

Abstract
The Mendelian disorders of the epigenetic machinery are genetic disorders that involve disruption of the various components of the epigenetic machinery (writers, erasers, readers, and remodelers) and are thus expected to have widespread downstream epigenetic consequences. Studying this group may offer a unique opportunity to learn about the role of epigenetics in health and disease. Among these patients, neurological dysfunction and, in particular, intellectual disability appears to be a common phenotype; however, this is often seen in association with other more specific features in respective disorders. The specificity of some of the clinical features raises the question whether specific cell types are particularly sensitive to the loss of these factors. Most of these disorders demonstrate dosage sensitivity as loss of a single allele appears to be sufficient to cause the observed phenotypes. Although the pathogenic sequence is unknown for most of these disorders, there are several examples where disrupted expression of downstream target genes accounts for a substantial portion of the phenotype; hence, it may be useful to systematically map such disease-relevant target genes. Finally, two of these disorders (Rubinstein-Taybi and Kabuki syndromes) have shown post-natal rescue of markers of the neurological dysfunction with drugs that lead to histone deacetylase inhibition, indicating that some of these disorders may be treatable causes of intellectual disability.
Epigenetic modifications are marks added to either the DNA itself or associated histone proteins, commonly considered to be inherited through mitosis (Russo et al. 1996). These can involve either the addition of a methyl group to a cytosine (cytosine methylation) or the addition of covalent modifications to histone tails. Here I will summarize the components that are currently known to maintain epigenetic modifications and the disorders that occur when these components are disrupted.
DNA cytosine methylation is deposited at individual CpG dinucleotides by three individual enzymes (DNMT1, DNMT3A, and DNMT3B) (Jurkowska et al. 2011) and read by proteins that contain methyl-binding domains (MBDs) such as MECP2 (Jørgensen and Bird 2002). Cytosine DNA methylation can be removed through the formation of intermediates (5-hydroxymethylcytosine, 5-formylcytosine, and 5-carboxylcytosine) by the Ten-Eleven Translocation (TET) enzymes (Tahiliani et al. 2009).
Similarly, the histone machinery consists of writers, readers, and erasers but also remodelers (Fahrner and Bjornsson 2014). Although the exact mechanism that maintains histone modifications through mitosis is unknown (Ptashne et al. 2013), there are considerable data associating the presence or absence of these marks with transcriptional activity and chromatin states. For instance, histone acetylation, a binary system (a tail is either acetylated or not), is exclusively present in open chromatin, but histone methylation, a quaternary system (unmethylated, mono-, di-, and tri-methylated), can be present in either open or closed chromatin depending on which lysine of the histone tail is modified. For instance, H3K4me3 is commonly observed in open chromatin at active promoters, whereas H3K27me3 is commonly observed in more closed chromatin conformations.
The histone machinery has several known reader domains that recognize modified histone tails. For instance, bromodomains bind to histone tails with acetylated lysines (Sanchez and Zhou 2009), whereas chromodomains and plant homeodomains (PHDs) bind to histone tails with methylated lysines (Baker et al. 2008; Eissenberg et al. 2012). In addition, ATP-dependent chromatin remodelers play a role in nucleosome maintenance (Clapier and Cairns 2009), a process integral to the deposition or removal of epigenetic modifications.
Here I focus on the emerging group of Mendelian disorders of the epigenetic machinery (Fahrner and Bjornsson 2014), all of which involve genetic mutations in components of the epigenetic machinery, making them likely to have significant downstream epigenetic consequences. Interestingly, these patients often demonstrate neurological dysfunction (Fahrner and Bjornsson 2014; Kleefstra et al. 2014), suggesting that precise epigenetic regulation may be critical for neuronal homeostasis. However, at the same time, it is important to keep in mind that many of these proteins have additional non-epigenetic roles. Going forward it will be important to elucidate how individual functions contribute to the observed phenotypes. A first step toward this goal is to look for shared phenotypes among the Mendelian disorders of the epigenetic machinery with the hope that this may help inform the role of epigenetics in health and disease.
Mendelian disorders of the epigenetic machinery: an emerging genetic etiology of intellectual disability
In the last decade, the field has witnessed an explosion in the discovery of mutations in the various components of the epigenetic machinery (Kleefstra et al. 2006; Ng et al. 2010; Gibson et al. 2012); mutations in many of these components have now been linked to a number of well-known causes of intellectual disability. Intellectual disability is generally defined as deficits of intellectual function and adaptive behavior that occur during the developmental period (see, e.g., http://aaidd.org). Although some investigators have used broader criteria of inclusion into prior groupings of epigenetic gene disorders (Berdasco and Esteller 2013; Kleefstra et al. 2014), here I have included only genes with known epigenetic domains that fulfill criteria as one of the four components of the epigenetic machinery apparatus (writer, eraser, reader, and remodeler). My rationale for these criteria was that dysfunction of any of these components would be expected to lead to epigenetic consequences; it would be relatively easy to define categories based on such domains, and since each of these categories contain multiple members, it would allow us to examine the characteristics of each group individually. However, this criterion excludes some important candidates that are not part of these large component groups (ZFP57, CTCF, SETBP1). Similarly, genes that are part of large deletions (contiguous gene syndromes) of many genes, each of which could contribute to the disease phenotype, were excluded even if one of the genes was a member of one of these categories (BAZ1B).
There are currently 44 such Mendelian disorders of the epigenetic machinery. The vast majority (93%) are associated with neurological dysfunction (Fig. 1, yellow and yellow hashed). Of the patients with neurological dysfunction, the majority are classified as having intellectual disability (Fig. 1, yellow) in general without further characterization. Therefore as a whole, this group serves as an emerging genetically determined cause of intellectual disability. Even though isolated intellectual disability is common in the population (1%–2%) and each of these syndromic disorders is relatively rare, together the Mendelian disorders of the epigenetic machinery form a sizeable fraction of genetically defined intellectual disability. Unbiased approaches, such as clinical exome sequencing, have also discovered a surprisingly large fraction of de novo mutations in chromatin genes in patients with isolated intellectual disability (Leduc et al. 2014). Many of the same genes are overrepresented in conditions with significant phenotypic overlap, such as autism (De Rubeis et al. 2014). The apparent overrepresentation of these ubiquitously expressed epigenetic factors in intellectual disability compared with other well-known neuronal components (ion channels, synaptic components) has come as a surprise, but this may suggest that neurons are particularly sensitive to epigenetic disruption or that some of these factors may have other undefined functions important for neuronal homeostasis. Similar to imprinted disorders, the other common phenotypic trait is disruption of normal growth; this can manifest as either growth retardation or overgrowth with the latter being exclusively seen in the category of disrupted writers (Fig. 1, red and green, respectively). Additional features frequently observed in many of these disorders include a wide variety of different limb malformations, ranging from nail abnormalities to severe brachydactyly (Fig. 1, orange), as well as immune dysfunction in some disorders (Fig. 1, purple). Other features observed in individual disorders can involve almost any organ system. It is interesting that although mutations in many of these genes have been found in a wide variety of cancers (Tsai and Baylin 2011), increased cancer predisposition does not appear to be the general rule (Kleefstra et al. 2014). However, these disorders are rare, and this may reflect a lack of long-term follow-up in some of these patients.
Figure 1.

Characteristic features of the Mendelian disorders of the epigenetic machinery. The most common phenotypic feature is intellectual disability (yellow). Other features include growth retardation (red), overgrowth (green), immune dysfunction (purple), and various limb abnormalities (orange). The components of the epigenetic machinery (horizontal labels) and genetic syndromes (vertical labels) are divided into four categories (writer, eraser, reader, or remodeler). The majority of these genes demonstrate dosage sensitivity (filled circle).
The majority of the mutated genes in these Mendelian disorders of the epigenetic machinery reside on autosomes (80%), with the rest being on the X Chromosome. However, within the eraser category, the genes on the X Chromosome are significantly overrepresented (71%, P = 0.005, Fisher's exact test) (Fig. 1), even after correction of four independent tests, compared with the rest of the group. In contrast, the writer category appears underrepresented on the X Chromosome (0%), but this is not significant after correction of four tests (P = 0.07, Fisher's exact test). It is worth pointing out that two of the X-linked genes from the eraser category escape X inactivation (KDM6A, KDM5C) and therefore potentially behave as autosomal genes. The reason for the overrepresentation of the genes in the eraser category on the X Chromosome remains an interesting question to be pursued.
What is the basis for the observed dosage sensitivity?
In most enzyme disorders, such as the inborn errors of metabolism, the loss of a single copy of an enzyme is tolerated, and patients with residual enzymatic activity generally have milder phenotypes than those with complete loss of an enzymatic function. About 80% of all Mendelian diseases attributed to enzyme deficiency demonstrate recessive inheritance (Jimenez-Sanchez et al. 2001) in contrast to only 14% of the Mendelian disorders of the epigenetic machinery (Fig. 1). This is true even though three out of four components of the epigenetic machinery involve enzymes (writer, eraser, and remodeler categories) (Fig. 1). If one looks only at those three categories, the vast majority (90%) of these enzyme disorders demonstrate only loss of a single allele. Haploinsufficiency is felt to be the predominant explanation for this dosage sensitivity, supported by the fact that most patients have a large number of loss-of-function mutations distributed throughout the gene (Ng et al. 2010), mutations that lead to nonsense mediated decay (Jones et al. 2012), or identical phenotypes observed in patients harboring either a loss-of-function mutation or a deletion of the region encompassing the gene (Williams et al. 2010; Fahrner and Bjornsson 2014). However, other mechanisms such as dominant-negative or gain-of-function mutations have not been excluded for many of these disorders. This raises questions about the molecular basis of this dosage sensitivity. One possible explanation relates to the fact that the epigenetic machinery is composed of complicated multiprotein complexes, so minor changes in amounts of various components could disrupt normal complex formation, thus contributing to dosage sensitivity. Another possible explanation is that the function of these enzymes is so critical that only the loss of single copy is tolerated, and even the loss of a single allele decreases fitness, making them less likely to be passed on compared with the other known enzyme disorders.
We have previously proposed that given the opposing activity of many of the components of the epigenetic machinery, the pathogenic sequence in these disorders involves an imbalance of chromatin states; i.e., a decrease in the amount of a particular enzyme leads to an imbalance in the opposing machinery, with a secondary effect on the expression on a subset of generally unknown disease-relevant target genes (Fahrner and Bjornsson 2014). This idea was prompted by considering the function of the two genes that cause Kabuki syndrome, KMT2D and KDM6A. These two causes both lead to Kabuki syndrome even though these individual components of the epigenetic machinery target two independent histone modifications, H3K4me3 and H3K27me3, respectively. However, despite the different targets, the overall effect on chromatin is predicted to be the same since KMT2D adds H3K4me3 (an open chromatin mark) and KDM6A removes H3K27me3 (a closed chromatin mark). Therefore, the final outcome is decreased open chromatin at a subset of target genes. This is further supported by the fact that a substantial fraction (at least a third) of the genes associated with these disorders are homologs of the Polycomb and Trithorax genes, and studies in both Drosophila and mouse models have demonstrated the importance of balance between these systems for the normal expression states of target genes. Specifically, mutations in either system have been shown to have opposing effects on the expression of a subset of target genes (Paro et al. 1995; Hanson et al. 1999). In recent years, investigators have shown that this regulation may be dynamic (Schwartz and Pirrotta 2008). For instance, in living Drosophila, the Polycomb group complexes appear to be undergoing constant exchange, allowing dynamic competition with other factors (Ficz et al. 2005). In addition, regions marked by either Polycomb or Trithorax also often demonstrate DNase hypersensitivity, suggesting the presence of an ongoing continuous process that disrupts nucleosomes at these regions (Mito et al. 2007). Keeping a subset of genes under “pressure” from two opposing systems may allow the cellular system to rapidly respond to environmental stimuli. The genes found as causative in the various Mendelian disorders of the epigenetic machinery appear to play a role in several dynamic processes such as the circadian rhythms and mTOR pathways, in which the system is directly responsive to an environmental stimulus (Mullegama et al. 2015).
Since epigenetic modifications vary stochastically even within populations of identical cells (Singer et al. 2014), the loss of a component of the epigenetic machinery may lead to a change in the probability of a particular chromatin state being observed in any given cell within a population of cells. In fact, early studies employing single-cell sequencing on primary fibroblasts reveal that many genes appear to only be expressed from a single allele in any given cell (Borel et al. 2015). If this observation holds true, the pathogenesis of some dominant disorders may be the consequence of having two distinct cellular populations: cells with or without a functioning copy of the gene in question. However, this observation may be an illusion that may stem from phenomena such as spontaneous burst of asynchronous expression, which has been described in some cellular systems (Cai et al. 2008; Crabtree and Graef 2008). In any case, the careful characterization of epigenetic abnormalities in relevant cell populations may be critical to understanding the pathophysiology of these disorders.
Are the classical Mendelian disorders of the epigenetic machinery just the tip of the iceberg?
Another notable feature among the Mendelian disorders of the epigenetic machinery is the breadth of the clinical phenotypes; this may explain why the discovery of genetic etiologies for many members had to await the advent of exome sequencing. For instance, in Kabuki syndrome, a disorder of histone methylation, individual patients have been described with a multitude of distinct cardiac malformations (Dentici et al. 2015), yet only a subset of patients has any given abnormality. This raises the possibility that the defect in the histone machinery is a predisposing factor that in combination with cis genetic variation at a subset of disease-relevant target genes determines the final outcome of this particular subphenotype (i.e., congenital heart disease) and therefore contributes to the broadening of the phenotype.
The view of the histone abnormality as a predisposing factor is supported by a recent study of patients with a wide variety of congenital heart disease found to have many different de novo mutations in a number of different histone-modifying genes (Zaidi et al. 2013). I was curious whether phenotypic breadth is a general feature of the Mendelian disorders of the epigenetic machinery compared with other disorders. Therefore, I took all available clinical synopses in OMIM (www.omim.org, March 2015) and binned disease entries based on number of described features and organ systems in each entry. When I did this, I observed a larger fraction of Mendelian disorders of the epigenetic machinery in high feature bins compared with other disorders (Fig. 2A). Similarly these disorders show a higher number of organ system entries within the OMIM clinical synopsis headings compared with other disorders (Fig. 2B). Although these findings are limited given the relatively few members of this group with available OMIM clinical synopsis, they support the idea that these disorders, on average, have unusual phenotypic breadth. Given the extent of the epigenetic machinery and the many components that have yet to be linked to a disease entity, I expect additional members to be discovered in the near future. However, it is also possible that a number of patients will be discovered that have less of the classically defined phenotype, a view supported by recent clinical sequencing efforts (Zaidi et al. 2013; Leduc et al. 2014). Furthermore, there have been reports of mosaic mutations in patients for some of these genes (Banka et al. 2013), which could profoundly impact penetrance and disease severity.
Figure 2.
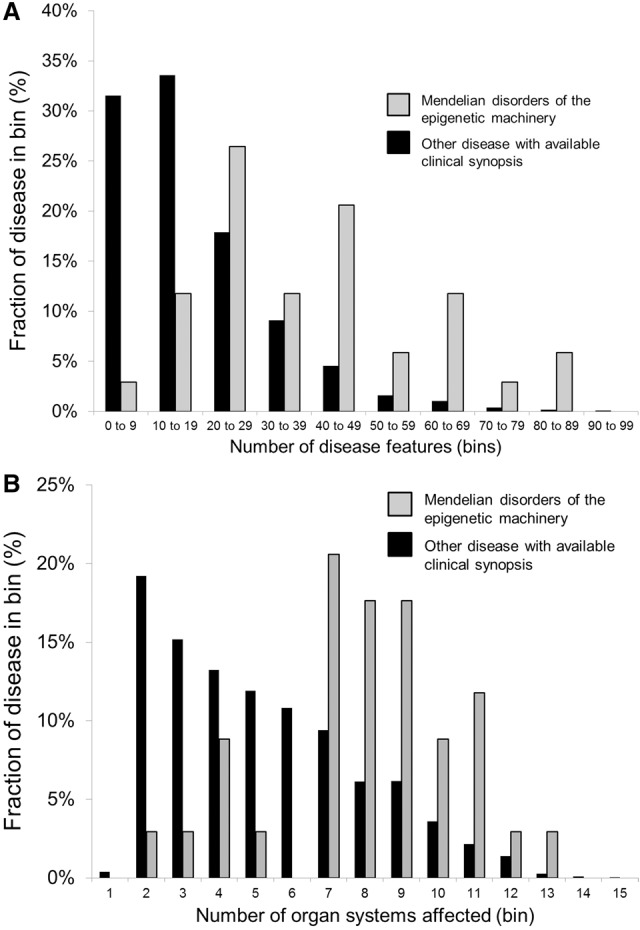
The Mendelian disorders of the epigenetic machinery exhibit phenotypic breadth. (A) When all phenotypic features are tallied in available OMIM clinical synopsis, the Mendelian disorders of the epigenetic machinery have a shift in the distribution toward higher bins, indicating phenotypic breadth compared with all other diseases with available clinical synopsis. (B) Similarly, there is shift in distribution toward higher number of organ systems affected in the Mendelian disorders of the epigenetic machinery compared with other diseases.
Mapping of disease-relevant target genes: the next frontier?
If misregulation of a subset of target genes is central to the pathogenesis of the Mendelian disorders of the epigenetic machinery (Fahrner and Bjornsson 2014), one should be able to identify downstream target genes, providing a unique window into the pathogenic sequence of these disorders. Given the non-lethality of all of these conditions, it is possible that the number of critical target genes is quite limited, and those defined so far support this notion (Table 1). Some disease-relevant target genes have already been linked to particular subphenotypes (Table 1). For instance, in ATR-X syndrome, researchers have determined that decreased expression of a downstream target genes locus (alpha-globin locus) in a relevant cell population (erythrocytes) confers the alpha-thalassemia subphenotype (Law et al. 2010). In this case, the decreased amounts of the ATRX protein lead to decreased expression of the alpha-globin locus. However, the impact of the decreased dose of ATRX protein is also dependent on the size of a variable number tandem repeat (VNTR) in the proximity of the alpha-globin locus, as individuals with a larger VNTR have more severe hemoglobin H disease, a quantitative measure of the severity of the thalassemia phenotype (Law et al. 2010). This suggests that to fully understand the transcriptional consequences of mutations in these trans-acting factors, one may need to integrate information of cis variation of the target gene locus as well. However, to do this, one has to identify the target genes, which will be greatly aided by the recent development of low cell epigenomic methods (Greenleaf 2015). However, it may sometimes be possible to identify target genes genetically as well since in some cases these phenotypes have more than one genetic etiology. When the second etiology does not involve an epigenetic component with overlapping function, it may represent a potential downstream target gene. In fact, there are several potential such examples, including Sotos syndrome, an overgrowth syndrome associated with cardinal features of intellectual disability and typical facial features but also congenital anomalies (Baujat and Cormier-Daire 2007). Recently, two siblings with a Sotos-like phenotype (intellectual disability, facial features, and overgrowth) were shown to harbor homozygous frameshift mutations in APC2 (Almuriekhi et al. 2015), making this an independent cause of a phenotype with great overlap with classical Sotos syndrome (Almuriekhi et al. 2015). The investigators further demonstrate that APC2 is a downstream target of the histone methyltransferase NSD1 (the cause of classical Sotos syndrome) and that a mouse model with loss of APC2 (Apc2−/−) has neuronal abnormalities that overlap those found in mice with decreased expression of Nsd1 (Almuriekhi et al. 2015). The investigators postulate that misregulation of APC2 secondary to defects of NSD1 leads to a subset of the phenotypes seen in Sotos syndrome but that other target genes may play a role in explaining the full phenotypic spectrum.
Table 1.
Potential downstream targets linked to subphenotypes
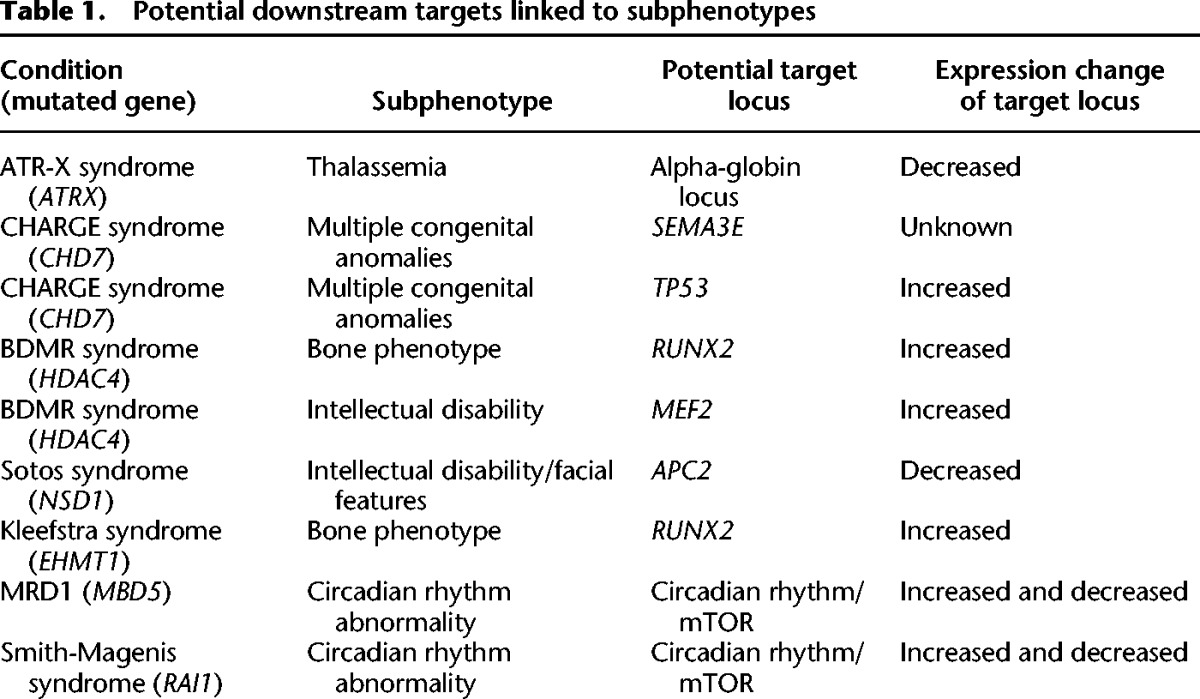
Similarly, in CHARGE syndrome, a disorder that has some phenotypic overlap with Kabuki syndrome (including post-natal growth retardation, hearing loss, cleft palate, horseshoe kidney, and immune dysfunction) (Schulz et al. 2014), there are two known genetic causes, one of which involves a component of the epigenetic machinery. In addition to CHD7 (a chromodomain helicase), mutations have also been found in semaphorin 3E (SEMA3E) (Lalani et al. 2004). One of the target genes of CHD7 is TP53, which is negatively regulated by CHD7 (Van Nostrand et al. 2014), and inappropriate activation of TP53 appears to give rise to many of the phenotypic features seen in CHARGE syndrome (Van Nostrand et al. 2014). Interestingly, in hypoxia, there is up-regulation of TP53, which secondarily leads to up-regulation of SEMA3E (Moriya et al. 2010). This raises the question whether the SEMA3E mutations found in patients with CHARGE are in fact activating mutations or whether this gene network forms some type of feedback loop.
Sometimes shared targets may explain overlapping phenotypes observed among individual Mendelian disorders of the epigenetic machinery. For example, both Kleefstra and brachydactyly-mental retardation (BDMR) syndromes appear to cause misregulation of the gene RUNX2 (Vega et al. 2004; Balemans et al. 2014), and both conditions lead to a particular limb abnormality (brachydactyly). Finally, sometimes the shared abnormalities appear to disrupt the function of target networks rather than single genes. For instance, three of the Mendelian disorders of the epigenetic machinery (MRD1, Smith-Magenis, and BDMR syndromes) have disordered sleep, which may be due to disruption of normal circadian rhythms as all these disorders have abnormalities of the mTOR and circadian gene expression networks (Mullegama et al. 2015). Further identification of target genes will surely help deepen our understanding of the pathogenic sequence of these disorders. Such explorations are establishing a new frontier in the field of epigenetics that takes advantage of emerging tools from genomics, epigenomics, and systems biology.
Do downstream epigenetic consequences explain the entire phenotype?
Downstream epigenetic consequences with concomitant target gene misregulation play a role in the pathogenic sequence of at least some of the Mendelian disorders of the epigenetic machinery. Mouse models of the Kabuki and Rubinstein-Taybi syndromes show global deficiency of the predicted deficient marks, histone H3K4me3 and histone acetylation, respectively (Valor et al. 2011; Bjornsson et al. 2014). In some cases, however, other epigenetic modifications may be affected as well. For instance, in cell lines from patients with ICF syndrome, a deficiency of one of the de novo methyltransferases (DNMT3B) leads to global DNA hypomethylation of about 700 genes (Jin et al. 2008) but also to histone abnormalities (decreased H3K27me3 and increased H3K9 acetylation) of the same genes. Therefore, a deficiency of one epigenetic modification may lead to secondary abnormalities of another epigenetic modification. Deficiency of the other de novo methyltransferase (DNMT3A) also suggests interplay among DNA methylation, histone modifications, and imprinting, as the phenotype observed with haploinsufficiency of DNMT3A (a component of the DNA methylation machinery) involves overgrowth (Tatton-Brown et al. 2014), a phenotype also shared among patients with defects of imprinting and with defects of components of the histone machinery (Fig. 1).
Are these disorders entirely due to transcriptional dysregulation secondary to abnormalities of one or more epigenetic modification, or do these proteins have additional non-histone functions? The histone acetylation machinery is a particularly informative example (Fig. 3). Here there may be a limited subset of target genes sensitive to the amount of histone acetyltransferases (CREBBP and EP300), only expressed when histone acetyltransferases are present in sufficient amounts to overcome the opposing histone deacetylases (HDACs) (Fig. 3, middle top). Alternatively, a gene that is normally repressed by HDAC inhibition such as MEF2, may become overexpressed with the loss of an HDAC such as HDAC4, as the opposing factors that favor chromatin opening may become dominant. In fact, for many of the known target genes, the observed gene expression abnormality is consistent with the predicted epigenetic abnormality (Table 1). For example, in the cases of the EHMT1 and HDAC4 genes whose normal function favors gene repression, their target genes (MEF2, RUNX2) show overexpression in cells that harbor mutations in these factors. Although direct epigenetic consequences offer a good starting point in the study of these disorders, there may be other functions that can contribute to the disease pathogenesis. Although HDAC4 is known to repress MEF2 (Miska et al. 1999), loss of HDAC4 does not lead to major changes in the global acetylation of proteins (Mielcarek et al. 2013), but HDAC4 may lead to repression through recruitment of other repressors (Ronan et al. 2013). Although loss of this protein is known to change the expression of several potential target genes (including RUNX2), the major cellular location of HDAC4 protein is cytoplasmic (Mielcarek et al. 2013), making it hard to exclude that the disease-relevant role of HDAC4 may lie in the cytoplasm. Similarly, the CREBBP and EP300 proteins have other roles in both the nucleus and cytoplasm (Fig. 3), such as modifying the activity of the TP53 protein in either compartment through acetylation and ubiquitination, respectively (Shi et al. 2009). Going forward, it will be important to clarify the roles in different cellular compartments. In addition, there may even be multiple nuclear roles. A particularly informative example is HDAC8, which, in addition to deacetylating histone tails, deacetylates the cohesin complex (Fig. 3). In fact, haploinsufficiency of HDAC8 has been shown to be one cause of Cornelia De Lange syndrome, a cohesinopathy (Deardorff et al. 2012), but all other known causes of this disorder involve factors involved in cohesion maintenance (Horsfield et al. 2012). Future studies will clarify the individual roles of HDAC8 as an epigenetic regulator and modulator of cohesin acetylation.
Figure 3.

Acetylation machinery has diverse functions. A subset of target genes may require the presence of both copies of either of the histone acetyltransferases “writers” (CREBBP and EP300). The full complement of histone acetyltransferases may ensure open chromatin and expression from some loci (top, middle). Alternatively, other genes may be dominated by HDACs that favor repressed chromatin (top, sides). However, HDAC8 also plays a role in removing the acetylation mark from cohesins and may also have other roles in cytoplasm. Similarly, CREBBP and EP300 regulate other proteins such as TP53 through acetylation in nucleus and ubiquitination in the cytoplasm. There are also other components that interact with the components of the epigenetic or cohesion machinery, such as RPS6KA3 and ESCO1, that also lead to intellectual disability.
Finally, sometimes the disease pathogenesis can be entirely unrelated to any function of the particular component. Up to now, it has remained unclear as to why mutations in the maintenance methyltransferase (DNMT1) would lead to an adult onset condition (progressive dementia), but a deficiency of the de novo DNA methyltransferases would lead to intellectual disability in childhood. However, investigators have recently shown that the mutant forms of the DNMT1 protein demonstrate cytoplasmic trafficking problems and end up in cytoplasm in aggresomes, leading to aggresome-induced autophagy, a pathophysiological process previously observed in other neurodegenerative disorders (Baets et al. 2015) and a potential mechanism for the late onset of the phenotype in question.
What are the relevant cell populations affected in the Mendelian disorders of the epigenetic machinery?
Some of the components of the epigenetic machinery are ubiquitously expressed, and although some of the phenotypes (such as intellectual disability) are shared among a number of different conditions (Fig. 1), other phenotypes are specific enough to have allowed clinical classification and diagnosis in genetics clinics for years. This therefore raises the question whether there are specific cell populations that are particularly sensitive to loss of these components of the epigenetic machinery at specific times during development. Here it may be necessary to explore target gene expression not only in the right cell type but also at the right developmental time. In addition to developmental phenotypes (multiple congenital anomalies), in some cases there appear to be ongoing defects that remain consequential in post-natal life. An example of the latter is the hippocampal memory defects seen in many of the mouse models with defects of the various components of the epigenetic machinery that correspond to many of the known syndromes. Hippocampal memory defects have been described in mouse models of Kabuki syndrome (Bjornsson et al. 2014), Kleefstra syndrome (Balemans et al. 2014), Rubinstein-Taybi syndrome (Alarcón et al. 2004; Korzus et al. 2004), Sotos syndrome (Almuriekhi et al. 2015), Weaver syndrome (Zhang et al. 2014), and several other syndromes. Defects of many of the components of the epigenetic machinery also lead to neurogenesis defects in the granule cell layer of the dentate gyrus (Lopez-Atalaya et al. 2011; Bjornsson et al. 2014; Zhang et al. 2014), defects of cortical neurogenesis (Harrison et al. 2012; Ritchie et al. 2014), and deficient synaptic plasticity (Levenson et al. 2006; Sando et al. 2012). This raises the question whether cells undergoing neurogenesis and synaptogenesis are particularly sensitive to subtle defects of the epigenetic machinery and downstream epigenetic abnormalities. Does this sensitivity reflect an inability to up-regulate a subset of relevant genes in specific populations, or does it reflect dysregulation of a process that could affect replication timing or the rate of entry of the cell cycle in many different cells (Clynes et al. 2014)? Given the recent advances in epigenomic methodologies using fewer and fewer cells (Greenleaf 2015), some of these questions are now within the grasp of investigators, and such analysis will hopefully help inform the pathogenic sequence of many of these conditions. However, many of these questions will probably be best answered using specific cell populations that can be robustly defined and isolated since epigenetic modifications are very dependent on cell type (Montaño et al. 2013), and variable mixtures of cells could give data that are hard to interpret.
Mendelian disorders of the epigenetic machinery: potentially treatable causes of intellectual disability?
Over the last decade, there have been a number of examples where a post-natal intervention improves some of the neurological abnormalities seen in mouse models with defective components of the epigenetic machinery (Alarcón et al. 2004; Korzus et al. 2004; Guy et al. 2007; Bjornsson et al. 2014). Several of these therapeutic strategies target the downstream epigenetic abnormalities either directly (Alarcón et al. 2004; Korzus et al. 2004) or indirectly (Bjornsson et al. 2014). For these particular examples, functional rescue was observed in various behavioral testing regimens, and in some, this was seen concomitantly with normalization of both epigenetic and neurogenesis defects (Bjornsson et al. 2014). These data suggest that some of these disorders may be potentially treatable causes of intellectual disability, an exciting possibility that will prompt additional studies both in patients of these disorders and in related conditions. Going forward, it will be of utmost importance to carefully phenotype these patients to see whether their intellectual disability can be classified in more detail, as such studies may both support findings from the mouse models and create potential outcome measures for future therapeutic trials. Furthermore, in any therapeutic trial, the epigenetic abnormality has the potential of being utilized as a biomarker both of disease state and of therapeutic efficiency. Once disease-relevant target genes are mapped, these will automatically become additional therapeutic targets downstream from the epigenetic abnormality, which may offer more specific therapeutic approaches than global modulation of the epigenome. Additional causes of intellectual disability also involve components that interact with the epigenetic machinery and may also lead to epigenetic dysregulation. For instance, RPS6KA3 (Fig. 3), mutations of which are associated with Coffin-Lowry syndrome, phosphorylates and thereby activates CREB1 (Xing et al. 1996). Some of these also lead to hippocampal memory defects (Morice et al. 2013). Therefore, these disorders may potentially extend the number of conditions that may be targets for therapeutic intervention.
Given the prominent disruption of hippocampal memory in many of the mouse models of these disorders, a major remaining question is whether neurogenesis defects and/or abnormalities of synaptic plasticity are a unifying pathophysiological process for the Mendelian disorders of the epigenetic machinery. If disruptions of these dynamic processes are found to be causative for the intellectual disability in these disorders, these processes could become independent therapeutic targets with great promise for a disease entity (intellectual disability) with few available therapeutic agents.
Competing interest statement
Third Rock Ventures, LLC. could be impacted by the research described in this publication. H.T.B. is a paid consultant to Third Rock Ventures, LLC. This arrangement has been reviewed and approved by the Johns Hopkins University in accordance with its conflict of interest policies.
Acknowledgments
I thank Joanna Amberger for help with OMIM clinical synopsis analysis; Dimitrios Avramopoulos for help with selection of statistical tests; Barbara Migeon, Jill Fahrner, Wendy Jones, Joel Benjamin, Genay Pilarowski, and Giovanni Carosso for critical reading of manuscript; and Catherine Kiefe for her assistance with creating and editing the figures. I also thank the anonymous reviewers for their many helpful comments. This work was supported by a grant to H.T.B. by the NIH Director's Early Independence Award (DP5OD017877).
Footnotes
Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.190629.115.
Freely available online through the Genome Research Open Access option.
References
- Alarcón JM, Malleret G, Touzani K, Vronskaya S, Ishii S, Kandel ER, Barco A. 2004. Chromatin acetylation, memory, and LTP are impaired in CBP+/− mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron 42: 947–959. [DOI] [PubMed] [Google Scholar]
- Almuriekhi M, Shintani T, Fahiminiya S, Fujikawa A, Kuboyama K, Takeuchi Y, Nawaz Z, Nadaf J, Kamel H, Kitam AK, et al. 2015. Loss-of-function mutation in APC2 causes Sotos syndrome features. Cell Rep 10: 1585–1589. [DOI] [PubMed] [Google Scholar]
- Baets J, Duan X, Wu Y, Smith G, Seeley WW, Mademan I, McGrath NM, Beadell NC, Khoury J, Botuyan MV, et al. 2015. Defects of mutant DNMT1 are linked to a spectrum of neurological disorders. Brain 138: 845–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker LA, Allis CD, Wang GG. 2008. PHD fingers in human diseases: disorders arising from misinterpreting epigenetic marks. Mutat Res 647: 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balemans MC, Ansar M, Oudakker AR, van Caam AP, Bakker B, Vitters EL, van der Kraan PM, de Bruijn DR, Janssen SM, Kuipers AJ, et al. 2014. Reduced euchromatin histone methyltransferase 1 causes developmental delay, hypotonia, and cranial abnormalities associated with increased bone gene expression in Kleefstra syndrome mice. Dev Biol 386: 395–407. [DOI] [PubMed] [Google Scholar]
- Banka S, Howard E, Bunstone S, Chandler KE, Kerr B, Lachlan K, McKee S, Mehta SG, Tavares AL, Tolmie J, et al. 2013. MLL2 mosaic mutations and intragenic deletion-duplications in patients with Kabuki syndrome. Clin Genet 83: 467–471. [DOI] [PubMed] [Google Scholar]
- Baujat G, Cormier-Daire V. 2007. Sotos syndrome. Orphanet J Rare Dis 2: 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdasco M, Esteller M. 2013. Genetic syndromes caused by mutations in epigenetic genes. Hum Genet 132: 359–383. [DOI] [PubMed] [Google Scholar]
- Bjornsson HT, Benjamin JS, Zhang L, Weissman J, Gerber EE, Chen YC, Vaurio RG, Potter MC, Hansen KD, Dietz HC. 2014. Histone deacetylase inhibition rescues structural and functional brain deficits in a mouse model of Kabuki syndrome. Sci Transl Med 6: 256ra135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borel C, Ferreira PG, Santoni F, Delaneau O, Fort A, Popadin KY, Garieri M, Falconnet E, Ribaux P, Guipponi M, et al. 2015. Biased allelic expression in human primary fibroblast single cells. Am J Hum Genet 96: 70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L, Dalal CK, Elowitz MB. 2008. Frequency-modulated nuclear localization bursts coordinate gene regulation. Nature 455: 485–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapier CR, Cairns BR. 2009. The biology of chromatin remodeling complexes. Annu Rev Biochem 78: 273–304. [DOI] [PubMed] [Google Scholar]
- Clynes D, Jelinska C, Xella B, Ayyub H, Taylor S, Mitson M, Bachrati CZ, Higgs DR, Gibbons RJ. 2014. ATRX dysfunction induces replication defects in primary mouse cells. PLoS One 9: e92915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree GR, Graef IA. 2008. Bursting into the nucleus. Sci Signal 1: pe54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, et al. 2014. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515: 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deardorff MA, Bando M, Nakato R, Watrin E, Itoh T, Minamino M, Saitoh K, Komata M, Katou Y, Clark D, et al. 2012. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesion acetylation cycle. Nature 489: 313–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dentici ML, Di Pede A, Lepri FR, Gnazzo M, Lombardi MH, Auriti C, Petrocchi S, Pisaneschi E, Bellacchio E, Capolino R, et al. 2015. Kabuki syndrome: clinical and molecular diagnosis in the first year of life. Arch Dis Child 100: 158–164. [DOI] [PubMed] [Google Scholar]
- Eissenberg JC. 2012. Structural biology of the chromodomain: form and function. Gene 496: 69–78. [DOI] [PubMed] [Google Scholar]
- Fahrner JA, Bjornsson HT. 2014. Mendelian disorders of the epigenetic machinery: tipping the balance of chromatin states. Annu Rev Genomics Hum Genet 15: 269–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficz G, Heintzmann R, Arndt-Jovin DJ. 2005. Polycomb group protein complexes exchange rapidly in living Drosophila. Development 132: 3963–3976. [DOI] [PubMed] [Google Scholar]
- Gibson WT, Hood RL, Zhan SH, Bulman DE, Fejes AP, Moore R, Mungall AJ, Eydoux P, Babul-Hirji R, An J, et al. 2012. Mutations in EZH2 cause Weaver syndrome. Am J Hum Genet 90: 110–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenleaf WJ. 2015. Assaying the epigenome in limited numbers of cells. Methods 72: 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J, Gan J, Selfridge J, Cobb S, Bird A. 2007. Reversal of neurological defects in a mouse model of Rett syndrome. Science 315: 1143–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson RD, Hess JL, Yu BD, Ernst P, van Lohuizen M, Berns A, van der Lugt NM, Shashikant CS, Ruddle FH, Seto M, et al. 1999. Mammalian Trithorax and Polycomb-group homologues are antagonistic regulators of homeotic development. Proc Natl Acad Sci 96: 14372–14377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison SJ, Nishinakamura R, Jones KR, Monaghan AP. 2012. Sall1 regulates cortical neurogenesis and laminar fate specification in mice: implications for neural abnormalities in Townes-Brocks syndrome. Dis Model Mech 5: 351–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horsfield JA, Print CG, Mönnich M. 2012. Diverse developmental disorders from the one ring: Distinct molecular pathways underlie the cohesinopathies. Front Genet 3: 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Sanchez G, Childs B, Valle D. 2001. Human disease genes. Nature 409: 853–855. [DOI] [PubMed] [Google Scholar]
- Jin B, Tao Q, Peng J, Soo HM, Wu W, Ying J, Fields CR, Delmas AL, Liu X, Qiu J, et al. 2008. DNA methyltransferase 3B (DNMT3B) mutations in ICF syndrome lead to altered epigenetic modifications and aberrant expression of genes regulating development, neurogenesis and immune function. Hum Mol Genet 17: 690–709. [DOI] [PubMed] [Google Scholar]
- Jones WD, Dafou D, McEntagart M, Woollard WJ, Elmslie FV, Holder-Espinasse M, Irving M, Saggar AK, Smithson S, Trembath RC, et al. 2012. De novo mutations in MLL cause Wiedemann-Steiner syndrome. Am J Hum Genet 91: 358–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jørgensen HF, Bird A. 2002. MeCP2 and other methyl-CpG binding proteins. Ment Retard Dev Disabil Res Rev 8: 87–93. [DOI] [PubMed] [Google Scholar]
- Jurkowska RZ, Jurkowski TP, Jeltsch A. 2011. Structure and function of mammalian DNA methyltransferases. Chembiochem 12: 206–222. [DOI] [PubMed] [Google Scholar]
- Kleefstra T, Brunner HG, Amiel J, Oudakker AR, Nillesen WM, Magee A, Geneviève D, Cormier-Daire V, van Esch H, Fryns JP, et al. 2006. Loss-of-function mutations in euchromatin histone methyltransferase 1 (EHMT1) cause the 9q34 subtelomeric deletion syndrome. Am J Hum Genet 79: 370–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleefstra T, Schenck A, Kramer JM, van Bokhoven H. 2014. The genetics of cognitive epigenetics. Neuropharmacology 80: 83–94. [DOI] [PubMed] [Google Scholar]
- Korzus E, Rosenfeld MG, Mayford M. 2004. CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron 42: 961–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalani SR, Safiullah AM, Molinari LM, Fernbach SD, Martin DM, Belmont JW. 2004. SEMA3E mutation in a patient with CHARGE syndrome. J Med Genet 41: e94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law MJ, Lower KM, Voon HP, Hughes JR, Garrick D, Viprakasit V, Mitson M, De Gobbi M, Marra M, Morris A, et al. 2010. ATR-X syndrome protein targets tandem repeats and influences allele-specific expression in a size-dependent manner. Cell 143: 367–378. [DOI] [PubMed] [Google Scholar]
- Leduc MS, Beuten J, Zhang J, Niu Z, Xia F, Landsverk M, Bekheirnia MR, Alcaraz W, Cui H, Walkiewicz MA, et al. 2014. Clinical whole exome sequencing provides insight on the prevalence of chromatin remodeling genes in the context of intellectual disability. In 2014 ACMG Annual Clinical Genetics Meeting: Abstract 290 American College of Medical Genetics and Genomics, Bethesda, MD. [Google Scholar]
- Levenson JM, Roth TL, Lubin FD, Miller CA, Huang IC, Desai P, Malone LM, Sweatt JD. 2006. Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J Biol Chem 281: 15763–15773. [DOI] [PubMed] [Google Scholar]
- Lopez-Atalaya JP, Ciccarelli A, Viosca J, Valor LM, Jimenez-Minchan M, Canals S, Giustetto M, Barco A. 2011. CBP is required for environmental enrichment-induced neurogenesis and cognitive enhancement. EMBO J 30: 4287–4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mielcarek M, Seredenina T, Stokes MP, Osborne GF, Landles C, Inuabasi L, Franklin SA, Silva JC, Luthi-Carter R, Beaumont V, et al. 2013. HDAC4 does not act as a protein deacetylase in the postnatal murine brain in vivo. PLoS One 8: e80849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miska EA, Karlsson C, Langley E, Nielsen SJ, Pines J, Kouzarides T. 1999. HDAC4 deacetylase associates with and represses the MEF2 transcription factor. EMBO J 18: 5099–5107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mito Y, Henikoff JG, Henikoff S. 2007. Histone replacement marks the boundaries of cis-regulatory domains. Science 315: 1408–1411. [DOI] [PubMed] [Google Scholar]
- Montaño CM, Irizarry RA, Kaufmann WE, Talbot K, Gur RE, Feinberg AP, Taub MA. 2013. Measuring cell-type specific differential methylation in human brain tissue. Genome Biol 14: R94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morice E, Farley S, Poirier R, Dallerac G, Chagneau C, Pannetier S, Hanauer A, Davis S, Vaillend C, Laroche S. 2013. Defective synaptic transmission and structure in the dentate gyrus and selective fear memory impairment in the Rsk2 mutant mouse model of Coffin–Lowry syndrome. Neurobiol Dis 58: 156–168. [DOI] [PubMed] [Google Scholar]
- Moriya J, Minamino T, Tateno K, Okada S, Uemura A, Shimizu I, Yokoyama M, Nojima A, Okada M, Koga H, et al. 2010. Inhibition of semaphorin as a novel strategy for therapeutic angiogenesis. Circ Res 106: 391–398. [DOI] [PubMed] [Google Scholar]
- Mullegama SV, Pugliesi L, Burns B, Shah Z, Tahir R, Gu Y, Nelson DL, Elsea SH. 2015. MBD5 haploinsufficiency is associated with sleep disturbance and disrupts circadian pathways common to Smith–Magenis and fragile X syndromes. Eur J Hum Genet 23: 781–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, McMillin MJ, Gildersleeve HI, Beck AE, Tabor HK, Cooper GM, Mefford HC, et al. 2010. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet 42: 790–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paro R. 1995. Propagating memory of transcriptional states. Trends Genet 11: 295–297. [DOI] [PubMed] [Google Scholar]
- Ptashne M. 2013. Epigenetics: core misconcept. Proc Natl Acad Sci 110: 7101–7103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie K, Watson LA, Davidson B, Jiang Y, Bérubé NG. 2014. ATRX is required for maintenance of the neuroprogenitor cell pool in the embryonic mouse brain. Biol Open 3: 1158–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronan JL, Wu W, Crabtree GR. 2013. From neural development to cognition: unexpected roles for chromatin. Nat Rev Genet 14: 347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo VEA, Martienssen RA, Riggs AD. 1996. Epigenetic mechanisms of gene regulation. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Sanchez R, Zhou MM. 2009. The role of human bromodomains in chromatin biology and gene transcription. Curr Opin Drug Discov Devel 12: 659–665. [PMC free article] [PubMed] [Google Scholar]
- Sando R III, Gounko N, Pieraut S, Liao L, Yates J III, Maximov A. 2012. HDAC4 governs a transcriptional program essential for synaptic plasticity and memory. Cell 151: 821–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz Y, Freese L, Mänz J, Zoll B, Völter C, Brockmann K, Bögershausen N, Becker J, Wollnik B, Pauli S. 2014. CHARGE and Kabuki syndromes: a phenotypic and molecular link. Hum Mol Genet 23: 4396–4405. [DOI] [PubMed] [Google Scholar]
- Schwartz YB, Pirrotta V. 2008. Polycomb complexes and epigenetic states. Curr Opin Cell Biol 20: 266–273. [DOI] [PubMed] [Google Scholar]
- Shi D, Pop MS, Kulikov R, Love IM, Kung AL, Grossman SR. 2009. CBP and p300 are cytoplasmic E4 polyubiquitin ligases for p53. Proc Natl Acad Sci 106: 16275–16280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer ZS, Yong J, Tischler J, Hackett JA, Altinok A, Surani MA, Cai L, Elowitz MB. 2014. Dynamic heterogeneity and DNA methylation in embryonic stem cells. Mol Cell 55: 319–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A. 2009. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324: 930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatton-Brown K, Seal S, Ruark E, Harmer J, Ramsay E, Del Vecchio Duarte S, Zachariou A, Hanks S, O'Brien E, Aksglaede L, et al. 2014. Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nat Genet 46: 385–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai HC, Baylin SB. 2011. Cancer epigenetics: linking basic biology to clinical medicine. Cell Res 21: 502–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valor LM, Pulopulos MM, Jimenez-Minchan M, Olivares R, Lutz B, Barco A. 2011. Ablation of CBP in forebrain principal neurons causes modest memory and transcriptional defects and a dramatic reduction of histone acetylation but does not affect cell viability. J Neurosci 31: 1652–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand JL, Brady CA, Jung H, Fuentes DR, Kozak MM, Johnson TM, Lin CY, Lin CJ, Swiderski DL, Vogel H, et al. 2014. Inappropriate p53 activation during development induces features of CHARGE syndrome. Nature 514: 228–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega RB, Matsuda K, Oh J, Barbosa AC, Yang X, Meadows E, McAnally J, Pomajzl C, Shelton JM, Richardson JA, et al. 2004. Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell 119: 555–566. [DOI] [PubMed] [Google Scholar]
- Williams SR, Aldred MA, Der Kaloustian VM, Halal F, Gowans G, McLeod DR, Zondag S, Toriello HV, Magenis RE, Elsea SH. 2010. Haploinsufficiency of HDAC4 causes brachydactyly mental retardation syndrome, with brachydactyly type E, developmental delays, and behavioral problems. Am J Hum Genet 87: 219–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing J, Ginty DD, Greenberg ME. 1996. Coupling of the RAS-MAPK pathway to gene activation by RSK2, a growth factor–regulated CREB kinase. Science 273: 959–963. [DOI] [PubMed] [Google Scholar]
- Zaidi S, Choi M, Wakimoto H, Ma L, Jiang J, Overton JD, Romano-Adesman A, Bjornson RD, Breitbart RE, Brown KK, et al. 2013. De novo mutations in histone-modifying genes in congenital heart disease. Nature 498: 220–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ji F, Liu Y, Lei X, Li H, Ji G, Yuan Z, Jiao J. 2014. Ezh2 regulates adult hippocampal neurogenesis and memory. J Neurosci 34: 5184–5199. [DOI] [PMC free article] [PubMed] [Google Scholar]