

Abstract
The past two decades have been marked by a surge in research to understand the microbial communities that live in association with the human body, in part stimulated by affordable, high-throughput DNA sequencing technology. In the context of the skin, this Perspective focuses on the current state of genomic- and metagenomic-based host–microbe research and future challenges and opportunities to move the field forward. These include elucidating nonbacterial components of the skin microbiome (i.e., viruses); systematic studies to address common perturbations to the skin microbiome (e.g., antimicrobial drugs, topical cosmetic/hygienic products); improved approaches for identifying potential microbial triggers for skin diseases, including species- and strain-level resolution; and improved, clinically relevant models for studying the functional and mechanistic roles of the skin microbiome. In the next 20 years, we can realistically expect that our knowledge of the skin microbiome will inform the clinical management and treatment of skin disorders through diagnostic tests to stratify patient subsets and predict best treatment modality and outcomes and through treatment strategies such as targeted manipulation or reconstitution of microbial communities.
Twenty years ago, our understanding of microbes was derived primarily from studies of what microorganisms could be isolated in culture, and our ideas about their roles in human disease were shaped by Koch's postulates. Dialog surrounding microbes was largely focused on eradication to prevent disease and promote cleanliness. Today it is increasingly recognized that the majority of microbes are not readily cultured under standard laboratory conditions and that, in fact, microbes function in the context of their communities. It is also now apparent that the microbial communities associated with our bodies are widely involved in the susceptibility and pathogenesis of human disease.
Access to next-generation sequencing technologies has greatly fueled the growing segment of biomedical research devoted to the human microbiome, the ecological community of microorganisms living in and on our bodies. Sequencing of the 16S ribosomal RNA (rRNA) gene is the predominant technique employed for surveying the bacterial composition of microbial communities. The sequence of this highly conserved gene encoding the small subunit of the ribosome is used for taxonomic identification and phylogenetic analysis of bacterial communities. Techniques pioneered by Norman Pace and colleagues allow for the amplification and sequencing of heterogeneous mixtures of 16S rRNA genes and the subsequent characterization of bacterial communities without reliance on culture-based techniques (Lane et al. 1985). Increasing in prevalence are studies that employ whole-metagenome shotgun sequencing techniques, thus surveying all microbial DNA sequences in a community without relying upon marker genes such as 16S rRNA. Since all genetic material is recovered and sequenced by this method, functional insight into the microbial communities is gained by analyzing the functional potential encoded in the genomes, including enrichment of metabolic pathways and genes encoding virulence and pathogenicity factors.
With the ability to affordably and quickly generate large data sets came the obvious need for analysis tools and databases to interpret the sequence data obtained from microbial community surveys. Alongside next-generation sequencing technologies, the bioinformatics face of microbiome research has also evolved, from a la carte tools for classification and diversity analysis to comprehensive all-encompassing packages that take the user from sequence quality control to graphical representation of the data, for example, QIIME (Caporaso et al. 2010) and mothur (Schloss et al. 2009). Another key impetus was the NIH Human Microbiome Project, implemented in 2007 with the objective to create resources that would catalyze microbiome research, including a reference data set of microbiomes from a large cohort of healthy individuals and a catalog of microbial reference genomes.
The past 20 years have marked a substantial transformation in our views of microbes and their community associations with human health and disease. As with any nascent field of research, there are not only significant challenges but also exciting opportunities for future research. Here, in the context of the skin, we present the current state of genomic- and metagenomic-based host–microbe research and future prospects for the field.
The ecology of the human skin
In 1954, Albert Kligman and Donald Pillsbury, two renowned dermatologists at the University of Pennsylvania who pioneered many of the seminal culture-based studies of the skin microbiota, stated, “a great deal more remains to be learned about the forces which control the bacterial ecology of the surface of the skin” (Pillsbury and Kligman 1954). Over 60 years later, the same statement continues to be true. With a more powerful “microscope” of DNA sequencing comes an improved ability to more comprehensively identify and classify constituents of the skin microbiota. A common theme among a number of 16S rRNA gene–based surveys and metagenomic shotgun sequencing studies to analyze the skin microbiome is that the microenvironment of the skin surface—largely defined by sebum, moisture content (eccrine and apocrine sweat), and hair follicle density—is highly associated with the bacterial community (Costello et al. 2009; Grice et al. 2009; Oh et al. 2014). These studies also generally agree in that a few dominant taxa are stably present in varying abundances depending on microenvironment, namely, the genera Propionibacterium, Corynebacterium, and Staphylococcus (Fig. 1), while the great majority of variability, both interpersonal and temporal, is present in the less abundant taxa that comprise the remainder. These particular genera are also readily cultured from the skin; however, culture-independent genomic and metagenomic approaches provide more precise quantification and can detect those species/strains that are less amenable to culture or may be outcompeted by other isolates in artificial culture settings.
Figure 1.
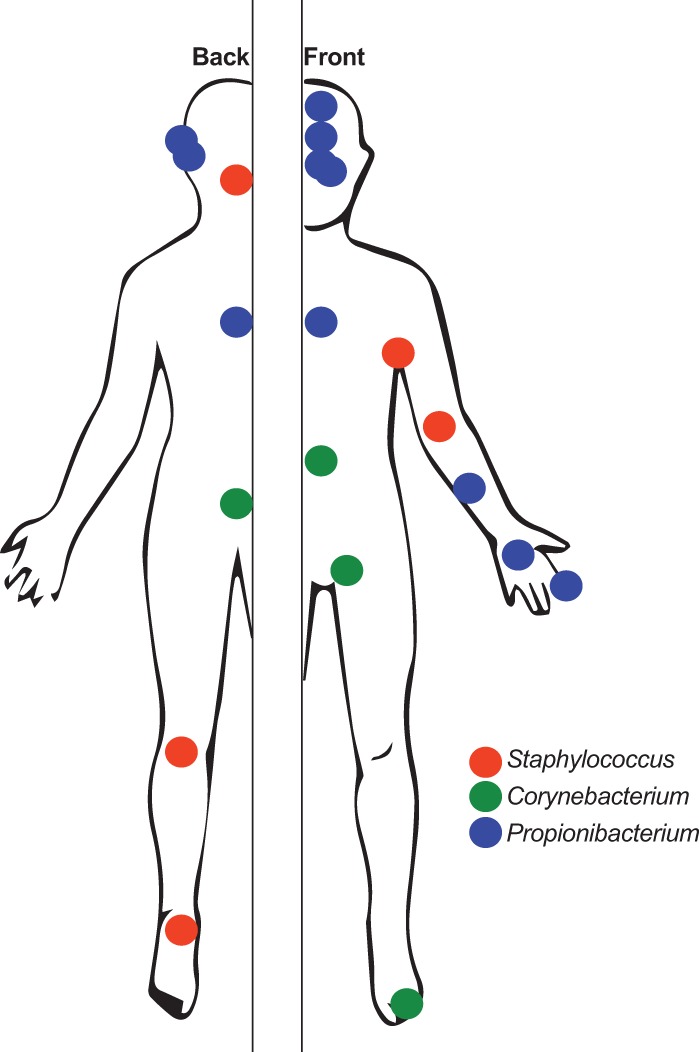
Topographical representation of the dominant types of bacteria present in the skin microbiome, based on 16S rRNA surveys. Shown are the three most common/abundant genera: Staphylococcus, Corynebacterium, and Propionibacterium. Data were gathered from Gao et al. (2007), Costello et al. (2009), Grice and Segre (2011), and The Human Microbiome Project Consortium (2012).
Fungi are also a prominent feature of the skin microbiome, the dominant organisms being the lipid-dependent yeast Malassezia (Findley et al. 2013). However, sites on the foot (plantar heel, toenail, and toe web) contain greater fungal diversity, including the Aspergillus, Cryptococcus, and Candida species. An area of future exploration will be bacterial–fungal interaction networks of the skin and how disrupting those networks can contribute to a dysbiotic state and predispose to skin diseases. For example, fungi are believed to, in part, mediate seborrheic dermatitis (dandruff) and some forms of atopic eczema, but the pathogenesis is incompletely understood (Saunders et al. 2012).
While it was assumed that microbes reside on the surface of the skin and within invaginations that open to the surface (e.g., sebaceous and sweat glands), an innovative approach demonstrated that microbial products are present in subepidermal compartments of the dermis and adipose tissue (Nakatsuji et al. 2013). Employing laser capture microdissection of these compartments followed by 16S rRNA sequencing, Nakatsuji et al. (2013) provided a potential mechanism by which microbes or their products may physically interact with immune cells in tissues previously believed to be sterile in order to exert an effect on the host. Indeed, colonization with a human commensal skin microbe, namely, Staphylococcus epidermidis, tunes T-cell homing and function in an IL-1–dependent manner in mice (Naik et al. 2012, 2015). On the other hand, deficiencies in immunity have been shown in humans and mice to result in altered cutaneous microbial communities (Chehoud et al. 2013; Oh et al. 2013). Thus it appears that cutaneous microbial communities are intimately linked with skin innate and adaptive immune functions.
Uncharted territory and emerging frontiers of the skin microbiome
The major segment of research devoted to understanding human host–microbe interactions has focused on the GI tract. While the GI microbiota indisputably is a key constituent of human health and new functions continue to be illuminated, the skin microbiota also offers many compelling reasons for further investigation. Our skin is a major interface with the outside environment, and we are reliant on the integrity of the skin barrier to protect us from invasion by pathogenic insult. Microbial triggers are hypothesized to, in part, mediate or exacerbate many dermatological disorders, but culture-based techniques have not elucidated their identity. There are technical advantages to sampling microbiota of the skin, including accessibility and minimal invasiveness. Study designs can take into account those spatial sites that have predilection for different skin disorders, and controls can often be collected from contralateral, unaffected skin sites. Here, we identify areas that are exciting opportunities for the future of skin microbiome research.
The skin virome
Viruses, including those that infect bacteria (bacteriophages), are significant components of microbial communities, though their study is complicated by the lack of a marker gene akin to the 16S rRNA gene. Lytic bacteriophages may modulate bacterial populations through predator–prey dynamics, while phages that enter lysogeny may participate in horizontal gene transfer events that confer pathogenicity, virulence, and antibiotic resistance to their hosts. Due largely to technical limitations associated with low bioburden on the skin, the viruses that inhabit the skin and interact with the bacterial microbiota are poorly characterized. Because viral genomes are orders of magnitude smaller than prokaryotic or eukaryotic genomes, they tend to become overwhelmed by larger genomes when metagenomic shotgun sequencing of whole-microbial communities is employed.
Metagenomic shotgun sequencing of purified virus-like particles has been applied to human systems, including gut and oral viromes, revealing important characteristics of those populations, including bacteriophage replication styles and diversity, hypervariable loci, and responses to perturbations (Reyes et al. 2010; Minot et al. 2011). In addition to technical challenges associated with isolating and purifying sufficient viral DNA or RNA for metagenomic sequencing, another major obstacle is the lacking availability of viral reference genomes, and especially those of bacteriophages. This is illustrated in Figure 2, where ∼85% of isolated and sequenced bacteriophages were grown on only three phyla. The absence of reliable reference genomes seriously limits the utility of reference-dependent approaches for analyzing viromes, and in most metagenomic virome studies, >90% of sequences are unidentifiable compared with references (Holmfeldt et al. 2013). However, reference-independent approaches, which do not rely upon taxonomic or functional assignment, for characterizing the viral “dark matter” can provide valuable information regarding viral community diversity and dynamics. One such approach, PHACCS (Phage Communities from Contig Spectrum), quantifies “virotypes” from contig assemblies to model viral community structure and estimate diversity (Angly et al. 2005). Increasing the number of sequenced viral genomes and further database development are necessary in order to improve methods for analyzing and characterizing viral communities.
Figure 2.

Phylogenetic tree of 35 major bacterial phyla. Highlighted in red are those phyla from which 85% of phages have been isolated and sequenced. Data from Holmfeldt et al. (2013).
In comparison to other body sites, very little is known about the skin virome, aside from what has been gleaned from studies that sequenced and analyzed the whole metagenome and identified and analyzed the viral fraction in the samples using reference-dependent approaches (Foulongne et al. 2012; Oh et al. 2014; Wylie et al. 2014). Common viruses identified by these studies include Propionibacterium and Staphylococcus phages, human papillomaviruses, and Merkel cell polyomaviruses (Foulongne et al. 2012; Oh et al. 2014; Wylie et al. 2014).
Genetic variation in microbial communities has rarely been studied, despite the important insights that can be gained regarding host–parasite interactions. The virome provides a unique opportunity to characterize patterns of variation in a natural community, due to the relatively small aggregate genome size of viruses, allowing for sequencing to a depth sufficient to assess genetic variability. In the gut virome, overall variation appears to be quite low, aside from hypervariable loci localized to predicted genes encoding tail-fibers and Ig-super family proteins (Minot et al. 2012). It is currently unknown if similar patterns of hypervariability exist in the skin virome despite the potential that genomic diversity generation contributes to mechanisms of host–parasite interactions.
Linking metabolomics and the skin microbiome
Understanding the chemical makeup of the host substrate can provide functional insights into microbial communities and their role in disease. Chemicals present on the skin provide nutrients and metabolic substrates for the microbiota, but in turn, the microbiota may also alter their chemical environment. For example, the skin commensal Propionibacterium acnes utilizes the triglycerides in sebum, cleaving them to free fatty acids that then serve to acidify the skin and also potentially promote adherence of the bacteria to the skin (Gribbon et al. 1993). As mentioned earlier, the characteristics of the topographical skin site sampled are strongly associated with the microbial communities, where sebaceous areas are strongly associated with Propionibacterium colonization, moist areas are associated with Staphylococcus and Corynebacterium, and dry areas tend to be highly diverse and transient. A recent study combined mass spectrometry with 16S rRNA microbiome sequencing to develop a 3D map of the skin surface (Bouslimani et al. 2015). As expected, Propionibacterium colocalized with molecules hypothesized to be products of sebum metabolism. A surprising finding was that the majority of the spectra (>80%) were not identifiable compared with references, indicating that the vast majority of molecules on the skin are uncharacterized. A major portion of those molecules that matched reference traces was derived from hygiene products applied to the skin. This indicates that daily skin care routines have a lasting effect on the chemical makeup of the skin surface and likely impact the skin microbiome, though few data exist in this area.
Wound healing is another area where metabolomics could be applied to further understand the context and functional significance of microbial communities. For example, targeted metabolomics revealed that diabetes alters the metabolic profiles of skin and wounds (Sood et al. 2015). In a mouse model of impaired diabetic wound healing, carnitine, glucose, and 3-nitrotyrosine were significantly increased in diabetic wounds compared with nondiabetic wounds. The presence and/or abundance of these metabolites may, in part, explain earlier studies that showed large differences in the microbiota colonizing diabetic wounds compared with nondiabetic wounds in the same mouse model (Grice et al. 2010), and further studies to characterize the wound-associated bacterial metabolic profiles could provide this information.
Microbe hunting in the skin
For some skin disorders, a role for microbial triggers and/or modulators has been proposed and investigated using genomic and metagenomic approaches. For example, flare states of the common childhood skin disorder atopic dermatitis (AD; “eczema”) are associated with increases in Staphylococcus aureus and decreases in overall skin microbial diversity (Kong et al. 2012). In a mouse model of AD (ADAM17-deficiency), S. aureus drives eczematous dermatitis (Kobayashi et al. 2015). Another example is acne vulgaris, in which involvement of P. acnes is proposed as a pathogenic factor. The relative abundance of P. acnes does not appear to be significantly different on acne skin compared with healthy skin, but genomic analysis demonstrated that certain strains of P. acnes are highly associated with acne (Fitz-Gibbon et al. 2013). Genomic analysis of these strains revealed genomic islands and a linear plasmid containing genes encoding potential pathogenicity factors.
However, the role of the microbiota is uncertain and less obvious in other dermatological disorders, and microbial triggers are sometimes hypothesized for disorders of unknown or uncertain etiology. For example, Borrelia burgdorferi infection is proposed to be associated with morphea (Weide et al. 2000); P. acnes and Mycobacterium have been proposed as infectious agents in sarcoidosis (Furukawa et al. 2009; Saidha et al. 2012; Eishi 2013). Bacterial involvement has also been proposed in hidradenitis suppurativa, though it is less clear if bacterial infection is a primary or secondary event (van der Zee et al. 2012).
A negative culture finding does not necessarily preclude the hypothesis of an infectious trigger or modulator of disease, since the limitations of cultures are now widely acknowledged. Identifying a causative organism can be challenging, especially for microorganisms such as viruses, whose genomes do not contain a readily amplified marker gene. Feng et al. (2008) applied an elegant “metatranscriptomic” approach to Merkel cell carcinoma (MCC), a rare but highly aggressive cutaneous malignancy. By employing “digital transcriptome subtraction” to select foreign (i.e., microbial) sequences in cDNA libraries derived from MCC, a fusion transcript of a viral gene and a human receptor tyrosine phosphatase was identified (Feng et al. 2008). Further analysis identified sequences of a previously undescribed polyoma virus, subsequently named Merkel cell polyomavirus (MCPyV), in association with tumors and less often with healthy tissues. Because MCPyV has been identified as a common feature of the healthy human skin microbiota, its role in MCC has been widely debated, and other factors are suggested to be at play, including the host immune system and viral mutation (Amber et al. 2013). Other skin cancers have been investigated with similar approaches in search of viral integration, but the results have largely been negative (Dimon et al. 2014). However, this type of sequencing and bioinformatics approach could potentially be useful in identifying microbial agents that trigger and/or modulate skin disease of unknown etiology. In a similar manner, RNA-seq of skin from patients with systemic sclerosis, an autoimmune disease, identified sequences from the environmental yeast Rhodotorula (Arron et al. 2014). There was no evidence of these sequences in RNA-seq data from normal skin. These findings raise a number of hypotheses to be tested, including cause-and-effect relationships between Rhodotorula and systemic sclerosis. By use of similar approaches, it is possible that microbial triggers could be identified for those diseases that have mystified dermatologists for decades.
Dissecting causation vs. consequence
Moving from descriptive data sets to the mechanism has been a major challenge for the overall field of microbiome research. The situation is not unlike that of genome-wide association studies, where lists of disease-associated variants are readily produced and available, but understanding the functional consequences of each variant is an ongoing challenge. If a particular microbe or microbial community is found to be associated with a disease, then the next logical step is to show that the microbe or microbial community causes the disease. Germ-free and gnotobiotic mice are the major tools used for functional validation of microbes and microbial communities in conferring a particular phenotype. Usually this involves isolating the microbe in the disease state, which can be a challenge in itself depending on the amenability of the microbe to culture and isolation. Then the isolated microbe is transferred to the germ-free mouse, and the phenotype of the mouse is monitored to see if the disease phenotype develops.
In some circumstances for skin-specific studies, the utility of these models can be called into question. Mouse skin is inherently histologically and biochemically different than human skin and, not surprisingly, naturally supports different populations of microbes. Mouse sebum is biochemically distinct from human sebum, containing vastly lower amounts of free fatty acids and practically no triglycerides, where human sebum is composed of >50% free fatty acids and triglycerides (Nikkari 1974). A likely result of this is that P. acnes, a microbe that produces lipases that cleave triglycerides to free fatty acids, is only a minor component of the mouse skin microbiome. Additionally, another key human skin commensal, S. epidermidis, does not naturally colonize murine skin, which typically hosts Staphylococcus xylosus and Staphylococcus saprophyticus. Most mouse strains have hair that needs to be removed to access the skin and the microbiota colonizing the skin, and this process alone may be disruptive to naturally occurring microbiota. With regards to cutaneous wound healing, the mouse heals by contraction more so than by re-epithelialization, which is in direct contrast to wound healing processes in human skin. Many skin diseases common in humans are not naturally recapitulated in mice, including acne, AD, and psoriasis, though models have been created that recapitulate key features. New models, such as organotypic three-dimensional skin models now used in cancer research (Ridky et al. 2010) may offer feasible, more biologically and clinically relevant, alternatives to the mouse for mechanistic experiments dissecting host–microbe interactions of the skin. These models are also not without caveat, as they do not recapitulate skin appendages and do not include adaptive immune responses.
Demonstration of causation in human populations is even more difficult, though recently there have been considerable pushes toward human-centric research, in light of findings that mouse models may poorly mimic human situations (Seok et al. 2013). Longitudinal study designs, for example, through the course of disease where there is relapse and remission or through treatment courses, have the potential to offer greater mechanistic insight than cross-sectional study designs. In light of temporal variability being a personalized feature of the human microbiome and the observation that the skin microbiome varies the most over time compared with other body habitats (Flores et al. 2014), the ideal clinical study design would incorporate longitudinal sampling to better understand disease pathogenesis. As illustrated in Figure 3, different microbiomic metrics can be monitored over time and overlaid with clinically relevant events to raise hypotheses regarding microbial causation of clinically relevant events and outcomes. Figure 3 illustrates an example from a patient with an acute traumatic wound whose wound microbiota was monitored at every clinical intervention and follow-up visit to the clinic. A complication in this patient, consisting of failure to heal and infection, was associated with increased levels of Moraxellaceae and low bacterial diversity.
Figure 3.

Example of longitudinal profiling of an acute traumatic injury (open fracture), overlaid with clinical metadata to identify potential microbiomic features associated with intervention (surgery) and outcomes. The y-axis represents longitudinal time points sampled. Swab samples were collected from the open fracture (prior to closure) or the surgical wound (after closure). (Top) Alpha diversity metrics; (middle) bacterial load; and (bottom) relative abundance of the top five most abundant bacterial taxa (EA Grice, unpubl.).
Approaches such as the recently described IgA-seq (Palm et al. 2014), where those bacteria highly coated with IgA are presumed to be pathogenic, may also help elucidate causation in human disease. Other techniques also utilizing flow sorting have identified active microbes in the human GI tract and their transcriptional responses to xenobiotics (Maurice et al. 2013). Similar principles could be applied to the skin microbiota, where it is unclear based on DNA sequence alone if the microbiota sampled are alive or dead.
Impact of perturbation
It is well known that antibiotics affect the gut microbiota and, in some cases where the individual is immunosuppressed, can also lead to life-threatening consequences such as infection with a multi-drug-resistant organism. Antibiotics are frequently prescribed by dermatologists, due to the high frequency and chronicity of cutaneous diseases, such as acne. However, systematic studies addressing how antimicrobials, oral or topical, affect the skin microbiome and the pervasiveness of the effects are lacking. Peripheral observations suggest a link, where treatment of acne with doxycycline is associated with Gram-negative folliculitis (Leyden et al. 1973). Data are also lacking regarding how the use of soaps, antiseptics, emollients, and cosmetics influences our skin microbiome. The skin takes the brunt of UV exposure, but how UV affects microbial communities is also unknown. Bacteriophages can be induced to replicate and lyse their host upon UV exposure, so an intriguing hypothesis is that UV exposure can change the composition of bacterial communities via predator–prey dynamics between bacteriophages and their hosts. Systematic studies of how these and other perturbations affect the skin microbiome are in high demand, as it becomes increasingly apparent that disruption of human microbiota may result in adverse outcomes.
The future of the microbiome and medicine
The accessibility and plasticity of the skin microbiome render it an ideal candidate for clinical diagnostic and therapeutic applications. Microbes are exquisitely responsive to their surroundings, a feature that could be taken advantage of for the development of clinical biomarkers for prognostic and predictive applications. Using a simple swab to sample the microbes, one can envision a clinical diagnostic test that would assign the risk of developing a particular skin disease or suffering a complication or nonideal outcome. For example, the microbes residing in a wound environment may offer clues about that wound's likelihood to go on to develop an infection-related complication. Those clues may be apparent in multiple dimensions of the microbiota, including microbial diversity, microbial load, or presence/absence or abundance of specific types of microbes. Identifying those patients at risk for developing a complication can inform management and treatment strategies to avoid a poor outcome. The microbiome could also be used to stratify patient populations and thus personalize treatment approaches.
Restoring microbial health on the skin is also an area of opportunity, but it needs to be informed by more thorough identification of which microbes are beneficial or harmful. The undeniable success of gut microbiome transplantation to treat Clostridium difficile infection (Austin et al. 2014) has raised the question if such approaches have utility in treating skin dysbiosis, for example, for the treatment of AD, which is characterized by an outgrowth of S. aureus during disease flares. Probiotic and prebiotic skin treatments are now being widely explored, but they could gain from more thorough investigation of which microbes are providing benefit.
Conclusion
The past two decades have illuminated the role of microbial communities in health and disease. It is expected that the field of microbiomics and metagenomics will continue to evolve alongside rapidly advancing sequencing technologies and improved bioinformatics tool development. 16S rRNA gene sequencing will likely continue to be a key approach for describing the composition and diversity of microbial communities. However, techniques such as metagenomic and metatranscriptomic sequencing will become more widely employed, given the rich data sets gained from these techniques and their potential for functional insights. Here we have identified challenges and opportunities for skin microbiome research that need to be addressed in order to move the field forward in the future, including: (1) increased understanding of the nonbacterial components of the skin microbiome such as the viruses; (2) integration of metabolomics data to better understand the environmental and functional context of microbial communities; (3) improved approaches for identifying potential microbial triggers for skin diseases; (4) systematic studies to address common perturbations to the skin microbiome, including antibiotics, antiseptics, cosmetics, hygienic products, and UV; and (5) improved, clinically relevant models for studying the functional and mechanistic roles of the skin microbiome. A reasonable goal for the next 20 years is that microbiomic and metagenomic science will routinely inform clinical management and treatment of skin disorders, through diagnostic tests to stratify patient subsets, predict best treatment modality and outcomes, and through treatment strategies such as targeted manipulation of microbial communities.
Acknowledgments
I thank Geoffrey Hannigan for assistance in preparing the figures.
Footnotes
Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.191320.115.
Freely available online through the Genome Research Open Access option.
References
- Amber K, McLeod MP, Nouri K. 2013. The Merkel cell polyomavirus and its involvement in Merkel cell carcinoma. Dermatol Surg 39: 232–238. [DOI] [PubMed] [Google Scholar]
- Angly F, Rodriguez-Brito B, Bangor D, McNairnie P, Breitbart M, Salamon P, Felts B, Nulton J, Mahaffy J, Rohwer F. 2005. PHACCS, an online tool for estimating the structure and diversity of uncultured viral communities using metagenomic information. BMC Bioinformatics 6: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arron ST, Dimon MT, Li Z, Johnson ME, A Wood T, Feeney L, G Angeles J, Lafyatis R, Whitfield ML. 2014. High Rhodotorula sequences in skin transcriptome of patients with diffuse systemic sclerosis. J Invest Dermatol 134: 2138–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin M, Mellow M, Tierney WM. 2014. Fecal microbiota transplantation in the treatment of Clostridium difficile infections. Am J Med 127: 479–483. [DOI] [PubMed] [Google Scholar]
- Bouslimani A, Porto C, Rath CM, Wang M, Guo Y, Gonzalez A, Berg-Lyon D, Ackermann G, Moeller Christensen GJ, Nakatsuji T, et al. 2015. Molecular cartography of the human skin surface in 3D. Proc Natl Acad Sci 112: E2120–E2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, et al. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chehoud C, Rafail S, Tyldsley AS, Seykora JT, Lambris JD, Grice EA. 2013. Complement modulates the cutaneous microbiome and inflammatory milieu. Proc Natl Acad Sci 110: 15061–15066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R. 2009. Bacterial community variation in human body habitats across space and time. Science 326: 1694–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimon MT, Wood HM, Rabbitts PH, Liao W, Cho RJ, Arron ST. 2014. No evidence for integrated viral DNA in the genome sequence of cutaneous squamous cell carcinoma. J Invest Dermatol 134: 2055–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eishi Y. 2013. Etiologic aspect of sarcoidosis as an allergic endogenous infection caused by Propionibacterium acnes. BioMed Res Int 2013: 935289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng H, Shuda M, Chang Y, Moore PS. 2008. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 319: 1096–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findley K, Oh J, Yang J, Conlan S, Deming C, Meyer JA, Schoenfeld D, Nomicos E, Park M, Becker J, et al. 2013. Topographic diversity of fungal and bacterial communities in human skin. Nature 498: 367–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitz-Gibbon S, Tomida S, Chiu BH, Nguyen L, Du C, Liu M, Elashoff D, Erfe MC, Loncaric A, Kim J, et al. 2013. Propionibacterium acnes strain populations in the human skin microbiome associated with acne. J Invest Dermatol 133: 2152–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores GE, Caporaso JG, Henley JB, Rideout JR, Domogala D, Chase J, Leff JW, Vazquez-Baeza Y, Gonzalez A, Knight R, et al. 2014. Temporal variability is a personalized feature of the human microbiome. Genome Biol 15: 531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foulongne V, Sauvage V, Hebert C, Dereure O, Cheval J, Gouilh MA, Pariente K, Segondy M, Burguiere A, Manuguerra JC, et al. 2012. Human skin microbiota: high diversity of DNA viruses identified on the human skin by high throughput sequencing. PLoS One 7: e38499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa A, Uchida K, Ishige Y, Ishige I, Kobayashi I, Takemura T, Yokoyama T, Iwai K, Watanabe K, Shimizu S, et al. 2009. Characterization of Propionibacterium acnes isolates from sarcoid and non-sarcoid tissues with special reference to cell invasiveness, serotype, and trigger factor gene polymorphism. Microb Pathog 46: 80–87. [DOI] [PubMed] [Google Scholar]
- Gao Z, Tseng CH, Pei Z, Blaser MJ. 2007. Molecular analysis of human forearm superficial skin bacterial biota. Proc Natl Acad Sci 104: 2927–2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribbon EM, Cunliffe WJ, Holland KT. 1993. Interaction of Propionibacterium acnes with skin lipids in vitro. J Gen Microbiol 139: 1745–1751. [DOI] [PubMed] [Google Scholar]
- Grice EA, Segre JA. 2011. The skin microbiome. Nat Rev Microbiol 9: 244–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grice EA, Kong HH, Conlan S, Deming CB, Davis J, Young AC, Bouffard GG, Blakesley RW, Murray PR, Green ED, et al. 2009. Topographical and temporal diversity of the human skin microbiome. Science 324: 1190–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grice EA, Snitkin ES, Yockey LJ, Bermudez DM, Liechty KW, Segre JA. 2010. Longitudinal shift in diabetic wound microbiota correlates with prolonged skin defense response. Proc Natl Acad Sci 107: 14799–14804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmfeldt K, Solonenko N, Shah M, Corrier K, Riemann L, Verberkmoes NC, Sullivan MB. 2013. Twelve previously unknown phage genera are ubiquitous in global oceans. Proc Natl Acad Sci 110: 12798–12803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Human Microbiome Project Consortium. 2012. Structure, function and diversity of the healthy human microbiome. Nature 486: 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Glatz M, Horiuchi K, Kawasaki H, Akiyama H, Kaplan DH, Kong HH, Amagai M, Nagao K. 2015. Dysbiosis and Staphylococcus aureus colonization drives inflammation in atopic dermatitis. Immunity 42: 756–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong HH, Oh J, Deming C, Conlan S, Grice EA, Beatson MA, Nomicos E, Polley EC, Komarow HD, Murray PR, et al. 2012. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res 22: 850–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane DJ, Pace B, Olsen GJ, Stahl DA, Sogin ML, Pace NR. 1985. Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses. Proc Natl Acad Sci 82: 6955–6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyden JJ, Marples RR, Mills OH Jr, Kligman AM. 1973. Gram-negative folliculitis: a complication of antibiotic therapy in acne vulgaris. Br J Dermatol 88: 533–538. [DOI] [PubMed] [Google Scholar]
- Maurice CF, Haiser HJ, Turnbaugh PJ. 2013. Xenobiotics shape the physiology and gene expression of the active human gut microbiome. Cell 152: 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minot S, Sinha R, Chen J, Li H, Keilbaugh SA, Wu GD, Lewis JD, Bushman FD. 2011. The human gut virome: inter-individual variation and dynamic response to diet. Genome Res 21: 1616–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minot S, Grunberg S, Wu GD, Lewis JD, Bushman FD. 2012. Hypervariable loci in the human gut virome. Proc Natl Acad Sci 109: 3962–3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik S, Bouladoux N, Wilhelm C, Molloy MJ, Salcedo R, Kastenmuller W, Deming C, Quinones M, Koo L, Conlan S, et al. 2012. Compartmentalized control of skin immunity by resident commensals. Science 337: 1115–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik S, Bouladoux N, Linehan JL, Han SJ, Harrison OJ, Wilhelm C, Conlan S, Himmelfarb S, Byrd AL, Deming C, et al. 2015. Commensal–dendritic-cell interaction specifies a unique protective skin immune signature. Nature 520: 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsuji T, Chiang HI, Jiang SB, Nagarajan H, Zengler K, Gallo RL. 2013. The microbiome extends to subepidermal compartments of normal skin. Nat Commun 4: 1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikkari T. 1974. Comparative chemistry of sebum. J Invest Dermatol 62: 257–267. [DOI] [PubMed] [Google Scholar]
- Oh J, Freeman AF, Park M, Sokolic R, Candotti F, Holland SM, Segre JA, Kong HH. 2013. The altered landscape of the human skin microbiome in patients with primary immunodeficiencies. Genome Res 23: 2103–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J, Byrd AL, Deming C, Conlan S, Program NCS, Kong HH, Segre JA, Program NCS. 2014. Biogeography and individuality shape function in the human skin metagenome. Nature 514: 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm NW, de Zoete MR, Cullen TW, Barry NA, Stefanowski J, Hao L, Degnan PH, Hu J, Peter I, Zhang W, et al. 2014. Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell 158: 1000–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillsbury DM, Kligman AM. 1954. Some current problems in cutaneous bacteriology. In Modern trends in dermatology (ed. MacKenna RMB), pp. 187–213. Butterworth, London. [Google Scholar]
- Reyes A, Haynes M, Hanson N, Angly FE, Heath AC, Rohwer F, Gordon JI. 2010. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature 466: 334–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridky TW, Chow JM, Wong DJ, Khavari PA. 2010. Invasive three-dimensional organotypic neoplasia from multiple normal human epithelia. Nat Med 16: 1450–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saidha S, Sotirchos ES, Eckstein C. 2012. Etiology of sarcoidosis: does infection play a role? Yale J Biol Med 85: 133–141. [PMC free article] [PubMed] [Google Scholar]
- Saunders CW, Scheynius A, Heitman J. 2012. Malassezia fungi are specialized to live on skin and associated with dandruff, eczema, and other skin diseases. PLoS Pathog 8: e1002701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, et al. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75: 7537–7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, et al. 2013. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci 110: 3507–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sood RF, Gu H, Djukovic D, Deng L, Ga M, Muffley LA, Raftery D, Hocking AM. 2015. Targeted metabolic profiling of wounds in diabetic and non-diabetic mice. Wound Repair Regen 23: 423–434. [DOI] [PubMed] [Google Scholar]
- van der Zee HH, Laman JD, Boer J, Prens EP. 2012. Hidradenitis suppurativa: viewpoint on clinical phenotyping, pathogenesis and novel treatments. Exp Dermatol 21: 735–739. [DOI] [PubMed] [Google Scholar]
- Weide B, Walz T, Garbe C. 2000. Is morphoea caused by Borrelia burgdorferi? A review. Br J Dermatol 142: 636–644. [DOI] [PubMed] [Google Scholar]
- Wylie KM, Mihindukulasuriya KA, Zhou Y, Sodergren E, Storch GA, Weinstock GM. 2014. Metagenomic analysis of double-stranded DNA viruses in healthy adults. BMC Biol 12: 71. [DOI] [PMC free article] [PubMed] [Google Scholar]