

Abstract
There are thousands of known associations between genetic variants and complex human phenotypes, and the rate of novel discoveries is rapidly increasing. Translating those associations into knowledge of disease mechanisms remains a fundamental challenge because the associated variants are overwhelmingly in noncoding regions of the genome where we have few guiding principles to predict their function. Intersecting the compendium of identified genetic associations with maps of regulatory activity across the human genome has revealed that phenotype-associated variants are highly enriched in candidate regulatory elements. Allele-specific analyses of gene regulation can further prioritize variants that likely have a functional effect on disease mechanisms; and emerging high-throughput assays to quantify the activity of candidate regulatory elements are a promising next step in that direction. Together, these technologies have created the ability to systematically and empirically test hypotheses about the function of noncoding variants and haplotypes at the scale needed for comprehensive and systematic follow-up of genetic association studies. Major coordinated efforts to quantify regulatory mechanisms across genetically diverse populations in increasingly realistic cell models would be highly beneficial to realize that potential.
The ultimate goal of genetic association studies is both to define the genetic architecture of complex traits and diseases and also to provide new insights into normal physiology and disease pathophysiology. Accomplishing that goal will require defining the causal variants that account for the observed associations, their mechanism of action, and their target genes. Success would have both near- and long-term benefits to health and science. In terms of health benefits, causal relationships between noncoding genetic variants and disease risk can be used to improve the prediction of disease onset and the design of prevention and early detection strategies. Subsequently determining the effects of causal variants on gene expression can prioritize downstream efforts to characterize causal genes and their role in disease etiology. That prioritization is particularly valuable when the target genes have an unknown function. This discovery pathway can ultimately lead to novel and potentially patient-specific therapeutic targets. In terms of scientific benefits, expanding the catalog of noncoding variants that are known to contribute to human traits is needed to determine general and transferrable principles about the genetic basis of complex human diseases. Recent conceptual and technical advances in genetics and genomics together have the potential to greatly improve our understanding of the noncoding genetic contributions to human traits. Although there are a wide variety of ways in which noncoding variants may affect phenotypes, we will focus specifically on variants that alter the activity of gene regulatory elements and, subsequently, the expression of target genes.
The plummeting cost of DNA sequencing has enabled parallel paradigm shifts in human genetics and genomics. For genetic studies, the major benefit has been access to all variants in an individual for association testing. That benefit has been predominantly realized by using whole-genome sequences of related populations to impute the alleles of variants that have not been directly genotyped (The 1000 Genomes Project Consortium 2012; Delaneau and Marchini 2014; Gudbjartsson et al. 2015; Horikoshi et al. 2015; Kuchenbaecker et al. 2015; Surakka et al. 2015) and by whole-exome sequencing (for examples and reviews, see Bamshad et al. 2011; Chong et al. 2015; de Bruin and Dauber 2015). Meanwhile, the first association studies that replace targeted genotyping with whole-genome sequencing are now starting to appear (Gaulton et al. 2013; Morrison et al. 2013; Taylor et al. 2015).
Even with perfect genotype information, there will remain a need for downstream functional studies to identify causal variants that contribute to human phenotypes. One major reason is that the resolution of genetic association is limited by patterns of recombination in the study population: Without recombination between a causal mutation and a nearby noncausal mutation, there is no ability to unambiguously determine which of the two contributes to phenotype with association alone. The ability to discriminate causal effects within those regions requires alternative strategies that effectively separate out the effects of variants that are close to one another on the same chromosome.
Concurrent with the first human population sequencing projects, large and coordinated genomics efforts completed the first comprehensive maps of the molecular state of the human genome and epigenome (The ENCODE Project Consortium 2012; Roadmap Epigenomics Consortium et al. 2015), and hundreds of similar studies have been completed in other biological systems (Fig. 1). The resulting data sets provide researchers with extensive catalogs of transcription factor binding and chromatin states across noncoding genomic regions in a wide diversity of cell types and environmental conditions. Integrating results from studies of genomic regulatory activity with genetic associations has shown initial promise for resolving causal variants of human phenotypes after genetic association, as demonstrated both by overall trends (Wang et al. 2010; Boyle et al. 2012; Ward and Kellis 2012; Zhou et al. 2015) and specific examples (Zhang et al. 2012; Corradin et al. 2014; Huang et al. 2014; Guo et al. 2015).
Figure 1.

Number of publications in the NCBI database matching the search queries for RNA-seq (circles) and ChIP-seq (squares). Queries were performed via the NCBI PubMed website (http://www.ncbi.nlm.nih.gov/pubmed/) on July 14, 2015. For RNA-seq and ChIP-seq, the exact query used was “RNA-seq OR RNAseq” and “ChIP-seq OR ChIPseq,” respectively.
Notwithstanding those initial successes, predicting the effect of noncoding genetic variation remains a foremost challenge for several reasons. First, regulatory activity across the genome varies dramatically between cell types and conditions (Thurman et al. 2012). Second, recent evidence suggests that few of the candidate regulatory elements defined by chromatin state and transcription factor binding have strong regulatory activity (Kwasnieski et al. 2014). Even with data supporting regulatory activity of an element, predicting the effects of genetic variants therein is complex and typically requires further empirical investigation. In this Perspective, we will describe recent advances at the interface between genetics and genomics that have improved the ability to identify regulatory mechanisms of disease. We particularly focus on emerging technologies that overcome some of the most eminent challenges and emphasize the need for collaborative studies between genetics and genomics investigators to realize the potential of those technologies.
The landscape of genetic association signals
As of February 2015, genome-wide association studies (GWAS) and other studies had demonstrated the association of more than 15,000 SNPs with a complex disease or trait (Welter et al. 2014). However, the mechanisms underlying these associations remain largely undefined. More generally, the underlying architecture of complex diseases and traits remains poorly defined. The common disease–common variant hypothesis (Gibson 2011) initially predicted that common variants present in all populations underlie phenotypic variation or disease risk and that, together, these variants have an additive or multiplicative effect on disease risk or trait variation. As initial genome-wide association studies failed to account for the observed narrow sense heritability of diseases and traits, alternative explanations have been proposed for the architecture of complex diseases and traits, including (1) a large number of small-effect common variants across the spectrum of allele frequency account for disease risk and quantitative trait variation; (2) a large number of large-effect rare variants underlie observed associations; or (3) a combination of genotypic, environmental, and epigenetic interactions account for the associations (Gibson 2011). It is likely that some combination of those different potential mechanisms accounts for the underlying architecture of complex diseases and traits as common, low frequency, and rare variants have all now been shown to be associated with complex diseases and traits (Fu et al. 2013; Morrison et al. 2013; Ratnapriya et al. 2014; Surakka et al. 2015).
The fundamental problem now faced by geneticists is that variants identified through genetic association studies are typically common SNPs that mark an associated locus rather than the variant that mechanistically contributes to the association. The reason is that alleles of variants that are close together in the genome are likely to be inherited together, a phenomenon known as linkage disequilibrium (LD). The small number of recombination events per human generation, the preferential occurrence of recombination events in certain genomic regions, the history of the population, and other influences all contribute to patterns of LD (for review, see Stumpf and McVean 2003). In the human genome, regions 10–100 kb in size within which causality cannot be inferred are typical (The 1000 Genomes Project Consortium 2012). For that reason, a small number of common variants can represent a large fraction of the genetic variation. Genetic association studies have taken advantage of that LD structure with great success by genotyping common variants rather than those most likely to cause the trait or disease.
The LD patterns that have made genome-wide association studies successful are also a major limiting factor in the identification of causal variants using statistical association alone. For example, a standard approach to identify a causal variant within a locus of genetic association is to first take advantage of different patterns of LD in different ancestry groups to narrow the boundaries of the association locus (for review, see Rosenberg et al. 2010; Edwards et al. 2013). Sequencing the narrowed locus in appropriate populations to identify all the genetic variation across the locus follows. At that point, the number of variants that could contribute to phenotype may number into the hundreds or thousands. Computational strategies to prioritize among the remaining variants based on genomics data and other features may help but, as described above, accurately predicting the functional impact of specific variants on the regulation of gene expression remains a largely unsolved problem.
Further complicating causal variant identification is the possibility that multiple as opposed to a single variant within an LD block may be functional and contribute to the observed association. Analogous to examples in which multiple coding variants in the same gene independently contribute to disease risk (Kotowski et al. 2006; Nejentsev et al. 2009; Rivas et al. 2011), Corradin and colleagues recently suggested a “multiple enhancer variant” (MEV) hypothesis (Corradin et al. 2014) based on investigation into six different autoimmune diseases. In that study, they provide evidence that multiple variants within an LD block impact the activity of multiple different enhancers, and those effects coordinately alter target gene expression. The MEV hypothesis is supported by case studies. In one example, we provided empirical evidence that regulatory variants spanning multiple enhancers within an LD block associated with maternal glucose levels during pregnancy have a coordinated allelic effect on expression of HKDC1 (Guo et al. 2015). Similar patterns were reported previously for the SOX9 region associated with prostate cancer risk (Zhang et al. 2012). The observation that multiple variants within an LD block can affect regulatory element activity and gene expression argues that testing single variants in isolation will be both an inefficient and potentially misleading approach for identifying causal variants. Instead, high-throughput strategies to systematically and comprehensively evaluate the function of variants and haplotypes present in a phenotype-associated locus are needed.
Genetic associations with gene expression reveal target genes
Studies to associate genetic variants with gene expression have generated extensive catalogs of expression quantitative trait loci (eQTLs) in diverse cell types and conditions (e.g., Gamazon et al. 2010; Lappalainen et al. 2013; Liang et al. 2013; The GTEx Consortium 2015). Known eQTLs are highly enriched in variants associated with traits and diseases (e.g., Nica et al. 2010; Nicolae et al. 2010; Torres et al. 2014), and those associations can mark candidate target genes for downstream mechanistic investigation (Cookson et al. 2009). That feature is especially useful when the target gene is not an obvious choice for follow-up because the gene is not in LD with the phenotype-associated variant or because there is not a clear biologic rationale for the association. For example, one of the most robust genetic associations is between genetic variants on Chromosome 16 and body mass index (Dina et al. 2007; Frayling et al. 2007; Scuteri et al. 2007). Despite localization of the associated variants in the first intron of the FTO gene and demonstration of a role for FTO in body weight regulation and fat mass in mouse models (Fawcett and Barroso 2010), recent eQTL analyses have suggested that the Iroquois-related homeobox 3 (IRX3) gene, located more than a megabase away from the most highly associated variant, may also be a causal gene (Smemo et al. 2014; Ronkainen et al. 2015). Mice deficient in Irx3 have obesity and diabetes-related traits, increasing confidence in this mechanistic connection (Smemo et al. 2014). Similar approaches have used eQTL analyses to reveal target genes in other studies (e.g., Teslovich et al. 2010; Innocenti et al. 2011; Hernandez et al. 2012; Farh et al. 2015), supporting the broad utility of the approach.
Expression QTLs have now been mapped for several different tissue types (Schadt et al. 2008; Dimas et al. 2009; Gibbs et al. 2010; Innocenti et al. 2011; Grundberg et al. 2012, 2013) and hormone responses (Maranville et al. 2011). Further studies have investigated the distribution of eQTLs across different tissues from the same individuals (Dimas et al. 2009; Nica et al. 2011; The GTEx Consortium 2015). Although those studies have revealed a substantial degree of shared eQTLs between tissues, the degree of sharing varies across tissues, and certain tissues such as brain appear to have an especially high degree of tissue-specific gene regulation (Hernandez et al. 2012; The GTEx Consortium 2015). The importance of tissue-specific eQTLs is supported by studies showing that GWAS results are specifically enriched for eQTLs in tissues that are relevant to the phenotype (Emilsson et al. 2008; Nica et al. 2010; Below et al. 2011; Brown et al. 2013; Torres et al. 2014). On the other hand, tissue-general eQTLs may be enriched for variants that have a function throughout the body and thus as a class may have a disproportionate effect on phenotypes. To the best of our knowledge, however, the relative contribution of tissue-general eQTLs to phenotypes has yet to be estimated. There is also growing evidence for substantial allelic heterogeneity in gene expression levels (Brown et al. 2013), in agreement with the previously described observations of multiple coordinated regulatory variants in disease loci (Zhang et al. 2012; Corradin et al. 2014; Guo et al. 2015). Taken together, increasing the diversity of primary tissues, cell types, and environments for which eQTLs have been mapped is likely to be highly valuable for identifying variants, genes, and tissues that contribute to phenotypes.
Genomic regulatory elements are highly enriched in phenotype-associated variants
Although eQTLs have demonstrated value in identifying target genes for genetic association studies, they too suffer from the same limitation that the most strongly associated variant may not be causal of the regulatory event. Comprehensive genomic measurements of the epigenetic and regulatory state of the genome, made possible by high-throughput sequencing, can overcome that limitation because LD does not limit their resolution. Many different assays have been developed to measure genomic components of gene regulation, and we will focus on two widely used approaches, chromatin accessibility mapping and ChIP-seq. The accessibility of chromatin to various enzymes such as DNase I is a well-established indicator of genomic regulatory activity. High-throughput sequencing-based assays such as DNase-seq and ATAC-seq exploit that principle to reveal comprehensive maps of chromatin accessibility across the human genome (Song and Crawford 2010; Thurman et al. 2012; Buenrostro et al. 2013). Similarly, the high-throughput sequencing version of chromatin immunoprecipitation, ChIP-seq (Johnson et al. 2007; Mikkelsen et al. 2007; Robertson et al. 2007), is now commonly used to identify binding sites for transcription factors and histone modifications associated with regulatory states of the human genome. ChIP-seq can localize a binding event or a modified histone to within 50 bp, and DNase-seq has a similar resolution.
There is now strong evidence that genetic variation within candidate regulatory elements identified with DNase-seq or with ChIP-seq contributes to human phenotypes. For example, several studies have found that phenotype-associated variants are enriched DNase- or ChIP-positive regions in a tissue-specific manner, and that tissue specificity can be used to implicate unexpected tissues in disease etiologies (Ernst et al. 2011; Maurano et al. 2012; Schaub et al. 2012; The ENCODE Project Consortium 2012; Parker et al. 2013; Pickrell 2014). Genetic variation in the same regions also accounts for a substantial and significantly enriched fraction of the heritability of complex human diseases (Gusev et al. 2014). Moreover, using an association approach similar to that used to identify eQTLs, several studies have identified genetic variants that are correlated with changes in chromatin accessibility, histone modifications, and DNA methylation; and they have shown that the identified variants explain a large fraction of eQTL associations (Degner et al. 2012; McVicker et al. 2013; Banovich et al. 2014). Together, those results suggest that variation in regulatory elements is a primary contributor to expression phenotypes. Those overall enrichments motivate a deeper investigation into the genetic architecture of gene regulation, with a particular focus on determining the specific variants that alter regulatory element activity.
Allele-specific genomic activity reveals candidate mechanisms of disease
The use of high-throughput sequencing as readout for DNase and ChIP assays not only improves detection of regulatory elements but also allows for the simultaneous observation of the genetic sequence of the identified elements. If one or more positions within the identified regulatory element are heterozygous in an individual, that feature can be leveraged to estimate the abundance of each allele in the assayed DNA. A significant deviation from the expected ratio based on that individual's genome indicates an allele-specific difference in the activity of that element. Allele-specific analyses were first used to investigate gene expression using a variety of approaches, including targeted sequencing, RT-qPCR, and microarrays (e.g., Singer-Sam et al. 1992; Yan et al. 2002; Bray et al. 2003; Lo et al. 2003; Pastinen et al. 2004; Gimelbrant et al. 2007; Ge et al. 2009; Adoue et al. 2014; for review, see Knight 2004; Pastinen 2010). Those studies typically found evidence for allele-specific expression of ∼10% of human genes. High-throughput sequencing of RNA advanced the field by making it possible to measure allele-specific gene expression genome-wide and agnostic of reference gene annotations (e.g., Degner et al. 2009; McManus et al. 2010; Pickrell et al. 2010; Reddy et al. 2012). Allele-specific analyses of DNase-seq and ChIP-seq data use similar strategies to reveal variants associated with chromatin state or transcription factor binding (Kasowski et al. 2010; McDaniell et al. 2010; Reddy et al. 2012). If the observations reflect direct local effects of genetic variation on gene regulation (Fig. 2A), then such allele-specific analyses can be used to identify individual causal variants within large LD blocks identified via association studies. On the other hand, long-range regulatory interactions may limit the ability to pinpoint individual genetic causes.
Figure 2.
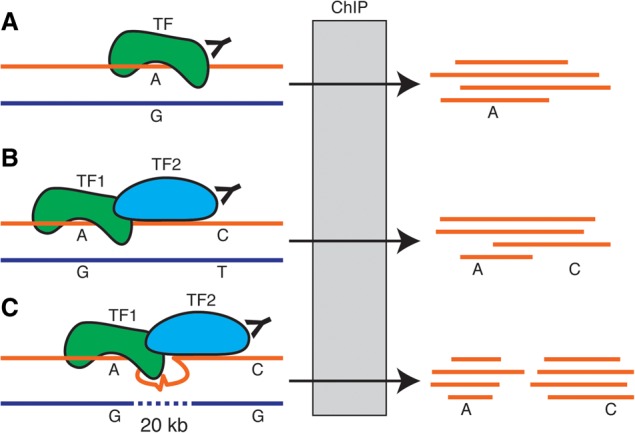
Mechanisms of allele-specific transcription factor occupancy. (A) Local effects occur when a genetic variant directly impacts the ability of a transcription factor to bind DNA. In this example, only the A allele is bound by the transcription factor and recovered by ChIP. (B) Genetic variants may lead to allele-specific binding of entire regulatory complexes. In this example, transcription factor TF1 binds the A but not the G allele. Because TF1 also recruits TF2 to the same regulatory complex, ChIP-seq for TF2 preferentially isolates the A and C alleles even though TF2 does not directly bind either variant. (C) Long-range interactions may also drive distal allele-specific effects. One potential mechanism is that TF1 and TF2 form a regulatory complex via DNA looping. Because occupancy of TF1 influences that of TF2, variants that impact TF1 binding lead to an allele-specific signal for TF2 occupancy.
Several findings indicate that allele-specific effects are indeed local events. The initial allele-specific ChIP-seq studies found that genetic variants with allele-specific transcription factor binding are enriched near the specific nucleotides bound by the transcription factor (Reddy et al. 2012). The inverse was also true: Variants without allele-specific binding were depleted near transcription factor binding sequences (Reddy et al. 2012). Moreover, the variants with the strongest effects typically altered the DNA sequences bound by transcription factors (Kasowski et al. 2010; Reddy et al. 2012). Allele-specific transcription factor binding and, to a lesser extent, chromatin state, are both heritable, indicating a clear genetic contribution (McDaniell et al. 2010; Reddy et al. 2012; Kasowski et al. 2013; Kilpinen et al. 2013). Although changes in the DNA sequence bound by the transcription factor explain the largest effects, most allele-specific effects are modest and cannot be explained by changes in the transcription factor binding sequence (Reddy et al. 2012). One potential explanation is that transcription factors often bind the human genome in complexes often referred to as cis-regulatory modules (for review, see Hardison and Taylor 2012). Observed allele-specific transcription factor binding may result from genetic variants that disrupt binding of other transcription factors in the same module (Fig. 2B). That model is supported by a strong degree of allele-specific coordination between multiple transcription factors and chromatin state at the same genomic locus (Reddy et al. 2012; The ENCODE Project Consortium 2012; McVicker et al. 2013; Soccio et al. 2015). On the other hand, observations of long-range coordination in allele-specific chromatin indicate that local effects of regulatory variants may affect distal sites on the same chromosome (Fig. 2C; Kilpinen et al. 2013). In that scenario, LD would still impair resolution of allele-specific analyses for identifying causal variants. Taken together, it is likely that allele-specific measurements of the regulatory state reflect a mixture of local and distal effects. Although the relative proportion of local and distal signals is not yet known, the contributions of local effects to the overall signal likely provide some ability to identify causal variants within regions of high LD.
There are several additional advantages of allele-specific analyses over association-based studies that motivate their increased use. Because the two alleles compete for regulatory factors in the same nucleus and in the same environment and because both alleles undergo the same sample processing steps, variation due to sample history or handling is unlikely to contribute to false positives. Allele-specific analyses also have a practical advantage in that, unlike for association studies, a large cohort of individuals is not needed to detect an allele-specific effect at an individual variant. In cases in which samples are rare or difficult to obtain, an allele-specific approach may therefore be the only viable path forward for identifying genetic associations with regulatory element activity. Finally, because comparisons are made between the two alleles present in the same individual, the power to detect an allele-specific effect of a heterozygous variant in that individual does not depend on the population frequency of the variant.
The corresponding limitations are that only heterozygous sites in an individual are informative, and there are additional analytical challenges over genetic association studies. The limitation to heterozygous sites means that homozygous individuals do not contribute to the power to detect an effect, and also that large cohorts are still needed to observe a rare variant. Ideally, it would be possible to combine allele-specific analyses with standard genetic association, and newly developed approaches to do so are a promising advance (van de Geijn et al. 2014). The primary additional analytical challenge is aligning short-read sequences in a manner that is not biased to the reference genome. Such alignment biases arise from numerous sources, including unobserved genetic variation and repetitive sequences (Degner et al. 2009; Stevenson et al. 2013). Alignment biases have been overcome previously by aligning sequences to personal genome sequences (McDaniell et al. 2010; Rozowsky et al. 2011; Reddy et al. 2012), prefiltering genomic regions prone to bias (Degner et al. 2009), and the development of variant-tolerant alignment algorithms (Wu and Nacu 2010). Once alignments are made, detecting a bias toward one allele requires statistical approaches to handle overdispersion in read-count data. Software packages for allele-specific alignment and statistical analysis have now been established, making those advances available to a wide diversity of researchers (Wu and Nacu 2010; Rozowsky et al. 2011; Skelly et al. 2011; van de Geijn et al. 2014). As for genetic association studies, allele-specific analyses rely on dense genotyping for each individual. However, recent advances to perform allele-specific analyses without supporting genome sequence information may remove that limitation (Harvey et al. 2015; Romanel et al. 2015). Such advances are major breakthroughs because they greatly reduce cost and complexity. For all of the aforementioned reasons, expanding studies to include allele-specific analyses is a promising strategy to improve the identification of causal regulatory variants in a diversity of tissues and cell types.
High-throughput measurement of regulatory element activity
One major outstanding challenge subsequent to the widespread adoption of DNase-seq and ChIP-seq is to reconcile the abundance of candidate regulatory elements identified. One likely explanation is that a small fraction of candidate regulatory elements are highly active and those elements affect the majority of gene regulation. Reporter-gene expression assays have a distinct advantage over eQTL associations and allele-specific genomic analyses because they directly measure the regulatory activity of a genomic sequence. Briefly, in a reporter assay, a candidate regulatory element is introduced into a plasmid that contains an easily observable reporter such as a fluorescent or chemiluminescent protein (Fig. 3A). The plasmid is then introduced into cells of interest by any of a variety of approaches. Once the plasmid enters the nucleus, transcription factors and RNA polymerases bind the plasmid and control reporter gene expression. Because reporter assays isolate regulatory elements from the surrounding genomic context, results are independent of adjacent elements that may be in LD. Critical for genetic studies, reporter assays can be used to estimate the effect of genetic variants on regulatory activity by comparing the activity of different alleles of the same regulatory element. For those reasons, reporter assays have been valuable for identifying individual regulatory variants that contribute to phenotype (Musunuru et al. 2010; Feng et al. 2013; Fogarty et al. 2014; Stadhouders et al. 2014; Guo et al. 2015).
Figure 3.

High-throughput reporter assays. (A) In a standard reporter assay, a candidate regulatory element is placed upstream of a reporter gene that is expressed from a constitutively active promoter. (B) In a high-throughput version of the same system, a random DNA sequence known as a molecular barcode is inserted into the 3′ UTR of the reporter gene, and a library of candidate regulatory elements are placed upstream of the promoter. Each individual candidate regulatory element is physically linked to a unique molecular barcode. Measuring the expression of each molecular barcode can then be used to estimate the activity of the associated regulatory element. (C) An alternative strategy is to clone the library of candidate regulatory elements directly into the 3′ UTR of the reporter gene. By that construction, the regulatory element controls its own expression, which can be measured with paired-end high-throughput sequencing. (D) The preceding strategy can be modified for genetic studies by cloning genetically diverse regulatory elements captured from donor genomes into the reporter gene. By that construction, each allele is expressed at a level that is directly related to its regulatory activity.
The major drawback to standard reporter assay systems for genetic screens is the throughput. Because readout is limited to a single reporter gene, assays must be individually constructed and assayed. Multiwell plates and automated liquid handling increase throughput substantially (e.g., Landolin et al. 2010; Whitfield et al. 2012), but not to the extent required to routinely comprehensively assay regulatory variants in an entire LD region identified by a genetic association study. To address the need to increase the scale of reporter assays, high-throughput versions have been developed in which regulatory activity is measured using high-throughput sequencing rather than by observing a fluorescent protein. One strategy is to construct a library of regulatory elements that are uniquely associated with DNA barcode sequences embedded in an otherwise ignored reporter gene (Fig. 3B). High-throughput sequencing of the expressed barcodes can then be used to estimate activity of the associated element (Kwasnieski et al. 2012; Melnikov et al. 2012; Patwardhan et al. 2012). Each of the initially published mammalian examples focused on evaluating the effects of genetic variants within a small set of previously defined regulatory elements. The CRE-seq assay developed by Kwasnieski et al. (2012) used high-throughput DNA synthesis to generate and assay more than 1000 genetic variants of a 52-bp rhodopsin promoter. Similarly, Melnikov et al. (2012) used DNA synthesis to generate and assay more than 27,000 variants of two 87-bp inducible enhancers. In both cases, the length of the regulatory elements assayed was limited by DNA synthesis. Patwardhan et al. (2012) used degenerate PCR rather than DNA synthesis to generate random mutants of three known liver enhancers. That approach enabled generation of more unique versions (more than 100,000) of longer regulatory elements (258–619 bp). Finally, a related strategy known as functional identification of regulatory elements within accessible chromatin (FIREWACh) assayed captured DNase hypersensitive regions rather then predefined regulatory elements. By combining DNase-seq with a reporter assay, Murtha and colleagues were able to agnostically quantify the activity of approximately 80,000 open chromatin sites in a single assay (Murtha et al. 2014).
For barcode-based approaches, each regulatory element to be assayed must be linked with a unique barcode in the assay. The STARR-seq approach uses a different library construction strategy that obviates that step. In STARR-seq, the reporter element itself is cloned into the 3′ untranslated region (UTR) of the reporter gene and serves as its own barcode (Fig. 3C; Arnold et al. 2013). The major advantage of STARR-seq is that the greatly simplified library construction makes the approach particularly amenable to assaying highly diverse libraries of randomly fragmented DNA. Specifically, in the initial demonstration of STARR-seq, libraries of more than 10 million unique regulatory fragments were assayed, and the median size of the regulatory fragments was ∼600 bp. That level of diversity was sufficient to agnostically assay the entire Drosophila melanogaster genome in multiple cell lines and six human bacterial artificial chromosomes ranging in size from 150 to 185 kb. As in the FIREWACh assay, combining capture of regulatory elements with STARR-seq has enabled focused investigation of genomic regions that are of interest because they are likely to be functional or because they are associated with disease (Vanhille et al. 2015; Vockley et al. 2015). The STARR-seq assay has now been used to investigate changes in regulatory activity across species (Arnold et al. 2014), in response to hormones (Shlyueva et al. 2014), and in combination with different promoters (Zabidi et al. 2015), highlighting the flexibility of the approach.
Comprehensive evaluation of human and mouse candidate regulatory elements identified using ChIP-seq, DNase-seq, and integrative techniques has revealed that only a small fraction of elements typically have a strong effect on gene expression (Kwasnieski et al. 2014; Murtha et al. 2014). Interestingly, with growing evidence that transcription factors tend to bind the genome in heterotypic and homotypic clusters, Smith and colleagues used a massively parallel reporter approach to show that heterotypic clusters of transcription factors are especially potent regulators of gene expression (Smith et al. 2013). Similarly, mutagenesis studies have shown that mutations in distal regulatory elements typically have modest effects on regulatory element activity (Melnikov et al. 2012; Patwardhan et al. 2012). Together, these results indicate that allele-specific DNase-seq and ChIP-seq will be useful to reduce the search space for causal variants, but that additional functional assays will be needed to identify individual causal regulatory variants.
Using high-throughput assays to measure the effects of noncoding variants in GWAS cohorts is one possible strategy that uses existing technology to identify causal variants underlying a genetic association result. As an initial example, we recently used STARR-seq to measure the activity of candidate regulatory elements captured from the genomes of 95 individuals from a recent genetic association study (Fig. 3D; Urbanek et al. 2013; Vockley et al. 2015). That population-scale approach allowed identification of functional regulatory variants within a genetically linked region of association. Because the capture was performed from donor genomes, all variants and haplotypes tested were found in the study population, including a substantial fraction of variants not found in existing databases. We expect that continued development and application of such high-throughput reporter assays to expanded populations is a promising strategy to connect genetics and genomics and thereby reveal causal variation within large genomic regions associated with disease.
As with any approach, reporter assays have limitations, some of which can be mitigated with improved study designs. Regulatory element activity may require additional contexts such as a specific promoter, genomic integration, or cellular environment. Those concerns can be largely addressed with experimental designs that include custom promoters (Zabidi et al. 2015), genomic integration (Dickel et al. 2014; Murtha et al. 2014), and strategies to assay libraries in vivo (Kwasnieski et al. 2012; Patwardhan et al. 2012; Smith et al. 2013). Genome and epigenome editing strategies are also emerging as complementary strategies to investigate regulatory element activity in vivo (Mendenhall et al. 2013; Yin et al. 2014; Hilton et al. 2015). However, there are many contexts for which tractable culture models do not yet exist, and continued development of more realistic models will remain invaluable to determine contributions of regulatory variation to disease.
Finally, although reporter assays are a promising strategy to identify causal regulatory variants, integration with results from other approaches, such as ChIP-seq and eQTL studies, will be needed to identify the responsible transcription factors and the causal genes, respectively. An additional exciting possibility is the integration with chromatin conformation assays such as ChIA-PET (Li et al. 2014), Hi-C (Belton et al. 2012), and variants thereof (Jäger et al. 2015) that can reveal physical interactions between causal variants and causal genes.
Informing future association studies by improving models of regulatory variants
Prioritizing variants that are most likely to have a phenotypic effect is a promising strategy for improving resolution within an associated locus (for review, see Cooper and Shendure 2011). Briefly, that strategy is commonly applied to genetic variants in coding regions, where gene annotations, codon sequences, and protein structure can guide analyses (Sunyaev et al. 2001; Ng and Henikoff 2003; Adzhubei et al. 2010, 2013; Price et al. 2010; Schwarz et al. 2010; Hu et al. 2013; Ionita-Laza et al. 2013). A current need is to improve understanding of the basic characteristics of the types of variants that most impact gene expression to support the development of analogous methods for noncoding genomic regions. Approaches to computationally predict the effects of genetic variants in noncoding regions have largely relied on evolutionary conservation in closely related species (e.g., Cooper et al. 2005; Siepel et al. 2005; Pollard et al. 2010). With a dramatic increase in empirical data describing the epigenetic and regulatory state of the genome, integrative strategies have also recently emerged that combine both conservation and empirical data to identify and predict the effects of noncoding variants (Lee et al. 2011; Ernst and Kellis 2012; Hoffman et al. 2012; Ward and Kellis 2012; Khurana et al. 2013; Kircher et al. 2014; Ritchie et al. 2014; Shihab et al. 2015). Such integrative approaches are limited by the empirical data available. As the high-throughput empirical approaches described above are further developed and applied, integrative strategies to predict the effects of noncoding variants are likely to immediately benefit. Meanwhile, the catalog of regulatory variants that are known to contribute to human phenotypes will be greatly expanded. That expansion is critical to support the development of additional guiding principles for the interpretation of noncoding regulatory variation. For example, certain classes of variants, such as rare variants or variants in regions with specific histone modifications, may be more likely to alter regulatory element activity. Another possibility is that the three-dimensional structure of the genome will help to model genetic effects on target gene expression. Early indications suggest that prioritization based on such models will be helpful, and the full extent of possibilities remains to be determined.
Discussion
The aforementioned approaches are only a few examples of the ways in which the integration of genomic and genetic analyses can inform our understanding of noncoding mechanisms of human phenotypes. Although by no means comprehensive, the preceding examples represent both the basic principles and common challenges of using genomic assays to inform a positive genetic association. Specifically, as LD is disrupted in experimental systems, it becomes easier to finely map causal variants. The sacrifice is that context-dependent regulation and the identity of target genes is typically lost. For that reason, we expect that the greatest value for mechanistic interpretation will be achieved when integration across multiple levels of resolution reveals both candidate causal variants and one or more target genes for downstream study.
This Perspective has focused on associations with complex phenotypes in which causal variants are difficult to ascertain. Similar strategies will likely benefit the investigation of rare Mendelian diseases, specifically in cases in which a causative coding mutation has not been found. Especially in cases of intermediate phenotypes, regulatory mutations are a plausible explanation for the missing diagnosis (Weedon et al. 2014). Identifying such cases will likely improve diagnosis and could reveal patients who may be candidates for novel treatments.
Much of the work described here involved efforts of major consortia focused specifically on genetic associations or on highly coordinated genomic studies. Moving forward, we believe that the greatest benefit for human health will be obtained through joint studies that integrate both genetic and genomic principles in their design. We have found that such highly interactive studies yield substantial mutual benefits. For example, performing high-throughput reporter assays on DNA from genetic association cohorts required access to unique DNA samples that had been collected over several years and expert knowledge about the cell models and conditions that are most relevant to the phenotype (Guo et al. 2015). Then, once regulatory variants are identified via genomic strategies, follow-up genotyping in an independent cohort is needed to confirm the effects and to establish genetic risk scores. Meanwhile, informed genetic association based on genomic evidence of activity will require the generation of new functional genomic data in relevant cell models to support that development. Joint research efforts by genetics and genomics teams dramatically lowers the bar for such cross-cutting activities and, for that reason, we believe that such approaches will be well-positioned to realize translational benefits of biomedical research in both the short and long term.
Acknowledgments
This work was funded by National Institutes of Health R01 grants DK099820 and DK097534.
Footnotes
Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.190603.115.
Freely available online through the Genome Research Open Access option.
References
- The 1000 Genomes Project Consortium. 2012. An integrated map of genetic variation from 1,092 human genomes. Nature 491: 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adoue V, Schiavi A, Light N, Almlöf JC, Lundmark P, Ge B, Kwan T, Caron M, Rönnblom L, Wang C, et al. 2014. Allelic expression mapping across cellular lineages to establish impact of non-coding SNPs. Mol Syst Biol 10: 754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. 2010. A method and server for predicting damaging missense mutations. Nat Methods 7: 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei I, Jordan DM, Sunyaev SR. 2013. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet 76: 7.20.1–7.20.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold CD, Gerlach D, Stelzer C, Boryń ŁM, Rath M, Stark A. 2013. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science 339: 1074–1077. [DOI] [PubMed] [Google Scholar]
- Arnold CD, Gerlach D, Spies D, Matts JA, Sytnikova YA, Pagani M, Lau NC, Stark A. 2014. Quantitative genome-wide enhancer activity maps for five Drosophila species show functional enhancer conservation and turnover during cis-regulatory evolution. Nat Genet 46: 685–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, Shendure J. 2011. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet 12: 745–755. [DOI] [PubMed] [Google Scholar]
- Banovich NE, Lan X, McVicker G, van de Geijn B, Degner JF, Blischak JD, Roux J, Pritchard JK, Gilad Y. 2014. Methylation QTLs are associated with coordinated changes in transcription factor binding, histone modifications, and gene expression levels. PLoS Genet 10: e1004663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Below JE, Gamazon ER, Morrison JV, Konkashbaev A, Pluzhnikov A, McKeigue PM, Parra EJ, Elbein SC, Hallman DM, Nicolae DL, et al. 2011. Genome-wide association and meta-analysis in populations from Starr County, Texas, and Mexico City identify type 2 diabetes susceptibility loci and enrichment for expression quantitative trait loci in top signals. Diabetologia 54: 2047–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belton JM, McCord RP, Gibcus JH, Naumova N, Zhan Y, Dekker J. 2012. Hi-C: a comprehensive technique to capture the conformation of genomes. Methods 58: 268–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, Karczewski KJ, Park J, Hitz BC, Weng S, et al. 2012. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res 22: 1790–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray NJ, Buckland PR, Owen MJ, O'Donovan MC. 2003. Cis-acting variation in the expression of a high proportion of genes in human brain. Hum Genet 113: 149–153. [DOI] [PubMed] [Google Scholar]
- Brown CD, Mangravite LM, Engelhardt BE. 2013. Integrative modeling of eQTLs and cis-regulatory elements suggests mechanisms underlying cell type specificity of eQTLs. PLoS Genet 9: e1003649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. 2013. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10: 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong JX, Buckingham KJ, Jhangiani SN, Boehm C, Sobreira N, Smith JD, Harrell TM, McMillin MJ, Wiszniewski W, Gambin T, et al. 2015. The genetic basis of Mendelian phenotypes: discoveries, challenges, and opportunities. Am J Hum Genet 97: 199–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson W, Liang L, Abecasis G, Moffatt M, Lathrop M. 2009. Mapping complex disease traits with global gene expression. Nat Rev Genet 10: 184–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GM, Shendure J. 2011. Needles in stacks of needles: finding disease-causal variants in a wealth of genomic data. Nat Rev Genet 12: 628–640. [DOI] [PubMed] [Google Scholar]
- Cooper GM, Stone EA, Asimenos G, NISC Comparative Sequencing Program, Green ED, Batzoglou S, Sidow A. 2005. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res 15: 901–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corradin O, Saiakhova A, Akhtar-Zaidi B, Myeroff L, Willis J, Cowper-Sal lari R, Lupien M, Markowitz S, Scacheri PC. 2014. Combinatorial effects of multiple enhancer variants in linkage disequilibrium dictate levels of gene expression to confer susceptibility to common traits. Genome Res 24: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruin C, Dauber A. 2015. Insights from exome sequencing for endocrine disorders. Nat Rev Endocrinol 11: 455–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degner JF, Marioni JC, Pai AA, Pickrell JK, Nkadori E, Gilad Y, Pritchard JK. 2009. Effect of read-mapping biases on detecting allele-specific expression from RNA-sequencing data. Bioinformatics 25: 3207–3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degner JF, Pai AA, Pique-Regi R, Veyrieras JB, Gaffney DJ, Pickrell JK, De Leon S, Michelini K, Lewellen N, Crawford GE, et al. 2012. DNase I sensitivity QTLs are a major determinant of human expression variation. Nature 482: 390–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaneau O, Marchini J, 1000 Genomes Project Consortium. 2014. Integrating sequence and array data to create an improved 1000 Genomes Project haplotype reference panel. Nat Commun 5: 3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickel DE, Zhu Y, Nord AS, Wylie JN, Akiyama JA, Afzal V, Plajzer-Frick I, Kirkpatrick A, Göttgens B, Bruneau BG, et al. 2014. Function-based identification of mammalian enhancers using site-specific integration. Nat Methods 11: 566–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimas AS, Deutsch S, Stranger BE, Montgomery SB, Borel C, Attar-Cohen H, Ingle C, Beazley C, Gutierrez Arcelus M, Sekowska M, et al. 2009. Common regulatory variation impacts gene expression in a cell type–dependent manner. Science 325: 1246–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dina C, Meyre D, Gallina S, Durand E, Korner A, Jacobson P, Carlsson LM, Kiess W, Vatin V, Lecoeur C, et al. 2007. Variation in FTO contributes to childhood obesity and severe adult obesity. Nat Genet 39: 724–726. [DOI] [PubMed] [Google Scholar]
- Edwards SL, Beesley J, French JD, Dunning AM. 2013. Beyond GWASs: illuminating the dark road from association to function. Am J Hum Genet 93: 779–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emilsson V, Thorleifsson G, Zhang B, Leonardson AS, Zink F, Zhu J, Carlson S, Helgason A, Walters GB, Gunnarsdottir S, et al. 2008. Genetics of gene expression and its effect on disease. Nature 452: 423–428. [DOI] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kellis M. 2012. ChromHMM: automating chromatin-state discovery and characterization. Nat Methods 9: 215–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. 2011. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473: 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farh KK, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, Shoresh N, Whitton H, Ryan RJ, Shishkin AA, et al. 2015. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 518: 337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett KA, Barroso I. 2010. The genetics of obesity: FTO leads the way. Trends Genet 26: 266–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q, Vickers KC, Anderson MP, Levin MG, Chen W, Harrison DG, Wilke RA. 2013. A common functional promoter variant links CNR1 gene expression to HDL cholesterol level. Nat Commun 4: 1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogarty MP, Cannon ME, Vadlamudi S, Gaulton KJ, Mohlke KL. 2014. Identification of a regulatory variant that binds FOXA1 and FOXA2 at the CDC123/CAMK1D type 2 diabetes GWAS locus. PLoS Genet 10: e1004633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, Perry JR, Elliott KS, Lango H, Rayner NW, et al. 2007. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 316: 889–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu W, O'Connor TD, Akey JM. 2013. Genetic architecture of quantitative traits and complex diseases. Curr Opin Genet Dev 23: 678–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamazon ER, Zhang W, Konkashbaev A, Duan S, Kistner EO, Nicolae DL, Dolan ME, Cox NJ. 2010. SCAN: SNP and copy number annotation. Bioinformatics 26: 259–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaulton K, Flannick J, Fuchsberger C, Kang HM, Burtt N, Ferrer J, Stitzel ML, Kellis M, McCarthy MI, Altshuler D, et al. 2013. Whole genome sequencing of 2,850 central-northern European type 2 diabetes cases and controls reveals insights into functional mechanisms underlying disease pathogenesis. In ASHG 63rd annual meeting, abstract no. 175. American Society of Human Genetics, Boston, MA. [Google Scholar]
- Ge B, Pokholok DK, Kwan T, Grundberg E, Morcos L, Verlaan DJ, Le J, Koka V, Lam KC, Gagné V, et al. 2009. Global patterns of cis variation in human cells revealed by high-density allelic expression analysis. Nat Genet 41: 1216–1222. [DOI] [PubMed] [Google Scholar]
- Gibbs JR, van der Brug MP, Hernandez DG, Traynor BJ, Nalls MA, Lai SL, Arepalli S, Dillman A, Rafferty IP, Troncoso J, et al. 2010. Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet 6: e1000952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson G. 2011. Rare and common variants: twenty arguments. Nat Rev Genet 13: 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimelbrant A, Hutchinson JN, Thompson BR, Chess A. 2007. Widespread monoallelic expression on human autosomes. Science 318: 1136–1140. [DOI] [PubMed] [Google Scholar]
- Grundberg E, Small KS, Hedman AK, Nica AC, Buil A, Keildson S, Bell JT, Yang TP, Meduri E, Barrett A, et al. 2012. Mapping cis- and trans-regulatory effects across multiple tissues in twins. Nat Genet 44: 1084–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundberg E, Meduri E, Sandling JK, Hedman AK, Keildson S, Buil A, Busche S, Yuan W, Nisbet J, Sekowska M, et al. 2013. Global analysis of DNA methylation variation in adipose tissue from twins reveals links to disease-associated variants in distal regulatory elements. Am J Hum Genet 93: 876–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The GTEx Consortium. 2015. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348: 648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudbjartsson DF, Helgason H, Gudjonsson SA, Zink F, Oddson A, Gylfason A, Besenbacher S, Magnusson G, Halldorsson BV, Hjartarson E, et al. 2015. Large-scale whole-genome sequencing of the Icelandic population. Nat Genet 47: 435–444. [DOI] [PubMed] [Google Scholar]
- Guo C, Ludvik AE, Arlotto ME, Hayes MG, Armstrong LL, Scholtens DM, Brown CD, Newgard CB, Becker TC, Layden BT, et al. 2015. Coordinated regulatory variation associated with gestational hyperglycaemia regulates expression of the novel hexokinase HKDC1. Nat Commun 6: 6069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusev A, Lee SH, Trynka G, Finucane H, Vilhjálmsson BJ, Xu H, Zang C, Ripke S, Bulik-Sullivan B, Stahl E, et al. 2014. Partitioning heritability of regulatory and cell-type-specific variants across 11 common diseases. Am J Hum Genet 95: 535–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardison RC, Taylor J. 2012. Genomic approaches towards finding cis-regulatory modules in animals. Nat Rev Genet 13: 469–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey CT, Moyerbrailean GA, Davis GO, Wen X, Luca F, Pique-Regi R. 2015. QuASAR: quantitative allele-specific analysis of reads. Bioinformatics 31: 1235–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez DG, Nalls MA, Moore M, Chong S, Dillman A, Trabzuni D, Gibbs JR, Ryten M, Arepalli S, Weale ME, et al. 2012. Integration of GWAS SNPs and tissue specific expression profiling reveal discrete eQTLs for human traits in blood and brain. Neurobiol Dis 47: 20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton IB, D'Ippolito AM, Vockley CM, Thakore PI, Crawford GE, Reddy TE, Gersbach CA. 2015. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol 33: 510–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman MM, Buske OJ, Wang J, Weng Z, Bilmes JA, Noble WS. 2012. Unsupervised pattern discovery in human chromatin structure through genomic segmentation. Nat Methods 9: 473–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horikoshi M, Mägi R, van de Bunt M, Surakka I, Sarin AP, Mahajan A, Marullo L, Thorleifsson G, Hägg S, Hottenga JJ, et al. 2015. Discovery and fine-mapping of glycaemic and obesity-related trait loci using high-density imputation. PLoS Genet 11: e1005230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Huff CD, Moore B, Flygare S, Reese MG, Yandell M. 2013. VAAST 2.0: improved variant classification and disease-gene identification using a conservation-controlled amino acid substitution matrix. Genet Epidemiol 37: 622–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q, Whitington T, Gao P, Lindberg JF, Yang Y, Sun J, Väisänen MR, Szulkin R, Annala M, Yan J, et al. 2014. A prostate cancer susceptibility allele at 6q22 increases RFX6 expression by modulating HOXB13 chromatin binding. Nat Genet 46: 126–135. [DOI] [PubMed] [Google Scholar]
- Innocenti F, Cooper GM, Stanaway IB, Gamazon ER, Smith JD, Mirkov S, Ramirez J, Liu W, Lin YS, Moloney C, et al. 2011. Identification, replication, and functional fine-mapping of expression quantitative trait loci in primary human liver tissue. PLoS Genet 7: e1002078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ionita-Laza I, Lee S, Makarov V, Buxbaum JD, Lin X. 2013. Sequence kernel association tests for the combined effect of rare and common variants. Am J Hum Genet 92: 841–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jäger R, Migliorini G, Henrion M, Kandaswamy R, Speedy HE, Heindl A, Whiffin N, Carnicer MJ, Broome L, Dryden N, et al. 2015. Capture Hi-C identifies the chromatin interactome of colorectal cancer risk loci. Nat Commun 6: 6178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DS, Mortazavi A, Myers RM, Wold B. 2007. Genome-wide mapping of in vivo protein-DNA interactions. Science 316: 1497–1502. [DOI] [PubMed] [Google Scholar]
- Kasowski M, Grubert F, Heffelfinger C, Hariharan M, Asabere A, Waszak SM, Habegger L, Rozowsky J, Shi M, Urban AE, et al. 2010. Variation in transcription factor binding among humans. Science 328: 232–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasowski M, Kyriazopoulou-Panagiotopoulou S, Grubert F, Zaugg JB, Kundaje A, Liu Y, Boyle AP, Zhang QC, Zakharia F, Spacek DV, et al. 2013. Extensive variation in chromatin states across humans. Science 342: 750–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana E, Fu Y, Colonna V, Mu XJ, Kang HM, Lappalainen T, Sboner A, Lochovsky L, Chen J, Harmanci A, et al. 2013. Integrative annotation of variants from 1092 humans: application to cancer genomics. Science 342: 1235587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilpinen H, Waszak SM, Gschwind AR, Raghav SK, Witwicki RM, Orioli A, Migliavacca E, Wiederkehr M, Gutierrez-Arcelus M, Panousis NI, et al. 2013. Coordinated effects of sequence variation on DNA binding, chromatin structure, and transcription. Science 342: 744–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. 2014. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46: 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight JC. 2004. Allele-specific gene expression uncovered. Trends Genet 20: 113–116. [DOI] [PubMed] [Google Scholar]
- Kotowski IK, Pertsemlidis A, Luke A, Cooper RS, Vega GL, Cohen JC, Hobbs HH. 2006. A spectrum of PCSK9 alleles contributes to plasma levels of low-density lipoprotein cholesterol. Am J Hum Genet 78: 410–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchenbaecker KB, Ramus SJ, Tyrer J, Lee A, Shen HC, Beesley J, Lawrenson K, McGuffog L, Healey S, Lee JM, et al. 2015. Identification of six new susceptibility loci for invasive epithelial ovarian cancer. Nat Genet 47: 164–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwasnieski JC, Mogno I, Myers CA, Corbo JC, Cohen BA. 2012. Complex effects of nucleotide variants in a mammalian cis-regulatory element. Proc Natl Acad Sci 109: 19498–19503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwasnieski JC, Fiore C, Chaudhari HG, Cohen BA. 2014. High-throughput functional testing of ENCODE segmentation predictions. Genome Res 24: 1595–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landolin JM, Johnson DS, Trinklein ND, Aldred SF, Medina C, Shulha H, Weng Z, Myers RM. 2010. Sequence features that drive human promoter function and tissue specificity. Genome Res 20: 890–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappalainen T, Sammeth M, Friedländer MR, ‘t Hoen PA, Monlong J, Rivas MA, Gonzàlez-Porta M, Kurbatova N, Griebel T, Ferreira PG, et al. 2013. Transcriptome and genome sequencing uncovers functional variation in humans. Nature 501: 506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D, Karchin R, Beer MA. 2011. Discriminative prediction of mammalian enhancers from DNA sequence. Genome Res 21: 2167–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Cai L, Chang H, Hong P, Zhou Q, Kulakova EV, Kolchanov NA, Ruan Y. 2014. Chromatin Interaction Analysis with Paired-End Tag (ChIA-PET) sequencing technology and application. BMC Genomics 15Suppl 12: S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang L, Morar N, Dixon AL, Lathrop GM, Abecasis GR, Moffatt MF, Cookson WO. 2013. A cross-platform analysis of 14,177 expression quantitative trait loci derived from lymphoblastoid cell lines. Genome Res 23: 716–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo HS, Wang Z, Hu Y, Yang HH, Gere S, Buetow KH, Lee MP. 2003. Allelic variation in gene expression is common in the human genome. Genome Res 13: 1855–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maranville JC, Luca F, Richards AL, Wen X, Witonsky DB, Baxter S, Stephens M, Di Rienzo A. 2011. Interactions between glucocorticoid treatment and cis-regulatory polymorphisms contribute to cellular response phenotypes. PLoS Genet 7: e1002162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, et al. 2012. Systematic localization of common disease-associated variation in regulatory DNA. Science 337: 1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniell R, Lee BK, Song L, Liu Z, Boyle AP, Erdos MR, Scott LJ, Morken MA, Kucera KS, Battenhouse A, et al. 2010. Heritable individual-specific and allele-specific chromatin signatures in humans. Science 328: 235–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus CJ, Coolon JD, Duff MO, Eipper-Mains J, Graveley BR, Wittkopp PJ. 2010. Regulatory divergence in Drosophila revealed by mRNA-seq. Genome Res 20: 816–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVicker G, van de Geijn B, Degner JF, Cain CE, Banovich NE, Raj A, Lewellen N, Myrthil M, Gilad Y, Pritchard JK. 2013. Identification of genetic variants that affect histone modifications in human cells. Science 342: 747–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnikov A, Murugan A, Zhang X, Tesileanu T, Wang L, Rogov P, Feizi S, Gnirke A, Callan CG Jr, Kinney JB, et al. 2012. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat Biotechnol 30: 271–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendenhall EM, Williamson KE, Reyon D, Zou JY, Ram O, Joung JK, Bernstein BE. 2013. Locus-specific editing of histone modifications at endogenous enhancers. Nat Biotechnol 31: 1133–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, et al. 2007. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448: 553–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison AC, Voorman A, Johnson AD, Liu X, Yu J, Li A, Muzny D, Yu F, Rice K, Zhu C, et al. 2013. Whole-genome sequence–based analysis of high-density lipoprotein cholesterol. Nat Genet 45: 899–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murtha M, Tokcaer-Keskin Z, Tang Z, Strino F, Chen X, Wang Y, Xi X, Basilico C, Brown S, Bonneau R, et al. 2014. FIREWACh: high-throughput functional detection of transcriptional regulatory modules in mammalian cells. Nat Methods 11: 559–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musunuru K, Strong A, Frank-Kamenetsky M, Lee NE, Ahfeldt T, Sachs KV, Li X, Li H, Kuperwasser N, Ruda VM, et al. 2010. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature 466: 714–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nejentsev S, Walker N, Riches D, Egholm M, Todd JA. 2009. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science 324: 387–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. 2003. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 31: 3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nica AC, Montgomery SB, Dimas AS, Stranger BE, Beazley C, Barroso I, Dermitzakis ET. 2010. Candidate causal regulatory effects by integration of expression QTLs with complex trait genetic associations. PLoS Genet 6: e1000895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nica AC, Parts L, Glass D, Nisbet J, Barrett A, Sekowska M, Travers M, Potter S, Grundberg E, Small K, et al. 2011. The architecture of gene regulatory variation across multiple human tissues: the MuTHER study. PLoS Genet 7: e1002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolae DL, Gamazon E, Zhang W, Duan S, Dolan ME, Cox NJ. 2010. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet 6: e1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker SC, Stitzel ML, Taylor DL, Orozco JM, Erdos MR, Akiyama JA, van Bueren KL, Chines PS, Narisu N, Black BL, et al. 2013. Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc Natl Acad Sci 110: 17921–17926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastinen T. 2010. Genome-wide allele-specific analysis: insights into regulatory variation. Nat Rev Genet 11: 533–538. [DOI] [PubMed] [Google Scholar]
- Pastinen T, Sladek R, Gurd S, Sammak A, Ge B, Lepage P, Lavergne K, Villeneuve A, Gaudin T, Brandstrom H, et al. 2004. A survey of genetic and epigenetic variation affecting human gene expression. Physiol Genomics 16: 184–193. [DOI] [PubMed] [Google Scholar]
- Patwardhan RP, Hiatt JB, Witten DM, Kim MJ, Smith RP, May D, Lee C, Andrie JM, Lee SI, Cooper GM, et al. 2012. Massively parallel functional dissection of mammalian enhancers in vivo. Nat Biotechnol 30: 265–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK. 2014. Joint analysis of functional genomic data and genome-wide association studies of 18 human traits. Am J Hum Genet 94: 559–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, Marioni JC, Pai AA, Degner JF, Engelhardt BE, Nkadori E, Veyrieras JB, Stephens M, Gilad Y, Pritchard JK. 2010. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature 464: 768–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. 2010. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 20: 110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Kryukov GV, de Bakker PI, Purcell SM, Staples J, Wei LJ, Sunyaev SR. 2010. Pooled association tests for rare variants in exon-resequencing studies. Am J Hum Genet 86: 832–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratnapriya R, Zhan X, Fariss RN, Branham KE, Zipprer D, Chakarova CF, Sergeev YV, Campos MM, Othman M, Friedman JS, et al. 2014. Rare and common variants in extracellular matrix gene Fibrillin 2 (FBN2) are associated with macular degeneration. Hum Mol Genet 23: 5827–5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy TE, Gertz J, Pauli F, Kucera KS, Varley KE, Newberry KM, Marinov GK, Mortazavi A, Williams BA, Song L, et al. 2012. Effects of sequence variation on differential allelic transcription factor occupancy and gene expression. Genome Res 22: 860–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie GR, Dunham I, Zeggini E, Flicek P. 2014. Functional annotation of noncoding sequence variants. Nat Methods 11: 294–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas MA, Beaudoin M, Gardet A, Stevens C, Sharma Y, Zhang CK, Boucher G, Ripke S, Ellinghaus D, Burtt N, et al. 2011. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat Genet 43: 1066–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. 2015. Integrative analysis of 111 reference human epigenomes. Nature 518: 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson G, Hirst M, Bainbridge M, Bilenky M, Zhao Y, Zeng T, Euskirchen G, Bernier B, Varhol R, Delaney A, et al. 2007. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods 4: 651–657. [DOI] [PubMed] [Google Scholar]
- Romanel A, Lago S, Prandi D, Sboner A, Demichelis F. 2015. ASEQ: fast allele-specific studies from next-generation sequencing data. BMC Med Genomics 8: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronkainen J, Huusko TJ, Soininen R, Mondini E, Cinti F, Mäkelä KA, Kovalainen M, Herzig KH, Järvelin MR, Sebert S, et al. 2015. Fat mass- and obesity-associated gene Fto affects the dietary response in mouse white adipose tissue. Sci Rep 5: 9233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg NA, Huang L, Jewett EM, Szpiech ZA, Jankovic I, Boehnke M. 2010. Genome-wide association studies in diverse populations. Nat Rev Genet 11: 356–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozowsky J, Abyzov A, Wang J, Alves P, Raha D, Harmanci A, Leng J, Bjornson R, Kong Y, Kitabayashi N, et al. 2011. AlleleSeq: analysis of allele-specific expression and binding in a network framework. Mol Syst Biol 7: 522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadt EE, Molony C, Chudin E, Hao K, Yang X, Lum PY, Kasarskis A, Zhang B, Wang S, Suver C, et al. 2008. Mapping the genetic architecture of gene expression in human liver. PLoS Biol 6: e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaub MA, Boyle AP, Kundaje A, Batzoglou S, Snyder M. 2012. Linking disease associations with regulatory information in the human genome. Genome Res 22: 1748–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. 2010. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 7: 575–576. [DOI] [PubMed] [Google Scholar]
- Scuteri A, Sanna S, Chen WM, Uda M, Albai G, Strait J, Najjar S, Nagaraja R, Orrú M, Usala G, et al. 2007. Genome-wide association scan shows genetic variants in the FTO gene are associated with obesity-related traits. PLoS Genet 3: e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shihab HA, Rogers MF, Gough J, Mort M, Cooper DN, Day IN, Gaunt TR, Campbell C. 2015. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics 31: 1536–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlyueva D, Stelzer C, Gerlach D, Yáñez-Cuna JO, Rath M, Boryń LM, Arnold CD, Stark A. 2014. Hormone-responsive enhancer-activity maps reveal predictive motifs, indirect repression, and targeting of closed chromatin. Mol Cell 54: 180–192. [DOI] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. 2005. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15: 1034–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer-Sam J, LeBon JM, Dai A, Riggs AD. 1992. A sensitive, quantitative assay for measurement of allele-specific transcripts differing by a single nucleotide. PCR Methods Appl 1: 160–163. [DOI] [PubMed] [Google Scholar]
- Skelly DA, Johansson M, Madeoy J, Wakefield J, Akey JM. 2011. A powerful and flexible statistical framework for testing hypotheses of allele-specific gene expression from RNA-seq data. Genome Res 21: 1728–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smemo S, Tena JJ, Kim KH, Gamazon ER, Sakabe NJ, Gomez-Marin C, Aneas I, Credidio FL, Sobreira DR, Wasserman NF, et al. 2014. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature 507: 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RP, Taher L, Patwardhan RP, Kim MJ, Inoue F, Shendure J, Ovcharenko I, Ahituv N. 2013. Massively parallel decoding of mammalian regulatory sequences supports a flexible organizational model. Nat Genet 45: 1021–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soccio RE, Chen ER, Rajapurkar SR, Safabakhsh P, Marinis JM, Dispirito JR, Emmett MJ, Briggs ER, Fang B, Everett LJ, et al. 2015. Genetic variation determines PPARγ function and anti-diabetic drug response. In Vivo Cell 162: 33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, Crawford GE. 2010. DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb Protoc 2010: pdb prot5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadhouders R, Aktuna S, Thongjuea S, Aghajanirefah A, Pourfarzad F, van Ijcken W, Lenhard B, Rooks H, Best S, Menzel S, et al. 2014. HBS1L-MYB intergenic variants modulate fetal hemoglobin via long-range MYB enhancers. J Clin Invest 124: 1699–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson KR, Coolon JD, Wittkopp PJ. 2013. Sources of bias in measures of allele-specific expression derived from RNA-sequence data aligned to a single reference genome. BMC Genomics 14: 536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpf MP, McVean GA. 2003. Estimating recombination rates from population-genetic data. Nat Rev Genet 4: 959–968. [DOI] [PubMed] [Google Scholar]
- Sunyaev S, Ramensky V, Koch I, Lathe W III, Kondrashov AS, Bork P. 2001. Prediction of deleterious human alleles. Hum Mol Genet 10: 591–597. [DOI] [PubMed] [Google Scholar]
- Surakka I, Horikoshi M, Magi R, Sarin AP, Mahajan A, Lagou V, Marullo L, Ferreira T, Miraglio B, Timonen S, et al. 2015. The impact of low-frequency and rare variants on lipid levels. Nat Genet 47: 589–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JC, Martin HC, Lise S, Broxholme J, Cazier JB, Rimmer A, Kanapin A, Lunter G, Fiddy S, Allan C, et al. 2015. Factors influencing success of clinical genome sequencing across a broad spectrum of disorders. Nat Genet 47: 717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, Pirruccello JP, Ripatti S, Chasman DI, Willer CJ, et al. 2010. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466: 707–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, et al. 2012. The accessible chromatin landscape of the human genome. Nature 489: 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres JM, Gamazon ER, Parra EJ, Below JE, Valladares-Salgado A, Wacher N, Cruz M, Hanis CL, Cox NJ. 2014. Cross-tissue and tissue-specific eQTLs: partitioning the heritability of a complex trait. Am J Hum Genet 95: 521–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbanek M, Hayes MG, Armstrong LL, Morrison J, Lowe LP, Badon SE, Scheftner D, Pluzhnikov A, Levine D, Laurie CC, et al. 2013. The chromosome 3q25 genomic region is associated with measures of adiposity in newborns in a multi-ethnic genome-wide association study. Hum Mol Genet 22: 3583–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Geijn B, McVicker G, Gilad Y, Pritchard J. 2014. WASP: allele-specific software for robust discovery of molecular quantitative trait loci. bioRxiv 10.1101/011221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhille L, Griffon A, Maqbool MA, Zacarias-Cabeza J, Dao LT, Fernandez N, Ballester B, Andrau JC, Spicuglia S. 2015. High-throughput and quantitative assessment of enhancer activity in mammals by CapStarr-seq. Nat Commun 6: 6905. [DOI] [PubMed] [Google Scholar]
- Vockley CM, Guo C, Majoros WH, Nodzenski M, Scholtens DM, Hayes MG, Lowe WL Jr, Reddy TE. 2015. Massively parallel quantification of the regulatory effects of non-coding genetic variation in a human cohort. Genome Res 25: 1206–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. 2010. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38: e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward LD, Kellis M. 2012. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res 40: D930–D934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weedon MN, Cebola I, Patch AM, Flanagan SE, De Franco E, Caswell R, Rodríguez-Seguí SA, Shaw-Smith C, Cho CH, Lango Allen H, et al. 2014. Recessive mutations in a distal PTF1A enhancer cause isolated pancreatic agenesis. Nat Genet 46: 61–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, Klemm A, Flicek P, Manolio T, Hindorff L, et al. 2014. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res 42: D1001–D1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield TW, Wang J, Collins PJ, Partridge EC, Aldred SF, Trinklein ND, Myers RM, Weng Z. 2012. Functional analysis of transcription factor binding sites in human promoters. Genome Biol 13: R50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TD, Nacu S. 2010. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 26: 873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Yuan W, Velculescu VE, Vogelstein B, Kinzler KW. 2002. Allelic variation in human gene expression. Science 297: 1143. [DOI] [PubMed] [Google Scholar]
- Yin H, Xue W, Chen S, Bogorad RL, Benedetti E, Grompe M, Koteliansky V, Sharp PA, Jacks T, Anderson DG. 2014. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat Biotechnol 32: 551–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabidi MA, Arnold CD, Schernhuber K, Pagani M, Rath M, Frank O, Stark A. 2015. Enhancer–core-promoter specificity separates developmental and housekeeping gene regulation. Nature 518: 556–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Cowper-Sal lari R, Bailey SD, Moore JH, Lupien M. 2012. Integrative functional genomics identifies an enhancer looping to the SOX9 gene disrupted by the 17q24.3 prostate cancer risk locus. Genome Res 22: 1437–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Li D, Zhang B, Lowdon RF, Rockweiler NB, Sears RL, Madden PA, Smirnov I, Costello JF, Wang T. 2015. Epigenomic annotation of genetic variants using the Roadmap Epigenome Browser. Nat Biotechnol 33: 345–346. [DOI] [PMC free article] [PubMed] [Google Scholar]