

Supplemental Digital Content is Available in the Text.
A functional mouse Comt allele, associated with pain behaviour, increases Comt expression by inserting an early polyadenylation signal and removing naturally occurring regulatory microRNA signals.
Keywords: Alternative splicing, miRNA, Molecular genetics, Transcription regulation, Comt
Abstract
A functional allele of the mouse catechol-O-methyltransferase (Comt) gene is defined by the insertion of a B2 short interspersed repeat element in its 3′-untranslated region (UTR). This allele has been associated with a number of phenotypes, such as pain and anxiety. In comparison with mice carrying the ancestral allele (Comt+), ComtB2i mice show higher Comt mRNA and enzymatic activity levels. Here, we investigated the molecular genetic mechanisms underlying this allelic specific regulation of Comt expression. Insertion of the B2 element introduces an early polyadenylation signal generating a shorter Comt transcript, in addition to the longer ancestral mRNA. Comparative analysis and in silico prediction of Comt mRNA potential targets within the transcript 3′ to the B2 element was performed and allowed choosing microRNA (miRNA) candidates for experimental screening: mmu-miR-3470a, mmu-miR-3470b, and mmu-miR-667. Cell transfection with each miRNA downregulated the expression of the ancestral transcript and COMT enzymatic activity. Our in vivo experiments showed that mmu-miR-667-3p is strongly correlated with decreasing amounts of Comt mRNA in the brain, and lentiviral injections of mmu-miR-3470a, mmu-miR-3470b, and mmu-miR-667 increase hypersensitivity in the mouse formalin model, consistent with reduced COMT activity. In summary, our data demonstrate that the Comt+ transcript contains regulatory miRNA signals in its 3′-untranslated region leading to mRNA degradation; these signals, however, are absent in the shorter transcript, resulting in higher mRNA expression and activity levels.
1. Introduction
Dysregulation of catecholamine signaling has been implicated in a variety of neurologic and neuropsychiatric disorders.20,27 Catechol-O-methyltransferase (COMT) is a critical enzyme in catechol catabolism. O-methylation of biologically active amines, such as epinephrine, norepinephrine, and dopamine, is a rate-limited step controlling bioavailability of these neurotransmitters.18,33 COMT has been implicated in a number of critical functions such as stress response,9,37 mood,60 and cognition.5 Furthermore, in recent years, the essential role of COMT activity in pain processes has become increasingly evident.22,49
Functional variability at the COMT gene locus has been identified in both the human population and laboratory mouse strains. It has been associated with responses to noxious stimuli,2,12,13 chronic pain conditions,11,13,53 intermediate pain phenotypes,11 and analgesics responding.56 In the human population, functional single nucleotide polymorphisms (SNPs) have been identified in the translated and untranslated regions (UTRs) of COMT mRNA. In the UTRs, an SNP situated 3′ to the coding region (3′-UTR) affects COMT transcript level,4 and an SNP situated in the 5′-UTR increases the efficiency of translation through relaxing tertiary mRNA structure.61 In the coding region, a number of variations have been established, representing several molecular genetic mechanisms. The nonsynonymous change of valine to methionine at codon 158 of COMT reduces protein stability,31 and the interaction between synonymous and nonsynonymous SNPs alters mRNA secondary structure leading to reduced translation efficiency.41 For SNPs in the promoter region of COMT gene locus, evidence for cis-acting influences on mRNA expression also has been observed.4,41
In mouse laboratory strains, the insertion of a type of transposon termed B2 short interspersed element (SINE) in the 3′-UTR of Comt has been shown to be associated with decreased stress response, decreased anxiety, and a decreased assortment of pain phenotypes mediated by increased mRNA and protein amounts.24,29,50 Overall, the presence of the B2 SINE element significantly decreased sensitivity to noxious stimuli measured by summation of 20 nociceptive assays,28 with largest contribution of acute spontaneous inflammatory nociception.2,50,67
Almost half of the laboratory mouse strains tested carry this insertion and thus demonstrate increased Comt level. It has been proposed that the B2 SINE insertion shortens the 3′-UTR of the mRNA by allowing utilization of an alternative polyadenylation signal (PAS), a sequence of nucleotides that signals the transcript to end with a poly (A) tail.24,29 However, the mechanisms underlying the connection between a shorter Comt mRNA 3′-UTR and increased mRNA and protein amounts have not been identified. Indeed, it is unclear whether the shorter transcript should be more abundant, and if so, what drives its higher level.
Here, we investigated and established the molecular genetic mechanisms contributing to the B2 SINE–dependent upregulation of Comt expression. We hypothesized that the 3′-UTR of the ancestral Comt mRNA (Comt+) contains regulatory signals responsible for mRNA degradation that are absent in the shorter alternative Comt mRNA isoform with the B2 SINE insertion.
2. Materials and Methods
2.1. Animal husbandry
All experiments adhered to guidelines set by the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals by the Canadian Council on Animal Care (CCAC) and were approved by the Institutional Animal Care and Use Committees at the University of North Carolina (UNC), the Genomics Institute of the Novartis Research Foundation (GNF), and McGill University. Mice at GNF and UNC-Chapel Hill were housed in specific pathogen-free colonies and were maintained on a 12-hour light–dark cycle. Standard high efficiency particulate air-filtered polycarbonate mouse cages housed between 2 and 4 mice. Every cage held a cotton nestlet on Bed-o-cob bedding. Water and food (Purina, USA) were available ad libitum. Mice at McGill were housed with their same-sex littermates (4 animals per cage) in standard shoebox cages, maintained in a temperature-controlled (20°C ± 1°C) environment (14:10-hour light/dark cycle), and fed (Harlan Teklad 8604) and watered ad libitum. Mice were assigned to experimental conditions in a randomized fashion within the cage.
2.2. Inbred strains
The 29 strains tested were 129S1/SvImJ, A/J, AKR/J, BALB/cByJ, BTBR T + tf/J, BUB/BnJ, C3H/HeJ, C57BL/6J, C57BR/cdJ, C58/J, CBA/J, CE/J, DBA/2J, FVB/NJ, I/LnJ, KK/HlJ, MA/MyJ, MRL/MpJ, NOD/LtJ, NON/LtJ, NZO/HlLTJ, NZW/LacJ, P/J, PL/J, RIIIS/J, SJL/J, SM/J, SWR/J, and WSB/EiJ.
2.3. Collection of specific brain regions for Affymetrix array data
Gene expression data were collected from the 29 mouse strains listed above as part of the GNF initiative. The data were collected at the GNF using the Affymetrix GeneChip 430v2 whole-genome mouse arrays. Mice of both sexes (8-10 weeks of age) were purchased from the Jackson Laboratory (Bar Harbor, ME) and habituated for 1 week before sacrifice by cervical dislocation without anesthesia. Dissections were performed between 0900 and 1130 hours. The regions dissected were prefrontal cortex, amygdala, hypothalamus, hippocampus, nucleus accumbens, striatum, and pituitary gland. Brain region dissections are described in more detail in Ref. 50.
2.4. Affymetrix array data: RNA, cDNA preparation, and gene expression analysis
After dissection, tissues were placed in RNase-free microcentrifuge tubes, set on dry ice, and stored at −80°C. Frozen tissue was pulverized to a fine powder (tissue pulverizer; Cole-Palmer, Vernon Hills, IL) in liquid nitrogen to obtain a homogeneous tissue sample. An aliquot was further homogenized in TRIzol (Life Technologies, Carlsbad, CA) with a rotor–stator homogenizer (Omni International, Kennesaw, GA). RNeasy columns (Qiagen, Hilden, Germany) were used to purify total RNA, and RNA integrity was assessed on a bioanalyzer (Agilent, Santa Clara, CA). Pools of high-quality RNA from 3 mice per strain, per sex, and per brain region were used to produce the cDNA required for 1 microarray. The Affymetrix 1-cycle target labeling kit was used to amplify and label the RNA used for each Affymetrix GeneChip 430v2 whole-genome mouse array. Probe sets 1449183_at and 1418701_at were used to analyze Comt hybridization pattern in the coding and 3′-UTR regions, respectively. Gene expression was analyzed using standard procedures,55 data were processed using the gcRMA algorithm,65 and raw data deposited under series accession GSE20160 in GEO (http://ncbi.nih.gov/geo). Z scores of the Comt transcript in the coding region and 3′-UTR region were calculated using the formula 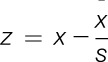 , where x = individual strain mean,
, where x = individual strain mean, 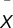 = group mean for all strains, and s = SD across strains.
= group mean for all strains, and s = SD across strains.
2.5. Cell constructs
Generation of the expression plasmids carrying the ancestral allele (pComt+) or the B2 SINE insertion (pComtB2i) has been described in detail previously.50 Briefly, expression plasmids have been generated in pCMV-SPORT6 by Dr Oskar Laur at the Custom Cloning Core Facility, Emory University. miExpress precursor microRNA (miRNA) expression clones were purchased from GeneCopoeia, Inc, Rockville, MD. The miRNA generating vectors used were cloned in pEZX-MR04 under a CMV promoter. The expression vectors were specific for mmu-miR3470a and mmu-miR-3470b. As the prediction software did not specify mmu-miR-667-3p or mmu-miR 667-5p, the precursor mmu-miR-667, which makes both transcripts, was ordered. pEZX without an insert was used as the empty vector control. All clones were verified by sequencing the entire cDNA insert in both directions. The full sequence of the clones can be found in Supplementary Table 1 (available online as Supplemental Digital Content at http://links.lww.com/PAIN/A116).
2.6. Cell transfection experiments: transfection, cDNA preparation, enzymatic, and gene expression analysis
2.6.1. Transfection
A mouse leukemic monocyte macrophage cell line, RAW 264.7, was transiently transfected into 24-well plates using TransIT-3T3 tranfection kit (Mirus, Madison WI) at a density of 50,000 cells per milliliter of media, in accordance with manufacture's recommendations. To account for transfection efficiency, a control plasmid, pSEAP2 (Clontech, Mountain View, CA), was kept at 10% of the total plasmid DNA transfected. Cells were harvested 18 to 24 hours after transfection.
2.6.2. cDNA preparation
TRIzol (Life Technologies) was used to isolate whole RNA from cell culture dishes, in accordance with the manufacturer's guidelines. Because expression constructs were added to the cell culture, it was necessary to reduce plasmid contamination from true cDNA signal. Turbo DNAse (Life Technologies) was used with the manufacturer's protocol in their rigorous DNAse treatment protocol. A no reverse transcriptase control sample was prepared at the same time for each cDNA synthesis. cDNA was synthesized using Superscript III (Life Technologies) in accordance with the manufacturer's instruction, using 500 ng total RNA per 20 μL reaction.
2.6.3. Gene expression analysis
All q-polymerase chain reaction (PCR) data were collected at UNC-Chapel Hill on an Applied Biosystems (Waltham, MA) 7900HT real-time PCR machine fitted with a 384-well reaction block (Life Technologies). Taqman and SYBR green assays were run in separate wells on the same plate. Polymerase chain reaction cycling conditions are as follows: 40 cycles of 15 seconds at 95°C followed by 60 seconds at 60°C, followed by a thermal melting curve to ensure a single dissociation peak. The Comt transcript was measured in the coding region using Taqman assay Mm00514377_m1, using TaqMan Universal PCR Master Mix. For the Comt 3′-UTR region, we designed primers in the region covered in the Affymetrix array probe set 1418701_at. Forward and reverse primers for the 3′-UTR amplimer are CTCACTAACTGGAAGAGGAGATTTTT and CATTCTTAAGATG AAATGTGGTATGA, respectively. SYBR Green Real-Time PCR Master Mix (Life Technologies) was used according to the manufacturer's recommendations. Data were analyzed using the comparative cycle threshold method,30 with the normalizing sample pComt+ or pComt+ × pmiR Empty Vector. Murine Mrpl19, Taqman assay Mm00452754_m1, was used as the housekeeping gene. For transfection efficiency, we measured the amount of Secreted Alkaline Phosphatase (SEAP) transcript with SYBR Green, using forward and reverse primers GCCGACCACTCCCACGTCTT and CCCGCTCTCGCTCTCGGTAA, respectively.
2.6.4. Enzymatic analysis
The enzymatic protocol follows Tsao et al.,62 which quantifies the amount of Comt enzymatic activity by the quantity of radiolabeled catechol extracted from a lysate. To account for transfection efficiency of the expression vectors into the cells, the media were sampled immediately before harvesting the cells and the SEAP assay was performed according to the manufacturer's recommendation. Samples with transfection efficiencies below 40% of the highest SEAP value were not used. After removing media, cells were washed with cold 1X PBS. Each well was covered by 65 μL of enzymatic buffer, composed of 10 mM Tris (pH 7.4), 1 mM MgCl2, and 1 μM DTT, and frozen overnight at −80°C. Plates were thawed a single time on ice, lysates collected, and cell lysis confirmed by visualization in the Cellometer (Nexcelom, Lawrence, MA). Lysates were normalized in enzymatic buffer for protein content using a BCA assay. Eighty microliters of an enzymatic master mix was added to 20 μL of normalized lysates in enzymatic buffer. The Master Mix is an enzymatic buffer with 10 μM catechol (Fisher Scientific, Waltham, MA), dissolved in H2O, and S-[methyl-3H]-adenosyl-l-methionine (10.0 Ci/mmol; PerkinElmer). The strip tubes were incubated in a PCR machine for 20 minutes at 37°C, and the reaction terminated with 100 μL of 1 M HCl, pipetted into each well with a multichannel pipette. Each 200 μL reaction was added to 10 mL scintillation fluid (Monoflow I; National Diagnostics Hessle, United Kingdom), vortexed briefly, and quantified using a scintillation counter. The range of the assay was determined in each experiment by a serial dilution of one of the lysates to generate a linear curve of scintillation values. The scintillation values were then normalized by the empirically determined SEAP multiplier.
2.6.5. Data analysis
Each experiment had between 2 and 4 technical replicates, and at least 3 experiments were conducted on 3 separate days. Results are expressed as mean ± SEM, from combining Z scores from individual experiments. Statistical significance was determined using GraphPad Prism, version 5.01 for Windows (GraphPad Software, San Diego, CA), 1-tailed, nonpaired, Student's t test.
2.6.6. Prediction of microRNA targets
Taking into account experimental evidence that AGO-bound target sites show strong structural accessibility,63 as well as miRNA target accessibility patterns from prior computational predictions,51 we considered predicted target sites located in the “open” regions of the mRNA, where mRNA folding stability is not low, that is, the local secondary structure might be easily unfolded and does not significantly influence target site accessibility.51,63 We analyzed RNA local secondary structures in full-length 3′-UTR, in the downstream region of the early PAS in ComtB2i transcripts, as well as in B2 SINE region and B1 repeat region, also situated within 3′-UTR of Comt+ mRNA. The local secondary structure was predicted by the Afold software,43 and the free energy of the target site accessibility was estimated as described earlier.51 The variability of RNA secondary structure predictions was minimized by using local RNA folding with a short window length (60 nucleotides) when estimating the accessibility of target sites.35 Other features that are important for optimization of miRNA-predicted targets are (1) the free energy of the duplex between miRNA and mRNA targets and (2) a stable duplex in the “seed” region containing a pairing pattern.58 The ThermoComposition software34 uses the following thresholds to predict stable miRNA–mRNA duplexes: (1) duplex free energy ≤−20 kcal/mol, (2) seed dG ≤ −10 kcal/mol, (3) seed length >8 nucleotides. In our selection algorithm, we considered recent experimental results on the location of optimal miRNA targets in 3′-UTRs.63 For unique sequences that are not in the repeat regions, we also used miRWalk program (http://mirwalk.uni-hd.de) to predict miRNA targets in the area of interest.
We analyzed sequence conservation of the predicted target sites between mouse and rat Comt 3′-UTRs using the rat genome downloaded from the University of California at Santa Cruz genome web browser (http://genome.ucsc.edu). The Owen sequence alignment tool25,43 was used to align the ancestral mouse Comt 3′-UTR sequences of the miRNA target sites to rat sequences.
2.7. Collection of hippocampus, hypothalamus, and amygdala for Comt and micro-RNA q-polymerase chain reaction
After generation of cDNA array expression results, no additional mRNA samples from the initial 29 mice strains were available for measuring mmu-miRs. We then limited our q-PCR experiments to the subset of 4 representative strains that were separately purchased from the Jackson Laboratory. Furthermore, based on the cDNA array expression analysis results, we concluded that the molecular effect of the B2 SINE element was the same for male and female mice (Fig. 2B). We thus carried out q-PCR experiments to test the correlation between relative Comt mRNA and mmu-miRs expression levels in male mice only, avoiding contribution of the female estrous cycle to expression of Comt, and thus reducing experimental SD.
Figure 2.

Comparison of expression levels of Comt+ and ComtB2i transcripts in vivo and in vitro. (A) The pComt+ construct has the entire ancestral sequence of Comt transcript (NM_001111063); the pComtB2i construct contains the same entire sequence with the B2 SINE insertion; the pComtB2i No 3′-UTR has the sequence after the major PAS within the B2 SINE removed. (B) In vivo Comt mRNA expression levels in the PFC of Comt+ and ComtB2i female and male animals measured in the coding and 3′-UTR regions. Z scores show that ComtB2i animals have significantly higher expression level than Comt+ animals when Comt is measured in the coding region, and significantly lower expression levels when Comt is measured in the 3′-UTR. (C) In vitro Comt mRNA expression levels in cells transfected with pComt+ or one of the 2 pComtB2i constructs measured in the coding and 3′-UTR regions. Z scores show that transfections with the pComtB2i construct result in higher expression level than with the pComt+ when Comt is measured in the coding region; in contrast with in vivo results, when Comt is measured in the 3′-UTR, transfection with the pComtB2i or the pComt+ construct results in similar expression levels. The pComtB2i No 3′-UTR mimics the shorter alternative ComtB2i transcript in the mouse generated by utilization of the alternative PAS, and transfection with it results in higher expression level than that of pComt+ when Comt is measured in the coding region. Significance is denoted as * for P < 0.05, ** for P < 0.01, and ***for P < 0.0001.
Ten-week-old male mice were housed and habituated at UNC-Chapel Hill as closely as possible to the protocol used at GNF. Data were collected from 16 individual 10-week-old male mice, 4 from each strain: AKR/J, BALB/cByJ, C3H/HeJ, and SJL/J. To increase the amount of available brain material, a region containing the hippocampus, hypothalamus, and amygdala (HHA) was dissected, and material from each mouse was kept separately in an attempt to capture intrastrain diversity in gene expression. Mice used for q-PCR experiments were sacrificed according to the guidelines set by the Institutional Animal Care and Use Committee (IACUC) at UNC-Chapel Hill.
2.8. Real-time polymerase chain reaction reagents and assays using HHA brain tissue
2.8.1. Reverse transcription polymerase chain reaction for Comt coding mRNA levels
All reagents were purchased from Life Technologies. For mouse brain RNA isolation, the HHA sample from each animal was collected and processed in the same manner as brain material for the Affymetrix array, up to the step of pulverizing the frozen tissue sample. An aliquot of frozen powdered material was further homogenized in TRIzol with a 16-gauge syringe, and total RNA was isolated according to the manufacturer's instructions. Total RNA was treated with Turbo DNase. Comt coding region cDNA was synthesized using the high-capacity cDNA reverse transcription kit, using 500 ng total RNA per 20 μL reaction, per the manufacturer's protocol. A control sample without reverse transcriptase (RT) enzyme was prepared at the same time for each cDNA synthesis, to ensure against amplification from plasmid or amplimer contamination. TaqMan Universal PCR Master Mix was used for q-PCR, as per the manufacturer's protocol. Data were analyzed using the comparative cycle threshold method,30 with data normalized to an arbitrarily selected animal. Comt transcript was measured in the coding region using Taqman assay Mm00514377_m1. Mouse Gapdh was included as an endogenous control.
2.8.2. Reverse transcription polymerase chain reaction for microRNA levels
All reagents were purchased from Life Technologies. For miRNA isolation from mouse brain, the remaining material from each frozen pulverized HHA sample was used with the mirVana miRNA isolation kit (Life Technologies) in accordance with the manufacturer's recommendations. Both miRNA and whole RNA extraction from brain were conducted as quickly as possible to avoid RNase degradation, and each extraction was taken from a single HHA aliquot per animal. To check RNA integrity, RNA preparations were run on an Agilent Bioanalyzer, as described previously. Taqman probes were purchased for quantification of mmu-miRs: 667-3p, 667-5p, 3470a, 3470b, and snoRNA202 as an endogenous control. TaqMan Universal PCR Master Mix without AmpErase UNG was used according to the manufacturer's protocol.
2.8.3. Data analysis
We determined the relative amount of Comt coding transcript levels in 15 animals (4 from strains AKR/J, C3H/HeJ, and SJL/J, and 3 from strain BALB/cByJ) normalized to an arbitrarily selected animal from the C3H/HeJ strain, which then had a Comt transcript value of 1.0. The fifteen other animals had Comt coding transcript values between 0.9 and 2.5 times than that of the arbitrarily selected animal. Normalization to the same arbitrarily selected animal was also conducted for the miRNA values, so that the amount of Comt transcript to amount of miRNA transcript the arbitrarily selected animal would always be 1.0 to 1.0. A linear regression between each miRNA of interest and amount of Comt transcript was then plotted using the GraphPad Prism program (version 6.03), which calculated the r2 goodness of fit and the P value for significance.
2.9. Expression pattern of microRNAs in the published data sets
The relative expression of mmu-miR-667-3p, -667-5p, -3470a, and -3470b, across mouse tissues and cells was estimated using the following published RNA-seq data sets: brain, testis, ovary, spleen, bone marrow, skin, salivary glands, whole body, embryo, and mouse NIH-3T3 fibroblastoma cell line.7
2.10. Lentiviral injection of microRNAs and formalin testing
Purified lentiviral particles for miRNA (mmu-miR-3470a, mmu-miR-3470b, and mmu-miR-667) were pseudotyped with VSV-G protein and generated using a standardized protocol with highly purified plasmids and EndoFectin-Lenti and TiterBoost reagents (GeneCopoeia Inc). The lentiviral transfer vector (pEZX-MR03) was cotransfected into 293Ta cells (catalog No. CLv-PK-01) with Lenti-Pac HIV packaging mix (catalog No.: HPK-LvTR-20). The lentivirus particles were purified and stored at -80°C in aliquots (purified particles). Full-length sequencing, restriction enzyme digestion, and PCR size validation using gene-specific and vector-specific primers were used to validate the lentiviral expression construct. Lentiviral particles were injected subcutaneously into the plantar surface of the left hind paw (12 μL total) using custom-designed small hub 30-gauge Hamilton needles at a concentration of 1.45 × 109 transduction units (TU) per milliliter. The TU of the lentiviral particles was estimated using the following formula: 1 TU = 100 copies of viral genomic RNA.
Mice of both sexes (outbred CD-1 [ICR:Crl]) were obtained from Charles River (Boucherville, QC, Canada). For formalin testing, mice were placed on a tabletop within Plexiglas cylinders (30 cm high and 15 cm diameter) and allowed to habituate. Then, 20 μL of 1% formalin was injected subcutaneously into the plantar surface of the left hind paw using a 100-μL microsyringe with a 30-gauge needle. Mice were then returned to the cylinders and left undisturbed for 60 minutes, with behaviors recorded using a digital video camera. Videos were later coded offline, where the first 10 seconds of every minute was monitored for the presence of licking/biting (positive sample) of the left hind paw for a total of 60 observations. The early phase was defined as the percentage of positive samples during the first 0 to 10 minutes after injection of formalin; the late phase as the percentage of positive samples during the period 10 to 60 minutes after injection. Formalin testing was conducted 21 days after injection of the lentiviral miRNAs.
3. RESULTS
3.1. Affymetrix microarray data reveal differential mRNA expression in the coding and 3′-UTR regions in the Comt+ and the ComtB2i mouse strains
Gene expression data were collected from 7 mouse brain regions (prefrontal cortex, amygdala, hypothalamus, hippocampus, nucleus accumbens, striatum, and pituitary gland) of the 29 mouse strains carrying either the ancestral (Comt+) or the B2 SINE insertion (ComtB2i) alleles. Consistent with previous reports, mouse strains carrying the ComtB2i allele showed higher levels of Comt mRNA than Comt+ animals when mRNA was measured in the coding region of the transcript27,56; the opposite expression profile was noted for the 3′-UTR region, where ComtB2i animals showed less mRNA than strains carrying the Comt+ allele (Fig. 1A, Supplementary Figures 1-4, available online as Supplemental Digital Content at http://links.lww.com/PAIN/A116). The B2 SINE insertion carries a strong PAS (Fig. 1B), AATAAA, which is duplicated to AATAAATAAA. The AATAAA is the most frequently used PAS in mouse genome (Fig. 1C), and considering the duplication of this signal, the probability of using this PAS is very high. Furthermore, the AATAAA PAS signal is followed by a less frequent PAS mouse signal, TTTAAA, both of which are in part of the B2 SINE sequence (Fig. 1D). The results of 3′RACE experiments suggested that these PASs are used in vivo.24 Utilization of these PASs leads to the expression of a substantially shorter Comt mRNA transcript than the ancestral one. The switched expression pattern was seen in all tested brain regions in both male and female mice (Fig. 1A, Supplementary Figures 1-4, available online as Supplemental Digital Content at http://links.lww.com/PAIN/A116) measured by the Affymetrix array, and this difference was statistically significant in virtually all 7 tested brain regions (Supplementary Table 2, available online as Supplemental Digital Content at http://links.lww.com/PAIN/A116). These results suggest that both full-length and shorter mRNA isoforms are expressed in ComtB2i mouse strains with higher prevalence of the latter, and the effect of the B2 SINE insertion is identical for both males and females. Our results also demonstrate lower overall Comt expression in female mice (Supplementary Table 2, available online as Supplemental Digital Content at http://links.lww.com/PAIN/A116), which is consistent with previous reports for both mice and humans,33,50 and is due to estrogen downregulation of Comt expression in females.66
Figure 1.
Microarray hybridization pattern of coding vs 3′-UTR regions for mouse Comt mRNA expressed by 29 Comt+ and ComtB2i mouse inbred lines. (A) The Affymetrix array reveals bimodal Comt expression hybridization patterns in coding (left) vs 3′-UTR (right) regions. Each bar represents the z score of 1 inbred line. Inbred mouse lines carrying the ancestral allele, Comt+ (white bars), have greater mRNA hybridization signal in the 3′-UTR than in the coding region, whereas strains carrying the ComtB2i allele (black bars) have the opposite hybridization profile in both female (top) and male (bottom) animals. The representative expression profile of prefrontal cortex is presented; see Supplementary Figures 1 to 4 for other brain regions. (B) Schematics of the ancestral (Comt+, top) and of the 2 alternative (ComtB2i, middle and bottom) allelic transcripts showing the coding and 3′-UTR array target regions, the B1 and B2 SINE transposons, and the ancestral (black star) and alternative (grey star) PASs. The alternative ComtB2i allele can generate both a full-length transcript containing B2 SINE transposon sequence (middle) and a shorter transcript that ends after the B2 SINE-inserted PAS (bottom). (C) Sequence of PAS hexamers and frequency found in the mouse genome, reported by Tian et al.59 (D) Sequence of the full-length ComtB2i transcript with marked B2 SINE and B1 transposons, as well as the ancestral and alternative PASs.
3.2. Comt expression constructs reveal polyadenylation signal selectivity
We designed Comt expression vectors with either the ancestral 3′-UTR (pComt+) or the 3′-UTR containing the B2 SINE insertion (pComtB2i; Fig. 2A). The pComtB2i constructs showed greater COMT enzymatic activity in different cell lines (Supplementary Figure 5, available online as Supplemental Digital Content at http://links.lww.com/PAIN/A116), which was in agreement with the previously reported higher Comt enzymatic activity from ComtB2i brain lysates.50 The mRNA levels in our cell transfection experiments reflected our in vivo observations to a certain extent. The in vivo results indicated lower mRNA levels in Comt+ animals when targeting the coding region, and higher mRNA levels when targeting the 3′-UTR region of Comt, as compared with ComtB2i animals, and measured by the overall Z score of Comt expression level across 7 different brain regions (Fig. 2B). The pattern of expression is virtually identical in both males and females, confirming that the effect of the B2 SINE insertion is the same for both males and females, regardless of baseline expression levels (Fig. 2B). The in vitro experiments were in the same direction, with a significant, although weaker, difference in mRNA levels: the pComtB2i construct showed higher mRNA levels when targeting the coding region but no significant difference in mRNA levels when targeting the 3′-UTR region compared with the pComt+ construct (Fig. 2C). The fold difference in the levels of mRNA in the coding region for pComtB2i vs pComt+ constructs was more modest than in ComtB2i vs Comt+ animals. We then hypothesized that the B2 SINE–inserted PAS was not used as efficiently in the in vitro system. To test if the shorter Comt mRNA is more stable than full size, we created a new construct, ComtB2i No 3′-UTR, in which we truncated the pComtB2i plasmid after the B2 SINE PAS (Fig. 2A). This construct showed significantly higher mRNA levels than the pComt+ construct in the coding region, as we predicted. The COMT enzymatic activity of this construct was also in line with mRNA levels (Supplementary Figure 5D, available online as Supplemental Digital Content at http://links.lww.com/PAIN/A116). Together, these data led us to propose the existence of regulatory elements in the 3′-UTR of Comt contributing to RNA stability.
3.3. Early polyadenylation signal eliminates microRNA target regions
To find miRNA targets that may have been lost by the early PAS in the ComtB2i transcripts, we analyzed the 3′-UTR Comt region immediately upstream of the later PAS of the full Comt transcript and downstream of the predicted PAS at the end of the B2 repeat region. We selected 3 miRNA targets from the list of top predicted targets: mmu-miR-667-3p, mmu-miR-667-5p, and mmu-miR-3470a/b (Fig. 3). All these targets are located in the predominantly unpaired regions. Both mature miRNAs, mmu-miR-667-3p and mmu-miR-667-5p, derived from original mmu-miR-667 precursor miRNA, are relatively unique in the mouse genome/transcriptome and could not be masked by a RepeatMasker search.52 Furthermore, mmu-miR-667 is the only candidate situated in the area of interest whose targets are in the list of top 3 predicted miRNA target sites with P < 0.001, as predicted by the miRWalk program (Supplementary Table 3, available online as Supplemental Digital Content at http://links.lww.com/PAIN/A116). miRNAs mmu-miR-3470a/b recognize the same site within the 3′-UTR of Comt mRNA, mapped in the rodent-specific B1 repeat region, and located close to the end of the longest known transcript (Fig. 3). Because mmu-miR-3470a and mmu-miR-3470b are located within B1 transposon sequence, their targets are extensively dispersed throughout the genome and cannot be considered unique on a transcriptome level. Because most of the repeat regions have the ability to interact and form highly stable duplexes, the free energy threshold is higher for repeats and different algorithms should be applied for searching these areas.23,36
Figure 3.

Predicted miRNA sites and profile of free energy of target accessibility in the 3′-UTR of the murine Comt gene. (A) Alignment of the 3 predicted murine miRNA target positions to the 3′-UTR of Comt+ and location of ancestral PAS (black star); (B) alignment of the 3 predicted murine miRNA target positions to the 3′-UTR of ComtB2i and location of the ancestral PAS (black star) and alternative PAS (gray star), when the full transcript is made in ComtB2i animals; (C) when the alternative PAS (gray star) is used in ComtB2i animals, the 3 predicted miRNA target sites for mmu-miR-667-3/-5 and mmu-miR-3470a/b are absent; (D) target locations for mmu-miR-667-3p (in red), mmu-miR-667-5p (in green), for mmu-miR-3470a or mmu-miR-3470b (in yellow) and their respective free energy (kcal/mol).
We next analyzed the conservation of miRNA targets. We only compared rodent genomes, because mmu-miR-667 is annotated only in mouse and rat genomes, and the target for mmu-miR-3470a/b is also located within the rodent-specific B1 transposon in the miRbase database. We found that the seed regions of mmu-miR-667-3p and -5p targets were substantially conserved between mouse and rat (Supplementary Figure 6, available online as Supplemental Digital Content at http://links.lww.com/PAIN/A116) in the 3′-UTR of the Comt+ transcript. Furthermore, the target of mmu-miR-3470a and mmu-miR-3470b is also extremely well conserved between mouse and rat because of its location in the B1 transposon (Supplementary Figure 6, available online as Supplemental Digital Content at http://links.lww.com/PAIN/A116).
Thus, we selected miRNAs mmu-miR-667-3p, mmu-miR-667-5p, mmu-miR-3470a, and mmu-miR-3470b as most reliable candidates to investigate which miRNA could differentially regulate full but not truncated Comt transcripts, taking into account that their target accessibility is high, display favorable free-energy duplex formation profiles within seed regions, exhibit high conservation, and are located within our area of interest.
3.4. Regulation of ancestral Comt by 3 microRNA in murine RAW cells
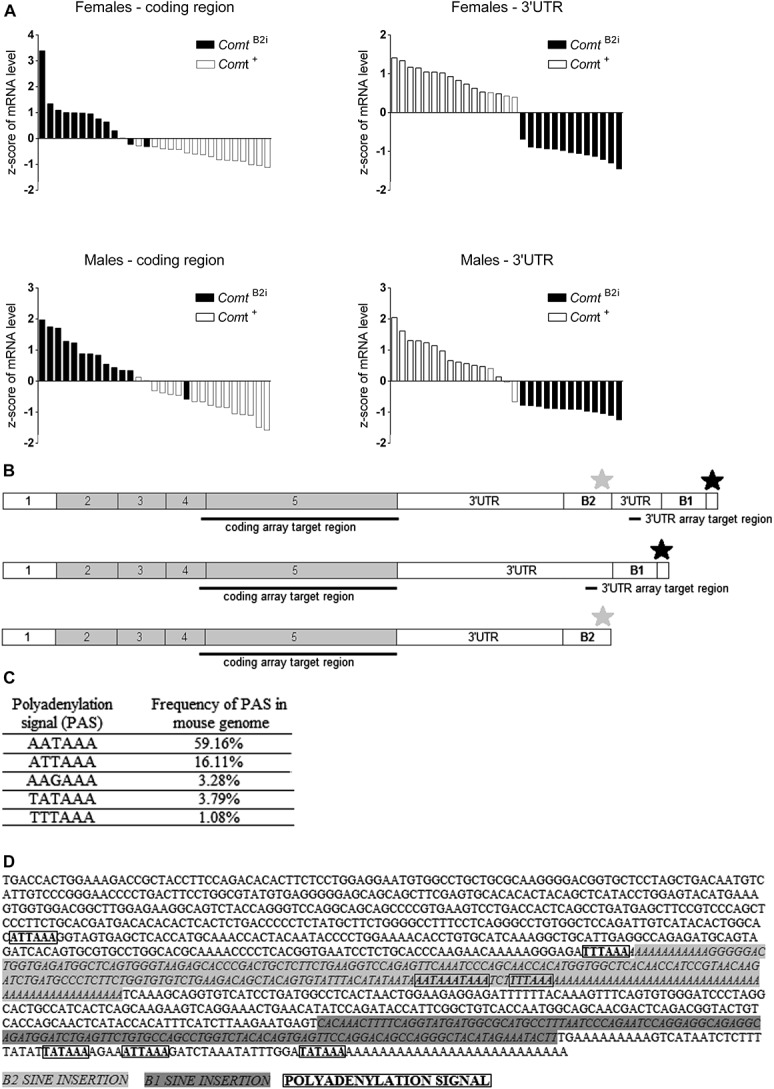
To provide evidence that ancestral Comt levels can be regulated by the identified miRNA targets absent in the truncated isoform, we transfected murine RAW cells with the ancestral pComt+ construct, a normalizing vector, and an expression vector containing the precursor for one of the 3 miRNAs. The expression vector for mmu-miR667 produces both species mmu-miR-667-3p and mmu-miR-667-5p. As determined by RT-PCR, Comt mRNA levels measured in the coding region were significantly decreased by the miRNA constructs pmiR 3470a (P < 0.0001) and pmir 3470b (P < 0.05) and were also decreased by pmiR 667, almost reaching significance (P = 0.0578) (Fig. 4A). Comt enzymatic activity was significantly decreased by all 3 miRNAs (Fig. 4B). These results demonstrate that both mRNA and enzymatic activity of murine ancestral but not truncated Comt can be downregulated by mmu-miR-667, mmu-miR-3470a, or mmu-miR-3470b miRNAs.
Figure 4.
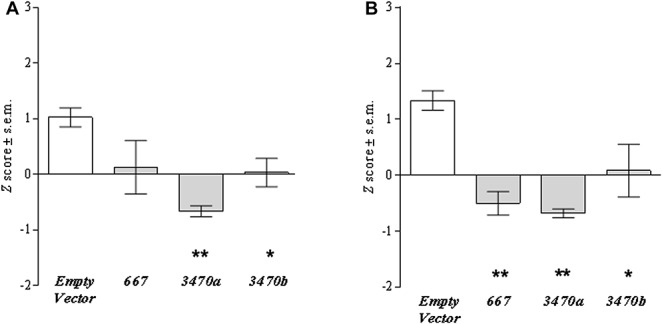
Effect of cell transfection with each miRNA expression construct on Comt mRNA expression levels and enzymatic activities. The pComt+ construct was cotransfected with pSEAP and either pmiR-Empty Vector, pmiR-667, pmiR-3470a, or pmiR-3470b; Comt mRNA expression was measured in the coding region, and SEAP activity was used for normalization of transfection efficiency. (A) Comt mRNA expression levels and (B) enzymatic activity in each transfected sample were compared with that of pComt+ cotransfected with pmiR-Empty Vector. Transfection with all miRNAs downregulated mRNA expression level and enzymatic activity, although the downregulation of mRNA expression by miR-667 did not reach significance (P = 0.0578). Significance is denoted as * for P < 0.05, ** for P < 0.01.
3.5. Correlation of endogenous microRNAs and Comt transcript levels in mouse brain
To provide evidence that ancestral Comt transcript level is regulated by the identified miRNA targets in vivo, we measured the transcript level of both the Comt coding region and each of the 3 miRNAs in individual mouse brain regions: hippocampus, hypothalamus, and amygdala. A new separate cohort of male AKR/J and BALB/cByJ (ComtB2i strains) and C3H/HeJ and SJL/J (Comt+ strains) mice were purchased and tested. We observed a significant negative correlation between mmu-miR-667-3p and Comt transcripts in ComtB2i and Comt+ animals (Fig. 5B). Neither mmu-miR-667-5p, mmu-miR-3470a, nor mmu-mi-3470b was significantly correlated with Comt transcript levels, although a negative slope was observed for all miRNAs (Fig. 5A, Supplementary Fig. 7A and B, available online as Supplemental Digital Content at http://links.lww.com/PAIN/A116). These results are in line with our in vitro findings on mmu-miR-667-3p and further demonstrate that brain tissues expressing higher levels of mmu-miR-667-3p express lower levels of Comt mRNA. Together with our in vitro experiments, these results suggest direct downregulation of Comt by mmu-miR-667-3p.
Figure 5.
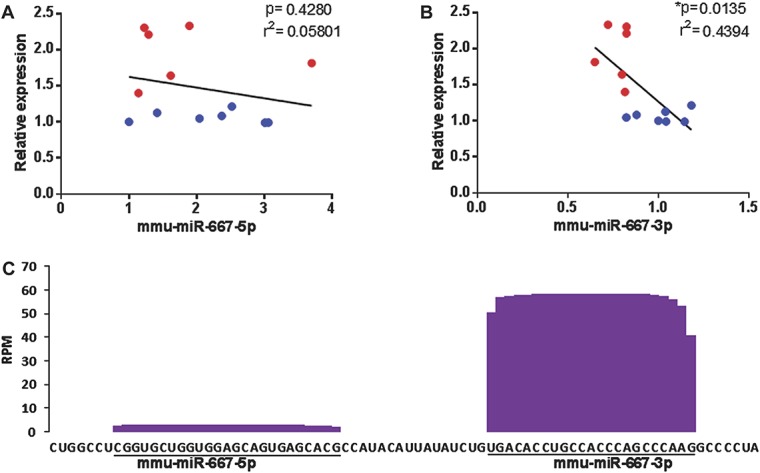
Correlation between relative mRNA expression levels of Comt and mmu-miR-667-3p or mmu-miR-667-5p. (A and B) RT-PCR results were generated on the independent cohort of 4 mouse strains: AKR/J and BALB/cByJ (ComtB2i strains), and C3H/HeJ and SJL/J (Comt+ strains). Relative Comt mRNA levels were measured in the coding region and plotted against relative miRNA levels from the same sample. Each data point represents a single mouse from the ComtB2i (red) or Comt+ (blue) strains. Results show (A) a strong negative correlation between increasing amounts of mmu-miR-667-3p and decreasing amounts of Comt transcript and (B) no correlation between amounts of mmu-miR-667-5p and Comt transcript. (C) RNA-seq read coverage of the mature mmu-miR-667-5p and mmu-miR-667-3p in mouse tissues and cell lines. The number of reads per million (RPM) of assessed mouse tissues (brain, testis, ovary, spleen, bone marrow, skin, and salivary glands) and cell line is presented.7
To investigate potential molecular processes underlying the association between mmu-miR-667-3p and Comt mRNA levels, but not mmu-miR-667-5p, mmu-miR-3470a, and mmu-miR-3470b, we estimated the relative abundance of all 4 miRNAs in different mouse tissues. We used a data set of differential tissue-specific miRNA expression available from Chiang et al.7 We observed that the mean relative expression of mmu-miR-667-3p miRNAs across all tissues, including brain, was 20 times higher than the mean relative expression of mmu-miR-667-5p (Supplementary Table 4, available online as Supplemental Digital Content at http://links.lww.com/PAIN/A116 and Fig. 5C). The relative expressions of mmu-miR-3470a and mmu-miR-3470b were 4 to 10 times lower than relative expression of mmu-miR-667-3p (Supplementary Tables 5 and 6, and Supplementary Figure 7C, available online as Supplemental Digital Content at http://links.lww.com/PAIN/A116). Although this difference is not as high as mmu-miR-667-5p, the location of mmu-miR-3470a/b targets within the B1 repeat region would further deplete their effective miRNA concentration because of its wide distribution throughout the mouse genome. Thus, mmu-miR-667-3p has a much higher relative expression, and therefore, can more efficiently influence relative level of Comt mRNA that is generally highly expressed in the brain.
3.6. Modulation of pain responses by lentiviral microRNA injections
To confirm that mmu-miR-3470a, mmu-miR-3470b, and mmu-miR-667 modulate pain responses in vivo, we subcutaneously injected the miRNAs into the plantar surface of the foot using lentiviral vectors. Outbred CD-1 mice were used because they do not have a B2 SINE insertion and thus have full-length 3′-UTR of Comt mRNA and thus targets for selected miRNAs. We chose to inject the virus peripherally because there is a substantial difference in Comt contribution to modulation of pain sensitivity through peripheral vs spinal adrenergic signalling. Nociceptive, inflammatory, and musculoskeletal pain seems to be driven by peripheral adrenergic receptor stimulation, and lower Comt activity is hyperlagesic.49
The complete time course for all formalin testing is shown in Fig. 6A. After formalin injection, mmu-miR-3470a was the only miRNA that significantly increased immediate licking behaviour (Fig. 6B). However, in the late tonic phase (10-60 minutes) of the formalin test, all 3 miRNAs significantly increased licking behaviour when compared with an injection of scrambled cDNA (Fig. 6B). This demonstrates that, at least peripherally mmu-miR-3470a, mmu-miR-3470b, and mmu-miR-667 increase hypersensitivity during tonic inflammatory pain states, which is consistent with reduced Comt activity.
Figure 6.
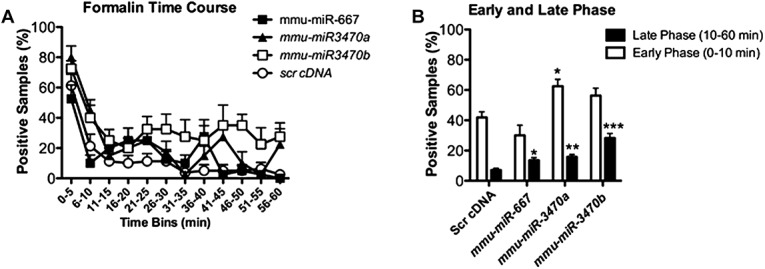
Intraplantar injection of mmu-miR-667, mmu-miR-3470a, and mmu-miR-3470b increases hypersensitivity on the formalin assay of nocifensive behavior. (A) The time course data for licking in the formalin test are presented for day 21 post-miRNA intraplantar injection. (B) The averaged data for the early (0-10 minutes) and late (10-60 minutes) phases of the formalin test are shown. Only mmu-miR-3470a increased licking during the early phase; however, all 3 miRNAs increased licking during the late phase of the assay when compared with scrambled cDNA (scr cDNA). Statistical analyses were performed using a 1-way ANOVA and compared all groups on either the early or late phase separately; n = 8 per group. Significance is denoted as * for P < 0.05, ** for P < 0.01, and *** for P < 0.0001.
4. Discussion
Investigation of functional genetic variants, such as the B2 SINE insertion, that regulate gene expression and function continues to be one of most challenging aspects of human genetic studies and related animal models. First, the discovery of truly functional genetic variants, but not their markers, assures that association patterns will be stable in future studies and allows for practical use of these genetic variants if needed, for example, in diagnostic testing. Second, a deep understanding of the exact order of molecular and cellular events connecting sequence alteration at a gene locus with alteration of protein function provides the ability to intervene in the process, leading to an ultimate understanding of the pathophysiology of genetic variability and ability to modify the pathological phenotypes.
Genetic variability in the COMT gene locus in both human and mouse has been reproducibly associated with multiple phenotypes. A reduced activity allele of human COMT64 has been implicated in an assortment of diseases and personality traits, such as obsessive–compulsive disorder,46 attention-deficit hyperactivity disorder,10,44,47 disproportionate anxiety response,14,15,17,21,39,54 mood,3 working memory,57 and cognition.16,32 In mouse laboratory strains, a B2 SINE insertion has been associated with stress response and anxiety behaviors.24,29,50 Furthermore, COMT seems to be one of the major contributors to variation in sensitivity to noxious stimuli and pathological pain states.2,11,22,40,56,67 Previously, the molecular genetic mechanisms of human COMT gene locus variability have been well studied,13,41,42 but the molecular genetic mechanisms whereby a SINE insertion in the 3′-UTR of mouse Comt affects the activity of the enzyme remained unknown.
De novo insertions of repeat elements into the human genome commonly result in negative outcomes.1 The contribution of repeat elements into alterations of gene function has not been as well characterized in mouse. It is estimated that 37.5% of the mouse genome is made up of transposable elements (SINEs, LINEs, and other interspersed repeat elements), with 1% being SINEs.8 The B2 SINE has been the most extensively studied, with publications listing its effects on transcription, alternative splicing, and evolution.6,26,38,48 However, the B2 SINE insertion in Comt is the first description of a repeat element upregulating gene expression, rather than downregulating it.
In studying the molecular genetic mechanism of the SINE-dependent Comt mRNA upregulation, we first tested whether the SINE insertion promotes alternative mRNA splicing by providing an early PAS in the Comt gene locus, and if this shorter mRNA is more stable. Analysis of cDNA array data showed that both Comt mRNAs, the full-length and shorter isoforms, are expressed in ComtB2i mouse strains, with preference for the shorter isoform. Our in vitro studies using expression vectors with cloned Comt cDNA with and without the SINE insertion (pComtB2i and pComt+ constructs, respectively) showed that the difference in the levels of shorter and longer isoforms expressed by pComtB2i construct is even smaller. However, the plasmid construct with no 3′-UTR after the SINE insertion shows very high stability. These data provide evidence that even when the SINE-derived earlier PAS is used only partially, the short mRNA isoform is much more stable and contributes significantly to the overall increase in mRNA levels of Comt in ComtB2i mouse strains.
Frequently, changes in mRNA levels reflect the impact of miRNAs on gene expression and indicate that destabilization of target mRNAs is the predominant reason for reduced protein output.19 These recent findings drove us to identify potential miRNA targets. Specifically, we searched for potential silencing signals in the sequence between the early PAS within the B2 SINE insertion and the later PAS in the Comt+ 3′-UTR using computational target prediction approaches.35,36,43,51
We then tested if ancestral Comt level can be regulated by the identified miRNA targets absent in the truncated Comt isoform both in vitro and in vivo. Our in vitro results demonstrated that both mRNA and enzymatic activity of murine ancestral Comt can be downregulated by mmu-miR-667, mmu-miR-3470a, or mmu-miR-3470b miRNAs. Interestingly, mmu-miR-667 showed much more robust downregulation of COMT protein level than mRNA level, suggesting that mmu-miR-667 may control not only the level of Comt mRNA stability but also the translational efficiency. Although the latter mechanism of miRNA-dependent gene expression regulation is not the main one, it has been observed previously. Recent studies on ribosome profiling demonstrated that approximately 16% of the decrease in protein production is not accounted for by lowered mRNA levels in both ectopic and endogenous miRNA regulatory interactions.19 Furthermore, it is interesting to note that transfection of all 3 miRNAs expression constructs leads to substantial biological outcomes on the transfected cells, such as intense cell growth. Whether the latter effect is related to an alteration in Comt levels or it is driven by other target(s) of these miRNAs is a subject for future studies.
We also correlated mRNA levels of Comt and each of the 4 miRNAs in individual mouse brains regions. Interestingly, only mmu-miR-667-3p showed a significant negative correlation with Comt levels. These data only partially support our in vitro results, which indicated that all 3 miRNA targets have functional effects by downregulating Comt mRNA level. However, taking into account our results on downstream estimation of endogenous expression levels of all miRNAs in mouse brain tissues, we would suggest that the absence of in vivo correlation between the levels of mmu-miR-667-5p, mmu-miR-3470a, mmu-miR-3470b miRNAs, and Comt mRNA is not evidence that these miRNA targets are not functional. Rather, it may mean that the Comt mRNA and miRNA levels were not in comparable ranges in the tested tissues and animals, so that these miRNAs could not regulate Comt mRNA at these conditions. There may be other tissues or conditions in which mmu-miR-667-5p, miR-3470a, and miR-3470b miRNAs would be able to regulate the level of Comt mRNA. Finally, our results of lentiviral injections of mmu-miR-3470a, mmu-miR-3470b, and mmu-miR-667 demonstrate that all 3 miRNAs increase hypersensitivity during tonic inflammatory pain states in the mouse formalin model, suggesting that in vivo induction of all tested miRNAs downregulates Comt mRNA level.
The identification of the molecular mechanism whereby an alternative allele that is common in laboratory mice regulates Comt function advances our understanding of basic biological processes and has implication for human genetics. First, it is clear that the molecular genetic mechanisms of COMT functional variation are highly diverse. All functional human COMT allelic variants affect COMT activity through a multitude of unique mechanisms, such as regulation of mRNA stability,4 protein stability,31 or secondary and tertiary mRNA structures.41,61 And yet, mouse functional genetic variation regulates Comt activity through another mechanism that has no reported analogy in humans. This suggests that functional molecular genetic studies will likely continue to be challenging and low throughput. Second, the presence of functional miRNA target sites in the 3′-UTR of mouse Comt warrants the search for functional miRNA target sites in the 3′-UTR of human COMT. In fact, our data show that even partial removal of the 3′-UTR carrying these targets yields substantial increases in Comt mRNA stability and protein amount. Considering that these regulatory sites are most likely to be conserved in evolution, they are of great significance and can play an important role in regulation of human COMT.
An additional translational component of this study lies in its contribution to the ongoing debate regarding the role of COMT in disorders such as anxiety and pain. Our mouse studies describe the molecular mechanism of a newly discovered allele of Comt function as an elegant proof-of-concept for diseases of catecholamine dysregulation. Evidence from monkey to human suggests that there is a strong correlation between the progressive increase in catecholamines with a concurrent increase in working memory, anxiety disorders, and pain conditions.45 The domesticated mice with the B2 SINE insertion are a reflecting mirror of the effects of low catecholamines on the same processes and represent a new animal model of the effects of genetic quantitative regulation of Comt and catechol load on Comt-dependent traits, including pain.
In summary, we demonstrated that the presence of the B2 SINE insertion allows utilization of an alternative PAS within a significant subpopulation of mRNA molecules, creating an alternative Comt mRNA with a shorter 3′-UTR. This alternatively spliced Comt mRNA does not carry targets for mmu-miR-667 and mmu-miR-3470a/b miRNAs, which downregulate both Comt mRNA stability and protein synthesis. Our study reveals a new molecular mechanism whereby Comt level and activity can be regulated through miRNAs and explains how the B2 SINE insertion eliminates naturally accruing degradation signals, leading to an increase in COMT protein levels.
Conflict of interest statement
The authors have no conflicts of interest to declare.
This work was supported by the National Research Service Award Institutional Research Training Program for the Next Generation of Oral Health Researchers (NextGen) [Grant No. T90-DE021986], by the Canadian Institutes of Health Research (CIHR) (Grant No. CERC08), by the National Institute of Dental and Craniofacial Research (Grant Nos. RO1-DE16558 and UO1-DE017018), by the National Institute of Neurological Disorders and Stroke (Grant No. PO1-NS045685 to L.D.), and by the Brazilian Federal Agency for the Support and Evaluation of Graduate Education (Grant No. CAPES/PDEE 0968-11-0).This research was also supported in part by the Intramural Research Program of the National Library of Medicine, NIH.
Acknowledgements
The authors thank Jean-Sebastien Austin for his help with tissue dissections and collection.
Appendix A. Supplemental Digital Content
Supplemental Digital Content associated with this article can be found online at http://links.lww.com/PAIN/A116.
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.painjournalonline.com).
References
- [1].Batzer MA, Deininger PL. Alu repeats and human genomic diversity. Nat Rev Genet 2002;3:370–9. [DOI] [PubMed] [Google Scholar]
- [2].Belfer I, Segall SK, Lariviere WR, Smith SB, Dai F, Slade GD, Rashid NU, Mogil JS, Campbell CM, Edwards RR, Liu Q, Bair E, Maixner W, Diatchenko L. Pain modality- and sex-specific effects of COMT genetic functional variants. PAIN 2013;154:1368–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bevilacqua L, Goldman D. Genetics of emotion. Trends Cogn Sci 2011;15:401–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bray NJ, Buckland PR, Williams NM, Williams HJ, Norton N, Owen MJ, O'Donovan MC. A haplotype implicated in schizophrenia susceptibility is associated with reduced COMT expression in human brain. Am J Hum Genet 2003;73:152–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Buckert M, Kudielka BM, Reuter M, Fiebach CJ. The COMT Val158Met polymorphism modulates working memory performance under acute stress. Psychoneuroendocrinology 2012;37:1810–21. [DOI] [PubMed] [Google Scholar]
- [6].Chernova T, Higginson FM, Davies R, Smith AG. B2 SINE retrotransposon causes polymorphic expression of mouse 5-aminolevulinic acid synthase 1 gene. Biochem Biophys Res Commun 2008;377:515–20. [DOI] [PubMed] [Google Scholar]
- [7].Chiang HR, Schoenfeld LW, Ruby JG, Auyeung VC, Spies N, Baek D, Johnston WK, Russ C, Luo S, Babiarz JE, Blelloch R, Schroth GP, Nusbaum C, Bartel DP. Mammalian microRNAs: experimental evaluation of novel and previously annotated genes. Genes Dev 2010;24:992–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Deininger PL. SINEs: short interspersed repeat elements in higher eucaryotes. In: Berg DE, Howe MM, editors. Mobile DNA. Washington, DC: American Society of Microbiology, 1989. p. 619–36. [Google Scholar]
- [9].Desbonnet L, Tighe O, Karayiorgou M, Gogos JA, Waddington JL, O'Tuathaigh CM. Physiological and behavioural responsivity to stress and anxiogenic stimuli in COMT-deficient mice. Behav Brain Res 2012;228:351–8. [DOI] [PubMed] [Google Scholar]
- [10].DeYoung CG, Getchell M, Koposov RA, Yrigollen CM, Haeffel GJ, af Klinteberg B, Oreland L, Ruchkin VV, Pakstis AJ, Grigorenko EL. Variation in the catechol-O-methyltransferase Val 158 Met polymorphism associated with conduct disorder and ADHD symptoms, among adolescent male delinquents. Psychiatr Genet 2010;20:20–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Diatchenko L, Fillingim RB, Smith SB, Maixner W. The phenotypic and genetic signatures of common musculoskeletal pain conditions. Nat Rev Rheumatol 2013;9:340–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Diatchenko L, Nackley AG, Slade GD, Bhalang K, Belfer I, Max MB, Goldman D, Maixner W. Catechol-O-methyltransferase gene polymorphisms are associated with multiple pain-evoking stimuli. PAIN 2006;125:216–24. [DOI] [PubMed] [Google Scholar]
- [13].Diatchenko L, Slade GD, Nackley AG, Bhalang K, Sigurdsson A, Belfer I, Goldman D, Xu K, Shabalina SA, Shagin D, Max MB, Makarov SS, Maixner W. Genetic basis for individual variations in pain perception and the development of a chronic pain condition. Hum Mol Genet 2005;14:135–43. [DOI] [PubMed] [Google Scholar]
- [14].Domschke K, Deckert J, O'Donovan MC, Glatt SJ. Meta-analysis of COMT val158met in panic disorder: ethnic heterogeneity and gender specificity. Am J Med Genet B Neuropsychiatr Genet 2007;144B:667–73. [DOI] [PubMed] [Google Scholar]
- [15].Drabant EM, Hariri AR, Meyer-Lindenberg A, Munoz KE, Mattay VS, Kolachana BS, Egan MF, Weinberger DR. Catechol O-methyltransferase val158met genotype and neural mechanisms related to affective arousal and regulation. Arch Gen Psychiatry 2006;63:1396–406. [DOI] [PubMed] [Google Scholar]
- [16].Egan MF, Goldberg TE, Kolachana BS, Callicott JH, Mazzanti CM, Straub RE, Goldman D, Weinberger DR. Effect of COMT Val108/158 Met genotype on frontal lobe function and risk for schizophrenia. Proc Natl Acad Sci U S A 2001;98:6917–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Evans J, Xu K, Heron J, Enoch MA, Araya R, Lewis G, Timpson N, Davies S, Nutt D, Goldman D. Emotional symptoms in children: the effect of maternal depression, life events, and COMT genotype. Am J Med Genet B Neuropsychiatr Genet 2009;150B:209–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Guldberg HC, Marsden CA. Catechol-O-methyl transferase: pharmacological aspects and physiological role. Pharmacol Rev 1975;27:135–206. [PubMed] [Google Scholar]
- [19].Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 2010;466:835–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Harrison PJ, Tunbridge EM. Catechol-O-methyltransferase (COMT): a gene contributing to sex differences in brain function, and to sexual dimorphism in the predisposition to psychiatric disorders. Neuropsychopharmacology 2008;33:3037–45. [DOI] [PubMed] [Google Scholar]
- [21].Hettema JM, An SS, Bukszar J, van den Oord EJ, Neale MC, Kendler KS, Chen X. Catechol-O-methyltransferase contributes to genetic susceptibility shared among anxiety spectrum phenotypes. Biol Psychiatry 2008;64:302–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kambur O, Mannisto PT. Catechol-O-methyltransferase and pain. Int Rev Neurobiol 2010;95:227–79. [DOI] [PubMed] [Google Scholar]
- [23].Kapitonov VV, Jurka J. A universal classification of eukaryotic transposable elements implemented in Repbase. Nat Rev Genet 2008;9:411–12; author reply 414. [DOI] [PubMed] [Google Scholar]
- [24].Kember RL, Fernandes C, Tunbridge EM, Liu L, Paya-Cano JL, Parsons MJ, Schalkwyk LC. A B2 SINE insertion in the Comt1 gene (Comt1(B2i)) results in an overexpressing, behavior modifying allele present in classical inbred mouse strains. Genes Brain Behav 2010;9:925–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kondrashov AS, Shabalina SA. Classification of common conserved sequences in mammalian intergenic regions. Hum Mol Genet 2002;11:669–74. [DOI] [PubMed] [Google Scholar]
- [26].Kress M, Barra Y, Seidman JG, Khoury G, Jay G. Functional insertion of an Alu type 2 (B2 SINE) repetitive sequence in murine class I genes. Science 1984;226:974–7. [DOI] [PubMed] [Google Scholar]
- [27].Lachman HM. Does COMT val158met affect behavioral phenotypes: yes, no, maybe? Neuropsychopharmacology 2008;33:3027–9. [DOI] [PubMed] [Google Scholar]
- [28].Lariviere WR, Wilson SG, Laughlin TM, Kokayeff A, West EE, Adhikari SM, Wan Y, Mogil JS. Heritability of nociception. III. Genetic relationships among commonly used assays of nociception and hypersensitivity. PAIN 2002;97:75–86. [DOI] [PubMed] [Google Scholar]
- [29].Li Z, Mulligan MK, Wang X, Miles MF, Lu L, Williams RW. A transposon in Comt generates mRNA variants and causes widespread expression and behavioral differences among mice. PLoS One 2010;5:e12181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001;25:402–8. [DOI] [PubMed] [Google Scholar]
- [31].Lotta T, Vidgren J, Tilgmann C, Ulmanen I, Melen K, Julkunen I, Taskinen J. Kinetics of human soluble and membrane-bound catechol O-methyltransferase: a revised mechanism and description of the thermolabile variant of the enzyme. Biochemistry 1995;34:4202–10. [DOI] [PubMed] [Google Scholar]
- [32].Malhotra AK, Kestler LJ, Mazzanti C, Bates JA, Goldberg T, Goldman D. A functional polymorphism in the COMT gene and performance on a test of prefrontal cognition. Am J Psychiatry 2002;159:652–4. [DOI] [PubMed] [Google Scholar]
- [33].Mannisto PT, Kaakkola S. Catechol-O-methyltransferase (COMT): biochemistry, molecular biology, pharmacology, and clinical efficacy of the new selective COMT inhibitors. Pharmacol Rev 1999;51:593–628. [PubMed] [Google Scholar]
- [34].Matveeva O, Nechipurenko Y, Rossi L, Moore B, Saetrom P, Ogurtsov AY, Atkins JF, Shabalina SA. Comparison of approaches for rational siRNA design leading to a new efficient and transparent method. Nucleic Acids Res 2007;35:e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Matveeva OV, Kang Y, Spiridonov AN, Saetrom P, Nemtsov VA, Ogurtsov AY, Nechipurenko YD, Shabalina SA. Optimization of duplex stability and terminal asymmetry for shRNA design. PLoS One 2010;5:e10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Matveeva OV, Nazipova NN, Ogurtsov AY, Shabalina SA. Optimized models for design of efficient miR30-based shRNAs. Front Genet 2012;3:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].McLean SA, Diatchenko L, Lee YM, Swor RA, Domeier RM, Jones JS, Jones CW, Reed C, Harris RE, Maixner W, Clauw DJ, Liberzon I. Catechol O-methyltransferase haplotype predicts immediate musculoskeletal neck pain and psychological symptoms after motor vehicle collision. J Pain 2011;12:101–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Michel D, Chatelain G, Mauduit C, Benahmed M, Brun G. Recent evolutionary acquisition of alternative pre-mRNA splicing and 3' processing regulations induced by intronic B2 SINE insertion. Nucleic Acids Res 1997;25:3228–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Mier D, Kirsch P, Meyer-Lindenberg A. Neural substrates of pleiotropic action of genetic variation in COMT: a meta-analysis. Mol Psychiatry 2010;15:918–27. [DOI] [PubMed] [Google Scholar]
- [40].Mogil JS. Pain genetics: past, present and future. Trends Genet 2012;28:258–66. [DOI] [PubMed] [Google Scholar]
- [41].Nackley AG, Shabalina SA, Tchivileva IE, Satterfield K, Korchynskyi O, Makarov SS, Maixner W, Diatchenko L. Human catechol-O-methyltransferase haplotypes modulate protein expression by altering mRNA secondary structure. Science 2006;314:1930–3. [DOI] [PubMed] [Google Scholar]
- [42].Nackley AG, Tan KS, Fecho K, Flood P, Diatchenko L, Maixner W. Catechol-O-methyltransferase inhibition increases pain sensitivity through activation of both beta2- and beta3-adrenergic receptors. PAIN 2007;128:199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ogurtsov AY, Shabalina SA, Kondrashov AS, Roytberg MA. Analysis of internal loops within the RNA secondary structure in almost quadratic time. Bioinformatics 2006;22:1317–24. [DOI] [PubMed] [Google Scholar]
- [44].Palmason H, Moser D, Sigmund J, Vogler C, Hanig S, Schneider A, Seitz C, Marcus A, Meyer J, Freitag CM. Attention-deficit/hyperactivity disorder phenotype is influenced by a functional catechol-O-methyltransferase variant. J Neural Transm 2010;117:259–67. [DOI] [PubMed] [Google Scholar]
- [45].Papaleo F, Crawley JN, Song J, Lipska BK, Pickel J, Weinberger DR, Chen J. Genetic dissection of the role of catechol-O-methyltransferase in cognition and stress reactivity in mice. J Neurosci 2008;28:8709–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Pooley EC, Fineberg N, Harrison PJ. The met(158) allele of catechol-O-methyltransferase (COMT) is associated with obsessive-compulsive disorder in men: case-control study and meta-analysis. Mol Psychiatry 2007;12:556–61. [DOI] [PubMed] [Google Scholar]
- [47].Qian Q, Wang Y, Zhou R, Li J, Wang B, Glatt S, Faraone SV. Family-based and case-control association studies of catechol-O-methyltransferase in attention deficit hyperactivity disorder suggest genetic sexual dimorphism. Am J Med Genet B Neuropsychiatr Genet 2003;118B:103–9. [DOI] [PubMed] [Google Scholar]
- [48].Roman AC, Benitez DA, Carvajal-Gonzalez JM, Fernandez-Salguero PM. Genome-wide B1 retrotransposon binds the transcription factors dioxin receptor and Slug and regulates gene expression in vivo. Proc Natl Acad Sci U S A 2008;105:1632–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Segall SK, Maixner W, Belfer I, Wiltshire T, Seltzer Z, Diatchenko L. Janus molecule I: dichotomous effects of COMT in neuropathic vs nociceptive pain modalities. CNS Neurol Disord Drug Targets 2012;11:222–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Segall SK, Nackley AG, Diatchenko L, Lariviere WR, Lu X, Marron JS, Grabowski-Boase L, Walker JR, Slade G, Gauthier J, Bailey JS, Steffy BM, Maynard TM, Tarantino LM, Wiltshire T. Comt1 genotype and expression predicts anxiety and nociceptive sensitivity in inbred strains of mice. Genes Brain Behav 2010;9:933–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Shabalina SA, Spiridonov AN, Ogurtsov AY. Computational models with thermodynamic and composition features improve siRNA design. BMC Bioinformatics 2006;7:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Smit AFA, Hubley R, Green P. RepeatMasker Open-4.0. Available at: http://repeatmasker.org. 2013–2015. Accessed May 2015. [Google Scholar]
- [53].Smith SB, Maixner DW, Greenspan JD, Dubner R, Fillingim RB, Ohrbach R, Knott C, Slade GD, Bair E, Gibson DG, Zaykin DV, Weir BS, Maixner W, Diatchenko L. Potential genetic risk factors for chronic TMD: genetic associations from the OPPERA case control study. J Pain 2011;12(11 suppl):T92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Stein DJ, Newman TK, Savitz J, Ramesar R. Warriors versus worriers: the role of COMT gene variants. CNS Spectr 2006;11:745–8. [DOI] [PubMed] [Google Scholar]
- [55].Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, Cooke MP, Walker JR, Hogenesch JB. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci U S A 2004;101:6062–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Tammimäki A, Männistö PT. Catechol-O-methyltransferase gene polymorphism and chronic human pain: a systematic review and meta-analysis. Pharmacogenet Genomics 2012;22:673–91. [DOI] [PubMed] [Google Scholar]
- [57].Tan HY, Chen Q, Goldberg TE, Mattay VS, Meyer-Lindenberg A, Weinberger DR, Callicott JH. Catechol-O-methyltransferase Val158Met modulation of prefrontal-parietal-striatal brain systems during arithmetic and temporal transformations in working memory. J Neurosci 2007;27:13393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Thomas M, Lieberman J, Lal A. Desperately seeking microRNA targets. Nat Struct Mol Biol 2010;17:1169–74. [DOI] [PubMed] [Google Scholar]
- [59].Tian B, Hu J, Zhang H, Lutz CS. A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res 2005;33:201–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Tost H, Alam T, Meyer-Lindenberg A. Dopamine and psychosis: theory, pathomechanisms and intermediate phenotypes. Neurosci Biobehav Rev 2010;34:689–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Tsao D, Shabalina SA, Gauthier J, Dokholyan NV, Diatchenko L. Disruptive mRNA folding increases translational efficiency of catechol-O-methyltransferase variant. Nucleic Acids Res 2011;39:6201–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Tsao D, Wieskopf JS, Rashid N, Sorge RE, Redler RL, Segall SK, Mogil JS, Maixner W, Dokholyan NV, Diatchenko L. Serotonin-induced hypersensitivity via inhibition of catechol O-methyltransferase activity. Mol Pain 2012;8:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Wan Y, Qu K, Zhang QC, Flynn RA, Manor O, Ouyang Z, Zhang J, Spitale RC, Snyder MP, Segal E, Chang HY. Landscape and variation of RNA secondary structure across the human transcriptome. Nature 2014;505:706–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Weinshilboum RM, Raymond FA. Inheritance of low erythrocyte catechol-o-methyltransferase activity in man. Am J Hum Genet 1977;29:125–35. [PMC free article] [PubMed] [Google Scholar]
- [65].Wu C, Delano DL, Mitro N, Su SV, Janes J, McClurg P, Batalov S, Welch GL, Zhang J, Orth AP, Walker JR, Glynne RJ, Cooke MP, Takahashi JS, Shimomura K, Kohsaka A, Bass J, Saez E, Wiltshire T, Su AI. Gene set enrichment in eQTL data identifies novel annotations and pathway regulators. PLoS Genet 2008;4:e1000070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Xie T, Ho SL, Ramsden D. Characterization and implications of estrogenic down-regulation of human catechol-O-methyltransferase gene transcription. Mol Pharmacol 1999;56:31–8. [DOI] [PubMed] [Google Scholar]
- [67].Young EE, Costigan M, Herbert TA, Lariviere WR. Heritability of nociception IV: neuropathic pain assays are genetically distinct across methods of peripheral nerve injury. PAIN 2014;155:868–80. [DOI] [PMC free article] [PubMed] [Google Scholar]