

Abstract
Eukaryotic genomes are packaged into an extensively folded state known as chromatin. Analysis of the structure of eukaryotic chromosomes has been revolutionized by development of a suite of genome-wide measurement technologies, collectively termed “epigenomics.” We review major advances in epigenomic analysis of eukaryotic genomes, covering aspects of genome folding at scales ranging from whole chromosome folding down to nucleotide-resolution assays that provide structural insights into protein-DNA interactions. We then briefly outline several challenges remaining and highlight new developments such as single-cell epigenomic assays that will help provide us with a high-resolution structural understanding of eukaryotic genomes.
The past two decades have seen dramatic advances in our ability to carry out genome-wide analysis of DNA and RNA populations, first spurred by microarray technology and continuing today with deep sequencing. These technical advances have revolutionized molecular biology, with early genomic studies focused on mRNA abundance, and later studies expanding genome-wide analyses into fields ranging from cancer genome sequencing to systematic dissection of protein structure and function using massively parallel mutagenesis, selection, and sequencing. One of the most productive of these areas has been the broad area of “epigenomics,” which is the use of genome-wide assays to interrogate global patterns of cytosine methylation, chromatin state, and RNA abundance. Although small RNAs (such as siRNAs and piRNAs) and DNA modifications (such as cytosine and adenine methylation) are central to many well-characterized epigenetic inheritance paradigms, this review will focus on epigenomic insights into the folding of the genome and thus will not cover DNA modifications or RNAs.
Below, we introduce the general methodologies for epigenomic analysis. Then, we discuss the structural organization of the eukaryotic genome, starting with 3C-based analyses of higher-order chromosome folding and domain organization, followed by nucleosome resolution chromosome organization (∼100- to 500-bp scale), and finally ending with insights afforded by nucleotide-resolution techniques.
Epigenomic methods
In general, epigenomics refers to the use of genome-wide measurement to characterize chromatin state and DNA modifications in eukaryotes. Such genome-wide measurements involve comprehensive analysis of nucleic acid populations, whether DNA or RNA, using DNA microarrays or so-called “deep sequencing” methods. The key differences between most epigenomic analyses thus involve the preparation of the nucleic acid population to be studied (Fig. 1).
Figure 1.
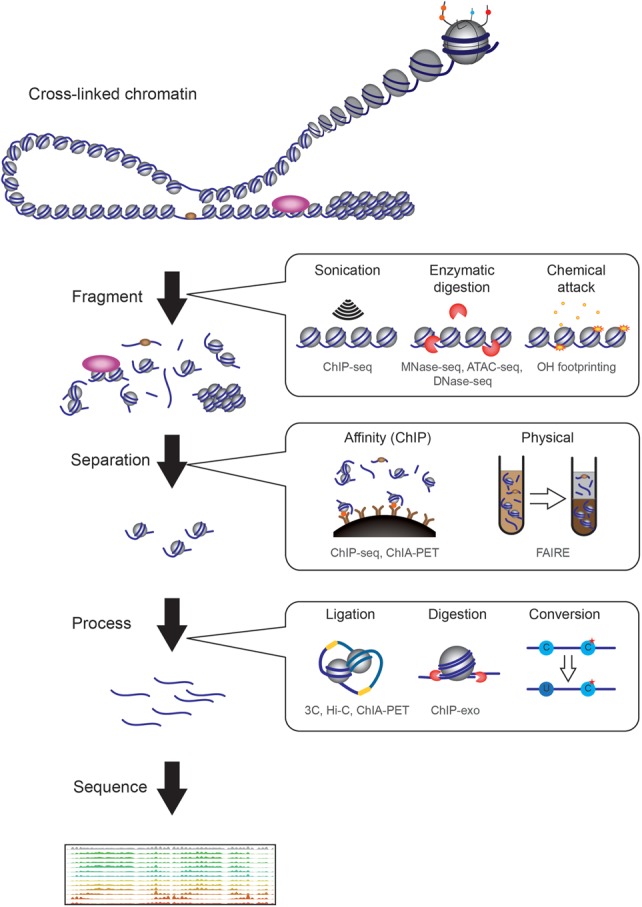
There are many broad classes of methods used in epigenomic studies prior to sequencing. We can roughly categorize methods according to three criteria: fragmentation, separation, and further processing. To interrogate chromatin, it has to be fragmented, with three broad classes of fragmentation method. First, physical fractionation methods, such as sonication, apply force to break chromatin. Second, nuclease-susceptibility methods such as MNase-seq, DNase-seq, and ATAC-seq separate regions of the genome based on their susceptibility to enzymatic attack, and typically separate “open” and accessible regions from more compact, often repressed, regions of the genome. Third, chemical susceptibility of chromatin is used in several different epigenomic methods, such as assays based on hydroxyl radical cleavage of the DNA backbone. After fragmentation, separation, either via affinity-based methods such as ChIP or physical methods such as solubility, can be used to enrich specific classes of chromatin. Finally, in some cases, further processing is used to reveal specific structural aspects of chromatin. Such methods include the proximity ligation used in the 3C family of techniques for assaying higher-order chromosome folding, or chemical conversion to reveal nucleotide modifications or precise DNA-protein crosslinks. Below each method, we list some of the assays that employ this method in gray text (e.g., ChIP, MNase-seq, etc.). Note that we do not broadly cover assays of cytosine methylation in this review, except insofar as access to methyltransferases is used as an assay for chromatin structure.
There are many broad classes of separation/fractionation methods used to generate DNA populations of interest for epigenomic studies. First, affinity-based methods, such as chromatin immunoprecipitation (ChIP), utilize antibodies specific for proteins or covalently modified peptides to isolate genomic DNA associated with the protein/modification in question. Second, nuclease-susceptibility methods such as MNase-seq, DNase-seq, and ATAC-seq are used to separate regions of the genome based on their susceptibility to enzymatic attack, and typically separate “open” and accessible regions from more compact, often repressed, regions of the genome. Third, chemical susceptibility of DNA and RNA is used in several different epigenomic methods, the most prevalent being the use of bisulfite treatment of DNA or RNA to distinguish between cytosine and 5-methylcytosine, but also including assays based on hydroxyl radical cleavage of the DNA backbone. Fourth, physical separation methods such as sedimentation of isolated chromatin (Gilbert et al. 2004), or partitioning between aqueous and phenol phases (Nagy et al. 2003), have been used to provide insight into packaging of genomic regions. Finally, in addition to separation methods, proximity ligation methods for analysis of chromosomal interactions, such as 3C and related techniques (Dekker et al. 2002), use chemical crosslinking to capture physical interactions between pieces of DNA (presumably mediated by proteins), providing a contact map for the folding of the genome.
It is worth noting that these different methods can be combined, as, for example, ChIP-exo consists of an affinity-based step to isolate DNA associated with a protein of interest, followed by exonuclease digestion to precisely footprint the protein's binding site on DNA (Rhee and Pugh 2011). Similarly, several methods leverage analysis of modified DNA, or of DNA cleavage sites, in cells carrying fusion proteins in which the protein to be mapped is genetically fused to a modifying enzyme (such as DNA adenine methyltransferase (van Steensel and Henikoff 2000), thereby substituting a genetic fusion in place of the affinity-based step to provide specificity.
Below, we discuss the structural organization of the eukaryotic genome, starting with higher order chromosome folding and domain organization, followed by nucleosome resolution chromosome organization, and ending with nucleotide-resolution techniques.
Chromosome folding
Two primary bodies of epigenomics literature provide insights into the large-scale folding of eukaryotic chromosomes and their relative locations within the nucleus. First, mapping of genomic regions associated with nuclear landmarks such as the nuclear lamina and nuclear pores provides a perspective on chromosomal territories that contact the nuclear periphery. Key insights from such mapping studies include the finding that active chromatin tends to associate with nuclear pores in yeast (Casolari et al. 2004), while megabase-scale domains of repressive chromatin termed lamina-associated domains (LADs) are intimately associated with the nuclear periphery in many organisms (Pickersgill et al. 2006). LADs are dynamic during development, with changes in lamina association being correlated with gene repression. Perhaps the most dramatic perturbation to LAD structure occurs in several examples of sensory epithelia, where physiological down-regulation of the lamin B receptor results in otherwise lamina-associated heterochromatin moving to the nuclear interior. In the case of the retina, this clump of heterochromatin in the nuclear interior has been proposed to play a role in allowing light to reach photoreceptors (Solovei et al. 2009), while in the olfactory epithelium, the central aggregation of normally peripheral heterochromatin appears to be required for repression of the majority of olfactory receptors and the selection of a single olfactory receptor for expression in each cell (Clowney et al. 2012).
A more comprehensive structural view of chromosome folding is provided by the 3C (Chromosome Conformation Capture) family of techniques, including 3C (Dekker et al. 2002), 4C (Simonis et al. 2006), 5C (Dostie et al. 2006), Hi-C (Lieberman-Aiden et al. 2009), and others. In these methods, chromatin is subject to crosslinking in vivo to capture interactions between chromosomal loci. The genome is then fragmented, typically with restriction enzymes, and DNA ligation is used to capture interactions between chromosomal loci that were in contact with one another in vivo. The abundance of ligation products between two chromosome regions is often interpreted as a measure of frequency/probability of contact, or proximity, between the pair of loci, providing a view of chromosome structure that is somewhat analogous to the view of protein structure generated by NMR techniques.
Several key insights into chromosome folding have been afforded by 3C-based methods. In many organisms, Hi-C reveals robust interactions between centromeres of different chromosomes, and similarly between various telomeres, consistent with the long-understood Rabl configuration of chromosomes in which centromeres cluster at the nuclear periphery due to their association with the spindle pole body (SPB), with chromosomes extending from the SPB to a point of telomeric attachment to the nuclear periphery (Duan et al. 2010; Feng et al. 2014; Marbouty et al. 2014). Beyond this genomic confirmation of the Rabl configuration, at relatively low resolution, Hi-C generally reveals two major compartments of active and repressed chromatin, with megabase-scale blocks of chromatin that preferentially interact with other blocks of the same type (Lieberman-Aiden et al. 2009; Feng et al. 2014). More recently, higher resolution Hi-C studies in many organisms reveal a common folding motif variously referred to as topologically associating domains (TADs), or chromosomally interacting domains (CIDs) (Dixon et al. 2012; Nora et al. 2012; Sexton et al. 2012; Le et al. 2013; Mizuguchi et al. 2014; Hsieh et al. 2015; Wang et al. 2015). These domains are chromosomal regions that exhibit relatively high levels of self-association, with low levels of interactions being observed between adjacent TADs, and can be conceptualized as relatively unstructured “globules” or “crumples” of chromatin separated by boundaries. Finally, 3C methods have been used extensively to study enhancer-promoter interactions and other forms of long-distance loops, with a recent ∼1-kb-resolution study in mammalian cells identifying ∼10,000 loops between CTCF binding sites (Rao et al. 2014). Enhancer-promoter interactions typically occur within the boundaries of a TAD/CID and do not cross boundaries between TADs (Symmons et al. 2014). Many other variants of 3C exist, including methods such as ChIA-PET, in which an initial affinity step is used to isolate chromatin associated with factors such as RNA polymerase II or CTCF—the reduction in genomic complexity inherent in such enrichment provides much higher-resolution 3C data (at a given sequencing depth) for the interactions of interest (Li et al. 2012).
The structural insights gleaned from 3C methods naturally raise questions about the principles underlying establishment of chromosomal domains, the functions of such domains, and how they relate to other aspects of chromosomal processes such as chromatin state (see below) and replication timing. Much progress is being made toward understanding the molecular basis for chromosome folding, as Hi-C maps have been generated in a variety of cell types, at interesting cell stages such as mitosis, and in an ever-expanding range of mutants. In contrast, the functional consequences of chromosomal domains remain challenging to investigate, although deletion of well-defined boundary elements and artificial loop insertion provide some tools for addressing the regulatory implications of TADs and other chromosomal domains.
Nucleosomes, beads on a string
The repeating packing unit of chromatin is the nucleosome (Kornberg and Lorch 1999), which consists of ∼147 bp of double-stranded DNA wrapped around a disk formed by the globular domains of eight histone proteins (two copies of histones H2A, H2B, H3, and H4) (Luger et al. 1997). This core structure is highly conserved throughout Eukaryota. Longer double-stranded DNA molecules are assembled into an array of nucleosomes, forming a structure often referred to as the “beads on a string.” At this scale, the structure of interest is the location of nucleosomes along the genome: Which ∼150-bp regions are wrapped around histones, and which DNA sequences form the relatively accessible “linker” DNA between nucleosomes? Much of what we know of nucleosome structure is based on differences in susceptibility of DNA to various fractionation processes, with nucleosomal DNA being protected from many enzymes and from chemical attack, and linker DNA being relatively accessible. We can divide genomic assays of nucleosome packing into two classes.
The first class of nucleosome-resolution assays tends to strongly enrich the relatively long linkers, known as nucleosome-depleted regions (NDRs), that are typically found at genomic regulatory elements such as promoters and enhancers. Nucleosome-depleted regions can be identified by elevated accessibility to enzymes such as the nuclease DNase I (Gross and Garrard 1988), transposases such as Tn5 (Buenrostro et al. 2013), or DNA methylases (Kelly et al. 2012). Alternatively, given the difference in protein occupancy between NDRs and bulk chromatin, physical fractionation (e.g., FAIRE) (Nagy et al. 2003) can be used to purify NDRs, as protein-enriched DNA partitions to phenol, leaving relatively protein-depleted DNA to partition to the aqueous phase.
The assays in the second class target the locations of individual nucleosomes. The most common tool for such analysis is micrococcal nuclease (MNase) digestion. This nuclease has preference for linker DNA over nucleosomal DNA (Noll and Kornberg 1977), and size separation of the DNA fragments remaining after MNase digestion results in a regular “ladder” with repeating unit size. Sequencing the resulting mononucleosome-sized fragments (MNase-seq) then provides a catalog of nucleosome-protected regions (Hughes and Rando 2014), revealing both locations of “well-positioned” nucleosomes that are present in the same location in most of the cell population, and locations of “fuzzy” regions where the nucleosome location varies between cells. Beyond the location and occupancy of nucleosomes, more nuanced details of nucleosomal organization can be gleaned from a thorough analysis of nuclease digestion products. Most notably, in addition to the stable nucleosomes revealed in typical MNase digestion reactions, a more labile subset of “fragile” nucleosomes is obtained only after relatively light MNase digestion of chromatin (Weiner et al. 2010; Xi et al. 2011). Moreover, while nucleosomes are by far the most widespread DNA-binding factors that interfere with MNase digestion, many other DNA-binding proteins provide some level of protection from nuclease digestion to the underlying sequence. In MNase-footprinting (Henikoff et al. 2011; Kent et al. 2011), the chromatin is digested, usually relatively lightly, followed by paired-end sequencing of fragments in a large size range (typically 40–400 bp). The resulting maps afford not only positions of nucleosomes and even relative nuclease access to the two edges of the nucleosome but also provide a broad survey of additional DNA-binding factors, most notably tightly bound transcription factors (TFs) that, in yeast, are known as “general regulatory factors.” Understanding the features that define strongly footprinting proteins remains an active area of analysis.
Application of these methods in a wide range of organisms (Mavrich et al. 2008; Shivaswamy et al. 2008; Valouev et al. 2008, 2011; Chodavarapu et al. 2010; Gilchrist et al. 2010; Tsankov et al. 2010) identifies several structural principles. First, active and poised promoters are nucleosome-depleted, as are active enhancers. Second, nucleosomes bordering regulatory elements are well positioned. Nucleosomes are positioned with decaying precision at greater distances, leading to a fuzzier nucleosome template further away (∼1000–2000 bp) from boundary nucleosomes. Curiously, in yeast, nucleosome delocalization is typically maximal at a point ∼2/3 of the way into coding regions, rather than at the midpoint of the gene (Vaillant et al. 2010). Third, highly transcribed genes tend to have lower nucleosome occupancy and less precise positioning, most likely due to action of RNA polymerase II and its associated factors. Finally, the density of the nucleosome template (average distance between two adjacent nucleosomes) can vary, mostly due to trans factors (Hughes et al. 2012).
Studies of nucleosome positioning in a variety of mutant backgrounds (see, e.g., van Bakel et al. 2013), along with insights gained from biochemical reconstitutions using histone proteins and purified DNA (Korber and Horz 2004; Sekinger et al. 2005; Kaplan et al. 2009; Zhang et al. 2011), provide insights into multiple forces that direct nucleosome positioning in vivo. First, the affinity of a given sequence for the histone octamer differs between DNA sequences based on their flexibility, with intrinsically stiff polyA/T tracts being disfavored for nucleosome incorporation and thereby “programming” nucleosome depletion. Second, nucleosome positions can be influenced by steric inhibition, either with adjacent nucleosomes or other DNA-binding proteins such as transcription factors. This can lead to local positioning cues having impacts on positions of nucleosomes some distance away, as, for example, proposed in the “statistical positioning” model for nucleosome positioning (Kornberg and Stryer 1988). Third, a broad class of regulatory factors, the ATP-dependent nucleosome remodelers, plays a key role in assembling, displacing, and evicting nucleosomes (Clapier and Cairns 2009). Finally, additional structural elements such as Heterochromatin Protein 1 (HP1) and Histone 1 (H1) can assemble multinucleosomal units with constrained structures. The combination of these forces defines both the location of nucleosome-depleted regions and boundaries, as well as the pressure to pack the remaining region, leading to the repeating template structure.
Chemical state of the nucleosome
The protein composition of the histone octamer is not uniform for all nucleosomes, as multiple variant isoforms exist of the core histone proteins, with variants of H2A and H3 being the most common (e.g., H2A.X and H2A.Z isoforms of H2A, H3.1 and H3.3 isoforms of H3). In addition, histone proteins can be chemically modified, with a prodigious variety of post-translational modifications occurring (acetylation, methylation, phosphorylation, ubiquitylation, and many more) at multiple residues in all of the histones (Tan et al. 2011). The enzymes responsible for these modifications are often highly residue-specific, as, for example, histone methyltransferases that modify lysine 4 on Histone 3 (H3K4) will not methylate nearby lysine residues (e.g., H3K9). These chemical modifications can alter the chemical and physical properties of the nucleosome but also serve to modulate binding by proteins that recognize the modified state (e.g., bind only to trimethylated H3K4) (Turner 1993; Jenuwein and Allis 2001). Thus, the beads on a string structure is not composed of uniform beads, as the chemical makeup of individual nucleosomes can, in principle, be extremely variable from nucleosome to nucleosome, with important consequences for genome function.
Mapping the nucleosome state is most commonly done by chromatin immunoprecipitation followed by sequencing (ChIP-seq). Crosslinked chromatin is fragmented, either mechanically or by MNase, and then incubated with antibody against a modification of interest. DNA fragments bound to the antibody are then extracted, sequenced, and mapped to the genome. The resulting occupancy map identifies genomic regions that are occupied by nucleosomes carrying the target modification. The accuracy of these assays depends on the quality of the antibody, its affinity for the target epitope, and cross-specificity to other epitopes (Egelhofer et al. 2011; Fuchs et al. 2011). Low affinity leads to low yields and a poor signal-to-background ratio. Cross-specificity can bias the results in a misleading manner, especially if the target modification state is rare compared to the off-target modification. The spatial precision depends on the fragmentation protocol, with larger fragments leading to a more spatially diffuse signal.
Large-scale efforts in histone state mapping in multiple model systems (Liu et al. 2005; Sinha et al. 2006; Filion et al. 2010; Ernst et al. 2011; Weiner et al. 2015) reveal a number of conserved aspects of chromatin structure. First, the process of transcription leaves a massive footprint on chromatin, with different histone modifications deposited by the initiation (5′ end of genes) and elongation (middle and 3′ ends) forms of RNA polymerase. Marks at the 5′ ends of genes include H3K4 di/trimethylation (H3K4me3), H3 and H4 tail hyperacetylation (H3K4/9/14/18/27ac and H4K5/8/12ac), H3.3 and H2A.Z variants, and H3K56 acetylation. The 5′ ends of poised genes have lower levels of histone tail acetylation but are marked with H3K4me3. Gene body nucleosomes are typically marked with H3K36me3 and H3K79me3 and are generally somewhat depleted of histone tail acetylations. Second, distal regulatory elements, such as enhancers, are marked with H3K4me1/2 but not H3K4me3 (Heintzman et al. 2007). Other histone marks distinguish repressed, poised, and active enhancers, with H3K27 methylation and acetylation marking repressed and active enhancers, respectively. Third, two major forms of repressive chromatin are associated with specific histone modifications. Classical heterochromatin (including telomeres and many repetitive sequences) is marked with H3K9 methylation, while genes repressed by Polycomb-group factors are marked with H3K27me3. H3K27-methylated nucleosomes can be either in a repressed state and lack other marks, or in a poised “bivalent” state in combination with the active mark H3K4me3 (Bernstein et al. 2006). Finally, nucleosomes associated with centromeres often contain the H3-like CENP-A protein, and during M phase, a broad pericentric domain of nucleosomes are marked with H3S10ph. Overall, there is a striking correspondence of histone state with the function of genomic regions, and because of this, mapping of histone modifications is an effective method to discover genes and regulatory elements (Guttman et al. 2009, 2010; Hon et al. 2009; Ernst and Kellis 2010).
In the context of chromosome folding, it has long been known that chemical modifications of the nucleosomes can alter chromatin fiber folding, as, for example, histone acetylation interferes with folding of nucleosome arrays into compact fibers in vitro (Shahbazian and Grunstein 2007). More complex interactions with chromosome folding are also likely, as modification-specific binding proteins or complexes often have the ability to bind to multiple distinct histone modifications, potentially resulting in bridging interactions between two or more nucleosomes. The integration of chemical modification maps into higher-order chromosome folding studies will be an area of great interest in the near future.
Subnucleosomal structures in chromatin
A shortcoming of the nuclease-based methods typically used for nucleosome mapping is imprecision in positioning data caused by variability in nuclease digestion of nucleosome ends. Several epigenomic methods have been developed which provide greater precision in mapping of DNA-bound factors, in some cases offering structural insights into DNA-protein complexes at a resolution that can be confirmed by comparison to crystal structures of the relevant factors. For example, extremely deep sequencing of nuclease- and transposase-susceptible genomic regions has proven to yield high resolution insights into nucleosome positioning and TF binding. This has been reported for DNase I digestions, where moderate depth mapping reveals nucleosome-depleted regulatory elements, but ultra-deep sequencing additionally reveals detailed footprints of certain transcription factor binding sites which can be reconciled with the pattern of protein-DNA contacts expected from crystal structures (Hesselberth et al. 2009). Similarly, deep sequencing of ATAC-seq libraries reveals transcription factor footprints and even exhibits 10.5-bp periodicity across nucleosomes (Buenrostro et al. 2013), potentially providing information on the rotational positioning of nucleosomes (e.g., where the DNA major groove faces toward the histones vs. toward the nucleoplasm).
For nucleosome mapping, a major improvement in resolution has been provided by the development of chemical mapping methodologies in which a cysteine is engineered into one of the core histones, enabling localized generation of hydroxyl radicals and precise cleavage of the nearby DNA backbone (Brogaard et al. 2012). Briefly, yeast are engineered to carry a mutant histone H4S47C, and after cell lysis, chromatin is treated with N-(1,10-phenanthroline-5-yl) iodoacetamide, which reacts with the H4 cysteine. Treatment of this adduct with copper and hydrogen peroxide results in generation of short-lived hydroxyl radicals, which diffuse very short distances before reacting with the DNA backbone or other molecules. Given the location of this H4 residue near the nucleosomal dyad axis, predominant cleavage sites occur at the +1 and +6 (and the −1 and −6) positions relative to the dyad. Deconvolution of the mix of cleavage sites then yields a map of precise locations of nucleosome dyads.
Chemical mapping of H4S47C nucleosomes generally confirms the basic patterns of nucleosome positioning across the yeast genome inferred from nuclease-based assays and yields additional insights into rotational positioning of nucleosomes—the precise sequences for which the DNA major groove faces toward vs. away from the histone octamer. First, for nucleosomes which exhibit apparent “overlap”—where dyads are measured within ∼70 nt of one another—dyads are preferentially separated by multiples of 10 nt, indicating that the multiple nucleosome positions that contribute to “fuzzy” nucleosomes (see above) all exhibit a consistent shared rotational setting. Analysis of nucleosomal DNA sequences from chemical cleavage maps also reveals a much stronger signal for dinucleotide periodicity across the nucleosome than previously appreciated, emphasizing the role for DNA sequence flexibility/curvature in establishing the rotational positioning of nucleosomes. Finally, and most interestingly, analysis of dyad to dyad distances for adjacent (nonoverlapping) nucleosomes revealed a strong preference for linker DNA to be 10n + 5 nt long. This finding has implications for secondary structure of chromatin, as it suggests that adjacent nucleosomes will be positioned approximately on opposite sides of the DNA double helix, potentially consistent with two-start models for 30-nm fiber (Yao et al. 1993).
A more general approach to improving the resolution of chromatin mapping assays is the ChIP-exo assay developed by Pugh and colleagues, in which material isolated by chromatin immunoprecipitation is treated with λ exonuclease prior to crosslink reversal and cloning of purified DNA (Rhee and Pugh 2011). The rationale behind this assay is that shearing and other chromatin fragmentation methods leave heterogeneous DNA ends on either side of the protein of interest. Exonuclease treatment is thus used to digest excess DNA surrounding the protein-DNA crosslink, providing a consistent DNA end a few nucleotides away from the crosslink site which cannot enter the exonuclease. ChIP-exo thus provides a far more precise footprint of proteins on DNA than do methods relying on shearing for chromatin fragmentation. For example, for the yeast TF Reb1, the known Reb1 binding motif occurs precisely at the center of ChIP-exo peaks with a standard deviation of <1 nt, while it is found within ∼25 nt of the center of a traditional ChIP-seq peak. Demonstrating the power of ChIP-exo as a tool for structural biology, comparison of protein-DNA crystal structures with ChIP-exo of the preinitiation complex (Rhee and Pugh 2012) and of the ATP-dependent chromatin remodeler Isw2 (Yen et al. 2012) shows that the pattern of ChIP-exo read starts for TFIIB and for Isw2 successfully identified DNA sites in close proximity to the relevant protein.
The most novel structural insight revealed by single-nucleotide-resolution epigenomic assays has been the recent discovery of asymmetric nucleosomes associated with the budding yeast genome. Nucleosome asymmetry was revealed by ChIP-exo analysis of several core histones and histone modifications (Rhee et al. 2014) and independently by analysis of H4S47C chemical cleavage maps (Ramachandran et al. 2015). Both of these studies reveal a surprising profusion of genomic regions characterized by asymmetric histone-DNA interactions, suggesting either hemi-nucleosomes comprised of single copies of the four histone proteins (which are typically present in two copies per nucleosome), or more likely, nucleosomes with a bulge of dissociated DNA on one side which precludes crosslinking to the histone proteins. These asymmetric nucleosomes occur at the 5′ ends of genes, and in general at locations exhibiting rapid replication-independent histone turnover. It will be interesting to determine whether asymmetric nucleosomes play functional roles in gene regulation or represent relatively transient assembly/disassembly intermediates.
The ability to assay structural aspects of chromosome packaging genome-wide has only been possible for a relatively short period of time, yet already has generated insights regarding nucleosome symmetry, rotational positioning, and DNA-protein contacts with various chromatin regulators. We anticipate that nucleotide-resolution epigenomic assays will continue to provide exciting and novel insights into chromatin structure in the coming years.
Emerging fields
Chromatin secondary structure
The studies and approaches described above provide insights into structural aspects of chromatin at length scales from ∼1 bp to several megabases. However, at present, there exists a “blind spot” in chromosome folding assays at the length scale between ∼200 bp and ∼1–2 kb. Specifically, 3C-based methods rely on fragmentation of the genome to assay chromatin fragments that can be crosslinked to one another. Typically, this fragmentation relies on restriction enzyme digestion, meaning that, for typical 3C and Hi-C experiments, the average fragment size is ∼4 kb. Even with four-cutter restriction enzymes, heterogeneous site locations across the genome and incomplete restriction result in a typical resolution of, at best, ∼1 kb. Thus, the length scale between 200 bp and ∼1–4 kb remains invisible with current chromosome folding assays. It is at this length scale that one would expect signal from secondary structures in chromatin, such as 30-nm fiber (Tremethick 2007), or short gene loops in budding yeast, and is thus a key area of interest for future studies in chromosome folding.
A number of methods potentially hold promise for uncovering details of chromosome folding at the 2- to 10-nucleosome length scale. We have recently developed a variant of the Hi-C protocol—“Micro-C”—in which micrococcal nuclease is used in place of restriction enzymes to fragment the genome, providing a mononucleosome-resolution chromosome folding assay (Hsieh et al. 2015). Intriguingly, this assay revealed abundant chromosomal interaction domains, similar to the TADs described in mammals and CIDs in Caulobacter crescentis, with strong interactions occurring through the domain but boundaries preventing interactions between adjacent TADs/CIDs. Curiously, CID length in different species is highly divergent when scaled by genomic distance but is relatively conserved when scaled by gene number—TADs in mammals and CIDs in yeast both encompass ∼1–5 genes, with this structure thus occurring over ∼1 Mb scales in mammals but only ∼5 kb in yeast. In terms of 30-nm fiber structure, no periodicity is observed in Micro-C ligation products (e.g., interactions between nucleosome N and nucleosomes N + 6, N + 12, N + 18, etc., as expected from solenoid models), although the use of the short-length crosslinker formaldehyde in 3C methods necessarily limits the ability to identify such longer-distance signals (Grigoryev et al. 2009). That said, Micro-C products between nucleosomes N and N + 2 were found to be as abundant as N/N + 1 products, generally supporting “two-start” fiber models such as zig-zag 30-nm fiber. Future studies with different length crosslinkers will be needed to further explore the finer-scale details of chromosome fiber structure.
Although Micro-C provides some insight into folding of the 30-nm fiber, no doubt other methods for assaying 30-nm fiber structure genome-wide are currently in development, and we believe this remains a key area for future structural investigations into chromosome folding.
Single-cell epigenomics and combinatorial epigenomics
Another general limitation of most extant epigenomic methods is that the vast majority of such assays are based on ensemble measurements in which the measurement of chromatin state reports on the average chromatin state for thousands or millions of cells. As decomposition of ensembles into individual entities has provided foundational insights into single-molecule biochemistry, single-cell gene regulation, and many other fields, it is expected that single-cell assays for chromatin will be required to illuminate several key questions in chromatin structure. In general, one may consider two broad methods of gaining insight into single-cell behavior of chromatin—computational deconvolution of ensemble measurements into constituent subpopulations (Houseman et al. 2015) and experimental analysis of single cells. We focus below on experimental approaches to single-cell epigenomics.
At the level of individual nucleosomes, the strong correlation observed between many histone modifications raises the question of whether histone modifications co-occur on the same nucleosome in a given cell, or whether “co-occurring” modifications occur at the same genomic location but in different cells throughout a population (e.g., cells at different cell-cycle phases, or due to cell-to-cell variation in expression). At the level of multiple nucleosomes, ensemble measurements leave open the question of which multinucleosome configurations are feasible or probable. In other words, if three adjacent nucleosomes all have an overall occupancy of ∼80%, do these nucleosomes always co-occur, with 20% of cells carrying a naked DNA template, or are these nucleosomes independent? Elegant EM studies from the Boeger lab find that upon activation of the PHO5 promoter, at most two of three promoter nucleosomes are evicted, but the configuration where all three are evicted is not observed, indicating that these eviction events are not independent (Brown et al. 2013; Brown and Boeger 2014). In another study, DNA methyltransferase was used to footprint nucleosomes in individual cells, with bisulfite sequencing (effectively a single-molecule technique) being used to assess nucleosome occupancy across a 500- to 600-bp region. Using this method, analysis of activated PHO5 and two other promoters reveals activated configurations in which the promoter nucleosomes are not ejected, but their position is shifted in a coordinated manner (Small et al. 2014). They also show that the frequency of different configurations changes upon transcriptional activation. Detailed single-cell studies of nucleosome configurations can increase our understanding of how nucleosome positioning impacts genomic processes. A promising recent advance is the development of single-cell ATAC-seq, which provides single-cell analysis of nucleosome depletion in metazoans and reveals subpopulations of cell states within a given cell type (Buenrostro et al. 2015; Cusanovich et al. 2015), although as ATAC-seq primarily provides information on nucleosome depletion, it does not address the questions of nucleosome configurational diversity raised above.
At the level of higher-order chromosome folding, the physical nature of structural domains such LADs and CIDs/TADs will be illuminated by single-cell techniques. A central question is whether these domains are constant in all cells or appear, possibly transiently, in smaller fraction of cells. For example, pioneering studies of LADs in single cells (Kind et al. 2013) showed that only ∼30% of LADs identified from ensemble measurements are actually attached to the nuclear periphery in any individual cell. A similar question arises from 3C-based assays, where any genomic locus that exhibits interactions with two or more other regions in ensemble measurements could, in principle, result from locus X interacting in a cluster (e.g., a “transcription factory”) with both locus Y and Z in the same cell, or a population of cells with distinct subsets of cells with either X-Y interactions or X-Z interactions. More concretely, the abundant interactions occurring throughout a megabase chromosomal domain such as a TAD/CID can be conceptualized as a globular domain in which all interacting pairs of chromosome fragments are in close proximity in a given cell, facilitating many interactions. Alternatively, a TAD/CID could also result from an ensemble population of individual cells with more extended chromatin in which a given cell might exhibit a single looping interaction within the domain of interest, but with many different loop structures occurring throughout the cell population. Initial results with single-cell Hi-C (Nagano et al. 2013) show a large amount of variability in terms of large-scale structure, although this is somewhat hard to interpret physically given the small number of interactions identified for any individual cell in this assay. Future studies should help reveal the single-cell details of such multilocus interactions.
Stated more broadly, single-cell epigenomics will facilitate insights into the combinatorial nature of chromatin—which combinations of marks and structures are possible, what mechanisms enforce such constraints, and whether these depend on global aspects of cell state (e.g., cell-cycle stage) or on more local factors. An alternative strategy to understand these questions is by combinatorial assays. For example, sequential ChIP (also known as re-ChIP) in which the output of one ChIP is subjected to another ChIP for a second epitope can determine whether two chromatin marks co-appear on the same chromatin fragment. Until recently such assays have been constrained by the small amount of material from the first ChIP. Recent advances in ChIP in low-volume conditions and multiplexing (Lara-Astiaso et al. 2014) make us optimistic that reliable combinatorial ChIP will be realizable in the near future and allow researchers to elucidate the constraints on combinations of marks.
Chromatin dynamics
Epigenomic assays capture snapshots of complex cellular states. Unlike the DNA sequence, which is relatively stable, chromatin can be highly dynamic, raising the question of the relevant time-scales that govern chromatin structures. This question is related to, but distinct from, the question of heterogeneity within a cell population. When we observe a “fuzzy” nucleosome in an ensemble measurement, does the location of the nucleosome change only at cell division (stable) or every few seconds (dynamic)? A wide range of time-scales is consistent with both the ensemble observation as well as the distribution of single-cell states. This question applies throughout the range of structures we review above, from long-range chromosomal interactions to nucleosome chemical modifications. Understanding the function of the structures observed will ultimately require knowing the relevant time-scales for the structures in question.
Insights into dynamics can be gained using live-cell fluorescence microscopy, either via observation of individual fluorophore-labeled chromosomal loci moving throughout the nucleus (Marshall et al. 1997; Heun et al. 2001), or via fluorescence recovery after bleaching (FRAP) studies on the time-scales of diffusion or replacement of proteins in large regions (McNally et al. 2000; Kimura and Cook 2001). At higher resolution, genetically encoded pulse chase schemes have been adapted to allow epigenomic analysis of chromatin dynamics. For example, nucleosome turnover rates have been characterized in budding yeast by inducing expression of epitope-tagged histones and analyzing the localization of the epitope of interest over a time course using ChIP-chip or ChIP-seq (Dion et al. 2007; Jamai et al. 2007; Rufiange et al. 2007), and a related metabolic labeling strategy has been used to similar ends in the fruit fly (Deal et al. 2010). These studies reveal replication-independent turnover at transcribed genes, with more rapid histone replacement occurring at regulatory regions (promoters and enhancers). Similar strategies have been applied to other proteins, including components of the basal transcription machinery (van Werven et al. 2009) and transcription factors (Lickwar et al. 2012).
Beyond such studies on protein replacement rates, there remains a pressing need to develop methodologies for studying the locus-specific dynamics of other aspects of chromatin structure. Is a highly H3K4-methylated nucleosome experiencing constant de- and remethylation, or is methylation stable once deposited? Are TADs unfolding and refolding over minute time-scales? These and many other questions await new measurement methods that explicitly address dynamic aspects of the epigenome.
Conclusions
Over the past decade, it has become a cliché that the deep sequencer is the microscope for modern chromatin researchers. Like many clichés, this analogy has some truth to it, as epigenomics methods provide a view of ensemble chromosome organization that approaches atomic resolution in some respects. Although heterogeneity in cell populations will always remain a significant confounding factor, it is clear that epigenomic assays of stringently selected cell populations will continue to provide structural insights into the folding of the chromosome, while single-cell methods have the promise to rigorously disentangle inevitable heterogeneity between individual cells. The coming decade promises continuing insights into the structure of eukaryotic genomes, and we anticipate that further investigations will continue to reveal the mechanistic principles responsible for chromosome folding and the functional importance of chromosomal substructures.
Acknowledgments
We thank Ronen Sadeh for critical comments on this manuscript. Work was supported in part by the National Institutes of Health (NIH) grant R01 GM079205 to O.J.R. and N.F., grant R01 HD080224 to O.J.R., European Research Council grant 340712 to N.F., and Israeli Science Foundation I-CORE on “Chromatin and RNA in Gene Regulation” to N.F.
Footnotes
Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.190165.115.
Freely available online through the Genome Research Open Access option.
References
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. 2006. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125: 315–326. [DOI] [PubMed] [Google Scholar]
- Brogaard K, Xi L, Wang JP, Widom J. 2012. A map of nucleosome positions in yeast at base-pair resolution. Nature 486: 496–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CR, Boeger H. 2014. Nucleosomal promoter variation generates gene expression noise. Proc Natl Acad Sci 111: 17893–17898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CR, Mao C, Falkovskaia E, Jurica MS, Boeger H. 2013. Linking stochastic fluctuations in chromatin structure and gene expression. PLoS Biol 11: e1001621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. 2013. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10: 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, Greenleaf WJ. 2015. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523: 486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casolari JM, Brown CR, Komili S, West J, Hieronymus H, Silver PA. 2004. Genome-wide localization of the nuclear transport machinery couples transcriptional status and nuclear organization. Cell 117: 427–439. [DOI] [PubMed] [Google Scholar]
- Chodavarapu RK, Feng S, Bernatavichute YV, Chen PY, Stroud H, Yu Y, Hetzel JA, Kuo F, Kim J, Cokus SJ, et al. 2010. Relationship between nucleosome positioning and DNA methylation. Nature 466: 388–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapier CR, Cairns BR. 2009. The biology of chromatin remodeling complexes. Annu Rev Biochem 78: 273–304. [DOI] [PubMed] [Google Scholar]
- Clowney EJ, LeGros MA, Mosley CP, Clowney FG, Markenskoff-Papadimitriou EC, Myllys M, Barnea G, Larabell CA, Lomvardas S. 2012. Nuclear aggregation of olfactory receptor genes governs their monogenic expression. Cell 151: 724–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusanovich DA, Daza R, Adey A, Pliner HA, Christiansen L, Gunderson KL, Steemers FJ, Trapnell C, Shendure J. 2015. Epigenetics. Multiplex single-cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 348: 910–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deal RB, Henikoff JG, Henikoff S. 2010. Genome-wide kinetics of nucleosome turnover determined by metabolic labeling of histones. Science 328: 1161–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J, Rippe K, Dekker M, Kleckner N. 2002. Capturing chromosome conformation. Science 295: 1306–1311. [DOI] [PubMed] [Google Scholar]
- Dion MF, Kaplan T, Kim M, Buratowski S, Friedman N, Rando OJ. 2007. Dynamics of replication-independent histone turnover in budding yeast. Science 315: 1405–1408. [DOI] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. 2012. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485: 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostie J, Richmond TA, Arnaout RA, Selzer RR, Lee WL, Honan TA, Rubio ED, Krumm A, Lamb J, Nusbaum C, et al. 2006. Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res 16: 1299–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Z, Andronescu M, Schutz K, McIlwain S, Kim YJ, Lee C, Shendure J, Fields S, Blau CA, Noble WS. 2010. A three-dimensional model of the yeast genome. Nature 465: 363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egelhofer TA, Minoda A, Klugman S, Lee K, Kolasinska-Zwierz P, Alekseyenko AA, Cheung MS, Day DS, Gadel S, Gorchakov AA, et al. 2011. An assessment of histone-modification antibody quality. Nat Struct Mol Biol 18: 91–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kellis M. 2010. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat Biotechnol 28: 817–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. 2011. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473: 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Cokus SJ, Schubert V, Zhai J, Pellegrini M, Jacobsen SE. 2014. Genome-wide Hi-C analyses in wild-type and mutants reveal high-resolution chromatin interactions in Arabidopsis. Mol Cell 55: 694–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filion GJ, van Bemmel JG, Braunschweig U, Talhout W, Kind J, Ward LD, Brugman W, de Castro IJ, Kerkhoven RM, Bussemaker HJ, et al. 2010. Systematic protein location mapping reveals five principal chromatin types in Drosophila cells. Cell 143: 212–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs SM, Krajewski K, Baker RW, Miller VL, Strahl BD. 2011. Influence of combinatorial histone modifications on antibody and effector protein recognition. Curr Biol 21: 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert N, Boyle S, Fiegler H, Woodfine K, Carter NP, Bickmore WA. 2004. Chromatin architecture of the human genome: Gene-rich domains are enriched in open chromatin fibers. Cell 118: 555–566. [DOI] [PubMed] [Google Scholar]
- Gilchrist DA, Dos Santos G, Fargo DC, Xie B, Gao Y, Li L, Adelman K. 2010. Pausing of RNA polymerase II disrupts DNA-specified nucleosome organization to enable precise gene regulation. Cell 143: 540–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoryev SA, Arya G, Correll S, Woodcock CL, Schlick T. 2009. Evidence for heteromorphic chromatin fibers from analysis of nucleosome interactions. Proc Natl Acad Sci 106: 13317–13322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross DS, Garrard WT. 1988. Nuclease hypersensitive sites in chromatin. Annu Rev Biochem 57: 159–197. [DOI] [PubMed] [Google Scholar]
- Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, et al. 2009. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458: 223–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, Garber M, Levin JZ, Donaghey J, Robinson J, Adiconis X, Fan L, Koziol MJ, Gnirke A, Nusbaum C, et al. 2010. Ab initio reconstruction of cell type–specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol 28: 503–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, et al. 2007. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 39: 311–318. [DOI] [PubMed] [Google Scholar]
- Henikoff JG, Belsky JA, Krassovsky K, MacAlpine DM, Henikoff S. 2011. Epigenome characterization at single base-pair resolution. Proc Natl Acad Sci 108: 18318–18323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesselberth JR, Chen X, Zhang Z, Sabo PJ, Sandstrom R, Reynolds AP, Thurman RE, Neph S, Kuehn MS, Noble WS, et al. 2009. Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat Methods 6: 283–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heun P, Laroche T, Shimada K, Furrer P, Gasser SM. 2001. Chromosome dynamics in the yeast interphase nucleus. Science 294: 2181–2186. [DOI] [PubMed] [Google Scholar]
- Hon G, Wang W, Ren B. 2009. Discovery and annotation of functional chromatin signatures in the human genome. PLoS Comput Biol 5: e1000566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houseman EA, Kelsey KT, Wiencke JK, Marsit CJ. 2015. Cell-composition effects in the analysis of DNA methylation array data: a mathematical perspective. BMC Bioinformatics 16: 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh TH, Weiner A, Lajoie B, Dekker J, Friedman N, Rando OJ. 2015. Mapping nucleosome resolution chromosome folding in yeast by Micro-C. Cell 162: 108–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL, Rando OJ. 2014. Mechanisms underlying nucleosome positioning in vivo. Annu Rev Biophys 43: 41–63. [DOI] [PubMed] [Google Scholar]
- Hughes AL, Jin Y, Rando OJ, Struhl K. 2012. A functional evolutionary approach to identify determinants of nucleosome positioning: a unifying model for establishing the genome-wide pattern. Mol Cell 48: 5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamai A, Imoberdorf RM, Strubin M. 2007. Continuous histone H2B and transcription-dependent histone H3 exchange in yeast cells outside of replication. Mol Cell 25: 345–355. [DOI] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. 2001. Translating the histone code. Science 293: 1074–1080. [DOI] [PubMed] [Google Scholar]
- Kaplan N, Moore IK, Fondufe-Mittendorf Y, Gossett AJ, Tillo D, Field Y, LeProust EM, Hughes TR, Lieb JD, Widom J, et al. 2009. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature 458: 362–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly TK, Liu Y, Lay FD, Liang G, Berman BP, Jones PA. 2012. Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome Res 22: 2497–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent NA, Adams S, Moorhouse A, Paszkiewicz K. 2011. Chromatin particle spectrum analysis: a method for comparative chromatin structure analysis using paired-end mode next-generation DNA sequencing. Nucleic Acids Res 39: e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H, Cook PR. 2001. Kinetics of core histones in living human cells: little exchange of H3 and H4 and some rapid exchange of H2B. J Cell Biol 153: 1341–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kind J, Pagie L, Ortabozkoyun H, Boyle S, de Vries SS, Janssen H, Amendola M, Nolen LD, Bickmore WA, van Steensel B. 2013. Single-cell dynamics of genome-nuclear lamina interactions. Cell 153: 178–192. [DOI] [PubMed] [Google Scholar]
- Korber P, Horz W. 2004. In vitro assembly of the characteristic chromatin organization at the yeast PHO5 promoter by a replication-independent extract system. J Biol Chem 279: 35113–35120. [DOI] [PubMed] [Google Scholar]
- Kornberg RD, Lorch Y. 1999. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell 98: 285–294. [DOI] [PubMed] [Google Scholar]
- Kornberg RD, Stryer L. 1988. Statistical distributions of nucleosomes: nonrandom locations by a stochastic mechanism. Nucleic Acids Res 16: 6677–6690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara-Astiaso D, Weiner A, Lorenzo-Vivas E, Zaretsky I, Jaitin DA, David E, Keren-Shaul H, Mildner A, Winter D, Jung S, et al. 2014. Immunogenetics. Chromatin state dynamics during blood formation. Science 345: 943–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le TB, Imakaev MV, Mirny LA, Laub MT. 2013. High-resolution mapping of the spatial organization of a bacterial chromosome. Science 342: 731–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Ruan X, Auerbach RK, Sandhu KS, Zheng M, Wang P, Poh HM, Goh Y, Lim J, Zhang J, et al. 2012. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell 148: 84–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lickwar CR, Mueller F, Hanlon SE, McNally JG, Lieb JD. 2012. Genome-wide protein-DNA binding dynamics suggest a molecular clutch for transcription factor function. Nature 484: 251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. 2009. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326: 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CL, Kaplan T, Kim M, Buratowski S, Schreiber SL, Friedman N, Rando OJ. 2005. Single-nucleosome mapping of histone modifications in S. cerevisiae. PLoS Biol 3: e328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. 1997. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389: 251–260. [DOI] [PubMed] [Google Scholar]
- Marbouty M, Cournac A, Flot JF, Marie-Nelly H, Mozziconacci J, Koszul R. 2014. Metagenomic chromosome conformation capture (meta3C) unveils the diversity of chromosome organization in microorganisms. Elife 3: e03318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall WF, Straight A, Marko JF, Swedlow J, Dernburg A, Belmont A, Murray AW, Agard DA, Sedat JW. 1997. Interphase chromosomes undergo constrained diffusional motion in living cells. Curr Biol 7: 930–939. [DOI] [PubMed] [Google Scholar]
- Mavrich TN, Ioshikhes IP, Venters BJ, Jiang C, Tomsho LP, Qi J, Schuster SC, Albert I, Pugh BF. 2008. A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome. Genome Res 18: 1073–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally JG, Muller WG, Walker D, Wolford R, Hager GL. 2000. The glucocorticoid receptor: rapid exchange with regulatory sites in living cells. Science 287: 1262–1265. [DOI] [PubMed] [Google Scholar]
- Mizuguchi T, Fudenberg G, Mehta S, Belton JM, Taneja N, Folco HD, FitzGerald P, Dekker J, Mirny L, Barrowman J, et al. 2014. Cohesin-dependent globules and heterochromatin shape 3D genome architecture in S. pombe. Nature 516: 432–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano T, Lubling Y, Stevens TJ, Schoenfelder S, Yaffe E, Dean W, Laue ED, Tanay A, Fraser P. 2013. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 502: 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy PL, Cleary ML, Brown PO, Lieb JD. 2003. Genomewide demarcation of RNA polymerase II transcription units revealed by physical fractionation of chromatin. Proc Natl Acad Sci 100: 6364–6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noll M, Kornberg RD. 1977. Action of micrococcal nuclease on chromatin and the location of histone H1. J Mol Biol 109: 393–404. [DOI] [PubMed] [Google Scholar]
- Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, et al. 2012. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485: 381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickersgill H, Kalverda B, de Wit E, Talhout W, Fornerod M, van Steensel B. 2006. Characterization of the Drosophila melanogaster genome at the nuclear lamina. Nat Genet 38: 1005–1014. [DOI] [PubMed] [Google Scholar]
- Ramachandran S, Zentner GE, Henikoff S. 2015. Asymmetric nucleosomes flank promoters in the budding yeast genome. Genome Res 25: 381–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. 2014. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159: 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee HS, Pugh BF. 2011. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell 147: 1408–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee HS, Pugh BF. 2012. Genome-wide structure and organization of eukaryotic pre-initiation complexes. Nature 483: 295–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee HS, Bataille AR, Zhang L, Pugh BF. 2014. Subnucleosomal structures and nucleosome asymmetry across a genome. Cell 159: 1377–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rufiange A, Jacques PE, Bhat W, Robert F, Nourani A. 2007. Genome-wide replication-independent histone H3 exchange occurs predominantly at promoters and implicates H3 K56 acetylation and Asf1. Mol Cell 27: 393–405. [DOI] [PubMed] [Google Scholar]
- Sekinger EA, Moqtaderi Z, Struhl K. 2005. Intrinsic histone-DNA interactions and low nucleosome density are important for preferential accessibility of promoter regions in yeast. Mol Cell 18: 735–748. [DOI] [PubMed] [Google Scholar]
- Sexton T, Yaffe E, Kenigsberg E, Bantignies F, Leblanc B, Hoichman M, Parrinello H, Tanay A, Cavalli G. 2012. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell 148: 458–472. [DOI] [PubMed] [Google Scholar]
- Shahbazian MD, Grunstein M. 2007. Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem 76: 75–100. [DOI] [PubMed] [Google Scholar]
- Shivaswamy S, Bhinge A, Zhao Y, Jones S, Hirst M, Iyer VR. 2008. Dynamic remodeling of individual nucleosomes across a eukaryotic genome in response to transcriptional perturbation. PLoS Biol 6: e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonis M, Klous P, Splinter E, Moshkin Y, Willemsen R, de Wit E, van Steensel B, de Laat W. 2006. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture–on-chip (4C). Nat Genet 38: 1348–1354. [DOI] [PubMed] [Google Scholar]
- Sinha I, Wiren M, Ekwall K. 2006. Genome-wide patterns of histone modifications in fission yeast. Chromosome Res 14: 95–105. [DOI] [PubMed] [Google Scholar]
- Small EC, Xi L, Wang JP, Widom J, Licht JD. 2014. Single-cell nucleosome mapping reveals the molecular basis of gene expression heterogeneity. Proc Natl Acad Sci 111: E2462–E2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solovei I, Kreysing M, Lanctot C, Kosem S, Peichl L, Cremer T, Guck J, Joffe B. 2009. Nuclear architecture of rod photoreceptor cells adapts to vision in mammalian evolution. Cell 137: 356–368. [DOI] [PubMed] [Google Scholar]
- Symmons O, Uslu VV, Tsujimura T, Ruf S, Nassari S, Schwarzer W, Ettwiller L, Spitz F. 2014. Functional and topological characteristics of mammalian regulatory domains. Genome Res 24: 390–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, Buchou T, Cheng Z, Rousseaux S, Rajagopal N, et al. 2011. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 146: 1016–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremethick DJ. 2007. Higher-order structures of chromatin: the elusive 30 nm fiber. Cell 128: 651–654. [DOI] [PubMed] [Google Scholar]
- Tsankov AM, Thompson DA, Socha A, Regev A, Rando OJ. 2010. The role of nucleosome positioning in the evolution of gene regulation. PLoS Biol 8: e1000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner BM. 1993. Decoding the nucleosome. Cell 75: 5–8. [PubMed] [Google Scholar]
- Vaillant C, Palmeira L, Chevereau G, Audit B, d'Aubenton-Carafa Y, Thermes C, Arneodo A. 2010. A novel strategy of transcription regulation by intragenic nucleosome ordering. Genome Res 20: 59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valouev A, Ichikawa J, Tonthat T, Stuart J, Ranade S, Peckham H, Zeng K, Malek JA, Costa G, McKernan K, et al. 2008. A high-resolution, nucleosome position map of C. elegans reveals a lack of universal sequence-dictated positioning. Genome Res 18: 1051–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valouev A, Johnson SM, Boyd SD, Smith CL, Fire AZ, Sidow A. 2011. Determinants of nucleosome organization in primary human cells. Nature 474: 516–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bakel H, Tsui K, Gebbia M, Mnaimneh S, Hughes TR, Nislow C. 2013. A compendium of nucleosome and transcript profiles reveals determinants of chromatin architecture and transcription. PLoS Genet 9: e1003479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B, Henikoff S. 2000. Identification of in vivo DNA targets of chromatin proteins using tethered dam methyltransferase. Nat Biotechnol 18: 424–428. [DOI] [PubMed] [Google Scholar]
- van Werven FJ, van Teeffelen HA, Holstege FC, Timmers HT. 2009. Distinct promoter dynamics of the basal transcription factor TBP across the yeast genome. Nat Struct Mol Biol 16: 1043–1048. [DOI] [PubMed] [Google Scholar]
- Wang C, Liu C, Roqueiro D, Grimm D, Schwab R, Becker C, Lanz C, Weigel D. 2015. Genome-wide analysis of local chromatin packing in Arabidopsis thaliana. Genome Res 25: 246–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner A, Hughes A, Yassour M, Rando OJ, Friedman N. 2010. High-resolution nucleosome mapping reveals transcription-dependent promoter packaging. Genome Res 20: 90–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner A, Hsieh TH, Appleboim A, Chen HV, Rahat A, Amit I, Rando OJ, Friedman N. 2015. High-resolution chromatin dynamics during a yeast stress response. Mol Cell 58: 371–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Y, Yao J, Chen R, Li W, He X. 2011. Nucleosome fragility reveals novel functional states of chromatin and poises genes for activation. Genome Res 21: 718–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Lowary PT, Widom J. 1993. Twist constraints on linker DNA in the 30-nm chromatin fiber: implications for nucleosome phasing. Proc Natl Acad Sci 90: 9364–9368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen K, Vinayachandran V, Batta K, Koerber RT, Pugh BF. 2012. Genome-wide nucleosome specificity and directionality of chromatin remodelers. Cell 149: 1461–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Wippo CJ, Wal M, Ward E, Korber P, Pugh BF. 2011. A packing mechanism for nucleosome organization reconstituted across a eukaryotic genome. Science 332: 977–980. [DOI] [PMC free article] [PubMed] [Google Scholar]