

Abstract
Somatic L1 retrotransposition events have been shown to occur in epithelial cancers. Here, we attempted to determine how early somatic L1 insertions occurred during the development of gastrointestinal (GI) cancers. Using L1-targeted resequencing (L1-seq), we studied different stages of four colorectal cancers arising from colonic polyps, seven pancreatic carcinomas, as well as seven gastric cancers. Surprisingly, we found somatic L1 insertions not only in all cancer types and metastases but also in colonic adenomas, well-known cancer precursors. Some insertions were also present in low quantities in normal GI tissues, occasionally caught in the act of being clonally fixed in the adjacent tumors. Insertions in adenomas and cancers numbered in the hundreds, and many were present in multiple tumor sections, implying clonal distribution. Our results demonstrate that extensive somatic insertional mutagenesis occurs very early during the development of GI tumors, probably before dysplastic growth.
Somatic mobilization of retroelements in the cancer genome has only recently been established as a widespread mutational phenomenon. In particular, Long INterspersed Element (LINE)-1 (L1)-mediated retrotransposition has been observed mostly in epithelial cancers. Somatic human-specific L1 (L1Hs) insertions are most abundant in these cancers, but L1-mediated Alu, SVA, and processed pseudogene insertions have also been detected (Iskow et al. 2010; Lee et al. 2012; Solyom et al. 2012; Ewing et al. 2013; Shukla et al. 2013; Cooke et al. 2014; Helman et al. 2014; Pitkanen et al. 2014; Tubio et al. 2014). L1s are autonomous mobile elements that comprise 17% of the human genome and retrotranspose by a “copy and paste” mechanism via an RNA intermediate. This process can lead to insertional mutagenesis and genetic instability (Goodier and Kazazian 2008). Potentially etiological L1 insertions have been reported in APC (Miki et al. 1992) and PTEN exons (Helman et al. 2014) in colorectal and endometrial cancer, respectively, and insertions of unknown significance have been found in numerous other cancer driver genes in a variety of malignancies (Iskow et al. 2010; Lee et al. 2012; Solyom et al. 2012; Ewing et al. 2013; Shukla et al. 2013; Cooke et al. 2014; Helman et al. 2014; Pitkanen et al. 2014; Tubio et al. 2014; Paterson et al. 2015). In a study of somatic retrotransposition during the evolution of prostate and lung cancer, Tubio et al. (2014) found evidence of insertions occurring during cancer development. Beyond this work, the timing of retrotransposition in cancer development has not been analyzed previously.
Results
Timing and distribution of somatic L1 insertions in GI cancers
Here, we studied the timing of L1Hs integration events in 30 tumors of different developmental stages from 18 GI cancer patients (Table 1). We studied DNA from four colorectal cancer patients previously diagnosed with colonic polyps (one hyperplastic polyp and four adenomas, one of which contained high-grade dysplasia), seven patients with invasive pancreatic carcinoma, one of whom also had a pancreatic intraepithelial neoplasia (PanIN, a precancerous lesion), and from seven patients with primary gastric cancer. Matched normal samples were of the same tissue type from which the tumors originated, and multiple metastases were also available in eight cases. Next-generation L1-resequencing (L1-seq) (Ewing and Kazazian 2010; Solyom et al. 2012) was carried out on DNA from dissected tissues. We also studied somatic L1Hs integration events in eight testicular germ cell tumors (TGCT) and matched blood of seven of these patients with familial TGCT. Sample characteristics and pathological data of each tumor type are described in Supplemental Table S2, sheets S2j, S2n, S2p, and S2t, respectively.
Table 1.
PCR-verified and Sanger-sequenced somatic L1 insertions
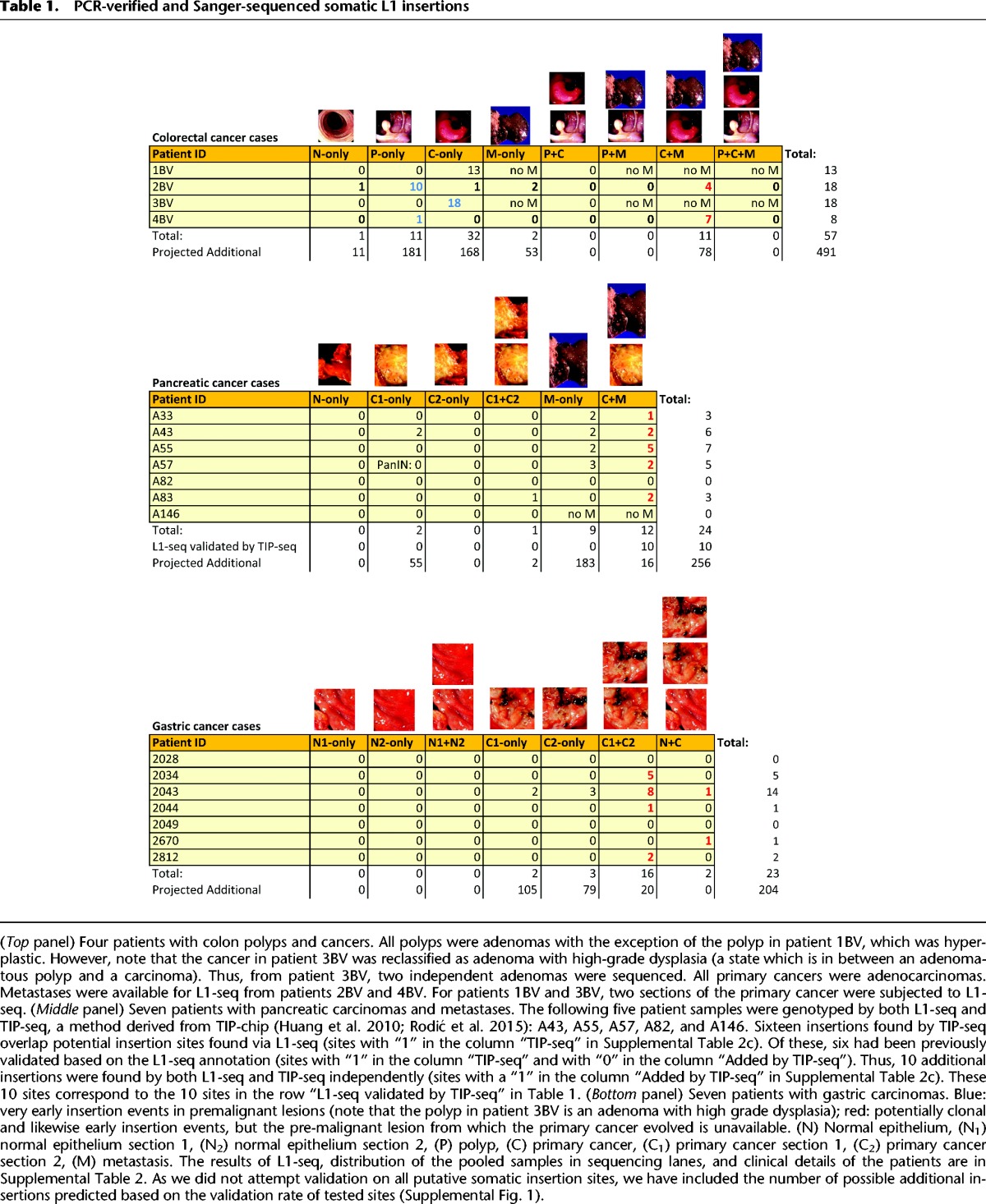
Altogether, 104 somatic heterozygous L1Hs insertions were validated by PCR and Sanger sequencing in the 18 GI cancer patients, while only one insertion validated in the seven TGCT patients (Table 1; Figs. 1, 2; Supplemental Data 1–4; Supplemental Table 2). Our major finding is that somatic L1 insertions occur in certain precancerous lesions. We also find that pancreatic and stomach cancer are permissive for L1 mobilization (Table 1; Fig. 2; Supplemental Table 2). Of 24 insertions validated in pancreatic cancers, 13 (54%) were present in two different sections of the primary cancers and in the matched liver metastases, signifying early occurring insertions. Similarly, of 23 insertions validated in gastric cancers, 18 (78%) were present in two independent tumor sections. In addition, out of the total of 57 validated insertions in colorectal cancer, we were able to analyze 43 in two to four sections of the same colorectal cancers and in two sections of the high-grade dysplastic adenoma of patient 3BV. Of these 43, 42 (98%) were present in all primary and metastatic cancer sections (Figs. 2, 3A). Surprisingly, of 57 validated somatic insertions in the colon tumors, 29 (50%) were detected exclusively in adenomas. As validation was attempted on a subset of insertions, we sought to obtain an estimate for how many insertions might validate across the entire data set based on peak characteristics and the empirical validation rate across various tissues. In total, we expect that hundreds of additional insertions would validate in these GI tumor cases (Table 1; Supplemental Fig. 1). However, with additional validations focused on smaller peak sizes (i.e., those represented by 10 or fewer read mappings), the expected number of insertions may increase dramatically, to well above 1000.
Figure 1.

Genomic distribution of L1 insertions in GI tumors. (A) The genomic distribution of reference (light blue histogram) (Supplemental Table 3) and putative nonreference L1 insertions (light red histogram) (Supplemental Table 4) in colon cancer cases is shown as a density plot binned into 10-Mbp intervals across the genome. Somatic insertions validated by PCR and capillary sequencing (Supplemental Table 2) are shown on the outside. The tissue distribution for somatic insertions is shown according to the following key (see Table 1 for counts and Supplemental Table 2 for further details on insertion sites): (C) primary cancer, (M) metastasis, (P) polyp, (N) normal colon. (B,C) Shown similarly for pancreatic cancer samples in B and gastric cancer samples in C.
Figure 2.

PCR and Sanger-sequencing validation scheme of L1-seq results. (A) Multistep PCR validation scheme and location of primers used. Insertions were primarily validated with conventional PCR at their 3′ junction using the L1Hs with the 3′ primer. Some insertions were also validated with nested PCR using the “G” primer with a nested 3′ primer. After the 3′ junction was located, we attempted to find the 5′ junction using the 5′ primer with L1 out primers. Triangles symbolize target site duplications (TSD). (B) PCR validation of clonal cancer-specific insertions. (Left panel) A primary colon cancer-and-metastasis-specific insertion (ins. E8). (Right panel) A primary pancreatic cancer-and-metastasis-specific insertion (ins. B7). The higher molecular weight bands visible above the tumor tissues of the empty site PCR products are the highly truncated L1 elements, as assessed by gel extraction and Sanger sequencing. (N) Normal, (P) polyp, (C) primary cancer, (C1) primary cancer section 1, (C2) primary cancer section 2, (M) metastasis, (FS) filled site PCR product (insertion allele), (ES) empty site PCR product (wild-type allele). (C) PCR validation of the normal colon-specific insertion “A5n” and the somatic normal-and-cancer-specific insertion “C1s” in stomach cancer. “A5n” is detectable exclusively using nested PCR in case 2BV, while the somatic L1 insertion in the stomach cancer of patient 2670 is detectable by both conventional and nested PCR and is also detectable in normal stomach using nested PCR. (NC) Normal colon, (NL) normal liver, (N1) normal stomach section 1, (N2) normal stomach section 2, (C1) primary cancer section 1, (C2) primary cancer section 2, (M1) metastasis section 1, (M2) metastasis section 2. O'GeneRuler 1 kb Plus DNA ladder was used (Thermo Scientific). (D) Reconstituted Sanger sequence of the 5′ truncated colorectal cancer-and-metastasis-specific ins. E8 from B. In blue, TSD (6 bp, alternatively, 7 bp due to microhomology at the 5′ junction); in green, highly truncated L1Hs (112/112-bp identity with L1RP, nt 5908–6019); in red, poly(A) tail.
Figure 3.

Representative examples of further somatic L1 insertions in colon cancer patient 2BV and gastric cancer patient 2043. (A) L1 insertions in colorectal cancer (all detectable by conventional PCR), from left to right: ins. D2 (polyp-specific, present exclusively in adenoma sample “5”); ins. A8n (cancer-and-metastasis-specific, present in three independent primary colorectal cancer sections and in three independent liver metastasis sections); ins. C7 (the only insertion in colon cancer cases that is detectable in only one tumor section, “6/1”); ins. G5 (a metastasis-specific insertion, present in only one of three sections of metastasis sample “7”). (N) Normal colon, (P) polyp, (C) primary colorectal cancer, (C1) primary colorectal cancer section 1, (C2) primary colorectal cancer section 2, (C3) primary colorectal cancer section 3, (M) metastasis, (M1) metastasis section 1, (M2) metastasis section 2, (M3) metastasis section 3, (NL) normal liver. (B) The fifth agarose gel shows the second somatic normal-and-cancer-specific L1 insertion in stomach cancer patient 2043 (detectable in cancer both by conventional and nested PCR, but detectable in normal stomach exclusively by nested PCR). (N1) Normal stomach section 1, (N2) normal stomach section 2, (C1) primary gastric cancer section 1, (C2) primary gastric cancer section 2. (C) Hematoxylin and eosin (H&E) staining revealed normal gastric mucosa in case of insertions A2s and C1s (Fig. 2C). (Top, left) Normal gastric mucosa from patient 2043; (top, right) gastric adenocarcinoma with intestinal differentiation from patient 2043. (Bottom, left) Normal gastric mucosa from patient 2670; (bottom, right) gastric adenocarcinoma with signet ring features from patient 2670. All photomicrographs were taken at 20× original magnification. H&E-stained slides were sectioned and stained in the Johns Hopkins Department of Pathology according to standard protocols. (D) In order to estimate the limits of detection of an insertion by conventional and seminested PCR, we performed a dilution series on DNA containing a heterozygous polymorphic germline insertion. Normal colon DNA containing a polymorphic L1 insertion of patient 4BV was mixed with normal colon DNA of patient 1BV, which did not contain this insertion, and served as competitor DNA. The amount of DNA used for conventional PCR in the case of 4BV DNA is shown in ng, which is constituted to a final amount of 25 ng using 1BV DNA. One microliter of each FS PCR product was used for seminested PCR. The detection limit of an insertion using conventional PCR appears to be 2.5 ng DNA against 22.5 ng competitor DNA (faint PCR band present). However, using seminested PCR, the detection limit is 25 pg DNA against 24.975 ng competitor DNA. Sanger sequencing confirmed that the lower molecular weight seminested PCR bands are nonspecific (seen in 1BV and in 4BV using ≤0.025 ng DNA for conventional PCR, and is marked by an asterisk), while the higher molecular weight band is the correct PCR product (marked by arrow). Thus, we detect one copy in 10 cells (very faint band) with conventional PCR, and one copy per 1000 cells with seminested PCR. Identical results were obtained for a second heterozygous polymorphic L1 and a different competitor DNA. O'GeneRuler 1 kb Plus DNA ladder was used (Thermo Scientific).
We observed evidence of clonal insertions in primary colorectal and pancreatic cancer-metastasis cases, where 23 of 27 (85%) somatic insertions in the primary cancers were present in their paired metastases. The clonal relationship of primary colon cancers and their metastases is corroborated by their comparable CNV patterns by SNP Array 6.0 data analysis (Supplemental Fig. 2). In contrast, the polyps always originated at some distance from their matched cancers, precluding the possibility that the sampled cancers arose from the sampled polyps. In agreement, no shared insertions have been validated between polyps and colon cancers (Table 1).
The fact that half of the total validated insertions (29/57) in colon tumors were found in precancerous lesions also implies that these insertions occurred earlier, either at the dysplastic stage or even in histologically normal cells. Indeed, we detected a somatic insertion in a histologically normal colon sample of patient 2BV exclusively with two-stage nested PCR, suggesting low abundance of the insertion. The L1 was not present in normal liver tissue, ruling out germline status or loss-of-heterozygosity (LOH) in the tumors (Fig. 2C). Importantly, we also detected two insertions that were abundantly present in two gastric cancer sections (detectable by both conventional and nested PCR) and were also present in two adjacent histologically normal gastric mucosa sections at low quantity (detectable exclusively by nested PCR) (Figs. 2C, 3B,C). These results suggest that, with the exception of metastasis-specific insertions, many—if not most—insertions occurred in apparently normal or very early pre-neoplastic cells and then became fixed during clonal outgrowth. Thus, we speculate that if one could obtain the normal parent cell that originally underwent malignant transformation, that cell would likely contain the somatic insertions present in the tumor.
In contrast, some low abundance insertions have been detected exclusively by nested PCR in multiple GI tumors, as well as the one somatic L1 insertion in a TGCT patient (Supplemental Table 2). Intriguingly, no insertions were verified in the hyperplastic polyp, an adenoma, a colon cancer, the PanIN, two pancreatic cancers, a lymph node metastasis, two gastric cancers, and seven TGCT tumors (Table 1). These latter findings suggest that either a subset of normal cells lacks somatic L1 insertions and some tumors arise from these cells, or we merely failed to detect insertions in some samples.
The presence of early-arising, clonally expanded insertions raises the possibility that some retrotranspositions are tumor-initiating events. Importantly, all analyzed cancers displayed widespread CNVs across their genome, as did the adenoma with high-grade dysplasia (“10/1”). In contrast, adenomas “5” and “12” were devoid of any large-scale CNVs (Supplemental Fig. 2b,d), suggesting a substantial shift in genetic instability occurring prior to or during the adenoma-carcinoma transition. Notably, adenoma “5” contained a large number of somatic L1 insertions, leading us to speculate that, in some cases, somatic retrotransposition may be more important in shaping the early tumor genome than large CNVs.
Insertions in known or candidate cancer driver genes
The insertions displayed hallmarks of retrotransposition, such as a poly(A) tail, various sizes of target site duplications (TSDs), 5′ truncations, and inversions (Supplemental Table 2). However, one full-length insertion (C9) was validated in a colorectal cancer and its paired metastasis, and one translocation event (C5) was validated in a liver metastasis of a primary pancreatic cancer. Microhomology between the L1 5′ junctions and the unique genomic regions frequently precluded the precise assessment of the 5′ junction, the size of the L1 and TSD.
Many of the insertions occurred in known or candidate cancer driver genes. For example, we verified a primary colorectal cancer-and-metastasis-specific intronic L1 insertion occurring within 1.9 kb of two exons of the CYLD gene. CYLD encodes a de-ubiquitinating enzyme mutated in familial cylindromatosis, Brooke–Spiegler syndrome, and multiple familial trichoepithelioma type 1. This gene is a known tumor suppressor that is also somatically mutated in various epithelial cancers and is represented in the COSMIC Cancer Gene Census (http://cancer.sanger.ac.uk/census). Another colon cancer had an insertion into the HDAC9 (histone deacetylase 9) gene which also exhibits a cancer-and-metastasis-specific distribution.
Notably, an exonic insertion into a protein coding region was validated in ELOVL4 (ELOVL fatty acid elongase 4) in both sections of a gastric cancer. Further, a 3′ UTR insertion was validated in SOX6 (sex determining region Y-box) with nested PCR in a single pancreatic cancer section. Another 3′ UTR insertion was detected in STX11 (whose encoded protein plays a role in intracellular transport) in both sections of the high-grade dysplastic adenoma. In another adenoma, an intronic L1 was detected in PANX1 (encodes for pannexin 1, a gap junction protein). We discovered a primary pancreatic cancer-and-metastasis-specific intronic integration event located between two APAF1 exons (apoptotic peptidase activating factor 1) 700 bp downstream and 1 kb upstream, respectively. APAF1 is a component of the apoptosome that is dysregulated in pancreatic ductal adenocarcinomas (Corvaro et al. 2007). In another pancreatic cancer patient, an intronic L1 in the GDNF gene was also present in both the primary cancer and its metastasis. GDNF is a glial cell line-derived neurotrophic factor and a ligand of RET, which has been suggested to participate in pancreatic cancer progression and invasion (Zeng et al. 2008). Intronic L1 insertions were likewise found in stomach tumors in the cancer driver candidate genes KLF12 (Kruppel-like factor 12), a known player in gastric cancer progression (Nakamura et al. 2009), and in CTNND2, which encodes for catenin delta 2.
Together with our results, cell adhesion and neuronal genes have repeatedly been reported to be excessively mutagenized by somatic L1 insertions in cancer (Iskow et al. 2010; Lee et al. 2012; Solyom et al. 2012; Ewing et al. 2013; Shukla et al. 2013; Cooke et al. 2014; Helman et al. 2014; Tubio et al. 2014), and some genes seem to act as hotspots for insertions. Intriguingly, CNTNAP2 (contactin associated protein-like 2, a member of the neurexin family with cell adhesion functions in the nervous system) has been recurrently mutagenized by L1 insertions in four lung and an endometrial carcinoma (Helman et al. 2014; Tubio et al. 2014). We also found two independent somatic L1 insertions in this gene in gastric cancer patient 2043. One of these insertions simultaneously targeted the MIR548I4 gene that is located within the primary transcript of CNTNAP2. Likewise, we found a somatic L1 insertion in a stomach cancer in RIMS2 (regulating synaptic membrane exocytosis 2). RIMS2 was found to be a target for insertional mutagenesis also by others (Lee et al. 2012) in head and neck squamous cell carcinoma, as well as in colon cancer. However, we did not observe any general enrichment of cancer driver or cell adhesion genes targeted by somatic L1 insertions when compared to germline insertions (data not shown).
L1 insertions and protein expression
We assessed the impact of somatic L1 insertions on the expression of the corresponding protein-coding genes by comparing protein abundance across the entire proteome of the polyp with the highest number of somatic L1 insertions (sample “10”) with that of its paired normal colon (sample “8”) using mass spectrometry analysis. Of the nine validated somatic insertions that were in protein coding regions in the polyp (Supplemental Table 2), two proteins—KIAA1217 and WARS2—were down-regulated in the adenoma >90% and >70%, respectively (Supplemental Fig. 3; Supplemental Table 5). Of 3025 proteins analyzed, 989 (32%) were down-regulated greater than or equal to twofold and 804 (26%) were down-regulated at least as much as WARS2. Among the 3025 proteins analyzed, only KIAA1217 and WARS2 were represented in nine genes with validated somatic L1 insertions. If one picks two genes that are expressed, one would expect both to be down-regulated 0.274 × 0.274 or 0.07 of the time. Thus, although it is quite possible that the intronic insertions in these two genes led to the decrease in their protein abundance, 7% of the time these two genes would be down-regulated by chance alone. Additional genetic, epigenetic, or post-transcriptional/post-translational changes affecting either allele cannot be ruled out. Thus, we cannot conclude from these results that the decrease in protein levels is due to the L1 insertions. Interestingly, KIAA1217 was previously insertionally mutagenized by an L1 in a colorectal cancer (Lee et al. 2012), and WARS2 was mutated by a processed pseudogene insertion in a chondrosarcoma (Cooke et al. 2014).
Discussion
We provide evidence that somatic retrotransposition events are an abundant source of endogenous mutagenesis in human GI tissues and that their presence in precancers is the likely result of clonally expanded normal/non-neoplastic precursor cells in which the insertions become fixed (Fig. 4). Of note, the clonal outgrowth of a cell containing a somatic insertion increases the ability to detect insertion events independent of these insertions being drivers of tumorigenesis. Our findings agree with a recent mathematical model predicting that at least half of the somatic mutations in cancers arising in self-renewing tissues originate prior to tumorigenesis (Tomasetti et al. 2013). Previously, we hypothesized that a larger fraction of insertions occurred late during tumorigenesis, but at that time, we did not study dissected cancers and precancers (Solyom et al. 2012).
Figure 4.
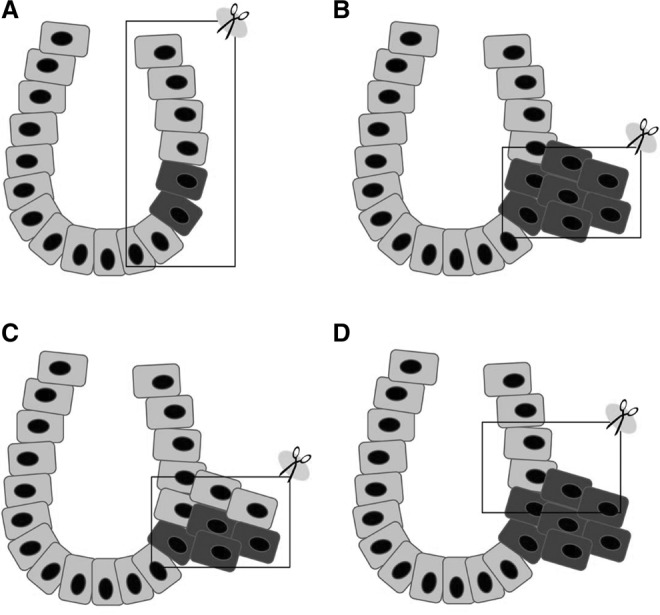
Distribution of somatic L1 insertions. (A) Normal colon crypt containing a few cells with L1 insertions (detectable only by nested PCR). (B) Colon tumor with early L1 insertions (detectable by conventional PCR). (C) L1 insertions occurring late during tumorigenesis (detectable only by nested PCR). (D) Colon tumor containing early L1 insertions with contaminating normal cells (detectable only by nested PCR). Note that by using nested PCR, it may be possible to misdiagnose contaminating or tumor-invading normal tissue-specific insertions (A) as tumor-specific (C or D). The frame represents the sampled tissue.
Our number of tumor-specific retrotransposon insertions is an underestimate because (1) only L1Hs, and not other types of retroelement insertions, were examined; (2) long 3′ transductions and insertions truncated 3′ to the diagnostic L1Hs nucleotides are missed by L1-seq; and (3) detection of insertions is bound by the sensitivity and specificity of L1-seq and PCR validation (Fig. 3D). Furthermore, the number of somatic normal-specific insertions was likely underestimated, as tumors are invariably contaminated by normal cells; thus, some normal tissue-specific insertions are likely scored as germline by L1-seq.
Although it has been accepted that classical mutations can cause cancer, functional studies on somatic tumor-specific retrotransposon insertions are lacking. Since retrotransposon insertions are large, abundant, and can be extremely disruptive to gene function, as evidenced by more than 100 germline disease-causing retroelement insertions in humans (Hancks and Kazazian 2012), they have the potential to initiate and aggravate tumorigenesis in somatic cells. Thus, the questions are: How large a fraction of these L1 insertions are drivers of dysplasia and/or subsequent cancer progression? How many contribute to genetic instability indirectly, e.g., by providing templates for homologous recombination, transcriptional interference, alternative splicing, epigenetic effects, or the generation of DNA double-strand breaks (Goodier and Kazazian 2008)? Since retrotransposition appears to be mostly a random process, somatic insertions are private mutations, and a portion of these could account for cancer cases for which causative recurrent mutations have not been identified.
Previously, evidence for L1 insertions in normal somatic cells has come from two sources. A number of studies have shown that somatic L1 insertions occur in neuronal development and are present in various sites in the human and mouse brain (Muotri et al. 2005; Coufal et al. 2009; Baillie et al. 2011; Evrony et al. 2012; Upton et al. 2015). Moreover, a small number of examples of L1, SVA, and processed pseudogene insertions have been reported to occur in early human development (van den Hurk et al. 2007; de Boer et al. 2014; Vogt et al. 2014). Now, we have definitive evidence for somatic L1 retrotransposition in normal colonic and gastric tissues.
Our results suggest the need for a shift of attention to insertion (mutation) timing, as it could be normal-appearing cells that harbor tumor-initiating genetic lesions. It would be extremely valuable if we could identify those cells that appear normal but are already molecularly committed to becoming dysplastic, particularly in patients at risk for nonresectable tumors. However, identification of these apparently normal cells by sequencing normal and tumor tissues will be problematic, since tumor-initiating normal-appearing cells likely become part of the tumor, and even microdissected tumors are contaminated with normal cells, making it difficult to distinguish insertions in tumor vs. contaminating normal cells. Although we did not find normal tissue-specific insertions (Fig. 4A) that were detectable by conventional PCR, such insertions may exist whose detection could depend on the amount of input DNA used for genotyping and the fraction of cells containing the insertion. This possibility raises questions regarding our TGCT insertion and insertions in previous studies using blood as the normal tissue (Lee et al. 2012; Ewing et al. 2013; Cooke et al. 2014; Helman et al. 2014; Tubio et al. 2014). That is to say, these insertions were not verified in the same normal tissue from which the tumor originated, or in other words, the nature of tumor-specificity versus simply normal tissue-specificity of the insertions is not known. Especially using nested PCR, one may misdiagnose some insertions as tumor-specific, when in reality they may be due to contamination from normal tissue. This problem arises because of the exquisite sensitivity of next-generation sequencing and is now detected by improved validation efforts (Supplemental Fig. 2d). However, the problem disappears when one finds somatic L1 insertions in primary cancers and their metastases to another tissue or organ, due to clonality. In future studies, it may be important to include normal samples of the same tissue type in which the tumor is located, use tissue microdissection, and single-cell sequencing. Especially using single-cell analysis, it will be interesting to learn whether somatic retrotransposition is widespread in many human tissues and whether the rate of somatic retrotransposition is increased in cancer (see Goodier 2014).
To summarize, numerous genes and intergenic regions are targeted by hundreds of somatic L1 insertions very early during GI, but not testicular tumorigenesis, indicating the preference of somatic retroelement movement in epithelial tumors. Our data indicate that somatic retrotransposition occurs very early during the development of most GI cancers. We suggest that many somatic insertions discovered in various cancers have their origin in a histologically normal cell and that one or more of the somatic retrotranspositions in that normal cell may lead to its selection for cancer development.
Methods
Human DNA samples
All samples were fresh-frozen. GI tissues were acquired from Johns Hopkins University, Baltimore (IRB# NA_00092914). Non-neoplastic tissues were dissected away from the neoplastic cells of colorectal, pancreatic, and stomach cancer patients to maximize neoplastic cell cellularity. DNA from these tissues was extracted using the AllPrep DNA/RNA Mini kit (Qiagen) after disruption and homogenization with a rotor-stator homogenizer. The TGCT samples with >75% tumor cells and matched peripheral blood were collected at Erasmus MC, The Netherlands. DNA-isolation of TGCTs and blood was done using the AllPrep DNA/RNA Mini kit (Qiagen).
L1-seq library construction, sequencing, and analysis
The sequencing libraries for L1Hs elements were constructed using L1-seq as previously described (Ewing and Kazazian 2010; Solyom et al. 2012). Briefly, this method amplifies the 3′ flanking regions of L1Hs elements using hemi-specific PCR. PCR-amplified L1Hs element insertion site junctions were TOPO-TA cloned (Invitrogen) and Sanger-sequenced for quality control of the library preparation and were subsequently sequenced on an Illumina HiSeq 2500 at the Johns Hopkins University Genetic Resources Core Facility High Throughput Sequencing Center. Sequence results were analyzed as previously described (Solyom et al. 2012). Python scripts used for the mapping and primary analysis are available in the Supplemental Material and at https://github.com/adamewing/l1seq. Overall mapping statistics and sensitivity estimates for each pooled library are presented in Supplemental Table 1. Reference insertions are defined as those with an L1Hs present in hg19/GRCh37 in the proper position (<500 bp from the peak) and orientation for a given cluster of aligned reads. Reference insertion sites are cataloged in Supplemental Table 4a–d. Nonreference germline insertions are defined as those present in every sample of the particular patient and represented by greater than 50 total reads, at least two unique read alignments, and not corresponding to a known L1Hs or L1PA element in the hg19/GRCh37 assembly. Putative nonreference insertions and overlap with previous studies in which nonreference L1 insertions have been cataloged are presented in Supplemental Table 3a–d.
Based on the validation results (see “PCR validation of the Illumina results”), successful and unsuccessful validations (validation failures) were used to predict the number of untested sites likely to validate using a conditional inference tree (Hothorn et al. 2006) as implemented by the “ctree” function in the “party” package available for the R statistical computing environment (R Core Team 2015). Conditional inference trees were generated separately for the colorectal, pancreatic, and stomach cancer cases (Supplemental Fig. 1). Validation status (1/0 for pass/fail) was used as the dependent variable, and the following peak characteristics were used as the independent variables: number of reads (maxcount), number of unique reads (maxuniq), span of alignments (width), mapping quality (mapq). The likelihood of a given insertion validating can be estimated by examining the partitions assigned by the conditional inference tree shown in Supplemental Figure 1 and following the branches of the tree to one of the terminal nodes. In order to obtain a rough estimate of the number of insertions that might validate from the total number of untested insertions in our data set, we applied this logic to all putative insertions with mapscore > 0.17 (average mappability score) and mapping quality of at least 12 (based on Bowtie 2 output mapping scores) (Langmead and Salzberg 2012). The cutoffs for untested pancreatic insertion sites were mapscore > 0.44 and mapping quality > 20.53. The cutoffs for untested gastric insertion sites were mapscore > 0.5 and mapping quality > 10. Cutoffs for each experiment differ, because they are based on the minimum values for mapscore and mapping quality for validated insertions in our data. These minimum values vary between experiments due to differing characteristics across libraries (Supplemental Table 1) and differences in the validation strategy across experiments (Supplemental Table 2f–s). The number of untested insertions falling into each validation bin (i.e., terminal node on the conditional inference tree) was multiplied by the likelihood for that bin, and then multiplied by the tissue-specific validation rate based on the combination of tissue types in which the insertion was detected. It should be noted as a caveat that validation was mostly performed on insertions deemed likely to validate based on peak and sample characteristics (e.g., high number vs. low number of reads, cancer-and-metastasis-specific vs. metastasis-only putative insertions), so extrapolation of smaller peak sizes is less reliable. Data used to generate conditional inference trees and untested sites are available as Supplemental Table 2, and results are shown in Table 1.
PCR validation of the Illumina results
A multistep PCR validation protocol was used to validate L1-seq reads and to retrieve 3′ and 5′ junctions. As the first step, L1 3′ ends together with flanking genomic regions were amplified using the same diagnostic L1Hs-specific “AC” di-nucleotide primer as used for L1-seq (GGGAGATATACCTAATGCTAGATGACAC) and a primer selected from the 3′ flanking region based on the reference genome sequence (3′ primer). PCR reactions were carried out in 12.5 µL 2× GoTaq Green master mix (Promega) in a total volume of 25 µL, with 0.8 µL of 3′ primer, 1.5 µL of L1Hs primer, and 25 ng DNA to amplify the filled site. The empty site (WT allele) was amplified with the same conditions, except that 1.5 µL of 3′ primer, 1.5 µL of 5′ primer (selected from the 5′ flanking region based on the reference genome sequence), and 12.5 ng DNA were used. Primers were 20 pmol/μL and their location is depicted in Figure 2A. Reactions were incubated for 2 min at 95°C, followed by 30 cycles of 30 sec at 95°C, 30 sec at 57°C, and 1.5 min at 72°C, followed by final extension of 5 min at 72°C on a Bio-Rad T100 Thermal Cycler. In some GI cases, nested PCR was used on 1 µL of filled site PCR product using the same L1Hs diagnostic “G” primer that was used for L1-seq (TGCACATGTACCCTAAAACTTAG), together with a 3′ nested primer for another 30 cycles. When no nested primer was available, seminested PCR was performed using the “G” primer together with the original 3′ primer. Long-range PCR to recover longer L1 insertions was performed with the Expand Long Template PCR System (Roche) according to the manufacturer's instructions in buffer 1, with 1 µL of 20 µM 3′ and 5′ primers each, and 25 ng DNA. 5′ junctions were PCR-amplified using the same conditions as for the 3′ junction, except that a primer hybridizing to the L1 5′ UTR was used (L1nt112out: GATGAACCCGGTACCTCAGA) together with the respective 5′ primer, and primer extension time was only 45 sec, or the “GTG” primer (reverse complement of the L1Hs-specific “AC” primer) was used with the 5′ primer using conventional or long-range PCR conditions. 3′ and 5′ primer sequences are included in Supplemental Table 2. PCR products were cut out of the gel, extracted with a QIAquick Gel Extraction kit (Qiagen) and Sanger-sequenced. See Supplemental Data 1–4 for Sanger sequence data. Insertions in pancreatic and stomach cancer cases were either PCR-validated from gDNA or from genome-amplified DNA that was produced using the REPLI-g Mini kit (Qiagen) with Multiple Displacement Amplification (MDA). Results on gDNA and MDA-amplified DNA were identical.
Human SNP Array 6.0 experiments
The following genomic DNAs from the four colorectal cancer patients were analyzed: 1BV “1” and “3/1”; 2BV “20” (normal liver), “5”, “6/1”, and “7”; 3BV “15” (another section of normal colon) and “10/1”; 4BV “11”, “12”, “13”, and “14”. Patient codes are explained in Supplemental Table 2, except for samples “20” and “15”, when a new sample was used, and their source is specified in parentheses. These DNAs were analyzed for concentration and quality with the Agilent 2100 Bioanalyzer (Agilent Technologies). The samples then underwent digestion, amplification, and hybridization to an Affymetrix Human SNP 6.0 Array, comprising over 1.8 million markers, as per the manufacturer's protocol. In short, DNA aliquots were digested with StyI Digestion Master mix, ligated to Sty primers and PCR-amplified. The process was repeated with NspI restriction enzyme, and the products were pooled and bead-purified. After quantification, the samples were fragmented and labeled with biotin. Samples were hybridized to the SNP arrays at 50°C for 16 h at 60 rpm, after which the arrays were transferred to the Affymetrix GeneChip Fluidics Station 450 for antibody staining and washing. The washed arrays were scanned with the Affymetrix GeneChip Scanner 3000 7G as per protocol, and data in the form of CEL and CHP files were extracted with the Affymetrix Genotyping Console (www.affymetrix.com). These raw data were imported for copy number analysis into the Partek Genomics Suite v6.6 platform (Partek Inc.). Partek's standard protocol was used to estimate copy number changes, for each of 1.8 million markers, for each patient's polyp, primary, and/or metastasis cancer samples as compared to that patient's control sample. Partek's Genomic Segmentation analysis was used to identify regions of copy number variation by looking for blocks of 20 or more genomic markers showing P-value thresholds of <0.001, minimal signal to noise ratios of 0.3, and a range of diploid copy number cut-off thresholds. For the macroscale CNV analysis, Partek was used to generate ideograms indicating CNV status, increase or decrease, at the various thresholds. Individual SNP genotypes were determined from the Affymetrix CHP files exported by their Genotyping Console using the Birdseed algorithm (Korn et al. 2008).
Quantitative proteomic analysis
For proteomic analysis, we processed a new section of sample “10” (tumor, highest number of somatic L1 insertions) and matched sample “15” (normal colon, no somatic L1 insertions detected). Although, the original sample “10” was first classified as a cancer, and subsequently as an adenoma with high-grade dysplasia, the new section used for proteomics was adenoma with low-grade dysplasia. Frozen OCT tissues were first cryo-pulverized in the presence of liquid nitrogen. Powderized tissue samples were then homogenized to extract proteins using a lysis buffer (4% SDS, 100 mM Tris, pH 7.6). Crude protein extracts were further sonicated before centrifugation at 2500g for 10 min at room temperature. Supernatant protein lysates were then transferred to a 1.5-mL tube for another centrifugation at 12,000g for 10 min. Cleared protein lysates were used for proteomic analysis. As described previously (Wisniewski et al. 2009; Kim et al. 2014), a total of 250 μg proteins were transferred to a 10-kDa filter unit, centrifuged at 12,000g for 10 min, reduced using 10 mM dithiothreitol at 60°C for 30 min, centrifuged to exchange to a urea buffer (8 M urea, 20 mM HEPES, pH 8.0), and alkylated with 10 mM iodoacetamide for 30 min in the dark. After centrifugation, urea concentration was diluted to <2 M, and proteins were digested with trypsin at an enzyme-to-protein ratio of 1:20 for 16 h at room temperature. Peptides were collected in filtrates by centrifugation at 12,000g, desalted using a Sep-Pak C18 cartridge, vacuum-dried, and labeled with TMT reagents (126, 127, 128, and 129) following the manufacturer's instructions. Four labeling reactions were carried out, including a replicate. Labeled peptides were mixed and fractionated using reverse phase liquid chromatography at pH 10 into 24 fractions. Each fraction was analyzed separately on a high-resolution Orbitrap Fusion mass spectrometer (Thermo) online connected with a high-pressure EASY-nLC 1000 liquid chromatography system (Thermo). Peptides were loaded onto a trap column and separated in a 250-nL/min nanoflow rate with a linear gradient of acetonitrile (7% to 35%). Precursor ions selected were fragmented by a higher energy C-trap dissociation method, and MS3 scans were sequentially acquired by utilizing synchronous precursor selection technology (Ting et al. 2011). Raw mass spectrometry data were analyzed on a Proteome Discoverer platform (version 1.4) using the Sequest database searching algorithm with the following parameters: up to 2 missed cleavages allowed, full trypsin digestion only considered, N-termini and lysine fully labeled with TMT reagent, oxidation at methionine allowed, peptide tolerance within 7 ppm, and fragment tolerance within 0.05 Da. Peptide identification was considered by applying peptide-spectrum matches with <1% false discovery rates. Quantification values were calculated by Proteome Discoverer.
Data access
The L1-seq data from this study have been submitted to the NCBI database of Genotypes and Phenotypes (dbGaP; http://www.ncbi.nlm.nih.gov/gap/) under study accession number phs000536.v3.p1. The SNP array data have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE63601. The proteomics data have been submitted to the PRoteomics IDEntifications database (PRIDE; http://www.ebi.ac.uk/pride/archive/) under accession number PXD001626.
Acknowledgments
We thank Alan F. Scott and David Mohr at the Genetic Resources Core Facility for the next-generation sequencing services, the Johns Hopkins Deep Sequencing and Microarray Core Facility and C. Conover Talbot, Jr. for microarray analysis, and the Synthesis and Sequencing Facility at Johns Hopkins University for Sanger sequencing. We also thank Kathleen H. Burns, Jared Steranka, and Nemanja Rodic for sharing TIP-seq data with us. We thank John L. Goodier for his great insights into the project and comments on the manuscript and Nusaiba Baker for excellent technical assistance. Research in the Kazazian laboratory is funded by grants from the National Institutes of Health (1R01GM099875 awarded to H.H.K., and 1P50GM107632 awarded to Jef D. Boeke), The Sol Goldman Pancreatic Cancer Research Foundation, and the Johns Hopkins GI Core Center. S.S. is supported by the 2013 AACR Basic Cancer Research Fellowship, Grant Number 13-40-01-SOLY. A.D.E. is supported by the Mater Foundation, R.H.H. by National Institutes of Health grant P50 CA62924, A.M.-M. by NIH grant F31CA180682, C.A.I.-D. by NIH grants CA140599 and CA179991, B.V. and K.W.K. by NIH grants RO1-CA43460, RO1-CA57345, The Virginia and D.K. Ludwig Fund for Cancer Research, Lustgarten Foundation for Pancreatic Cancer Research, and The Sol Goldman Center for Pancreatic Cancer Research.
Author contributions: H.H.K. and S.S. designed the study, and S.S. wrote the manuscript. A.D.E. performed bioinformatic and statistical analysis of L1-seq data. S.S., A.G., F.M., G.A., and G.P. PCR-validated the L1-seq results. K.W.K., B.V., and K.E.R. provided the samples of the colorectal cancer patients, C.A.I.-D. and A.M.-M. provided the pancreatic cancer patient samples, K.E.R. provided the samples of the gastric cancer patients, and L.H.J.L. and A.J.M.G. provided DNA of testicular germ cell tumor patients. L.D.W. and D.X. with R.H.H. dissected the samples of the GI cancer patients. R.A.A. evaluated H&E-stained gastric samples for histology. S.S. isolated DNA from the GI cancer cases. M.-S.K. and A.P. performed proteomic experiments, and S.S.M. and A.P. analyzed the proteomics data using bioinformatics.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.196238.115.
References
- Baillie JK, Barnett MW, Upton KR, Gerhardt DJ, Richmond TA, De Sapio F, Brennan PM, Rizzu P, Smith S, Fell M, et al. 2011. Somatic retrotransposition alters the genetic landscape of the human brain. Nature 479: 534–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke SL, Shlien A, Marshall J, Pipinikas CP, Martincorena I, Tubio JM, Li Y, Menzies A, Mudie L, Ramakrishna M, et al. 2014. Processed pseudogenes acquired somatically during cancer development. Nat Commun 5: 3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corvaro M, Fuoco C, Wagner M, Cecconi F. 2007. Analysis of apoptosome dysregulation in pancreatic cancer and of its role in chemoresistance. Cancer Biol Ther 6: 209–217. [DOI] [PubMed] [Google Scholar]
- Coufal NG, Garcia-Perez JL, Peng GE, Yeo GW, Mu Y, Lovci MT, Morell M, O'Shea KS, Moran JV, Gage FH. 2009. L1 retrotransposition in human neural progenitor cells. Nature 460: 1127–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer M, van Leeuwen K, Geissler J, Weemaes CM, van den Berg TK, Kuijpers TW, Warris A, Roos D. 2014. Primary immunodeficiency caused by an exonized retroposed gene copy inserted in the CYBB gene. Hum Mutat 35: 486–496. [DOI] [PubMed] [Google Scholar]
- Evrony GD, Cai X, Lee E, Hills LB, Elhosary PC, Lehmann HS, Parker JJ, Atabay KD, Gilmore EC, Poduri A, et al. 2012. Single-neuron sequencing analysis of L1 retrotransposition and somatic mutation in the human brain. Cell 151: 483–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing AD, Kazazian HH Jr. 2010. High-throughput sequencing reveals extensive variation in human-specific L1 content in individual human genomes. Genome Res 20: 1262–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing AD, Ballinger TJ, Earl D; Broad Institute Genome Sequencing and Analysis Program and Platform, Harris CC, Ding L, Wilson RK, Haussler D. 2013. Retrotransposition of gene transcripts leads to structural variation in mammalian genomes. Genome Biol 14: R22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodier JL. 2014. Retrotransposition in tumors and brains. Mob DNA 5: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodier JL, Kazazian HH Jr. 2008. Retrotransposons revisited: the restraint and rehabilitation of parasites. Cell 135: 23–35. [DOI] [PubMed] [Google Scholar]
- Hancks DC, Kazazian HH Jr. 2012. Active human retrotransposons: variation and disease. Curr Opin Genet Dev 22: 191–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helman E, Lawrence MS, Stewart C, Sougnez C, Getz G, Meyerson M. 2014. Somatic retrotransposition in human cancer revealed by whole-genome and exome sequencing. Genome Res 24: 1053–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hothorn T, Hornik K, Zeileis A. 2006. Unbiased recursive partitioning: a conditional inference framework. J Comput Graph Stat 15: 651–674. [Google Scholar]
- Huang CR, Schneider AM, Lu Y, Niranjan T, Shen P, Robinson MA, Steranka JP, Valle D, Civin CI, Wang T, et al. 2010. Mobile interspersed repeats are major structural variants in the human genome. Cell 141: 1171–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iskow RC, McCabe MT, Mills RE, Torene S, Pittard WS, Neuwald AF, Van Meir EG, Vertino PM, Devine SE. 2010. Natural mutagenesis of human genomes by endogenous retrotransposons. Cell 141: 1253–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R, Madugundu AK, Kelkar DS, Isserlin R, Jain S, et al. 2014. A draft map of the human proteome. Nature 509: 575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn JM, Kuruvilla FG, McCarroll SA, Wysoker A, Nemesh J, Cawley S, Hubbell E, Veitch J, Collins PJ, Darvishi K, et al. 2008. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat Genet 40: 1253–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg S. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E, Iskow R, Yang L, Gokcumen O, Haseley P, Luquette LJ III, Lohr JG, Harris CC, Ding L, Wilson RK, et al. 2012. Landscape of somatic retrotransposition in human cancers. Science 337: 967–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki Y, Nishisho I, Horii A, Miyoshi Y, Utsunomiya J, Kinzler KW, Vogelstein B, Nakamura Y. 1992. Disruption of the APC gene by a retrotransposal insertion of L1 sequence in a colon cancer. Cancer Res 52: 643–645. [PubMed] [Google Scholar]
- Muotri AR, Chu VT, Marchetto MC, Deng W, Moran JV, Gage FH. 2005. Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature 435: 903–910. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Migita T, Hosoda F, Okada N, Gotoh M, Arai Y, Fukushima M, Ohki M, Miyata S, Takeuchi K, et al. 2009. Kruppel-like factor 12 plays a significant role in poorly differentiated gastric cancer progression. Int J Cancer 125: 1859–1867. [DOI] [PubMed] [Google Scholar]
- Paterson AL, Weaver JM, Eldridge MD, Tavare S, Fitzgerald RC, Edwards PA; OCCAMs Consortium. 2015. Mobile element insertions are frequent in oesophageal adenocarcinomas and can mislead paired-end sequencing analysis. BMC Genomics 16: 473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitkanen E, Cajuso T, Katainen R, Kaasinen E, Valimaki N, Palin K, Taipale J, Aaltonen LA, Kilpivaara O. 2014. Frequent L1 retrotranspositions originating from TTC28 in colorectal cancer. Oncotarget 5: 853–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2015. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org/. [Google Scholar]
- Rodić N, Steranka JP, Makohon-Moore A, Moyer A, Shen P, Sharma R, Kohutek ZA, Huang CR, Ahn D, Mita P, et al. 2015. Retrotransposon insertions in the clonal evolution of pancreatic ductal adenocarcinoma. Nat Med 10.1038/nm.3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla R, Upton KR, Munoz-Lopez M, Gerhardt DJ, Fisher ME, Nguyen T, Brennan PM, Baillie JK, Collino A, Ghisletti S, et al. 2013. Endogenous retrotransposition activates oncogenic pathways in hepatocellular carcinoma. Cell 153: 101–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solyom S, Ewing AD, Rahrmann EP, Doucet T, Nelson HH, Burns MB, Harris RS, Sigmon DF, Casella A, Erlanger B, et al. 2012. Extensive somatic L1 retrotransposition in colorectal tumors. Genome Res 22: 2328–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting L, Rad R, Gygi SP, Haas W. 2011. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat Methods 8: 937–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasetti C, Vogelstein B, Parmigiani G. 2013. Half or more of the somatic mutations in cancers of self-renewing tissues originate prior to tumor initiation. Proc Natl Acad Sci 110: 1999–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubio JM, Li Y, Ju YS, Martincorena I, Cooke SL, Tojo M, Gundem G, Pipinikas CP, Zamora J, Raine K, et al. 2014. Mobile DNA in cancer. Extensive transduction of nonrepetitive DNA mediated by L1 retrotransposition in cancer genomes. Science 345: 1251343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton KR, Gerhardt DJ, Jesuadian JS, Richardson SR, Sanchez-Luque FJ, Bodea GO, Ewing AD, Salvador-Palomeque C, van der Knaap MS, Brennan PM, et al. 2015. Ubiquitous L1 mosaicism in hippocampal neurons. Cell 161: 228–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Hurk JA, Meij IC, Seleme MC, Kano H, Nikopoulos K, Hoefsloot LH, Sistermans EA, de Wijs IJ, Mukhopadhyay A, Plomp AS, et al. 2007. L1 retrotransposition can occur early in human embryonic development. Hum Mol Genet 16: 1587–1592. [DOI] [PubMed] [Google Scholar]
- Vogt J, Bengesser K, Claes KB, Wimmer K, Mautner VF, van Minkelen R, Legius E, Brems H, Upadhyaya M, Hogel J, et al. 2014. SVA retrotransposon insertion-associated deletion represents a novel mutational mechanism underlying large genomic copy number changes with non-recurrent breakpoints. Genome Biol 15: R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski JR, Zougman A, Nagaraj N, Mann M. 2009. Universal sample preparation method for proteome analysis. Nat Methods 6: 359–362. [DOI] [PubMed] [Google Scholar]
- Zeng Q, Cheng Y, Zhu Q, Yu Z, Wu X, Huang K, Zhou M, Han S, Zhang Q. 2008. The relationship between overexpression of glial cell-derived neurotrophic factor and its RET receptor with progression and prognosis of human pancreatic cancer. J Int Med Res 36: 656–664. [DOI] [PubMed] [Google Scholar]