

Abstract
Chromatin immunoprecipitation followed by next-generation DNA sequencing (ChIP-seq) is a widely used technique for identifying transcription factor (TF) binding events throughout an entire genome. However, ChIP-seq is limited by the availability of suitable ChIP-seq grade antibodies, and the vast majority of commercially available antibodies fail to generate usable data sets. To ameliorate these technical obstacles, we present a robust methodological approach for performing ChIP-seq through epitope tagging of endogenous TFs. We used clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9-based genome editing technology to develop CRISPR epitope tagging ChIP-seq (CETCh-seq) of DNA-binding proteins. We assessed the feasibility of CETCh-seq by tagging several DNA-binding proteins spanning a wide range of endogenous expression levels in the hepatocellular carcinoma cell line HepG2. Our data exhibit strong correlations between both replicate types as well as with standard ChIP-seq approaches that use TF antibodies. Notably, we also observed minimal changes to the cellular transcriptome and to the expression of the tagged TF. To examine the robustness of our technique, we further performed CETCh-seq in the breast adenocarcinoma cell line MCF7 as well as mouse embryonic stem cells and observed similarly high correlations. Collectively, these data highlight the applicability of CETCh-seq to accurately define the genome-wide binding profiles of DNA-binding proteins, allowing for a straightforward methodology to potentially assay the complete repertoire of TFs, including the large fraction for which ChIP-quality antibodies are not available.
Chromatin immunoprecipitation followed by next-generation DNA sequencing (ChIP-seq) is one of the most widely used and powerful methods for mapping regulatory elements and analyzing transcription factor (TF) function (The ENCODE Project Consortium 2007, 2012; Johnson et al. 2007). However, the measurement of genome-wide TF binding requires high-quality, validated antibodies that do not cross-react with other DNA-binding proteins for each transcription factor and that work in the ChIP assay (Landt et al. 2012). Notably, estimates from thousands of tests indicate that fewer than 10% of tested antibodies are suitable for ChIP-seq analyses (our unpublished observations and from additional ENCODE [Encyclopedia of DNA Elements] Consortium data). The addition of epitope tags on TFs of interest and the subsequent use of ChIP-seq grade epitope tag antibodies is a method for potentially circumventing this obstacle, because a single high-quality antibody can be used for all experiments.
The adaptation of the clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 system for genome editing in mammalian systems allows for the direct manipulation of endogenous genomic sequences in a simple and multiplexed manner (Cong et al. 2013; Jinek et al. 2013; Mali et al. 2013; Doench et al. 2014). CRISPR technology has been applied for a variety of genetic manipulations, including gene disruptions through nonhomologous end joining (Cong et al. 2013; Mali et al. 2013), homologous recombination (Wang et al. 2013; Yang et al. 2013), and modulation of gene regulation (Maeder et al. 2013; Perez-Pinera et al. 2013). Here we provide an additional approach that adapts CRISPR genome editing for epitope tagging of endogenous DNA-binding proteins for ChIP-seq experimentation.
Distinct tagging approaches have been developed, but these methods lack key features required for generating accurate DNA-binding interactomes. For instance, although TF-tagged transgene constructs have been used (Mazzoni et al. 2011; Najafabadi et al. 2015), this strategy can lead to artificial expression patterns as the TF is typically under the control of a nonnative promoter in nonnative endogenous sequence context. To circumvent some of these concerns, bacterial artificial chromosome (BAC) recombineering (Zhang et al. 1998, 2000) has also been performed to place epitope tags at the 3′ end of genes in BAC clone constructs harboring a TF gene (Poser et al. 2008; Kittler et al. 2013). This approach has also been utilized in mouse models (Zhou et al. 2004), and subsequent studies have performed ChIP assays using antibodies for these epitope tags (Pilon et al. 2011). However, there are also several notable limitations with this BAC-mediated approach. Despite covering hundreds of kilobases of sequence, only BACs spanning an entire TF gene locus can be used, which may further preclude large TF genes for tagging. Additionally, BACs may not harbor all promoter-distal regulatory elements required for proper TF gene expression. Indeed, some regulatory elements are located >1-megabase away from their corresponding target gene (Lettice et al. 2003). Moreover, highly efficient transfection and integration of an intact BAC construct into mammalian cells can present technical hurdles (Montigny et al. 2003), while the additional presence of sequence variants between exogenous BAC sequences and the synonymous endogenous locus in cells may add confounding biological effects on TF expression and/or function.
Here we provide a simple and direct approach for performing ChIP-seq using endogenous TF proteins that have been epitope tagged. Our strategy capitalizes on the recent advances of CRISPR/Cas9 genome engineering technology. We demonstrate that our method is simple, specific, and robust, requires minimal manipulation, and can be further applied to a variety of DNA-binding proteins across distinct cell types.
Results
Overview of CETCh-seq method
We took advantage of CRISPR/Cas nuclease activity to direct double-strand DNA breaks at the 3′ end of endogenous TF loci, followed by the integration of a Flag epitope that can be utilized in downstream ChIP-seq assays. We call our method CRISPR epitope tagging ChIP-seq or CETCh-seq. We engineered a Flag epitope tag ChIP (pFETCh) plasmid donor construct containing three Flag epitope sequences, followed by a self-cleaving 2A peptide sequence (P2A) and neomycin resistance gene (Fig. 1A). We further flanked this construct with homology arm sequences upstream of and downstream from an endogenous TF stop codon. During homologous recombination, this experimental design will lead to the excision of the endogenous DNA-binding protein stop codon and the concomitant integration of the 3×Flag-P2A-neomycin in frame with the endogenous protein coding sequence (Fig. 1A). CRISPR guide RNAs (gRNAs) are designed to direct Cas9 nuclease activity near stop codons of endogenous DNA-binding proteins, and cotransfection of a plasmid expressing both gRNA and Cas9 with the pFETCh donor plasmid leads to the efficient recombination of the donor construct. The use of a P2A self-cleaving linker sequence within integrated, in-frame donor constructs results in the cotranscription of Flag-tagged DNA-binding protein with the neomycin resistance gene and the subsequent translation of two unique proteins through amino acid peptide cleavage and ribosomal skipping at the P2A sequence (Fig. 1A; Szymczak et al. 2004; Kim et al. 2011). Transfected cells are grown under neomycin selection using G418 and expanded as a stable polyclonal cell population for subsequent experimentation (Fig. 1A). Examples of our CETCh-seq data sets and comparisons to standard ChIP-seq approaches that utilize antibodies directly targeting DNA-binding proteins are given in Figure 1B.
Figure 1.
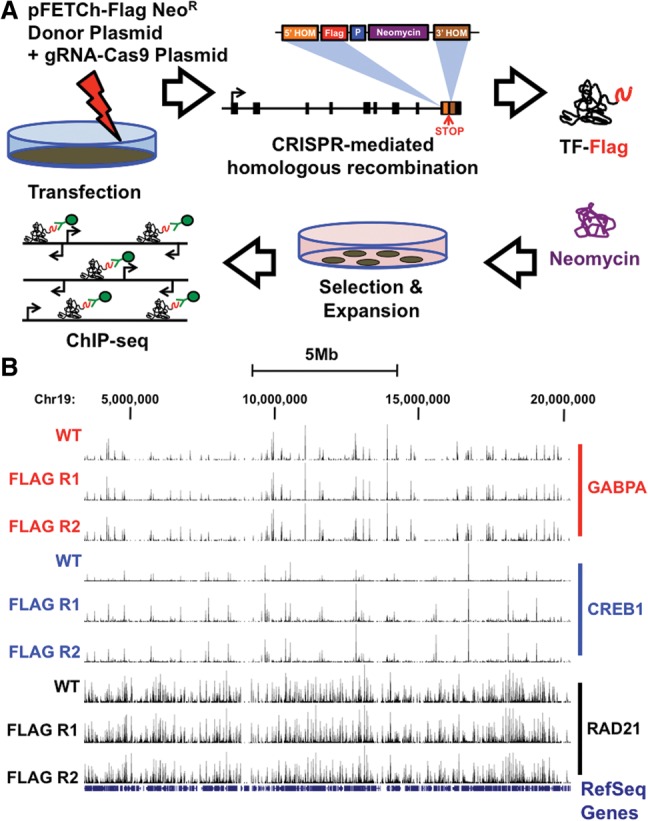
Overview of CETCh-seq experimental method. (A) A schematic of the CETCh-seq approach is displayed. Cells are transfected with plasmids containing the Cas9 nuclease, gRNAs, and epitope tag donor constructs, leading to the homologous integration of the Flag tag, P2A linker sequence, and neomycin resistance gene at the 3′ end of the transcription factor in place of the endogenous transcription factor stop codon. Flag-tagged transcription factor and neomycin resistance genes are cotranscribed. Subsequently, a tagged transcription factor and a neomycin resistance protein are generated due to the P2A linker sequence. Cells are selected and colony-forming units are expanded for ChIP-seq experimentation using a Flag antibody. (B) HepG2 DNA-binding protein read enrichment tracks on the UCSC Genome Browser are given. The names of transcription factors are given. WT denotes transcription factor antibody experiments, while Flag Rep 1 and Flag Rep 2 are technical replicates using Flag antibodies for CETCh-seq experiments.
CETCh-seq genome editing analyses in HepG2 cells
We tested our CETCh-seq method by designing our donor constructs with homology arms targeting sequences flanking stop codons of five DNA-binding proteins (RAD21, CREB1, ATF1, NR1H2, and GABPA) in HepG2 hepatocellular carcinoma cell lines. We chose these DNA-binding proteins carefully as they span a wide range of expression levels in HepG2 cell lines (RAD21 = 40.09, CREB1 = 12.84, NR1H2 = 7.59, ATF1 = 5.99, and GABPA = 2.42, denoted in reads per kilobase per million mapped reads [RPKM]) (Supplemental Table 1). Due to a lack of ATF1 interactome information in HepG2 cells and an inability to identify a ChIP-seq grade ATF1 antibody, we used the ATF1 transcription factor to further evaluate the efficacy of CETCh-seq for analyzing DNA-binding proteins that are intractable to standard ChIP-seq approaches. We utilized ∼925-bp homology arms (both 5′ and 3′), on average, for CRISPR-mediated homologous recombination (580- to 1618-bp range in arms) (Supplemental Tables 2, 3). We attempted to design two gRNAs to target the Cas9 near the endogenous stop codons (cut site ±15 bp from the pre-stop codon) of these distinct DNA-binding protein genes. To assess gRNA design, we utilized both off target and efficiency scores (Hsu et al. 2013; Doench et al. 2014) and preferred to target the 3′ untranslated region as opposed to the open reading frame to avoid creation of insertions and deletions in untagged alleles. For RAD21 and ATF1, we could only identify one suitable gRNA, while for CREB1, NR1H2, and GABPA, we engineered two unique gRNAs near stop codons that we cotransfected together with the homology donor plasmid. A complete list of the gRNA and homology arm sequences is given in Supplemental Table 2.
We expanded transfected cell populations and maintained cells under neomycin (G418) selection during cell culture experimentation. In order to indirectly assess homologous recombination efficiency, we estimated the number of independent colony-forming units (CFUs). Notably, we identified a wide range of CFUs (RAD21 ≥ 100, CREB1 ≤ 25, NR1H2 = 2, ATF1 ≥ 50, and GABPA ≤ 10). Apart from homologous recombination, these data may further reflect different efficiencies for distinct gRNAs (Doench et al. 2014). However, the number of CFUs did not correlate well with gRNA targeting efficiencies (see Supplemental Table 2 for on-target scores), supporting the notion that these data may reflect different rates of homologous recombination and/or that our current predictive understanding of Cas9 genome editing still remains rudimentary.
We performed PCR assays to validate the proper insertion of the Flag donor cassette (Fig. 2A; see Supplemental Fig. 1 for gel images; see Supplemental Table 3 for primer sequences). This initial validation supported the proper integration of constructs at the 5′ and 3′ ends of four out of the five TFs (Supplemental Fig. 1); PCR from the Flag-tagging NR1H2 experiment failed to amplify. As a secondary confirmation of proper homologous recombination, we performed Western blot and immunoprecipitation (IP) Western blot experiments (Fig. 2B; Supplemental Fig. 2). By utilizing a Flag antibody for these Western blot experiments and by comparing these data with experiments utilizing antibodies targeting the DNA-binding protein directly (for Western blots) or using a mock IP, these results would further determine if our selected HepG2 cells are expressing Flag-tagged DNA-binding proteins. In support of PCR data, we identified protein bands at the predicted sizes for all tested DNA-binding proteins that passed PCR validation, pointing to the presence of a properly tagged TF protein in our cell populations.
Figure 2.

HepG2 ChIP-seq reproducibility and gene expression analysis. (A) PCR validation of ATF1 homologous recombination. From left to right, the gel image displays ladder, 5′ end, and 3′ end homology PCR products. Ladder band sizes are given at the left. Correct homologous recombination generates 1626-bp (5′ end) and 1433-bp (3′ end) gel bands. (B) IP Western blot validation of epitope-tagged ATF1. An ∼40-kDa protein band (marked by red arrow) corresponding to the predicted size of ATF1 is visible in IP Western blots using a Flag antibody but is absent in a control IgG pulldown. (C) DNA-binding protein read enrichment tracks on the UCSC Genome Browser are shown at distinct genetic loci. Data are given for both CETCh-seq (Tag, lower panels) and standard ChIP-seq using transcription factor antibodies (Ctrl, upper panels). The transcription factor name is displayed at the left of each image. For ATF1, technical CETCh-seq replicates are displayed (Rep1 and Rep2). (D) RAD21 rank correlations of normalized sequence read counts between CRISPR-modified HepG2 cells (Modified cells) and wild-type HepG2 cells (WT), both using a RAD21 antibody (top). (Bottom) RAD21 rank correlations of normalized sequence read counts between CRISPR-modified HepG2 cells using a RAD21 TF antibody (Modified cells) and CETCh-seq results of tagged RAD21 using Flag antibodies (Flag). Average rank correlations for all Flag and RAD21 replicate pairwise comparisons are given in the top left corner of each plot. (E) RNA-seq gene RPKM comparisons between CRISPR-modified HepG2 cells (Modified cells RPKM) and wild-type HepG2 cells (WT RPKM) are plotted for RAD21 (top) and ATF1 (bottom) experiments. Rank correlations and the location of tagged transcription factor RPKM values on each graph are displayed.
We next Sanger-sequenced cloned sequences spanning the 5′ and 3′ homologous recombination sites for all DNA-binding proteins and identified proper integration events (Supplemental Table 4). We further sequenced PCR amplicons spanning the 3′ homologous recombination sites for RAD21, CREB1, ATF1, and GABPA Flag-tagged HepG2 cell populations (Supplemental Figs. 3–6). Notably, these amplicons identified accurate homologous recombination events for all DNA-binding proteins. However, we also identified some noise in the electropherogram traces for a subset of DNA-binding proteins, suggesting a subset of alleles may harbor small insertions or deletions. To further determine the background genetic alterations, namely, nonhomologous end joining (NHEJ) disruptions, which in theory could disrupt the coding sequence and potentially the function of the untagged TFs, we deep sequenced the gRNA target loci. These loci would only represent the alleles with Cas9 genome editing at our integration site at sequences that failed homologous recombination and were untagged. We performed sequencing on PCR fragments that spanned both 5′ and 3′ integration sites after selection on a MiSeq next-generation machine. Our data show a NHEJ rate of ∼10%–15% at nonrecombined loci (Supplemental Fig. 7), suggesting that in the selected cell population, most alleles underwent proper homologous recombination or were unmodified. Additionally, we did not detect any NHEJ events at the target sites for NR1H2, indicating that ineffective gRNAs were likely the cause of few CFUs and the inability to confirm integration by PCR or Western blot validation.
CETCh-seq analyses of DNA-binding protein interactomes
We next analyzed our CETCh-seq DNA-binding interactome results. CETCh-seq data generated high-quality results, with strong quality, normalized strand coefficient (NSC) and relative strand correlation (RSC) scores (Landt et al. 2012) in all experiments (Supplemental Table 5). To be thorough in our characterization, we also assessed DNA-binding profiles for NR1H2, a TF that failed PCR and protein validation. Supporting our validation screen, we did not identify binding events using a Flag antibody for NR1H2-targeted cells. These data highlight the specificity of the Flag antibody and our overall Flag epitope tagging approach. Moreover, our results suggest that future studies can utilize a simple PCR validation to screen cells prior to more extensive downstream experimentation (Western blot and ChIP-seq).
We further compared our CETCh-seq data with standard ChIP-seq data from unmodified (wild-type) HepG2 cells that utilized antibodies targeting each DNA-binding protein (Fig. 2C; Supplemental Table 5; Supplemental Figs. 8–12). As a qualitative metric strategy, we assessed the overlap between binding sites identified with DNA-binding protein antibodies from standard ChIP-seq assays and Flag antibodies from CETCh-seq. CETCh-seq data sets were highly coincident with DNA-binding protein antibody-based data sets, with 85% of binding sites, on average, being shared between assays (range 74%–94%) (Supplemental Table 5). Importantly, CETCh-seq binding events were enriched for the identical canonical motif for each DNA-binding protein as was identified through standard ChIP-seq assays (Supplemental Fig. 8). For ATF1, a TF that lacked data in HepG2 cells, CETCh-seq identified the identical motif as has an independent research group (Guo et al. 1997).
As a quantitative metric of binding, we further calculated normalized sequencing read counts at the complete set of sites obtained from CETCh-seq and standard ChIP-seq data and generated Spearman rank correlations (Fig. 2C; Supplemental Fig. 9). All CETCh-seq data sets were highly concordant with standard ChIP-seq data (RAD21 rho = 0.80, CREB1 rho = 0.86 and GABPA rho = 0.90) and notably, the extent of these correlations was further comparable to similar analyses between traditional ChIP-seq replicates that targeted the DNA-binding protein directly in wild-type HepG2 cells (RAD21 rho = 0.84, CREB1 rho = 0.93 and GABPA rho = 0.94) (Supplemental Fig. 9). A summary of the number of binding sites and the extent of overlap is given in Supplemental Table 5.
To assess CETCh-seq reproducibility, we also performed technical replicates for all DNA-binding proteins tested (Supplemental Fig. 10). A summary of these replicate CETCh-seq data is tabulated in Supplemental Table 5. Notably, the identical canonical binding motif was highly enriched across replicate CETCh-seq experiments for the same DNA-binding protein. We next determined the extent of binding site concordance between replicate CETCh-seq experiments through qualitative and quantitative approaches. Supporting the specificity of our assay, CETCh-seq technical replicates exhibited high reproducibility; on average, 91% of sites were shared between technical replicates (Supplemental Table 5) and strong rank correlations were also observed (rho = 0.98–0.92) (Supplemental Fig. 10). We also confirmed a high concordance of these replicate CETCh-seq experiments with standard ChIP-seq assays for applicable DNA-binding proteins (RAD21, CREB1, and GABPA) harboring these additional independent ChIP-seq data sets (Supplemental Table 5). These data support the reproducibility of the CETCh-seq assay, including for TFs (ATF1) that lack a suitable ChIP-seq-grade antibody.
We next generated a biological replicate for Flag-tagged CREB1 to evaluate CETCh-seq performance across distinct cell culture experiments (Supplemental Fig. 11). This biological replicate was generated from an independent transfection experiment and subsequent HepG2 cell selection. Similar to the technical replicates for this tagged TF, the second biological replicate generated was concordant with both the first biological CETCh-seq replicate (83% sites shared, rho = 0.89) (Supplemental Fig. 11; Supplemental Table 5) as well as with ChIP-seq data that used an antibody targeting CREB1 directly (84% sites shared, rho = 0.9) (Supplemental Fig. 11; Supplemental Table 5).
Finally, we endeavored to determine whether technical artifacts are present in our CETCh-seq assay. We performed Flag ChIP-seq experiments in wild-type HepG2 cells to identify potential Flag antibody signatures that correspond to false-positive binding events. Supporting the high specificity of the Flag antibody and our overall assay, no binding sites were identified. These data are further supported by our inability to detect binding events in our NR1H2 CETCh-seq experiment mentioned above. We also performed ChIP-seq using antibodies targeting our DNA-binding proteins in CRISPR-modified cell lines. In line with minimal confounding effects of CRISPR genome editing on DNA-binding integrity across our population of selected cells, our standard ChIP-seq results in CRISPR-modified HepG2 cells were highly comparable with ChIP-seq data in wild-type HepG2 cells (rho = 0.86 on average) (Supplemental Fig. 12). Notably, our ChIP-seq binding sites in CRISPR-modified HepG2 cells were also highly coincident with CETCh-seq binding events (rho = 0.91) (Fig. 2D; Supplemental Fig. 12).
Transcriptome analysis in CRISPR-modified cells
For our CETCh-seq experiments, we selected for and used polyclonal cell populations to restrict the effect of any off-target effects to a minority of cells, as these effects have been documented (Hsu et al. 2013; Sander and Joung 2014) for CRISPR genome editing. To evaluate how well these CRISPR-modified polyclonal cell populations represent the unmodified wild-type HepG2 cell line, we performed massively parallel sequencing on mRNA (RNA-seq) to identify alterations to the HepG2 transcriptome. We performed RNA-seq on wild-type HepG2 cells and CRISPR-modified HepG2 cell lines for RAD21 and ATF1 Flag-tagged TFs (Fig. 2E). We calculated gene RPKM and correlated between wild-type and modified HepG2 cells. These RNA-seq analyses exhibit strong rank correlation (rho = 0.97 for RAD21 and rho = 0.98 for ATF1), suggesting that our population of CRISPR-modified cells does not harbor large off-target effects that impact the global transcriptome. We further assessed the expression levels of tagged TFs in each RNA-seq experiment. Notably, RAD21 and ATF1 Flag-tagged TFs were not outliers on the plot, highlighting a lack of pronounced TF dysregulation from the homologous recombination of the Flag-P2A-neomycin donor construct or from neomycin selection. To further validate minimal changes to the cellular transcriptome, we determined the extent of differentially regulated genes in CRISPR-modified HepG2 cells and wild-type HepG2 cells (Anders and Huber 2010). We compared RNA-seq data obtained from RAD21 and ATF1 epitope-tagged HepG2 cells with data from wild-type HepG2 cells. We also generated an additional wild-type HepG2 RNA-seq biological replicate experiment to assess differences between wild-type HepG2 cells. Our data shows a paucity of significant P-values for all comparisons compared to expectations (Supplemental Fig. 13). Importantly, even without FDR correction, ATF1 and RAD21 were not identified as significantly differentially regulated (nonadjusted P-value > 0.05) in tagged data sets. Collectively, these results support the notion that our epitope tagging strategy does not generate large regulatory alterations.
We also utilized our RNA-seq data to identify our 3′ end tags, as well as the integrity of the 3′ untranslated region. By aligning RNA-seq-derived reads onto exons and Flag tags of RAD21 and ATF1 transcripts, we identified a substantial proportion of expressed transcripts in CRISPR-modified cell populations that harbored the Flag tag (Supplemental Fig. 14). We also assessed sequence integrity of endogenous RNA-derived sequences upstream of and downstream from 5′ and 3′ homologous recombination sites, respectively (Supplemental Figs. 15, 16). The resulting alignments indicate that the vast majority of reads aligned to the reference genome, supporting the notion that our CRISPR genome editing generated efficient and proper homologous recombination in our polyclonal HepG2 cell population.
Applicability of CETCh-seq in distinct cell types and species
The CETCh-seq approach is widely applicable and can be used to assay TFs across distinct cell types. To demonstrate the robustness of our method, we performed CETCh-seq experimentation in breast adenocarcinoma MCF7 cells. We targeted the RAD21, CREB1, and ATF1 DNA-binding proteins in MCF7 cells using our previous gRNAs and pFETCh constructs (Supplemental Table 2). PCR and Western blot validation experiments both supported the correct homologous recombination of RAD21 with the Flag-tag construct (Fig. 3A,B; Supplemental Table 3). However, we failed to detect proper PCR and protein products for CREB1 and ATF1 TFs. In light of the fact that the CREB1 and ATF1 gRNAs and homology arm pFETCh constructs were successful in HepG2 cells, these data may reflect lower homologous recombination efficiencies in MCF7 using lipid-based transfection strategies. Alternatively, potential DNA sequence mismatches between HepG2 and MCF7 cells may preclude efficient homologous recombination, as five out of six independent homology arms were generated by PCR amplification of HepG2 genomic DNA. For our PCR-validated RAD21 experiments, we further Sanger-sequenced cloned sequences spanning the 5′ and 3′ homologous recombination sites and identified proper integration events (Supplemental Table 4). We also sequenced PCR amplicons spanning the 3′ homologous recombination sites for RAD21 and validated proper homologous site integration (Supplemental Fig. 17).
Figure 3.

MCF7 ChIP-seq reproducibility and gene expression analysis. (A) PCR validation of RAD21 homologous recombination. From left to right, the gel image displays ladder, 5′ end, and 3′ end homology PCR products. Ladder band sizes are given at the left. Correct homologous recombination generates 1363-bp (5′) and 2278-bp (3′) gel bands. (B) Western blot validation of epitope-tagged RAD21. An ∼130-kDa protein band (marked by red arrow) corresponding to the predicted full-length size of RAD21 is visible in Western blots using a Flag antibody in CRISPR-modified cells but is absent in wild-type HepG2 cells. (C) DNA-binding protein read enrichment tracks on the UCSC Genome Browser are shown at distinct genome distance intervals. Data is given for CETCh-seq RAD21 biological replicates (BR1 and BR2, lower panels) and standard ChIP-seq using a RAD21 antibody (Ctrl, upper panels). (D) MCF7 RAD21 rank correlations of normalized sequence read counts between wild-type MCF7 cells (WT) using a RAD21 antibody and CETCh-seq Flag-tagged RAD21 (Flag) data sets (top). (Bottom) MCF7 RAD21 rank correlations of normalized sequence read counts between CETCh-seq biological replicates (Flag BR1 and Flag BR2). Average rank correlation values for all Flag replicate pairwise comparisons are given in the top left corner of each plot.
We subsequently compared the CETCh-seq data sets with standard RAD21 antibody-based ChIP-seq data in MCF7 cells. For RAD21 CETCh-seq data, we further performed both technical and biological replicate experimentation (Fig. 3; Supplemental Figs. 18–21). Importantly, we identified the identical binding motif for all control and CETCh-seq data sets (Supplemental Fig. 18). Through qualitative and quantitative strategies, we further observed a high concordance between ChIP-seq data generated by using a RAD21 antibody and data from CETCh-seq experiments; 88% of RAD21 sites are shared (Fig. 3C; Supplemental Table 5), and both data sets exhibit a high rank correlation (rho = 0.89), consistent with ChIP-seq replicate correlations (Fig. 3D; Supplemental Fig. 19). Technical CETCh-seq RAD21 replicates further exhibited high correlation in MCF7 cells (Supplemental Fig. 19), and these observations were also consistent through assessment of a biological replicate CETCh-seq experiment for RAD21 (Supplemental Table 5; Supplemental Fig. 20).
We next evaluated potential technical artifacts in the MCF7 CRISPR-modified cells. We performed ChIP-seq with RAD21 antibodies in CRISPR-modified MCF7 cells. Similar to our observations in HepG2 cells, MCF7-modified cell RAD21-binding integrity was maintained, as RAD21 antibody-based ChIP-seq results in CRISPR-modified MCF7 cells were in accordance with data generated from wild-type MCF7 cells (rho = 0.9) (Supplemental Fig. 21), and these RAD21 data sets were further coincident with CETCh-seq data (rho = 0.85) (Supplemental Fig. 21).
To finally assess the feasibility of our approach in primary cells from a distinct species, we performed CETCh-seq in murine embryonic stem (ES) cells, as these cells can be further utilized to generate live transgenic mice for potential in vivo CETCh-seq experimentation (Savic et al. 2013). We targeted the Gabpa TF using 800-bp 5′ and 3′ homology arms (Supplemental Tables 2, 3), as well as two mouse-sequence-derived gRNAs (Supplemental Table 2). A high degree of accurate homologous recombination was supported through Sanger sequencing of PCR amplicons (Supplemental Table 3) spanning the 3′ homologous recombination site (Supplemental Fig. 22). CETCh-seq DNA-binding data were of high quality and were further highly enriched for the canonical GABPA motif (Supplemental Table 5; Supplemental Fig. 23). Importantly, ChIP-seq using a Flag antibody in unmodified mouse ES cells failed to identify binding sites. Collectively, these data support the feasibility of CETCh-seq in distinct cell lines and across distinct mammalian species.
Discussion
ChIP-seq is a routinely used functional genomic assay for identifying regulatory elements involved in gene regulation and genome function (Furey 2012; Sakabe et al. 2012). However, the approach is highly limited by the need to identify ChIP-grade antibodies, which makes it particularly difficult for a large fraction of human TFs. To increase the lexicon of annotated DNA-binding proteins and ameliorate these technical restrictions, we applied and systematically evaluated CETCh-seq, a method that uses CRISPR genome editing to place epitope tags at the 3′ ends of the endogenous genes and the subsequent identification of DNA-binding interactomes through the use of an antibody targeting the epitope tag on TF proteins. Our data demonstrate the usefulness of CETCh-seq for tagging DNA-binding proteins that are refractory to standard ChIP-seq analysis.
We demonstrate that CETCh-seq shows high specificity and adaptability as we successfully tagged four TFs spanning a large range of expression levels in HepG2 cells (RPKM ranging from 2 to 40). Moreover, all data sets for the tagged DNA-binding proteins were enriched for the same DNA-binding motifs as those obtained in ChIP-seq experiments with TF antibodies. CETCh-seq also exhibited high reproducibility, as both technical and biological replicates displayed strong concordance. We confirmed that our method generates minimal alterations to the cellular transcriptome and also demonstrate that our strategy is robust, as we successfully tagged DNA-binding proteins in HepG2 and MCF7 human cell lines, as well as mouse ES cells. The latter experiment suggests that CETCh-seq can be further applied within in vivo systems. Notably, our data also suggest that a PCR validation using primers for correctly targeted 5′ and 3′ homology arms is an accurate method for identifying successful experiments, allowing for a simple screen prior to more extensive downstream experimentation. Interestingly, in light of the concordance with standard ChIP-seq assays for antibodies that have been validated by ENCODE, our data further alludes to a low level of cross-reactivity with additional DNA- and/or chromatin-binding proteins for these antibodies (The ENCODE Project Consortium 2012).
Although we obtained high-quality ChIP-seq data with our tagging strategy, we note that this may not hold true for all DNA-binding proteins. For instance, the secondary structure of distinct DNA-binding proteins may preclude the use of a 3′ tag or even the use of a TF tagged with a Flag epitope by transgene integration. However, our construct can be modified to tag TF genes at the 5′ end, and alternative epitope tags can be substituted for the Flag tag. The reliance of the endogenous TF promoter and regulatory landscape for selectable neomycin resistance gene co-expression may restrict tagging, particularly for low- or nonexpressed DNA-binding protein genes. Despite these concerns, our results demonstrate that our tagging platform works across a wide range of TF expression levels, and further optimizations to cellular selection procedures may mitigate these concerns. Moreover, our vector can be further modified to place a neomycin resistance gene under the control of an independent promoter, and an additional negative cell selection step would be required to control for random integration events. The neomycin resistance cassette can be further floxed, and Cre recombinase can be used to excise the selectable marker to reduce alterations and potential confounding effects at the endogenous locus. Similar constructs exist for Drosophila (Bottcher et al. 2014), but their use in mammalian systems and for ChIP-seq assays needs to be more thoroughly assessed.
An added concern is the potential sequence restriction imposed by the need to identify suitable gRNAs near stop codons of endogenous DNA-binding proteins. We utilized an ∼60-bp window of sequence centered on the stop codon (20 bp upstream and 40 bp downstream) to identify gRNAs. Although the use of gRNAs within the 3′ untranslated sequence within a few nucleotides of the TF gene stop codon is preferred, the use of gRNAs targeting the upstream exonic sequence is feasible through the additional use of 5′ homology arm constructs with introduced synonymous sequence variants at the gRNA site in order to prevent Cas9-mediated recleavage events at correctly recombined alleles. The remaining upstream portion of the 5′ homology arm that shares perfect homology with the endogenous locus should be long enough to facilitate efficient homologous recombination. We utilized this approach for the ATF1 TF (Supplemental Table 2). In addition, the ongoing generation of additional Cas9 systems with unique protospacer adjacent motif (PAM) sites will enable greater flexibility in gRNA design for CETCh-seq experimentation (Esvelt et al. 2013; Kleinstiver et al. 2015).
We further stress that each cell type will need proper transfection and cellular selection optimizations, including optimizing gRNA and pFETCh construct concentrations, as we did identify a decrease in CETCh-seq success in MCF7 cells, despite the use of identical gRNAs and pFETCh constructs. For cells that are difficult to transfect, higher efficiency approaches such as nucleofection, rather than lipid-based transfection approaches, would be preferred. Moreover, the generation of homology arms directly amplified using the appropriate cell line genome DNA, or alternatively, using DNA sequence information from the appropriate cell line for the synthetic synthesis of DNA fragment homology arms such as genome Block sequence fragments (gBlocks), should be considered. This idea is also supported by differences in cell-type CETCh-seq success rates, as the majority of independent homology arms (five out of six) were generated using PCR amplicons from HepG2 genomic DNA. Consequently, potential sequence variations between HepG2 and MCF7 cells may have contributed to reduced recombination efficiencies in MCF7 cells.
Next-generation DNA sequencing technologies have ushered in a functional genomic era in biological research. These high-throughput methodologies can assess gene regulation and genome function in a broad genome-wide manner, allowing for a previously unattainable level of resolution. Despite these technologies, we have only interrogated a small subset of eukaryotic DNA-binding protein interactomes through ChIP-seq techniques. In light of the aforementioned technical hurdles associated with standard TF antibody-based ChIP-seq, we believe that the CETCh-seq method described can circumvent these limitations, expanding the number of TFs assayed and advancing our overall understanding of TF function and genome architecture.
Methods
Construct engineering and homology arm cloning
The 3×Flag-P2A-Neomycin epitope tagging donor construct (pFETCh-Donor, Addgene plasmid #63934) was synthesized (Blue Heron) and subcloned into pHSG299 (Clontech). Homology arms for individual transcription factors were PCR amplified and/or ordered as synthetic dsDNA genomic blocks (IDT, gBlocks), and assembly of the final donor plasmid was achieved with Gibson Assembly (New England BioLabs). CRISPR gRNAs were cloned into pSpCas9(BB)-2A-GFP (PX458), which was a kind gift from Feng Zhang (Addgene plasmid # 48138). Oligos for gRNAs near 3′ end stop codons were identified and ordered through Integrated DNA Technologies (IDT) and cloned downstream from a U6 promoter element as previously described (Ran et al. 2013). Our pFETCh donor plasmid (Addgene plasmid # 63934) and all cloned Cas9/gRNA and homology arm donor plasmids are available on Addgene. A complete list of gRNAs and homology arm sequences, including how each homology arm was generated, is given in Supplemental Table 2.
Cell culture and transfection
HepG2 and MCF7 cells were grown under recommended growth conditions. Cells were transfected at ∼75% confluence using FUGENE reagent (Promega, E2311), selected using G418 (Invitrogen, 10131035), and expanded for ChIP-seq, RNA-seq, Western blot, and PCR experimentation. FUGENE transfection for HepG2 and MCF7 cells was performed in six-well plates using recommended conditions and concentrations. Nucleofection (Lonza) was done for mouse ES cells using a P3 kit with the Nucleofector 4D. pFETCh constructs and Cas9/gRNA plasmids were cotransfected for all CETCh-seq experiments. For a subset of our targeted DNA-binding proteins, we also transfected two gRNAs in parallel (Supplemental Table 2). HepG2 cells were initially selected with 400 µg per mL G418 until cells reached a confluence of 10%–20%. G418 selection was subsequently reduced to 200 µg per mL. MCF7 cells were selected using 200 µg per mL of G418. Mouse ES cells were selected using 25 µg per mL of G418. We estimated the number of colony-forming units by visual inspection of six-well plates after 2–3 wk of G418 selection. Cells were maintained under selection as a polyclonal pool for the generation of cell stocks and prior to harvest for validation and functional genomic experimentation.
Homologous recombination validation
DNA from CRISPR-modified cell pellets was isolated using the DNeasy Blood and Tissue kit (Qiagen, 69504). Primers internal to the tag construct and specific to each TF locus were ordered through IDT (Supplemental Table 3). Protein from cell pellets was extracted and analyzed on the ProteinSimple Wes machine (ProteinSimple) for standard Western blot and immunoprecipitation Western blot experiments. For IP Western blots, Flag antibody was used for IP and for blotting. Protein extraction was performed on frozen wild-type and CRISPR-modified HepG2 and MCF7 cell pellets. PCR amplicons for 5′ and 3′ end homologous recombination sites were cloned and submitted for Sanger sequencing. In addition, for 3′ homologous recombination sites, PCR amplicons were directly submitted for Sanger sequencing. For the identification of nonhomologous end joining events at unmodified alleles, PCR amplicons using primers at endogenous sequences spanning 5′ and 3′ homologous recombination sites were submitted for next-generation sequencing on a MiSeq Illumina sequencing machine for 150-bp single-end sequencing. For subsequent analyses, FASTQ files were converted to FASTA and collapsed using FastX-Toolkit. Reads were scored as unmodified or containing insertions or deletions by alignment to target sites.
ChIP-seq experimentation
ChIP-seq experimentation was performed as previously outlined (Reddy et al. 2009). We utilized Flag (Sigma, F1804), RAD21 (abcam, ab992), CREB1 (Santa Cruz Biotechnology, sc-240), and GABPA (Santa Cruz Biotechnology, sc-28312) antibodies for all ChIP-seq experiments. ChIP-seq libraries were run on an Illumina HiSeq 2500 next-generation sequencer. Technical replicates were done with independent chromatin immunoprecipitation experimentations from the same transfected cell pool. Biological replicates represent independent cellular transfection experiments.
RNA-seq experimentation
Wild-type HepG2 cells were cultured under standard growth conditions, while CRISPR-modified cells were cultured under G418 selection. Cells were pelleted and stored at −80°C. For RNA preparation, the Norgen Total RNA Preparation kit (Norgen Biotek, 17200) was used to isolate mRNA. cDNA synthesis was performed using SuperScript II reverse transcriptase (Invitrogen, 18064-014), while next-generation libraries were prepared using the Nextera DNA Sample Prep kit (Illumina, FC-121-1031). RNA-seq libraries were run on an Illumina HiSeq 2000 next-generation sequencer using paired-end 50-bp sequencing.
Data analysis
Peaks were identified using the MACS peak caller (Zhang et al. 2008). Position weight matrices for transcription factor binding motifs were identified using MEME (Bailey et al. 2006). For quantitative correlation analyses, normalized sequence read depths (reads per million aligned) were calculated at binding sites identified across all experiments for each independent transcription factor, including replicates (Flag-tag ChIP-seq, wild-type cell TF antibody-based ChIP-seq, and CRISPR-modified cell TF antibody-based ChIP-seq). For this analysis we analyzed a 100-bp sequence at each binding site centered on the binding site peak summit. Binding sites were merged across identical transcription factor experiments and normalized read depths were calculated at all merged sites. Spearman rank correlations were determined for all pairwise comparisons. RNA-seq RPKM were tabulated, and Spearman rank correlations were calculated across CRISPR-tagged cells and unmodified HepG2 cells for RAD21 and ATF1 data sets. RNA-seq data were further analyzed using DESeq (Anders and Huber 2010) to identify differentially regulated genes.
Data access
All ChIP-seq and RNA-seq data sets have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE72082.
Acknowledgments
This work was supported by National Institutes of Health grant U54 HG006998-0 (to R.M.M.). We thank Amy Ridgeway for key technical support, Candice Coppola for assistance with sequencing and analysis of NHEJ, and Timothy Reddy, Barbara Wold, Ross Hardison, and Ali Mortazavi for helpful suggestions and advice.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.193540.115.
References
- Anders S, Huber W. 2010. Differential expression analysis for sequence count data. Genome Biol 11: R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Williams N, Misleh C, Li WW. 2006. MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res 34: W369–W373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottcher R, Hollmann M, Merk K, Nitschko V, Obermaier C, Philippou-Massier J, Wieland I, Gaul U, Forstemann K. 2014. Efficient chromosomal gene modification with CRISPR/cas9 and PCR-based homologous recombination donors in cultured Drosophila cells. Nucleic Acids Res 42: e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Hartenian E, Graham DB, Tothova Z, Hegde M, Smith I, Sullender M, Ebert BL, Xavier RJ, Root DE. 2014. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat Biotechnol 32: 1262–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. 2007. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447: 799–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esvelt KM, Mali P, Braff JL, Moosburner M, Yaung SJ, Church GM. 2013. Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat Methods 10: 1116–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furey TS. 2012. ChIP-seq and beyond: new and improved methodologies to detect and characterize protein-DNA interactions. Nat Rev Genet 13: 840–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo B, Stein JL, van Wijnen AJ, Stein GS. 1997. ATF1 and CREB trans-activate a cell cycle regulated histone H4 gene at a distal nuclear matrix associated promoter element. Biochemistry 36: 14447–14455. [DOI] [PubMed] [Google Scholar]
- Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, et al. 2013. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31: 827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J. 2013. RNA-programmed genome editing in human cells. Elife 2: e00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DS, Mortazavi A, Myers RM, Wold B. 2007. Genome-wide mapping of in vivo protein-DNA interactions. Science 316: 1497–1502. [DOI] [PubMed] [Google Scholar]
- Kim JH, Lee SR, Li LH, Park HJ, Park JH, Lee KY, Kim MK, Shin BA, Choi SY. 2011. High cleavage efficiency of a 2A peptide derived from porcine teschovirus-1 in human cell lines, zebrafish and mice. PLoS One 6: e18556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittler R, Zhou J, Hua S, Ma L, Liu Y, Pendleton E, Cheng C, Gerstein M, White KP. 2013. A comprehensive nuclear receptor network for breast cancer cells. Cell Rep 3: 538–551. [DOI] [PubMed] [Google Scholar]
- Kleinstiver BP, Prew MS, Tsai SQ, Topkar VV, Nguyen NT, Zheng Z, Gonzales AP, Li Z, Peterson RT, Yeh JJ, et al. 2015. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 523: 481–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, et al. 2012. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res 22: 1813–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lettice LA, Heaney SJ, Purdie LA, Li L, de Beer P, Oostra BA, Goode D, Elgar G, Hill RE, de Graaff E. 2003. A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum Mol Genet 12: 1725–1735. [DOI] [PubMed] [Google Scholar]
- Maeder ML, Linder SJ, Cascio VM, Fu Y, Ho QH, Joung JK. 2013. CRISPR RNA-guided activation of endogenous human genes. Nat Methods 10: 977–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. 2013. RNA-guided human genome engineering via Cas9. Science 339: 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzoni EO, Mahony S, Iacovino M, Morrison CA, Mountoufaris G, Closser M, Whyte WA, Young RA, Kyba M, Gifford DK, et al. 2011. Embryonic stem cell-based mapping of developmental transcriptional programs. Nat Methods 8: 1056–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montigny WJ, Phelps SF, Illenye S, Heintz NH. 2003. Parameters influencing high-efficiency transfection of bacterial artificial chromosomes into cultured mammalian cells. Biotechniques 35: 796–807. [DOI] [PubMed] [Google Scholar]
- Najafabadi HS, Mnaimneh S, Schmitges FW, Garton M, Lam KN, Yang A, Albu M, Weirauch MT, Radovani E, Kim PM, et al. 2015. C2H2 zinc finger proteins greatly expand the human regulatory lexicon. Nat Biotechnol 33: 555–562. [DOI] [PubMed] [Google Scholar]
- Perez-Pinera P, Kocak DD, Vockley CM, Adler AF, Kabadi AM, Polstein LR, Thakore PI, Glass KA, Ousterout DG, Leong KW, et al. 2013. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat Methods 10: 973–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilon AM, Ajay SS, Kumar SA, Steiner LA, Cherukuri PF, Wincovitch S, Anderson SM, Mullikin JC, Gallagher PG, Hardison RC, et al. 2011. Genome-wide ChIP-Seq reveals a dramatic shift in the binding of the transcription factor erythroid Kruppel-like factor during erythrocyte differentiation. Blood 118: e139–e148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poser I, Sarov M, Hutchins JR, Heriche JK, Toyoda Y, Pozniakovsky A, Weigl D, Nitzsche A, Hegemann B, Bird AW, et al. 2008. BAC TransgeneOmics: a high-throughput method for exploration of protein function in mammals. Nat Methods 5: 409–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. 2013. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8: 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy TE, Pauli F, Sprouse RO, Neff NF, Newberry KM, Garabedian MJ, Myers RM. 2009. Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res 19: 2163–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakabe NJ, Savic D, Nobrega MA. 2012. Transcriptional enhancers in development and disease. Genome Biol 13: 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander JD, Joung JK. 2014. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol 32: 347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savic D, Gertz J, Jain P, Cooper GM, Myers RM. 2013. Mapping genome-wide transcription factor binding sites in frozen tissues. Epigenetics Chromatin 6: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymczak AL, Workman CJ, Wang Y, Vignali KM, Dilioglou S, Vanin EF, Vignali DA. 2004. Correction of multi-gene deficiency in vivo using a single ‘self-cleaving’ 2A peptide-based retroviral vector. Nat Biotechnol 22: 589–594. [DOI] [PubMed] [Google Scholar]
- Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R. 2013. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153: 910–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R. 2013. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 154: 1370–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Buchholz F, Muyrers JP, Stewart AF. 1998. A new logic for DNA engineering using recombination in Escherichia coli. Nat Genet 20: 123–128. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Muyrers JP, Testa G, Stewart AF. 2000. DNA cloning by homologous recombination in Escherichia coli. Nat Biotechnol 18: 1314–1317. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9: R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Ren JX, Ryan TM, Higgins NP, Townes TM. 2004. Rapid tagging of endogenous mouse genes by recombineering and ES cell complementation of tetraploid blastocysts. Nucleic Acids Res 32: e128. [DOI] [PMC free article] [PubMed] [Google Scholar]