt method and all samples were set up as duplicates.
Flow cytometric analysis
Proliferation was assessed by measuring the S phase within the cell cycle following propidium iodide (PI) incorporation as previously described [18]. Fifteen thousand events were gated on PI intensity. Data were acquired using the BD CFlow® Plus software and re-analysis was performed using the FCS Express 4 RUO software.
3-(4,5)-dimethylthiazol-2-yl-2,5-diphenyltetrazolium bromide (MTT) cell viability assay
Two hundred and fifty thousand cells per well were cultured in 12-well plates. Twenty-four hours later, cells were treated with either vehicle (ethanol), MDV3100 (10 μM), CCG-1423 (10 μM) or a combination of MDV3100 and CCG-1423 for 48 hours. Cell viability was assessed by MTT cell staining as previously described [18].
Sample Collection/Tissue Microarray Construction
CRPC metastases TMA
Human tissue microarrays were constructed consisting of 65 soft tissue metastases and 120 bone metastases from 42 patients with advanced PCa. Samples were obtained from patients who died of metastatic CRPC and who signed written informed consent for a rapid autopsy to be performed ideally within 2 hours of death, under the aegis of the Prostate Cancer Donor Program at the University of Washington [19]. Two replicate 1 mm cores of soft tissue metastases and bone metastases were taken from every case where available [20]. The tissue microarrays were assembled using the Beecher Instruments Tissue-ArrayerTM (Beecher Instruments, Silver Spring, MD).
Radical prostatectomies TMA
A TMA was constructed from a population-based cohort of 341 PCa patients who underwent open radical prostatectomy between 1998 and 2006 at the Department of Urology, Skåne University Hospital, Malmö, Sweden using a previously described protocol [21]. From each patient, benign and malignant cores in duplicate were mounted in a total of 13 paraffin blocks. Biochemical recurrence (BCR) was defined as a blood PSA level of at least 0.2 ng/ml with a subsequent confirmatory value.
Immunohistochemical (IHC) Analysis
Antigen retrieval of the deparaffinised tissue sections was performed using a PT-Link module (DAKO) at 95°–99°C for 20 min in a citric acid buffer (0.01M, pH 6.0). Slide staining was performed using a DAKO autostainer Link 48 according to the manufacturer instructions. Tonsil sections were used as positive controls for SRF staining. Prior to this study, both SRF and AR antibodies were subjected to western blot analysis which confirmed antibody specificity [1].
Scoring of SRF and AR Protein Expression and Statistical Analysis
Some unusable cores were found in the TMAs due to the tissue cores being missing, cancer necrosis, or insufficient cancer cells. These cores were excluded from the study. Nuclear immunoreactivity for SRF in soft tissue metastases and bone metastases was assessed by two independent observers (GOH and EK); nuclear immunoreactivity for SRF and AR in androgen-naïve prostatectomies was also assessed by two independent observers (MP and AF). Immunostaining was assessed using a nuclear score for SRF and AR, created by multiplying each intensity level (0, no staining, 1, faint but clearly detectable staining, 2, moderate staining and 3, strong staining) by the corresponding percentage of positive epithelial cells.
For the purpose of statistical analysis, the nuclear scores of SRF and AR were then further divided into two groups: negative (immunohistochemical score <100) and positive (immunohistochemical score >100). Pearson correlation was performed on 2X2 contingency tables using IBM SPSS 20 for Windows®.
Computational Model
Mathematica was used to develop and run the model simulations. The model equations used to generate Figure 6 are presented below. Additional details can be found in the Supplementary Information.
The model input is DHT.
Results
SRF response to androgen stimulation is dependent on AR expression
To assess whether SRF transcriptional activity was responsive to androgen stimulation, parental LNCaP cells and their castrate-resistant subline LNCaP Abl were treated with increasing concentrations of DHT (1, 10, 100 nM) for 24 hours. Following DHT treatment, luciferase reporter assays showed a significant dose-dependent increase in SRF transcriptional activity in both cell lines. However this induction was 10 to 15 fold higher in the Abl cells compared with the parental cell line (Figure 1A). To confirm that SRF transcriptional activity was responsive to androgens, we stimulated the cells with DHT and simultaneously treated them with 10 μM of the potent AR inhibitor MDV3100. Luciferase assays showed a significant decrease in SRF transcriptional activity following MDV3100 treatment in both parental and Abl cells (p<0.001 and p<0.01 respectively) (Figure 1B). Similar results were obtained using bicalutamide (data not shown).
Figure 1. SRF transcriptional activity in response to androgen stimulation and inhibition.
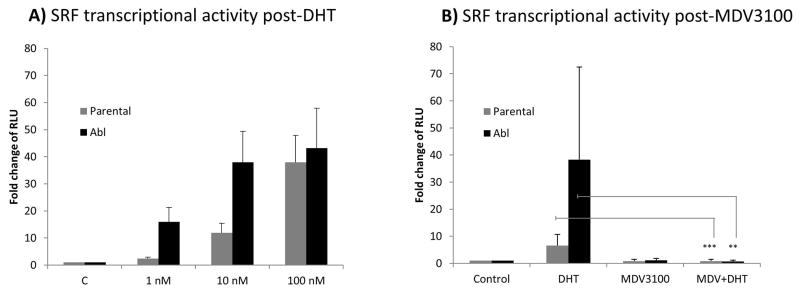
Cells were seeded in 12 well plates at a density of 5×105 cells per well. The following day they were transiently transfected with the reporter construct pGL4.34 driven by a SRF responsive element (CArG box) that drives the transcription of the luciferase reporter gene luc2P. Twenty four hours post-transfection cells were treated with increasing concentrations of DHT (1, 10 and 100 nM) (panel A) or 10 μM of MDV3100 alone, 10 nM of DHT alone, MDV3100 and DHT in combination (panel B). Reporter gene activity was measured 24hours after treatment. Columns, mean values obtained from three independent experiments in triplicate; bars, SD. Mean values were compared using t-test assuming equal variances. ** p<0.01; *** p<0.001. RLU: relative luciferase units.
As the mechanisms underlying disease progression are associated with AR hypersensitivity [22] and previous studies in myoblasts have linked SRF with AR [10], we next investigated the role of AR in the induction of SRF transcriptional activity in response to DHT. In order to explore whether SRF induction in response to androgen stimulation was dependent on AR, we silenced AR expression by small-interference RNA (siRNA) (Figure 2A) and looked at SRF transcriptional activity and protein expression. As shown in figure 2B, AR silencing resulted in a significant reduction of SRF transcriptional activity following DHT treatment in both parental and Abl cell lines (p<0.05 in parental LNCaP and p<0.0001 in Abl cells). While no change in SRF mRNA expression was detected following AR silencing (data not shown), Western blotting showed down-regulation of SRF protein expression following AR siRNA (Figure 2C), which would suggest a post-translational regulation as the mechanism by which SRF transcriptional activity is affected in response to AR silencing. To strengthen the luciferase assays results we assessed the mRNA expression of endogenous Twist (Figure 2D), which we had previously identified as a predicted SRF transcriptional target, based on TF binding motives databases ([1], data not shown). In the parental LNCaP cells, TaqMan analysis showed that Twist mRNA expression was significantly decreased (p<0.05) following AR silencing in the presence of DTH, in accordance with the luciferase assay. In the Abl cells however, while in the absence of androgens Twist mRNA expression was significantly decreased (p<0.05) following AR silencing, in the presence of DHT, a significant increase in Twist mRNA expression (p<0.05) was detected following AR siRNA.
Figure 2. SRF transcriptional activity is dependent on AR.

A) Cells were seeded in 6 well plates at a density of 250,000 cells per well. The following day they were transiently transfected with a pool of siRNAs specific for AR or non- specific control siRNAs. Following 48 hours, total protein extracts were prepared for western blotting and β-actin was used as loading control. Representative images from three independent experiments are shown. B) Cells were seeded in 12 well plates at a density of 5×105 cells per well. The following day they were transiently transfected with a pool of siRNAs specific for AR or non- specific control siRNAs. Twenty four hours post-siRNA transfection, cells were transfected again with the reporter construct driven by an SRF responsive element. The following day cells were either left untreated or treated with 10 nM DHT. Reporter gene activity was measured 24 hours after treatment. Columns, mean values obtained from three independent experiments in triplicate; bars, SD. Mean values were compared using t-test assuming equal variances. * p<0.05, ** p<0.01, **** p<1 E-04. C) Cells were seeded in 6 well plates at a density of 250,000 cells per well. The following day they were transiently transfected with a pool of siRNAs specific for AR or non- specific control siRNAs. Twenty four hours post-siRNA transfection, cells were either left untreated or treated with 10 nM DHT. Total protein extracts were prepared for western blotting 24 hours after treatment. β-actin was used as loading control. Representative images from three independent experiments are shown. Densitometry is shown on the right panel. Columns, mean values obtained from three independent experiments in triplicate; bars, SD. D) Cells were seeded and treated as in panel C. Forty-eight hours after DHT treatment, RNA was isolated and cDNA was synthesised. The cDNA was subsequently used as template for gene-specific real time quantitative reverse transcription PCR (qRT-PCR). Columns, mean values obtained from three independent experiments in triplicate; bars, SD. Mean values were compared using t-test assuming equal variances. * p<0.05 E-F) PC346 DCC cells were seeded in 12 well plates at a density of 5×105 cells per well. The following day they were transiently transfected either with a pEGFP-C1-AR plasmid or with the empty vector (EV). Twenty four hours post-siRNA transfection, cells were transfected again with either the reporter construct driven by an AR responsive element (panel D) or with the reporter construct driven by a SRF responsive element (panel E). The following day cells were treated with increasing concentrations (0.01, 0.1, 1 nM) of DHT. Reporter gene activity was measured 24 hours after treatment. Black columns, EV; grey columns, pEGFP-C1-AR plasmid. Columns, mean values obtained from three independent experiments in triplicate; bars, SD. Mean values were compared using t-test assuming equal variances. * p<0.05, **** p<1 E-04.
To further support our hypothesis that SRF transcriptional activity was dependent on AR protein expression, we transfected a pEGFP-C1-AR plasmid in the AR negative cell line PC346 DCC, in order to ectopically express AR in these cells. As shown in figure 2E, AR over-expression was successful with the cells transfected with the pEGFP-C1-AR plasmid responding to DHT stimulation compared with the empty vector (EV) control cells which did not respond. We next looked at SRF transcriptional activity following AR over-expression. While cells transfected with the EV did not show increased SRF transcriptional activity in response to DHT stimulation, cells transfected with the pEGFP-C1-AR plasmid showed a significant induction of SRF transcriptional activity post-DHT (Figure 2F). Taken together these results clearly demonstrate that SRF transcriptional activity is dependent on AR protein expression.
AR transcriptional activity is affected by SRF protein expression
To explore whether SRF dependency on AR was reciprocal, we down-regulated SRF expression by siRNA (Figure 3A) and looked at AR transcriptional activity. At the baseline level, luciferase reporter assays showed no significant change in AR transcriptional activity following SRF silencing in both parental and Abl cell lines. However, following DHT stimulation, a significant increase in AR transcriptional activity was shown in the Abl cells (p<0.05) but not in the parental LNCaP (Figure 3B). Western blotting analysis showed a moderate increase in AR expression following SRF silencing in both parental and Abl cells, before and after DHT treatment (Figure 3C). These data show that AR transcriptional activity is affected by SRF in castration-resistant cells. The increase in AR transcriptional activity following SRF down-regulation by siRNA is indicative of a negative feedback loop between SRF and AR in castration-resistant Abl LNCaP cells.
Figure 3. AR transcriptional activity is affected by SRF.

A) Cells were seeded in 6 well plates at a density of 250,000 cells per well. The following day they were transiently transfected with a pool of siRNAs specific for SRF or non- specific control siRNAs. Following 48 hours, total protein extracts were prepared for western blotting and β-actin was used as loading control. Representative images from three independent experiments are shown. B) Cells were seeded in 12 well plates at a density of 5×105 cells per well. The following day they were transiently transfected with a pool of siRNAs specific for SRF or non- specific control siRNAs. Twenty four hours post-siRNA transfection, cells were transfected again with the reporter construct driven by an AR responsive element. The following day cells were either left untreated or treated with 10 nM DHT. Reporter gene activity was measured 24 hours after treatment. Columns, mean values obtained from three independent experiments in triplicate; bars, SD. Mean values were compared using t-test assuming equal variances. * p<0.05. C) Cells were seeded in 6 well plates at a density of 250,000 cells per well. The following day they were transiently transfected with a pool of siRNAs specific for SRF or non- specific control siRNAs. Twenty four hours post-siRNA transfection, cells were either left untreated or treated with 10 nM DHT. Total protein extracts were prepared for western blotting 24 hours after treatment. β-actin was used as loading control. Representative images from three independent experiments are shown. Densitometry is shown on the right panel. Columns, mean values obtained from three independent experiments in triplicate; bars, SD.
Correlation between SRF and AR expression in prostate cancer tissues
To translate our in vitro findings to clinical tissues from patients, we assessed SRF and AR expression by immunohistochemistry (Figure 4) and looked at their correlation, using two TMAs representative of the androgen-sensitive and castration-resistant stages of the disease. Staining of the TMA containing tissue samples from androgen-naïve prostatectomies (341 patients) showed a positive correlation between SRF and AR expression in both benign (p<0.001, R=0.414) (Table 1) and tumour cores (p<0.001, R=0.362) (Table 2). Staining of the TMA containing metastatic tumours (from bone and soft tissue sites) from 42 patients who died of CRPC, had previously been performed for SRF[5] and AR [20]. Pearson correlation tests showed a negative correlation between SRF and AR expression in these castrate-resistant patients (P=0.01, R=−0.208) (Table 3).
Figure 4. Protein expression assessed by IHC on clinical tissues from patients with prostate cancer.

A) SRF IHC staining. B) AR IHC staining. Examples of negative (1), weak (2), moderate (3) and strong (4) nuclear staining are shown for both SRF and AR (40X magnifications).
Table 1.
Two -way contingency table comparing SRF expression vs. AR expression in benign cores from radical prostatectomies
| SRF vs. AR Expression in Benign Cores from Radical Prostatectomies | AR Score | |||
|---|---|---|---|---|
| Negative | Positive | Total | ||
|
| ||||
| SRF Score | Negative | 190 (32.2%) | 400 (67.8%) | 590 (100%) |
| Positive | 1 (3.8%) | 25 (96.2%) | 26 (100%) | |
| Total | 191 (31%) | 425 (69%) | 616 (100%) | |
|
| ||||
| Value | Sig (2-sided) | |||
|
| ||||
| Pearson Correlation | 0.414 | 0.000 | ||
Table 2.
Two-way contingency table comparing SRF expression vs. AR expression in cancercores of radical prostatectomies
| SRF vs. AR Expression in Cancer Cores from Radical Prostatectomies | AR Score | |||
|---|---|---|---|---|
| Negative | Positive | Total | ||
|
| ||||
| SRF Score | Negative | 51 (12.2%) | 368 (87.8%) | 419 (100%) |
| Positive | 3 (2.9%) | 101 (97.1%) | 104 (100%) | |
| Total | 54 (10.3%) | 469 (89.7%) | 523 (100%) | |
|
| ||||
| Value | Sig (2-sided) | |||
|
| ||||
| Pearson Correlation | 0.362 | 0.000 | ||
Table 3.
Two-way contingency table comparing SRF expression vs. AR expression in CRPC matastases
| SRF vs. AR Expression in CRPC Matastases | AR Score | |||
|---|---|---|---|---|
| Negative | Positive | Total | ||
|
| ||||
| SRF Score | Negative | 13 (14.3%) | 78 (85.7%) | 91 (100%) |
| Positive | 19 (31.7%) | 41 (68.3%) | 60 (100%) | |
| Total | 32 (21.2%) | 119 (78.8%) | 151 (100%) | |
|
| ||||
| Value | Sig (2-sided) | |||
|
| ||||
| Pearson Correlation | −0.208 | 0.01 | ||
Impact of SRF inhibition on cellular viability and proliferation compared to AR inhibition
We have recently shown that SRF inhibition significantly decreases cell proliferation in parental and Abl LNCaP cells [1]. Here we wanted to assess the impact of SRF inhibition on cellular viability and proliferation in comparison to inhibiting AR, the current treatment strategy for locally advanced and metastatic disease. LNCaP Abl cells were used in these experiments since they represent the cellular model closer to castrate-resistant patients. Cellular proliferation was measured as the S phase of the cell cycle following propidium iodide (PI) staining and flow cytometry. While SRF silencing with siRNA caused a significant decrease in cellular proliferation (p<0.05), no difference was displayed post AR inhibition. Moreover, the combined knock-down of AR and SRF did not show any difference in cell proliferation when compared with SRF knock-down alone (Figure 5A). Cell viability was assessed next following inhibition of SRF and AR using small molecule inhibitors. SRF was inhibited using CCG-1423 as previously published [1], while AR was inhibited using MDV3100 (Figure S1). Cell viability assays showed that both inhibitors caused a similar decrease in cell viability. However, the combination of the two drugs showed a significantly higher decrease in cell viability (CCG-1423 vs. combination p=0.001; MDV3100 vs. combination p<0.001) (Figure 5B).
Figure 5. Impact of SRF inhibition on cellular viability and proliferation.

A) Cells were seeded in 6 well plates at a density of 250,000 cells per well. The following day they were transiently transfected with a pool of siRNAs specific for AR, SRF or non- specific control siRNAs. Following 48 hours, total protein extracts were prepared for western blotting and β-actin was used as loading control. Representative images from three independent experiments are shown. B) Cells were seeded in 6 well plates at a density of 250,000 cells per well. The following day they were transiently transfected either with a non- specific control siRNAs, a pool of siRNAs specific for SRF, a pool of siRNAs specific for AR or a combination of SRF-siRNA and AR- siRNA. Following 48 hours, cells were tripsinised and stained with PI prior to FACS analysis. Proliferation was assessed by measuring the S phase within the cell cycle. C) Cells were seeded in 12 well plates at a density of 5×105 cells per well. The following day they were treated either with vehicle (DMSO), 10 μM of CCG-1423, 10 μM of MDV3100 or a combination of CCG-1423 and MDV3100. Following 48 hours, cellular viability was measured by MTT assay. Columns, mean values obtained from three independent experiments in triplicate; bars, SD. Mean values were compared using t-test assuming equal variances. * p<0.05; ** p<0.01; **** p<1 E-04.
Computational modelling of the proposed mechanism of SRF action
In order to summarise our findings on the relationship between SRF and AR in LNCaP cells in a simplified model, we constructed a computational model to simulate the interactions between SRF and AR under various conditions. Based on our experimental data, the model was designed assuming a positive stimulation of AR on SRF and a negative feedback between SRF and AR (see Supplementary information for detail on model equations and assumptions) (Figure 6A). The model consists of a set of ordinary differential equations which are formulated based on a more detailed kinetic reaction scheme of the AR-SRF network (see Supplementary information for detail on model equations and assumptions) (Figure 6B). Simulations of this simple model agreed very well with our experimental data (Figure 6C–F). Specifically, figure 6C (computational model, CM) corresponds to figure 1A (experimental data, ED), figure 6D (CM) with figure 2A (ED), figure 6E (CM) with figure 2D (ED), figure 6G (CM) with figure 3B (ED, Abl cells) and figure 6H (CM) with figure 3C (ED, Abl cells). The agreement of the experimental data with this computational model suggests that the negative feedback between SRF and AR is likely to be the core regulatory mechanism underlying the observed data.
Figure 6. Computational model and simulations of the proposed mechanism of SRF action.

A) A simplified diagram showing the existence of a negative feedback mechanism between SRF and AR under DHT stimulation. B) An assumed kinetic scheme describing the mechanistic reactions of the AR-SRF network that capture the negative feedback regulation (see Supplementary Information for details). C) Model simulations of SRF activity comparing scramble and AR knock-down using siRNA. D–E) Model simulations of AR activity and expression level comparing scramble and SRF knock-down using siRNA.
Discussion
Despite CRPC ability to survive in an androgen-depleted environment, AR remains a valid therapeutic target in advanced disease, which justifies the use of abiraterone acetate and Enzalutamide (MDV3100) in this setting [11]. However, reports of patients failing these treatments are starting to emerge [23], highlighting the need for additional novel and more effective therapeutic targets. Due to the fact that a significant proportion of CRPCs remain sensitive to ligand activation of AR [11], perhaps novel therapeutic targets should be identified among the numerous co-regulators and secondary transcription factors involved in AR action. We have recently identified SRF as a promising target for CRPC treatment [1]. In the current study we have gone on to show evidence supporting SRF association with AR and the potential use of SRF as a therapeutic target which works synergistically with AR inhibition.
Firstly we demonstrated that SRF transcriptional activity is responsive to androgen stimulation, which led us to investigate whether this was driven by AR. Our data showed that the Abl cells were much more sensitive to androgen stimulation compared with the parental LNCaP cells, which is in line with previous reports [12] and in agreement with androgen hypersensitivity as an important mechanism for surviving in an androgen-depleted environment [22]. Not only were the LNCaP Abl cells sensitive to androgen stimulation but SRF transcriptional activity was also decreased following Enzalutamide (MDV3100) and bicalutamide (data not shown) treatment in these cells. The fact that the LNCaP Abl cells are grown in CCS serum would support the concept that CRPC cells have the ability to produce their own androgens [24]. Supporting this, pathway analysis of the transcriptomics profiling which led to this study, identified steroid metabolism as one of the pathways associated with the differential gene expression observed ([1], data not shown).
We next wanted to assess whether SRF activation following androgen stimulation had the potential to affect AR transcriptional activity and expression. Vlahopoulos and colleagues had previously shown that AR was specifically recruited by serum response elements only in the presence of SRF in C2C12 myoblats, suggesting a protein-protein interaction between SRF and AR in these cells [10]. Here we showed that SRF transcriptional activity is dependent on AR protein expression, using two approaches: down-regulation of AR in LNCaP parental and Abl cells using siRNA and over-expression of AR in the AR-negative PC346 DCC cell line [13]. These results clearly demonstrated that SRF protein levels and transcriptional activity are dependent on AR protein expression, in line with a previous study showing that AR knock-down by siRNA prevented androgen induction of the SRF transcriptional targets, CNN2 and SDK1 [8]. In agreement with this, we have shown that AR silencing using siRNA interferes with the androgen induction of Twist-1, which we had previously identified as a predicted transcriptional target of SRF[1]. Interestingly, these experiments have highlighted a different behaviour in the parental LNCaP compared to the Abl cells. While in the parental LNCaP cells AR silencing prevented androgen induction of Twist-1, in the Abl cells there was a differential response depending on the absence or presence of androgen stimulation. In the absence of androgens Abl cells behaved like the parental LNCaP; however in the presence of DHT Twist-1 transcription was increased by AR silencing rather than being inhibited. While further studies are needed to elucidate this differential behaviour of Twist-1 in the Abl cells, we could speculate that Twist-1 transcription could be either stimulated or repressed depending on SRF levels. In other words there might be a certain threshold of SRF activation after which Twist-1 transcription is inhibited rather than enhanced.
Several studies reported that alterations in AR co-regulators can modulate AR activity when androgen levels are decreased [25–29]; we questioned whether this could be the case also for SRF. Our experiments showed that AR transcriptional activity is affected by SRF protein expression, suggesting a role for SRF as an AR co-regulator. The fact that SRF knock down by siRNA has only a minor effect on AR protein expression suggests that SRF may act as a modulator of AR post-translational modifications rather than directly modulating its transcription/translation. Modulation of AR activity by post-translational modifications can occur through several mechanisms including: protein stability, interaction with other proteins and cellular localisation [30]. Studies are ongoing in our laboratory to determine the molecular mechanisms by which SRF down-regulation affects AR protein expression and activity.
The increase in AR activity post SRF down-regulation is indicative of a negative feedback loop between SRF and AR. This negative feedback loop was confirmed to be clinically relevant in CRPC bone metastases which showed a negative correlation between SRF and AR. In contrast, a positive correlation between SRF and AR was demonstrated in radical prostatectomies which represent patients who would still be sensitive to androgen. This switch between a positive to a negative correlation between SRF and AR during prostate cancer progression would support the need for additional therapeutic targets in addition to AR in later stages of the disease. The importance of the negative feedback loop as the core regulatory mechanism underlying the observed data was highlighted by a computational model based on our experimental data. Based on this model, the negative feedback between SRF and AR may be responsible for the ability of these cells to grow in an androgen-depleted environment, where lower SRF transcriptional activity would drive up-regulation of AR with the consequential hypersensitivity to androgens. However, our model also predicts that AR up-regulation will in turn drive SRF transcriptional activity through which cells will survive due to SRF role in promoting cell survival and proliferation. Therefore concomitant inhibition of SRF and AR would seem a reasonable therapeutic approach to overcome up-regulation of both targets in CRPC. In order to test SRF as a potential therapeutic target, the impact of its inhibition on cellular viability and proliferation was compared with inhibiting AR, which is the main therapeutic target currently in clinical use. Inhibition of SRF using siRNA was more effective in decreasing cell proliferation compared to AR knock-down, suggesting that SRF may be a more effective target for CRPC. These results were partially confirmed by using small molecule inhibitors targeting SRF and AR. In the LNCaP Abl cells both inhibitors showed the same effect in decreasing cellular viability. Moreover, while the combined knock-down of SRF and AR did not show a further decrease in cellular proliferation compared with the single knock-down of SRF and AR, the use of CCG-1423 and Enzalutamide (MDV3100) in combination showed a synergistic effect in decreasing cell viability in both cell lines. The apparent discrepancy between the siRNA and the small molecule inhibitors can be explained by the fact that down-regulation of protein expression by siRNA cannot guarantee complete protein silencing, while by acting directly on the transcriptional activity of SRF and AR [31, 32], both CCG-1423 and MDV3100 have a more effective inhibition of SRF and AR respectively. Overall, both siRNA and small molecule inhibitors experiments pointed to SRF as an interesting target for CRPC; however the small molecule inhibitors are more clinically relevant since MDV3100 is already used in the clinic for CRPC patients and CCG-1423 has already been tested in in vivo models [32].
Conclusion
In conclusion this study has shown evidence of a cross-talk between AR and SRF in advanced prostate cancer. At the core of this cross-talk a negative feedback loop between SRF and AR was demonstrated in vitro and in clinical samples as well as being supported by a computational model. Our data highlight the importance of SRF in CRPC and support SRF as a promising therapeutic target in combination with current treatments.
Supplementary Material
Acknowledgments
Funding is acknowledged from the Irish Cancer Society under grant CRF12PRE, the Science Foundation Ireland, Strategic Research Cluster award to Molecular Therapeutics for Cancer Ireland (award 08/SRC/B1410) and Marie Curie (FP-7) Industry-Academia Partnerships and Pathways who fund this research under the “FAST-PATH” program. L.K.N was supported by Science Foundation Ireland under Grant No. 06/CE/B1129.
We thank the patients and their families who were willing to participate in the Prostate Cancer Donor Program. The Prostate Cancer Donor Program is supported by the Pacific Northwest Prostate Cancer SPORE (P50CA97186), the PO1 NIH grant (PO1CA085859), and the Richard M. LUCAS Foundation. The authors would like to thank Prof. Helmut Klocker from Innsbruck University for the LNCaP Abl cell line, Dr. Wytske van Weerden from Erasmus EC for the PC346 DCC cell line and Dr. Michael Mancini for the pEGFP-C1-AR plasmid.
References
- 1.Prencipe M, Madden SF, O’Neill A, O’Hurley Gillian, Culhane Aedin, O’Connor Darran, Klocker H, Kay EW, Gallagher WM, Watson WR. Identification of transcription factors associated with castration-resistance: is the Serum Responsive Factor a potential therapeutic target? Prostate. 2013;73(7):743–53. doi: 10.1002/pros.22618. [DOI] [PubMed] [Google Scholar]
- 2.Sun Q, Chen G, Streb JW, Long X, Yang Y, Stoeckert CJ, Jr, Miano JM. Defining the mammalian CArGome. Genome research. 2006;16(2):197–207. doi: 10.1101/gr.4108706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vickers ER, Kasza A, Kurnaz IA, Seifert A, Zeef LA, O’donnell A, Hayes A, Sharrocks AD. Ternary complex factor-serum response factor complex-regulated gene activity is required for cellular proliferation and inhibition of apoptotic cell death. Molecular and cellular biology. 2004;24(23):10340–51. doi: 10.1128/MCB.24.23.10340-10351.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heemers HV, Regan KM, Dehm SM, Tindall DJ. Androgen induction of the androgen receptor coactivator four and a half LIM domain protein-2: evidence for a role for serum response factor in prostate cancer. Cancer Res. 2007;67(21):10592–9. doi: 10.1158/0008-5472.CAN-07-1917. [DOI] [PubMed] [Google Scholar]
- 5.O’Hurley G, Prencipe M, Lundon D, O’Neill A, Boyce S, O’Grady A, Gallagher WM, Morrissey C, Kay EW, Watson W. The analysis of Serum Response Factor expression in bone and soft tissue Prostate Cancer metastases. Prostate. 2014;74:306–313. doi: 10.1002/pros.22752. [DOI] [PubMed] [Google Scholar]
- 6.Yu W, Feng S, Dakhova O, Creighton CJ, Cai Y, Wang J, Li R, Frolov A, Ayala G, Ittmann M. FGFR-4 Arg(3)(8)(8) enhances prostate cancer progression via extracellular signal-related kinase and serum response factor signaling. Clinical cancer research. 2011;17(13):4355–66. doi: 10.1158/1078-0432.CCR-10-2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heemers HV, Schmidt LJ, Sun Z, Regan KM, Anderson SK, Duncan K, Wang D, Liu S, Ballman KV, Tindall DJ. Identification of a clinically relevant androgen-dependent gene signature in prostate cancer. Cancer Res. 2011;71(5):1978–88. doi: 10.1158/0008-5472.CAN-10-2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Verone AR, Duncan K, Godoy A, Yadav N, Bakin A, Koochekpour S, Jin JP, Heemers HV. Androgen-responsive Serum Response Factor target genes regulate prostate cancer cell migration. Carcinogenesis. 2013;34(8):1737–46. doi: 10.1093/carcin/bgt126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schmidt LJ, Duncan K, Yadav N, Regan KM, Verone AR, Lohse CM, Pop EA, Attwood K, Wilding G, Mohler JL, Sebo TJ, Tindall DJ, Heemers HV. RhoA as a mediator of clinically relevant androgen action in prostate cancer cells. Mol Endocrinol. 2012;26(5):716–35. doi: 10.1210/me.2011-1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vlahopoulos S, Zimmer WE, Jenster G, Belaguli NS, Balk SP, Brinkmann AO, Lanz RB, Zoumpourlis VC, Schwartz RJ. Recruitment of the androgen receptor via serum response factor facilitates expression of a myogenic gene. J Biol Chem. 2005;280(9):7786–92. doi: 10.1074/jbc.M413992200. [DOI] [PubMed] [Google Scholar]
- 11.Massard C, Fizazi K. Targeting continued androgen receptor signaling in prostate cancer. Clin Cancer Res. 2011 Jun 15;17(12):3876–83. doi: 10.1158/1078-0432.CCR-10-2815. [DOI] [PubMed] [Google Scholar]
- 12.Culig Z, Hoffmann J, Erdel M, Eder IE, Hobisch A, Hittmair A, Bartsch G, Utermann G, Schneider MR, Parczyk K, Klocker H. Switch from antagonist to agonist of the androgen receptor bicalutamide is associated with prostate tumour progression in a new model system. Br J Cancer. 1999;81(2):242–51. doi: 10.1038/sj.bjc.6690684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marques RB, Erkens-Schulze S, de Ridder CM, Hermans KG, Waltering K, Visakorpi T, Trapman J, Romijn JC, van Weerden WM, Jenster G. Androgen receptor modifications in prostate cancer cells upon longterm androgen ablation and antiandrogen treatment. Int J Cancer. 2005;117:221–229. doi: 10.1002/ijc.21201. [DOI] [PubMed] [Google Scholar]
- 14.Marques RB, van Weerden WM, Erkens-Schulze S, de Ridder CM, Bangma CH, Trapman J, Jenster G. The human PC346 xenograft and cell line panel: a model system for prostate cancer progression. Eur Urol. 2006;49:245–257. doi: 10.1016/j.eururo.2005.12.035. [DOI] [PubMed] [Google Scholar]
- 15.Prencipe M, Fitzpatrick P, Gorman S, Tosetto M, Klinger R, Furlong F, Harrison M, O’Connor D, Roninson IB, O’Sullivan J, McCann A. Cellular senescence induced by aberrant MAD2 levels impacts on paclitaxel responsiveness in vitro. Br J Cancer. 2009;101:1900–1908. doi: 10.1038/sj.bjc.6605419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stenoien DL, Cummings CJ, Adams HP, Mancini MG, Patel K, DeMartino GN, Marcelli M, Weigel NL, Mancini MA. Polyglutamine-expanded androgen receptors form aggregates that sequester heat shock proteins, proteasome components and SRC-1, and are suppressed by the HDJ-2 chaperone. Hum Mol Genet. 1999;8(5):731–41. doi: 10.1093/hmg/8.5.731. [DOI] [PubMed] [Google Scholar]
- 17.Annette M, Dirac G, Bernards René. The Deubiquitinating Enzyme USP26 Is a Regulator of Androgen Receptor Signaling. Mol Cancer Res. 2010;8(6):844–54. doi: 10.1158/1541-7786.MCR-09-0424. [DOI] [PubMed] [Google Scholar]
- 18.O’Neill AJ, Prencipe M, Dowling C, Fan Y, Mulrane L, Gallagher WM, O’Connor D, O’Connor R, Devery A, Corcoran C, Rani S, O’Driscoll L, Fitzpatrick JM, Watson RW. Characterisation and manipulation of docetaxel resistant prostate cancer cell lines. Mol Cancer. 2011;10:126. doi: 10.1186/1476-4598-10-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roudier MP, True LD, Higano CS, Vesselle H, Ellis W, Lange P, Vessella RL. Phenotypic heterogeneity of end-stage prostate carcinoma metastatic to bone. Human pathology. 2003;34(7):646–53. doi: 10.1016/s0046-8177(03)00190-4. [DOI] [PubMed] [Google Scholar]
- 20.Zhang X, Morrissey C, Sun S, Ketchandji M, Nelson PS, True LD, Vakar-Lopez F, Vessella RL, Plymate SR. Androgen receptor variants occur frequently in castration resistant prostate cancer metastases. PloS one. 2011;6(11):e27970. doi: 10.1371/journal.pone.0027970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tassidis H1, Brokken LJ, Jirström K, Ehrnström R, Pontén F, Ulmert D, Bjartell A, Härkönen P, Wingren AG. Immunohistochemical detection of tyrosine phosphatase SHP-1 predicts outcome after radical prostatectomy for localized prostate cancer. Int J Cancer. 2010;126(10):2296–307. doi: 10.1002/ijc.24917. [DOI] [PubMed] [Google Scholar]
- 22.Pienta KJ, Bradley D. Mechanisms underlying the development of androgen-independent prostate cancer. Clin Cancer Res. 2006;12 (6):1665–1671. doi: 10.1158/1078-0432.CCR-06-0067. [DOI] [PubMed] [Google Scholar]
- 23.Yuan X, Cai C, Chen S, Chen S, Yu Z, Balk SP. Androgen receptor functions in castration-resistant prostate cancer and mechanisms of resistance to new agents targeting the androgen axis. Oncogene. 2013 doi: 10.1038/onc.2013.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, True LD, Nelson PS. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumour growth. Cancer Res. 2008;68:4447–4454. doi: 10.1158/0008-5472.CAN-08-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heinlein C, Chang C. Androgen receptor (AR) coregulators: an overview. Endoc Rev. 2002;23:175–200. doi: 10.1210/edrv.23.2.0460. [DOI] [PubMed] [Google Scholar]
- 26.Kaur R, Yuan X, Lu ML, Balk SP. Increased PAK6 expression in prostate cancer and identification of PAK6 associated proteins. Prostate. 2008;68:1510–1516. doi: 10.1002/pros.20787. [DOI] [PubMed] [Google Scholar]
- 27.Mohler JL, Gregory CW, Ford O, III, Kim D, Weaver CM, Petrusz P, Wilson EM, French FS. The androgen axis in recurrent prostate cancer. Clin Cancer Res. 2004;10:440–448. doi: 10.1158/1078-0432.ccr-1146-03. [DOI] [PubMed] [Google Scholar]
- 28.Taplin ME, Balk SP. Androgen receptor: a key molecule in the progression of prostate cancer to hormone independence. J Cell Biochem. 2004;91:483–490. doi: 10.1002/jcb.10653. [DOI] [PubMed] [Google Scholar]
- 29.Yuan X, Balk SP. Mechanisms mediating androgen receptor reactivation after castration. Urol Oncol. 2009;27:36–41. doi: 10.1016/j.urolonc.2008.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coffey K, Robson CN. Regulation of the androgen receptor by post-translational modifications. J Endocrinol. 2012;215(2):221–37. doi: 10.1530/JOE-12-0238. [DOI] [PubMed] [Google Scholar]
- 31.Evelyn CR, Wade SM, Wang Q, Wu M, Iñiguez-Lluhí JA, Merajver SD, Neubig RR. CCG-1423: a small-molecule inhibitor of RhoA transcriptional signaling. Mol Cancer Ther. 2007;6(8):2249–60. doi: 10.1158/1535-7163.MCT-06-0782. [DOI] [PubMed] [Google Scholar]
- 32.Jin W, Goldfine AB, Boes T, Henry RR, Ciaraldi TP, Kim EY, Emecan M, Fitzpatrick C, Sen A, Shah A, Mun E, Vokes V, Schroeder J, Tatro E, Jimenez-Chillaron J, Patti ME. Increased SRF transcriptional activity in human and mouse skeletal muscle is a signature of insulin resistance. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.