

Myxococcus xanthus development requires CsgA, a member of the short-chain alcohol dehydrogenase (SCAD) family of proteins. Boynton and Shimkets show that CsgA and SocA oxidize the 2′-OH glycerol moiety on cardiolipin and phosphatidylglycerol to produce diacylglycerol, dihydroxyacetone, and orthophosphate. SCADs that prevent neurodegenerative disorders, such as Drosophila Sniffer and human HSD17B10, oxidize cardiolipin with similar kinetic parameters.
Keywords: Myxococcus xanthus, CsgA, Drosophila Sniffer, HSD10, phospholipase, cardiolipin
Abstract
Myxococcus xanthus development requires CsgA, a member of the short-chain alcohol dehydrogenase (SCAD) family of proteins. We show that CsgA and SocA, a protein that can replace CsgA function in vivo, oxidize the 2′-OH glycerol moiety on cardiolipin and phosphatidylglycerol to produce diacylglycerol (DAG), dihydroxyacetone, and orthophosphate. A lipid extract enriched in DAGs from wild-type cells initiates development and lipid body production in a csgA mutant to bypass the mutational block. This novel phospholipase C-like reaction is widespread. SCADs that prevent neurodegenerative disorders, such as Drosophila Sniffer and human HSD10, oxidize cardiolipin with similar kinetic parameters. HSD10 exhibits a strong preference for cardiolipin with oxidized fatty acids. This activity is inhibited in the presence of the amyloid β peptide. Three HSD10 variants associated with neurodegenerative disorders are inactive with cardiolipin. We suggest that HSD10 protects humans from reactive oxygen species by removing damaged cardiolipin before it induces apoptosis.
Myxococcus xanthus displays a unique multicellular lifestyle when faced with amino acid limitation. Cells move in travelling waves or ripples (Shimkets and Kaiser 1982), form evenly spaced multicellular aggregates known as fruiting bodies (Xie et al. 2011), and differentiate into dormant spores within these aggregates (Holkenbrink et al. 2014). Several differentiating cell types are produced, leading to programmed cell death (PCD), sporulation, and production of peripheral rods (O'Connor and Zusman 1991; Boynton et al. 2013). Some mutations that abolish multicellular development can be transiently restored in the presence of wild-type cells (Hagen et al. 1978). Extracellular complementation is due to either intercellular signaling or metabolic cross-feeding.
Perhaps the most interesting extracellular complementation group is produced by loss of the csgA gene (MXAN_RS06255), which fully eliminates development (Hagen and Shimkets 1990). Increasing levels of csgA expression induce successive developmental behaviors (Li et al. 1992). Low expression levels (10%–20% of final developmental levels) induce rippling. Higher levels of expression then entrain cell aggregation into fruiting bodies. Sporulation is achieved when csgA is expressed at the peak concentration, completing the developmental program. Addition of exogenous, purified CsgA can restore aggregation at low concentrations and sporulation at higher concentrations (Kim and Kaiser 1991).
In the current model for extracellular complementation of csgA mutants, often referred to as C-signaling, full-length p25 (kilodalton) CsgA is processed to a p17 form that is exported to the cell surface where it binds a receptor on an adjacent cell (Lobedanz and Sogaard-Andersen 2003). Proteolytic processing of CsgA is mediated by serine protease PopC, and a popC mutant has abrogated development (Rolbetzki et al. 2008). However, the current model fails to account for many critical pieces of data that are inconsistent with it. For example, CsgA has features associated with enzymatic function, such as a Rossmann fold, shown to bind NAD+ in vitro, and conserved catalytic residues typical of short-chain alcohol dehydrogenases (SCADs), shown to be essential for development in vivo (Lee et al. 1995). Membrane fractionation indicated that CsgA is an inner membrane protein (Simunovic et al. 2003), not an outer membrane protein as previously reported (Lobedanz and Sogaard-Andersen 2003). Extensive proteomic studies have likewise never placed CsgA in the outer membrane or on the cell surface (Kahnt et al. 2010; Bhat et al. 2011), and no receptor has ever been identified. The p17 form lacks the N-terminal NAD+-binding domain, and genomic replacement of a truncated csgA gene encoding an 18.1-kDa protein abolishes development (Lobedanz and Sogaard-Andersen 2003). Finally, up-regulation of SocA, a distantly related homolog of CsgA with only 28% amino acid identity, restores development (Lee and Shimkets 1994, 1996). Since SocA must be substantially up-regulated to rescue development in vivo, it was suggested that rescue is due to SocA having substrate overlap. SocA was shown to oxidize the phospholipid lysophosphatidylethanolamine (lyso-PE) in vitro (Avadhani et al. 2006).
CsgA homologs are found in eukaryotes, and genetic studies have provided compelling evidence that they prevent neurodegenerative disorders. A unifying feature of these neurodegenerative disorders is a marked increase in reactive oxygen species (ROS) (Cobb and Cole 2015). Lipid peroxyl radicals are among the most dangerous products of ROS, since they autocatalyze their own production (Gardner 1989). Once a lipid radical is formed, it perpetuates production by creating a new radical on a neighboring fatty acid at the site of bis-allylic hydrogens. Formation of new radicals terminates only when two radicals react to produce a fatty acyl peroxide. An insertion in the csgA homolog sniffer from Drosophila produces oxidative stress-related neurodegeneration that includes enhanced apoptosis and reduced locomotor fitness (Botella et al. 2004). Sniffer can reduce a byproduct of lipid peroxidation, (E)-4-oxonon-2-enal (4ONE), to (E)-4-hydroxynon-2-enal (4HNE) in vitro, which is less neurotoxic (Martin et al. 2011). However, 4HNE is still a potent electrophile with deleterious effects on proteins and nucleic acids, suggesting that Sniffer has another role in neuroprotection.
Another interesting CsgA homolog is the human SCAD 17β-hydroxysteroid dehydrogenase type 10 (HSD10), the product of the HSD17B10 gene located on chromosome Xp11.2. HSD10 is imported into the mitochondria by a pathway involving Parkin, a protein whose function is abrogated in patients with Parkinson's disease (Bertolin et al. 2015). A wide variety of enzymatic activities have been associated with HSD10 (Yang et al. 2014), and their clinical significance has been examined extensively. Mutations in the HSD17B10 gene have been reported in 19 families (Zschocke 2012). In the classical infantile form of the disease caused by mutation p.R130C, symptoms emerge ∼6–18 mo after birth, and males show progressive neurodegenerative disease, leading to death a few years later. Substantially up-regulated levels of HSD10 are also found in hippocampal synaptic mitochondria of βAPP transgenic mice (He et al. 2002) and human patients with Alzheimer's disease (Kristofikova et al. 2009), suggesting a correlation with this neurodegenerative disorder. HSD10 (also known as amyloid β [Aβ] peptide-binding alcohol dehydrogenase) is a strong binding partner of the Aβ peptide, the principle component of the extracellular plaques indicative of Alzheimer's disease (Yan et al. 1997). Binding of Aβ to HSD10 can be detected at nanomolar concentrations, and micromolar concentrations inhibit HSD10 activity (Borger et al. 2013). Altered HSD10 levels have also been observed in patients with multiple sclerosis (Kristofikova et al. 2009).
Here we show that CsgA, SocA, HSD10, and Sniffer have a novel phospholipase C-like activity. They oxidize the 2′-OH in the head group of cardiolipin (CL) and phosphatidylglycerol (PG). The enzymatic product is unstable and fragments into diacylglycerol (DAG), dihydroxyacetone (DHA), and orthophosphate (Pi). We propose that developing Myxococcus cells use this reaction to convert phospholipids into triacylglycerol (TAG) as the cells shorten during sporulation. Drosophila and humans instead use this reaction to remove damaged CL before it causes apoptosis.
Results
Expression and purification of CsgA yields an active enzyme
To facilitate production of p25 CsgA, the csgA gene (MXAN_RS06225; MXAN_1294) was codon-optimized for Escherichia coli (Supplemental Fig. S1) and cloned with an N-terminal six-histidine tag. For the p17 form, only the 168 C-terminal codons were used. Purified recombinant protein (Supplemental Fig. S2A) rescues csgA mutant (LS2442) development to produce spore-filled fruiting bodies comparable in size and number with those produced by wild-type DK1622 cells (Supplemental Fig. S2B). Our p17 failed to restore development in the same assay.
Enzymatic activity of the recombinant enzyme was examined with a modified version of the SocA assay (Avadhani et al. 2006). Since a known substrate of SocA is lyso-PE, whole-cell lipids were extracted with chloroform and methanol. This extract was used as a CsgA substrate, and activity was measured as the conversion of NAD+ to NADH (Fig. 1A). CsgA displays enzymatic activity in the presence of these lipids. Removal of exogenous NAD+ resulted in a small amount of observed activity, presumably due to copurified NAD+. No activity is generated unless the enzyme is present. As expected, p17 CsgA displays no activity in this assay, since it lacks the NAD+-binding domain. As an additional negative control, CsgA bearing the active site mutation K155R (Lee et al. 1995) was examined and is inactive. Enzymatic reactions with CsgA did not take place in the presence of NADP+ (Supplemental Fig. S3), unlike SocA, which was previously reported to show no cofactor preference (Avadhani et al. 2006).
Figure 1.

CsgA (p25) and SocA oxidize lipid substrates containing a glycerol moiety with a 2′-OH. (A) CsgA enzymatic activity in the presence of lipid extracts from developing M. xanthus cells. Activity was detected for p25 CsgA in the presence of lipids and NAD+ (•) but not in the absence of lipids (▾). A slight increase of activity was detected in the presence of lipids alone (○), possibly due to a small amount of copurified NAD+. No activity was seen for p17 CsgA, as expected, since it lacks the NAD+ binding pocket (Δ). A putative noncatalytic mutant, K155R, also displayed no activity (□). (B) CsgA activity in the presence of synthetic lipids. CsgA was assayed in the presence of several synthetic C14:0 lipids to identify substrates. CL (□) and PG (▪) have the highest activity. Lesser activity was observed with lyso-PE (Δ), sn-1-monoacylglycerol (MAG) (•), and 1,3-DAG (▾). Activity was not seen with 1,2 DAG (○). (C) SocA activity in the presence of these same lipids. The highest activity was observed with lyso-PE (Δ). Activity was also seen with MAG (•), 1,3-DAG (▾), CL (□), and PG (▪). Activity was again undetected in the presence of 1,2 DAG (○). (D) Initial rates for CsgA and SocA show that CsgA activity is an order of magnitude higher for CL and PG than SocA. (E) Kinetic analyses of CsgA (•) and SocA (○) using CL as a substrate. (F) Kinetic analyses of CsgA (•) and SocA (○) using PG as a substrate. Error bars indicate standard deviation of three replicates.
CsgA and SocA share overlapping substrate specificities
To determine what lipids are potential substrates, we further assessed CsgA activity using a variety of synthetic C14:0 partial glycerides (Fig. 1B). Recombinant p25 CsgA oxidizes sn-1-monoacylglycerol (MAG), sn-1,3-DAG, and lyso-PE, but not sn-1,2-DAG (structures depicted in Supplemental Fig. S4). These data point to the glycerol 2′-OH being the CsgA target. CL and PG both contain this moiety and were examined as possible substrates, as CL is abundant in Myxococcus where it is found almost exclusively in the inner membrane (Orndorff and Dworkin 1980; Lorenzen et al. 2014). Both CL and PG are excellent substrates for CsgA (Fig. 1B). The initial rates with C14:0 CL and PG are about fivefold higher than those of the partial glycerides (Fig. 1D). When substrate is in excess, complete conversion of 100 μM NAD+ to NADH is observed, unlike reactions with the partial glycerides that reach ∼50% conversion as expected for a reversible reaction. The CL and PG reactions thus appear to be irreversible and could not be driven in the opposite direction with NADH.
Since SocA can replace CsgA in vivo, their substrate specificity should overlap if enzymatic activity is responsible for bioactivity. While the previously reported SocA substrate lyso-PE gave the most activity, catalysis was also observed with sn-1-MAG, sn-1,3-DAG, PG, and CL substrates (Fig. 1C,D). As with CsgA, no activity was observed with sn-1,2-DAG. Kinetic analyses for both enzymes are shown for CL (Fig. 1E) and PG (Fig. 1F) substrates. Kinetic parameters are compared in Table 1. Of note is the difference in affinity constants between CsgA and SocA for CL (62 μM and 603 μM, respectively) and PG (57 μM and 324 μM, respectively). CsgA is roughly an order of magnitude more efficient than SocA at using PG and CL. As controls, both p17 and K155R CsgA showed no activity in kinetic assays (Table 1).
Table 1.
Kinetic parameters of enzymes
Oxidation of CL or PG constitutes a novel phospholipase C-like reaction
To determine the consequence of the activity exhibited by these enzymes, the products of reactions using CsgA with C14:0 CL and PG were determined. Hydrophobic products were first extracted in chloroform and subjected to liquid chromatography/mass spectrometry and electron spectroscopic imaging (LC/MS-ESI). C14:0 DAG was identified in both reactions. A representative peak can be seen in Supplemental Figure S5A as a sodium adduct with mass 535.4202 [M + Na]+. DAG was not identified in reactions lacking enzyme (Supplemental Fig. S5B). The remaining soluble products were Pi and DHA. Pi is produced at a ratio of two per CL converted and one per PG converted relative to the amount of NADH produced (Fig. 2A,B, respectively). For both substrates, one molecule of DHA was found per substrate oxidized (Fig. 2A,B). Since it is possible that glycerol could be generated by hydrolysis, glycerol was assayed and not detected.
Figure 2.

Enzymatic activity with CL and PG constitutes a novel lipase-like reaction yielding DAG, DHA, and Pi. (A) Quantification of soluble products from a CsgA enzymatic reaction using CL as a substrate. For each molecule of CL turned over, one molecule of NADH (•) is produced as well as one DHA molecule (▾) and two Pi (○). Error bars indicate standard deviation of three replicates. (B) Quantification of soluble products from a reaction using PG as a substrate. For each molecule of PG turned over, one NADH (•) is produced along with one DHA (▾) and one Pi (○) molecule. (C) Proposed enzymatic reaction mechanism for CL. Catalytic activity removes the hydrogen from the 2′-OH to form a ketone intermediate that undergoes nucleophilic attack by water. Rearrangement releases two DAG moieties and forms an unstable cyclic head group. The cyclic molecule is further hydrated to form DHA bisphosphate. Further hydration produces free Pi and DHA. Oxidation of PG is proposed to occur similarly but with single DAG and Pi products.
These products suggest a novel phospholipase C-like reaction (Fig. 2C). In the proposed reaction mechanism, CL is first oxidized by CsgA to form a ketone intermediate. The aqueous environment and neighboring phosphates leave the molecule susceptible to hydration at the 2′ position of the glycerol moiety. The resulting rearrangement removes the two DAG side chains. A proposed cyclic intermediate derived from the head group is rearranged in the presence of water to form DHA bisphosphate. Pi appears to be released through another hydration step. The PG reaction is proposed to occur in a similar fashion.
Addition of a partial glyceride extract restores lipid body production
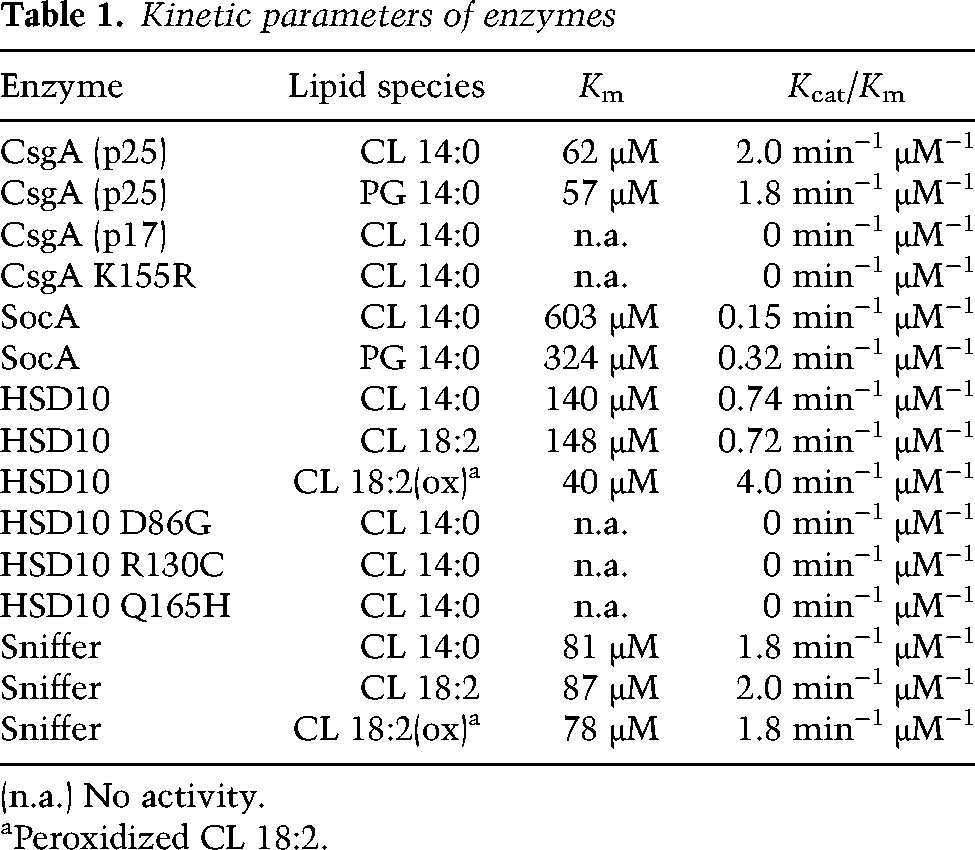
Myxococcus cells generate lipid bodies directly from their membrane lipids as cells shorten to become spores (Bhat et al. 2014). csgA mutants fail to synthesize these structures, and cells remain long. Lipid bodies are principally composed of TAG species, and one in particular is known to induce sporulation of an esg mutant (Hoiczyk et al. 2009). We sought to determine whether CsgA catalysis of CL and PG could be a limiting step in the conversion of phospholipids to TAGs. The rationale behind this idea is that feeding a csgA mutant the product of its reaction should bypass the mutational block. However, addition of commercial C14:0 DAG or DAG generated from enzymatic conversion of C14:0 CL failed to stimulate development of a csgA/socA double mutant (LS3931). Since fatty acid composition may be important to bioactivity and since DAGs with the unusual fatty acids found in Myxococcus are not commercially available, we extracted a lipid fraction containing partial glycerides (MAGs and DAGs) from developing cells 24 h post-induction. Supplementation of LS3931 with this lipid fraction restored development (Fig. 3). Fruiting bodies of a size similar to that of wild type appeared by 24 h, and sporulation occurred at levels greater than half of wild type compared with ∼1% in the unsupplemented mutant (Fig. 3A). Addition of the lipid extract also restored lipid body production. Representative cells can be seen in Figure 3B, stained with the lipophilic fluorescent dye Nile red. Lipid bodies are seen as red aggregates within cells. Quantification of this complementation was assessed by comparing the average lipid body composition of 30 cells at each lipid concentration (Fig. 3C). Lipid body induction displays sigmoidal kinetics over a narrow concentration range. Complementation could not be achieved with lipid extracts from vegetative cells or LS3931, indicating that it is developmentally specific.
Figure 3.
Partial glycerides produced by Myxococcus restore development to a csgA mutant. (A) Bioactive partial glycerides were purified from Myxococcus cells at 24 h post-development and characterized using a bioassay in which development of csgA was restored. LS3931 cells supplemented with this lipid fraction form fruiting bodies and sporulate. Sporulation data are presented below each strain name as a percentage of wild-type spores ± the standard deviation, an average of three independent counts. Bar, 200 μm. (B) These lipids also restore lipid body production in csgA socA mutant cells (LS3931) to wild-type (DK1622) levels. Lipid bodies inside cells are indicated by Nile red staining. Bar, 10 μm. (C) Quantification of lipid body production relative to wild-type amounts. Lipid bodies were quantified by average Nile red intensity normalized to cell length. Error bars indicate standard deviation of three replicates.
CL oxidation is a widespread SCAD activity
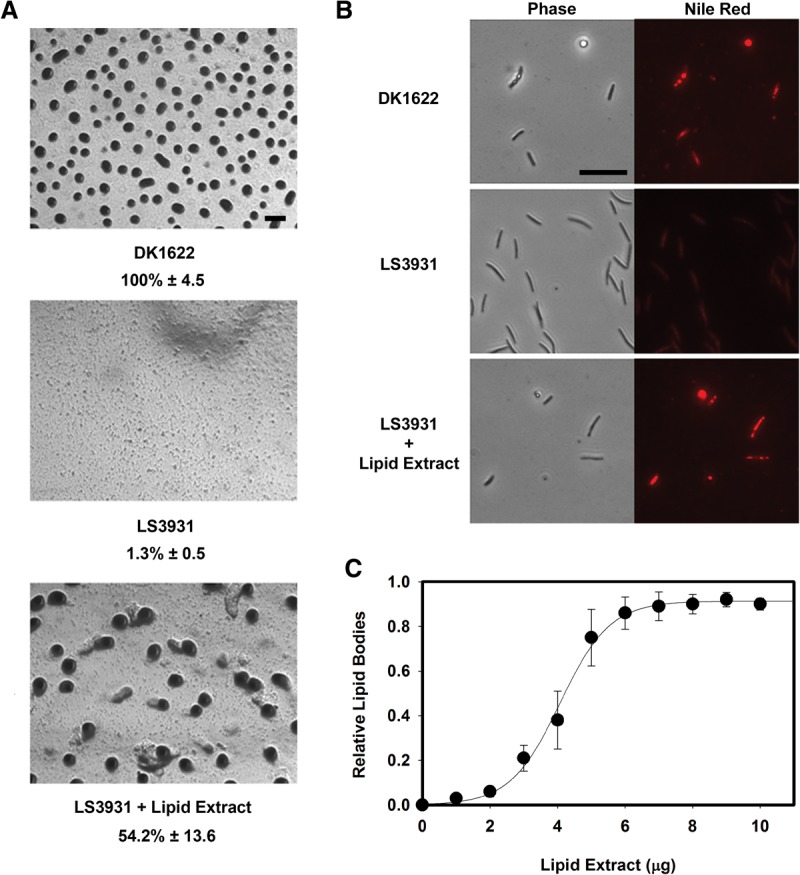
Representative homologs of CsgA and SocA localize into three distinct clades among SCAD enzymes (Fig. 4A). The first group, which includes SocA, is annotated as 3-oxoacyl-acyl carrier protein reductases based on homology with E. coli FabG, an essential enzyme required for fatty acid biosynthesis. Other members of this group include HetN, a protein of unknown function required for heterocyst differentiation in Anabaena and Nostoc (Black and Wolk 1994; Liu and Chen 2009) and NodG, which is essential for nodulation of legumes by Rhizobium (Lopez-Lara and Geiger 2001). Interestingly, HetN contains a signaling motif separate from its catalytic site (Higa et al. 2012) that is not conserved in other homologs. The second clade contains enzymes annotated as hydroxysteroid dehydrogenases in humans, Comamonas, and Streptomyces, including HSD10. The final clade includes CsgA and Drosophila Sniffer.
Figure 4.
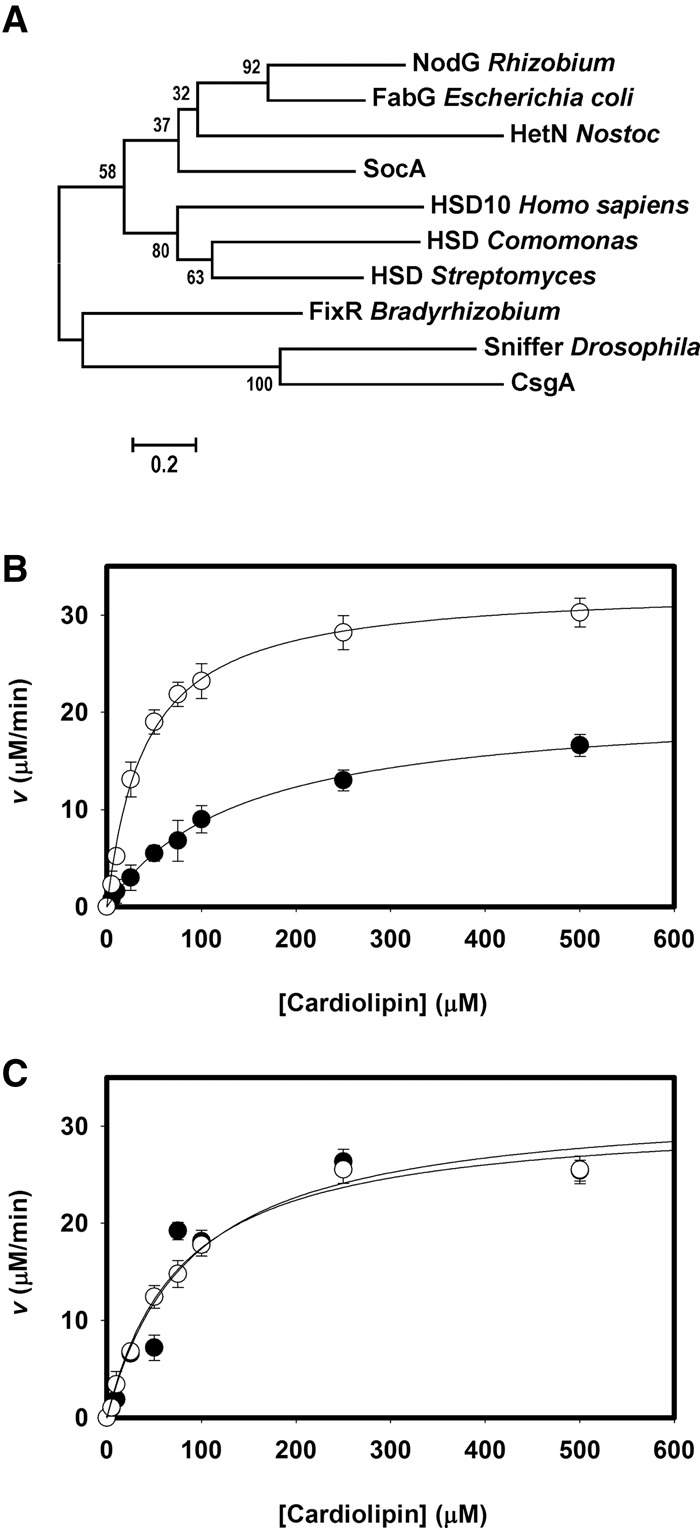
CsgA represents a class of enzymes capable of CL oxidation, some of which may be important during oxidative stress by removing CL peroxides. (A) Cladogram showing CsgA homologs clustering into three groups, with either SocA, HSD10, or CsgA. (B) Kinetic analyses of HSD10 using CL 18:2 (•) or CL 18:2 peroxide (○) as a substrate. (C) Kinetic analyses of Drosophila sniffer using CL 18:2 (•) or CL 18:2 peroxide (○) as a substrate. Error bars indicate standard deviation of three replicates.
Both Sniffer and HSD10 oxidize CL in vitro with NAD+ (Table 1). Sniffer displays kinetics similar to CsgA, while HSD10 activity is intermediate between CsgA and SocA. These results point to a diverse class of SCADs in both prokaryotes and eukaryotes capable of oxidizing CL. Three HSD10 mutants known to cause mitochondrial disease in humans—D86G, R130C, and Q165H—are all unable to oxidize CL. Sniffer can replace NAD+ with NADP+ as a cofactor during oxidation of CL, while HSD10 cannot (Supplemental Fig. S3).
CL peroxidation is required for mitochondrial-induced apoptosis. Since HSD10 and Sniffer are thought to be neuroprotective and since mutations result in mitochondrial disrepair, we assayed these enzymes for catalysis using CL substrates containing oxidized fatty acids. In the presence of O2, polyunsaturated fatty acids form lipid radicals. CL bearing four tetralinoleoyl (C18:2) fatty acids was oxidized by exposure to air, verified by colorimetric assay, and assessed as a potential substrate. HSD10 activity increases greatly in the presence of the oxidized C18:2 (CLox) species compared with unoxidized fatty acids (Fig. 4B). The catalytic efficiency under these conditions was twice that of CsgA due to greater affinity for the substrate (Table 1). HSD10 displayed activity with the unoxidized C18:2 species equivalent to that seen with the C14:0 species. Sniffer was also examined in the same manner (Fig. 4C), but no difference was seen, both yielding kinetics similar to that of the C14:0 form (Table 1). Sniffer does not appear to prefer one state over another, but the efficiency for all CL substrates is comparable with that of CsgA.
The Aβ peptide inhibits oxidation of CL by HSD10
Some previously characterized HSD10 activities are inhibited upon binding of the Aβ peptide. Aβ binds tightly to HSD10 and is thought to cause inhibition by altering the active site conformation (Yan et al. 2007). To determine whether inhibition of HSD10 occurs with CLox, we assayed HSD10 activity in the presence of the Aβ peptide (amino acids 1–42) (Fig. 5A). Addition of 10 μM Aβ greatly reduced enzymatic activity and displayed noncompetitive inhibition. Inhibitory kinetics were determined over a course of Aβ concentrations, and the inhibitor constant (Ki) was determined to be 2.5 μM (Fig. 5B), in agreement with previous reports of HSD10 inhibition using a soluble substrate (Muirhead et al. 2010).
Figure 5.
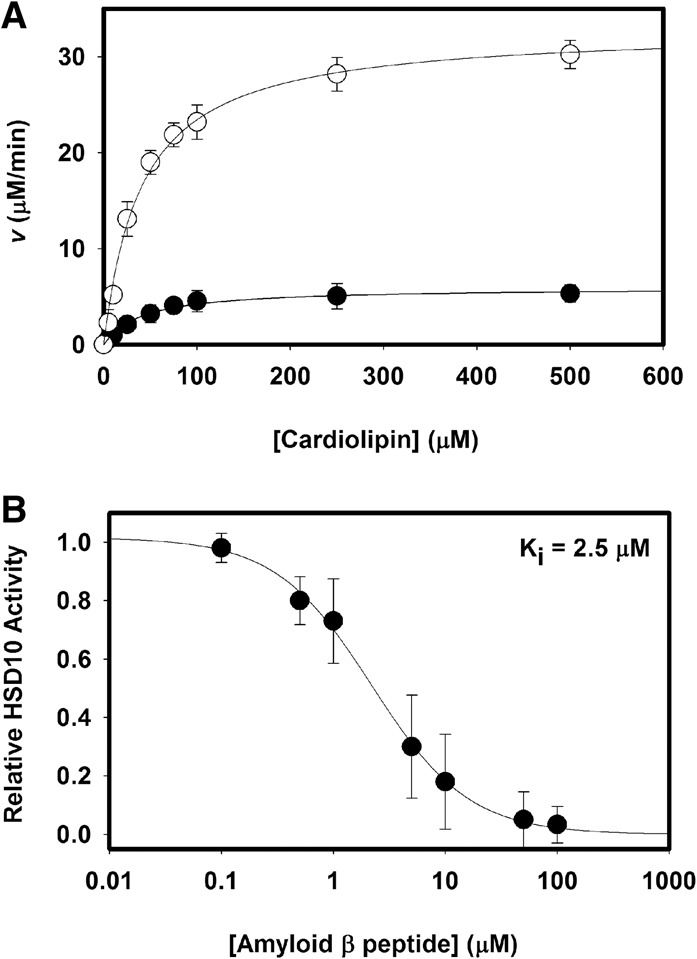
The enzymatic activity of HSD10 is inhibited by Aβ. (A) Kinetic activity of HSD10 with the substrate CLox in the presence (•) or absence (○) of 10 mM Aβ (1–42) peptide. (B) Aβ inhibition kinetics as measured by apparent Vmax changes relative to wild type. Aβ displayed noncompetitive inhibition on HSD10 activity with a Ki = 2.5 µM. Error bars indicate standard deviation of three replicates.
Discussion
We describe a widespread and novel phospholipase C-like enzyme activity with some members of the SCAD family. A single proton abstraction from the 2′-OH of the CL or PG head group leads to the release of Pi, DAG, and DHA. As an added benefit, cells recover one NADH molecule in comparison with a conventional phospholipase C reaction. This activity appears to be essential for the development of M. xanthus. This enzymatic reaction seems to be repurposed in eukaryotes, where it is likely to be important in mitochondrial function.
CsgA is required for lipid body synthesis in Myxococcus
Myxococcus cells lacking CsgA fail to enter multicellular development. These mutants have an unusual property, however, by which mixture with wild-type cells efficiently restores mutant development. The nature of what is transferred from the wild type to stimulate complementation is a matter of debate. The current model suggests that extracellular complementation is achieved through cell surface recognition of a truncated CsgA protein (Lobedanz and Sogaard-Andersen 2003). We propose a new model built around the enzymatic activity of CsgA and show that SocA and CsgA have common substrates in vitro, albeit with different kinetic parameters. CsgA has the highest catalytic efficiency with CL and PG, components of the Myxococcus inner membrane (Orndorff and Dworkin 1980), where CsgA has been shown to reside (Simunovic et al. 2003). The SocA affinity constant (Km) for CL is nearly 10-fold higher than that of CsgA, leading to a 13-fold reduction in catalytic efficiency (Table 1). This lower catalytic efficiency of SocA may explain the requirement for SocA up-regulation in order to replace CsgA.
During development, wild-type M. xanthus cells produce lipid bodies, while csgA mutants do not. We show that an extract of partial glycerides, including DAGs from developing wild-type cells, is capable of restoring lipid body production to csgA cells. From these results, we propose that TAGs are constructed in two steps. CL and PG are converted to DAGs by removal of the head group with CsgA. Next, an acyltransferase adds the final fatty acid to complete the TAG (Fig. 6A). Production of DAGs within a phospholipid membrane can induce negative curvature to initiate vesicle formation in eukaryotes (Szule et al. 2002). CsgA could initiate vesicle formation of lipid bodies in Myxococcus by a similar mechanism.
Figure 6.
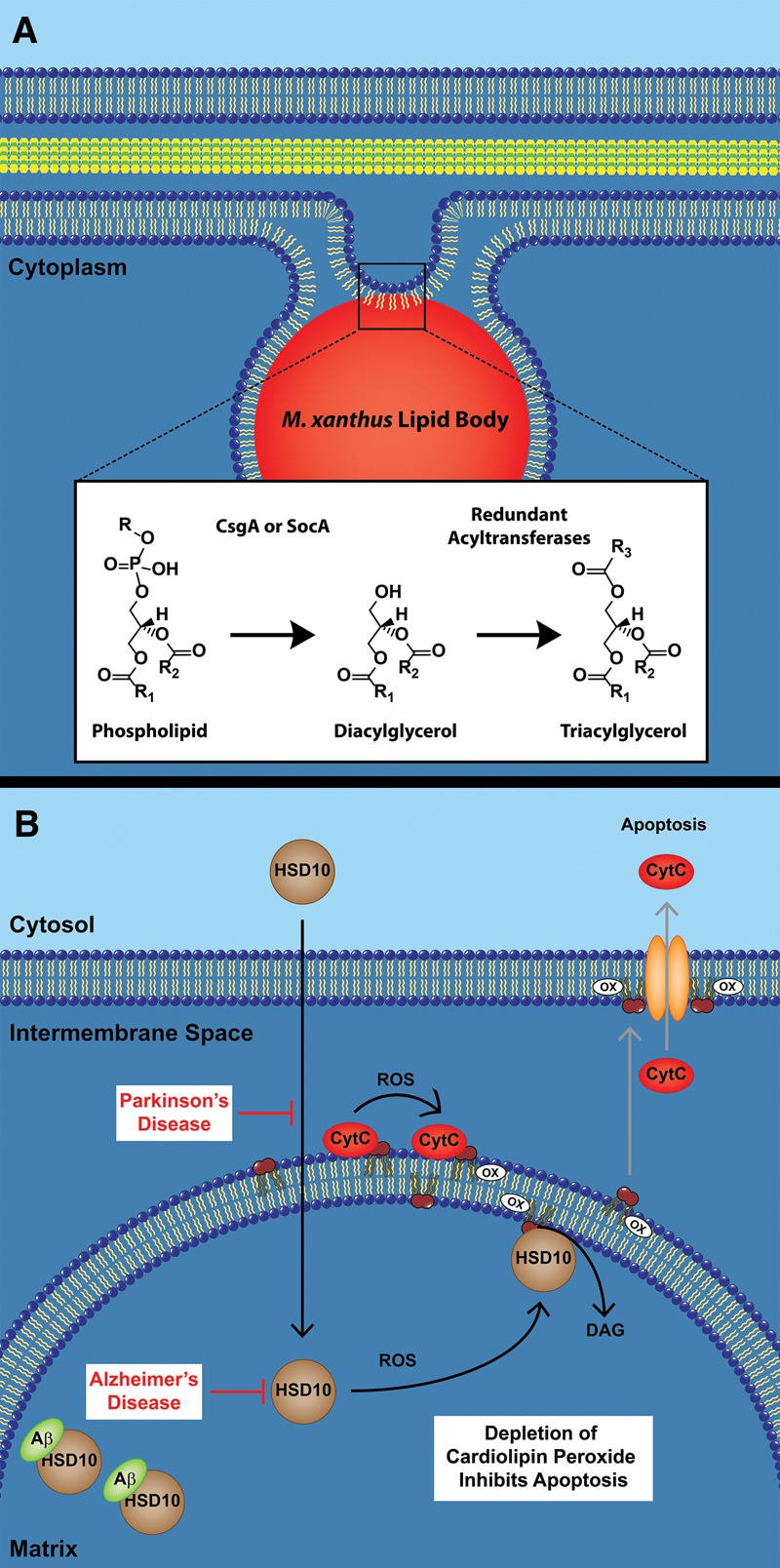
Proposed functions of CsgA and HSD10 in Myxococcus and humans. (A) M. xanthus lipid bodies are synthesized from existing membrane phospholipids. This reaction is proposed to occur directly from the breakdown of PG and CL into DAGs by CsgA. An as yet uncharacterized acyltransferase could then convert the DAGs into TAGs. (B) In the presence of ROS created from oxidative stress, HSD10 is recruited to the inner mitochondrial membrane where CytC simultaneously converts CL to CLox. HSD10 removes CLox before it is exported to the outer membrane, and CytC is released, inhibiting apoptosis. In Parkinson's disease, HSD10 levels within the mitochondria are greatly diminished. In Alzheimer's disease, HSD10 is inactivated within the matrix by the binding of the Aβ peptide. In both instances, oxidative damage occurs, leading to apoptosis.
HSD10 is a multifunctional enzyme involved in mitochondrial disease
Reduction or loss of HSD10 in Xenopus, mice, or humans diminishes mitochondrial integrity and enhances apoptotic death (Rauschenberger et al. 2010). HSD10 was initially described in isoleucine catabolism (Zschocke 2012), and the original hypothesis was given that patients with HSD17B10 mutations suffered from buildup of pathway intermediates. This particular activity is not directly correlated with human disease, however, since an isoleucine-restricted diet did not relieve symptoms. Furthermore, the R130C and D86G mutations associated with severe symptoms still produced 64% and 30%, respectively, of wild-type activity, while Q165H, which causes milder clinical symptoms, has <3% activity. HSD10 is also capable of steroid metabolism, where allopregnanolone (ALLOP), a potent ligand of the GABAA receptor, is oxidized to the neuro-inactive molecule 5α-dihydroprogesterone (Melcangi and Panzica 2014). HSD10 could play a role in brain development and cognitive function, but ALLOP metabolism has not yet been studied in patients with HSD10 deficiency.
A dramatically different model for the human disease state involves HSD10 as an essential component of the RNase P complex (Holzmann et al. 2008). This complex is required for 5′ cleavage of mitochondrial tRNAs from the polycistronic precursor transcript and contains the proteins MRPP1, MRPP2 (HSD10), and MRPP3, each of which is essential for correct processing in vitro. Mutation or knockdown of HSD17B10 impairs this step and can result in loss of mitochondrial integrity (Deutschmann et al. 2014). Expression of the HSD10 mutant Q165H, which has nearly complete loss of NAD+ binding, appears to alleviate this loss of function, indicating that dehydrogenase activity is unnecessary (Rauschenberger et al. 2010). More detailed results with other mutant alleles support the idea that RNA processing and tRNA methylation are compromised but leave open the idea that dehydrogenase activity is important (Chatfield et al. 2015; Vilardo and Rossmanith 2015).
It is against this backdrop of alternate HSD10 activities that we introduce a novel and unexpected function for HSD10 as a CL-specific phospholipase C-like enzyme. This activity may be relevant to the mitochondrial disease state experienced by carriers of HSD10 mutations. HSD10 has excellent kinetic constants with CL compared with other known substrates. On the other hand, all three mutant proteins lack activity on CL despite the fact that Q165H supports better neurological development than the other two. It appears that more work will be necessary to understand the precise role of this activity in familial neurodegenerative disorders.
HSD10 may mediate CL homeostasis
Mitochondria are massive oxygen consumers that provide vast amounts of energy but leave toxic waste products in their wake. Between 0.15% (St-Pierre et al. 2002) and 2% (Chance et al. 1979) of mitochondrial oxygen is converted into the potent ROS hydrogen peroxide. We propose that HSD10 is part of a previously unrecognized pathway to reduce the ROS burden of stressed cells and ameliorate the damage.
In eukaryotic cells, CL is found predominantly in the mitochondria, where it induces negative curvature to help form the highly folded cristae. Proper formation of cristae requires extensive deacylation and acylation of CL (Mejia and Hatch 2015). Within the cristae, CL stabilizes the respiratory complexes involved in electron transfer (Zhang et al. 2002). Perturbation of these complexes increases production of ROS (Dudek et al. 2013) leading to peroxidation of CL and apoptosis. The major contributor to CL peroxidation is cytochrome c (CytC). During respiration, CytC functions to transfer an electron from respiratory complex III to respiratory complex IV. The CytC structure in this role is well-studied and involves a heme moiety covalently attached by two thioether linkages. In a separate function, CytC can also bind CL and induces a disordered structure that has presented a challenge to study but has also led to an appreciation that these changes provoke the transition from electron carrier to peroxidase (Muenzner and Pletneva 2014). Once bound to CytC, the polyunsaturated fatty acids of CL are oxidized in the presence of hydrogen peroxide (Kagan et al. 2005, 2009).
The formation of CLox is essential for release of proapoptotic factors such as CytC from the mitochondria into the cytosol (Petrosillo et al. 2009). For apoptosis to occur, CLox must first be translocated from the inner membrane to the outer membrane (Garcia Fernandez et al. 2002). While controlled apoptosis is necessary to produce form and function in developing organisms (for example, the digitation of fingers and toes during embryogenesis), rampant apoptosis causes necrosis. In the latter case, the CytC–CL complex performs a seemingly subversive function. We suggest to the contrary that CytC peroxidase performs a valuable, regulated response to ROS by using HSD10 to remove CLox before oxidative stress progresses to apoptosis (Fig. 6B). This is a dangerous proposition given the autocatalytic nature of the lipid oxidation pathway and can be easily derailed by diminished HSD10 activity or abundance. In addition, HSD10 would create DAGox, a product that can activate cell signaling pathways outside of the mitochondria, such as protein kinase C, a well-known enzyme that blocks further effects of oxidative stress (Gopalakrishna and Jaken 2000; Cosentino-Gomes et al. 2012). DAGox is known to activate these pathways similarly and in some cases in greater magnitude than DAG (Kambayashi et al. 2007; Takekoshi et al. 2014).
Alzheimer's disease, Parkinson's disease, and Drosophila sniffer have increased ROS
A unifying theme in HSD10 and Sniffer disease states is increased levels of oxidative stress. In Drosophila, the P-element insertion into the X-chromosomal sniffer gene causes reduced life span, neurodegeneration, and sluggish walking that deteriorates with age (Botella et al. 2004). sniffer is distinct from the Drosophila version of HSD17B10, called scully, which appears to be an essential gene (Torroja et al. 1998). The sniffer phenotype worsens in a hyperoxic environment and is relieved upon overexpression of sniffer, suggesting oxidative stress as the major determinant. The cellular location of Sniffer is unknown, but if it is located in the mitochondria and in proximity to CLox, it could provide protective value through removal of this radical. It might also convert CytC–CL into the soluble and energetically favorable form of CytC.
In humans, HSD10 is normally found in the brain and cerebral spinal fluid, where it becomes elevated in Alzheimer's disease patients (Yang et al. 2014). HSD10 binds Aβ and is suspected of direct involvement in Alzheimer's disease neurodegeneration (Yan et al. 1997). Interestingly, Aβ binding does not exhibit a deleterious effect on HSD10 involvement in the RNase P complex, suggesting that the leading theory of HSD10 function in mitochondria is not involved in Alzheimer's disease (Vilardo and Rossmanith 2013). Our work shows that Aβ binding inhibits the CL phospholipase activity of HSD10 despite the substantially larger and more complex hydrophobic substrate in comparison with other previous known substrates. These results suggest that the Alzheimer's disease state could be at least partially due to inactivation of HSD10, which would be expected to block removal of CLox and stimulate apoptosis (Fig. 6B).
HSD10 may play a somewhat analogous role in some forms of Parkinson's disease. Nearly 40% of the Parkinson's disease cases beginning before age 45 are due to PARK2 mutations. These mutations prevent ubiquitylation of HSD10, leading to reduced import into the mitochondria (Bertolin et al. 2015). Disruption of HSD10 import would effectively remove its ability to deplete CLox levels and could lead to rampant apoptosis (Fig. 6B). Thus, one could consider Parkinson's and Alzheimer's as diseases where HSD10 is depleted or inactivated, respectively. Mitochondrial dysfunction due to ROS has been proposed to play a major role in these two diseases (Nunomura et al. 2006; Schapira 2006). In a broad sense, oxidative stress is correlated with all age-related degenerative disorders. Unfortunately, it has been consistently ignored in mechanistic studies that attempt to discern a path to the disease state.
Conclusions
CsgA, Sniffer, and HSD10 have similar enzyme activities on CL but different purposes in their respective organisms. The Myxococcus saturated and monounsaturated fatty acids are not a ready target for peroxidation, so this organism has little need to repair/replace lipids whose fatty acids have become oxidized. However, CsgA phospholipase C activity appears to provide a direct path to shape change that accompanies sporulation. Our work predicts that CsgA does so by initiating the conversion of excess CL and PG into DAGs that can be converted to TAGs. Future experiments can be devoted to identifying the molecular complexes at work during sporulation, such as the conversion and relocation of membrane phospholipids into lipid body TAGs. Specific probes can be developed to quantify the carbon flux through these lipid metabolic pathways.
Humans have robust pathways for making lipid bodies and would have no need to derive lipid bodies from existing phospholipids except under dire nutritional conditions. Fatty acids are fed into mitochondria and oxidized as the principle energy source. Here, HSD10 appears to be a beneficial gatekeeper that removes damaged CL and in so doing blocks the apoptotic pathway. Our results suggest that quantification of CLox and perhaps DAGox in normal and disease states, particularly those molecules in proximity to CytC, should figure prominently into metrics and mechanisms regarding the maintenance of healthy cells and the development of neurodegenerative disorders.
Materials and methods
Bacterial strains, plasmids, and recombinant proteins
All strain and plasmids used in this study are listed in Supplemental Table S1. M. xanthus and E. coli strains were grown in CYE (1.0% Bacto Casitone, 0.5% Difco yeast extract, 10 mM 3-[N-morpholino] propanesulfonic acid at pH 7.6, 0.1% MgSO4) and LB, respectively, and supplemented with 50 µg mL−1 kanamycin or 50 µg mL−1 ampicillin where appropriate. M. xanthus and E. coli were grown at 32°C and 37°C, respectively.
pTOB10 contains the full-length csgA gene (MXAN_RS06225; MXAN_1294) codon optimized for E. coli and synthesized by Genscript bearing 5′ NheI and 3′ HindIII restriction sequences (Supplemental Fig. S1). The gene was ligated into the respective sites of the commercial vector pTrcHisB (Invitrogen) to create the p25 expression plasmid pTOB12. For the p17 expression vector pTOB24, the appropriate sequence was obtained from pTOB10 through PCR with 5′ NheI and 3′ HindIII flanking restriction sequences and ligated into pTrcHisB. The beginning of the p17 sequence is denoted in Supplemental Figure S1 and is taken from the previously proposed start site for the processed protein (Lobedanz and Sogaard-Andersen 2003). For the CsgA K155R mutant expression plasmid, pTOB57 was constructed via QuickChange PCR (Agilent Technologies) using primers designed by PrimerX (http://www.bioinformatics.org/primerx) to change codon AAA to AGA.
Expression and purification of recombinant proteins
All recombinant CsgA proteins were expressed as 6xHis-tagged in E. coli grown overnight in 1 L of Circlegrow medium (MP Biomedicals) and then purified from inclusion bodies (IBs) resulting in a yield of ∼5 mg of protein per liter of culture. Cells were lysed by sonication in solubilization buffer (50 mM Tris-buffered MOPS at pH 8.0, 100 mM KCl, 1% sodium cholate) and centrifuged at 100,000g for 30 min. The resulting pellet was resuspended in solubilization buffer containing 1% Triton X-100 and again harvested at 100,000g for 30 min to enrich IB purity. This pellet was resuspended in 40 mL of TE buffer (100 mM Tris-HCl at pH 8.0, 1 mM EDTA) and centrifuged at 1100g for 5 min. Supernatant was collected and centrifuged at 12,000g for 3 min to pellet IBs. IBs were washed three times with 40 mL of TE buffer containing 2% Triton X-114. The final pellet was resuspended in 5 mL of TE containing 1.5% sodium lauryl sulfate (SLS) and dialyzed overnight in solubilization buffer. Protein was then diluted in 50 mL of solubilization buffer and purified as previously describe using HisPur cobalt resin (Thermo Scientific) (Boynton et al. 2013). SocA, HSD10 variant, and Sniffer expression was induced by 1 mM IPTG, and proteins were purified directly as soluble proteins. Cells were lysed in solubilization buffer and directly applied to cobalt affinity columns as above. The purity and integrity of all proteins were analyzed by SDS-PAGE and Coomassie blue staining. Protein analysis and quantification was carried out using UV-Vis spectrometry and the predicted extinction coefficients for each protein.
Development of M. xanthus
For the development of Myxococcus cells, cultures were first grown in CYE to a density of 5 × 108 cells per milliliter. Cells were then harvested by centrifugation at 10,000 rpm for 10 min and resuspended to a final concentration of 5 × 109 cells per milliliter. The resulting suspension was plated onto TPM agar (10 mM Tris-HCl, 8 mM MgSO4, 1 mM K-PO4 at pH 7.6, 1.5% agar) to initiate development and incubated for appropriate periods at 32°C. For quantification of spores, spots were taken at 5 d post-starvation, resuspended in 1 mL of dH2O, and incubated for 1 h at 55°C followed by brief sonication to kill vegetative cells. Spores were counted directly using a Petroff-Hauser chamber.
For submerged culture, cultures were grown to a concentration of 5 × 108 cells per milliliter in CYE and then diluted to a calculated density of 1 × 106 cells per milliliter. One milliliter of these cells was added to a 15-mm diameter well of a 24-well culture dish and incubated for 24 h at 32°C to induce biofilm. CYE was aspirated off, and the biofilm was washed gently with 1 mL of MOPS buffer (10 mM MOPS at pH 7.2, 1 mM CaCl2, 4 mM MgCl2) to remove residual growth medium. This was again aspirated and replaced with 1 mL of MOPS buffer, and the cells were incubated for 48 h at 32°C to initiate development. For complementation, recombinant protein was added at appropriate concentrations directly to buffer prior to incubation.
Enzyme assays
CsgA activity was assayed in a manner similar to that previously described for the CsgA homolog SocA (Avadhani et al. 2006). One-milliliter reaction mixtures consisted of TE buffer (10 mM Tris-HCl at pH 8.0, 1 mM EDTA) containing 0.1% (w/v) sodium cholate, 200 nM CsgA, 100 μM NAD+, and varying amounts of substrate. Activity was measured as the rate of NADH formation over 10 min via absorbance at 340 nm. For kinetics, NAD+ was increased to 1 mM, and data were fitted to the following equation:
where Vmax is the apparent maximum rate, and Km is the apparent affinity constant. Whole-cell lipid extracts were obtained from developing M. xanthus DK1622 cells using the Bligh-Dyer method (Bligh and Dyer 1959) and reconstituted at 5 mg mL−1. Synthetic substrates were obtained from Avanti Lipids and contained C14:0 side chains with the exception of C18:2 CL obtained from Sigma-Aldrich. For peroxidation of C18:2 CL, lipid was exposed to air for 48 h and detected using the PeroxiDetect kit from Sigma-Aldrich. For Aβ inhibition studies, HSD10 enzyme was first incubated with the Aβ (1–42) peptide for 2 h prior to enzyme assay. Synthetic human Aβ (1–42) peptide was obtained from Tocris, dissolved in DMSO to a concentration of 1 M, and sonicated before being diluted to appropriate concentrations in buffer containing enzyme.
Enzymatic product identification
DAG was identified by collecting the organic phase of a completed reaction through chloroform/methanol (2:1) extraction. Samples were dried under nitrogen, resuspended in methanol containing 100 μM sodium acetate, and identified with mass spectrometry using a Shimadzu LCMS-IT-TOF instrument. Peaks were recorded as Na+ adducts. From the soluble phase, Pi was quantified using a detection system from Innova Biosciences. DHA was quantified by absorbance at 470 nm by first adding 0.5 mL of 6% α-napthol and then 0.2 mL of 40% potassium hydroxide (Turner et al. 1942). Glycerol was assayed at 570 nm by the coupled enzymatic glycerol assay kit from Sigma-Aldrich.
Complementation and lipid body restoration
To assess complementation of LS3931 with partial glycerides, lipids were extracted from cells 24 h into development using the Bligh-Dyer method (Bligh and Dyer 1959). This extract was separated by solid-phase separation using LC-Si to obtain the partial glyceride fraction as previously described (Hamilton and Comai 1988). Partial glycerides were further purified by thin-layer chromatography (TLC) on silica gel using toluene/chloroform/methanol (85:15:5). A band with an Rf value of 0.55 was extracted and dried under nitrogen. For complementation, various amounts (by weight) of lipids dissolved in DMSO were added 1:10 to cells prior to development. After 24 h, complementation was indicated by fruiting bodies, and cells were stained with 5 μg mL−1 Nile red to view lipid bodies as previously described (Bhat et al. 2014). Lipid bodies were quantified by averaging the fluorescent intensity of individual cells normalized to cell length.
Supplementary Material
Acknowledgments
We thank Chengli Zong and Andre Venot for their help with mass spectrometry, Robert Phillips for his thoughtful insight into the proposed reaction mechanism, Hans-Joerg Martin for generously providing the sniffer plasmid, Albert Amberger for generously providing HSD10 wild-type and mutant plasmids, Anthony Shimkets for purifying the lipids, and Christopher Cotter for critical reading of the manuscript. The research reported here was supported by the National Institute of General Medical Science of the National Institutes of Health under award number R01GM095826 and the National Science Foundation under award number MCB-1411891.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.268482.115.
References
- Avadhani M, Geyer R, White DC, Shimkets LJ. 2006. Lysophosphatidylethanolamine is a substrate for the short-chain alcohol dehydrogenase SocA from Myxococcus xanthus. J Bacteriol 188: 8543–8550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolin G, Jacoupy M, Traver S, Ferrando-Miguel R, Saint Georges T, Grenier K, Ardila-Osorio H, Muriel MP, Takahashi H, Lees AJ, et al. 2015. Parkin maintains mitochondrial levels of the protective Parkinson's disease-related enzyme 17-β hydroxysteroid dehydrogenase type 10. Cell Death Differ 10.1038/cdd.2014.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat S, Zhu X, Patel RP, Orlando R, Shimkets LJ. 2011. Identification and localization of Myxococcus xanthus porins and lipoproteins. PLoS One 6: e27475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat S, Boynton TO, Pham D, Shimkets LJ. 2014. Fatty acids from membrane lipids become incorporated into lipid bodies during Myxococcus xanthus differentiation. PLoS One 9: e99622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black TA, Wolk CP. 1994. Analysis of a Het- mutation in Anabaena sp. strain PCC 7120 implicates a secondary metabolite in the regulation of heterocyst spacing. J Bacteriol 176: 2282–2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bligh EG, Dyer WJ. 1959. A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37: 911–917. [DOI] [PubMed] [Google Scholar]
- Borger E, Aitken L, Du H, Zhang W, Gunn-Moore FJ, Yan SS. 2013. Is amyloid binding alcohol dehydrogenase a drug target for treating Alzheimer's disease? Curr Alzheimer Res 10: 21–29. [PMC free article] [PubMed] [Google Scholar]
- Botella JA, Ulschmid JK, Gruenewald C, Moehle C, Kretzschmar D, Becker K, Schneuwly S. 2004. The Drosophila carbonyl reductase sniffer prevents oxidative stress-induced neurodegeneration. Curr Biol 14: 782–786. [DOI] [PubMed] [Google Scholar]
- Boynton TO, McMurry JL, Shimkets LJ. 2013. Characterization of Myxococcus xanthus MazF and implications for a new point of regulation. Mol Microbiol 87: 1267–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chance B, Schoener B, Oshino R, Itshak F, Nakase Y. 1979. Oxidation-reduction ratio studies of mitochondria in freeze-trapped samples. J Biol Chem 254: 4764–4771. [PubMed] [Google Scholar]
- Chatfield KC, Coughlin CR II, Friederich MW, Gallagher RC, Hesselberth JR, Lovell MA, Ofman R, Swanson MA, Thomas JA, Wanders RJ, et al. 2015. Mitochondrial energy failure in HSD10 disease is due to defective mtDNA transcript processing. Mitochondrion 21: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb CA, Cole MP. 2015. Oxidative and nitrative stress in neurodegeneration. Neurobiol Dis 10.1016/j.nbd.2015.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosentino-Gomes D, Rocco-Machado N, Meyer-Fernandes JR. 2012. Cell signaling through protein kinase C oxidation and activation. Int J Mol Sci 13: 10697–10721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutschmann AJ, Amberger A, Zavadil C, Steinbeisser H, Mayr JA, Feichtinger RG, Oerum S, Yue WW, Zschocke J. 2014. Mutation or knock-down of 17β-hydroxysteroid dehydrogenase type 10 cause loss of MRPP1 and impaired processing of mitochondrial heavy strand transcripts. Hum Mol Genet 23: 3618–3628. [DOI] [PubMed] [Google Scholar]
- Dudek J, Cheng IF, Balleininger M, Vaz FM, Streckfuss-Bomeke K, Hubscher D, Vukotic M, Wanders RJ, Rehling P, Guan K. 2013. Cardiolipin deficiency affects respiratory chain function and organization in an induced pluripotent stem cell model of Barth syndrome. Stem Cell Res 11: 806–819. [DOI] [PubMed] [Google Scholar]
- Garcia Fernandez M, Troiano L, Moretti L, Nasi M, Pinti M, Salvioli S, Dobrucki J, Cossarizza A. 2002. Early changes in intramitochondrial cardiolipin distribution during apoptosis. Cell Growth Differ 13: 449–455. [PubMed] [Google Scholar]
- Gardner HW. 1989. Oxygen radical chemistry of polyunsaturated fatty acids. Free Radic Biol Med 7: 65–86. [DOI] [PubMed] [Google Scholar]
- Gopalakrishna R, Jaken S. 2000. Protein kinase C signaling and oxidative stress. Free Radic Biol Med 28: 1349–1361. [DOI] [PubMed] [Google Scholar]
- Hagen TJ, Shimkets LJ. 1990. Nucleotide sequence and transcriptional products of the csg locus of Myxococcus xanthus. J Bacteriol 172: 15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagen DC, Bretscher AP, Kaiser D. 1978. Synergism between morphogenetic mutants of Myxococcus xanthus. Dev Biol 64: 284–296. [DOI] [PubMed] [Google Scholar]
- Hamilton JG, Comai K. 1988. Rapid separation of neutral lipids, free fatty acids and polar lipids using prepacked silica Sep-Pak columns. Lipids 23: 1146–1149. [DOI] [PubMed] [Google Scholar]
- He XY, Wen GY, Merz G, Lin D, Yang YZ, Mehta P, Schulz H, Yang SY. 2002. Abundant type 10 17 β-hydroxysteroid dehydrogenase in the hippocampus of mouse Alzheimer's disease model. Brain Res Mol Brain Res 99: 46–53. [DOI] [PubMed] [Google Scholar]
- Higa KC, Rajagopalan R, Risser DD, Rivers OS, Tom SK, Videau P, Callahan SM. 2012. The RGSGR amino acid motif of the intercellular signalling protein, HetN, is required for patterning of heterocysts in Anabaena sp. strain PCC 7120. Mol Microbiol 83: 682–693. [DOI] [PubMed] [Google Scholar]
- Hoiczyk E, Ring MW, McHugh CA, Schwar G, Bode E, Krug D, Altmeyer MO, Lu JZ, Bode HB. 2009. Lipid body formation plays a central role in cell fate determination during developmental differentiation of Myxococcus xanthus. Mol Microbiol 74: 497–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holkenbrink C, Hoiczyk E, Kahnt J, Higgs PI. 2014. Synthesis and assembly of a novel glycan layer in Myxococcus xanthus spores. J Biol Chem 289: 32364–32378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzmann J, Frank P, Loffler E, Bennett KL, Gerner C, Rossmanith W. 2008. RNase P without RNA: identification and functional reconstitution of the human mitochondrial tRNA processing enzyme. Cell 135: 462–474. [DOI] [PubMed] [Google Scholar]
- Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, Osipov AN, Belikova NA, Kapralov AA, Kini V, et al. 2005. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol 1: 223–232. [DOI] [PubMed] [Google Scholar]
- Kagan VE, Bayir HA, Belikova NA, Kapralov O, Tyurina YY, Tyurin VA, Jiang J, Stoyanovsky DA, Wipf P, Kochanek PM, et al. 2009. Cytochrome c/cardiolipin relations in mitochondria: a kiss of death. Free Rad Biol Med 46: 1439–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahnt J, Aguiluz K, Koch J, Treuner-Lange A, Konovalova A, Huntley S, Hoppert M, Sogaard-Andersen L, Hedderich R. 2010. Profiling the outer membrane proteome during growth and development of the social bacterium Myxococcus xanthus by selective biotinylation and analyses of outer membrane vesicles. J Proteome Res 9: 5197–5208. [DOI] [PubMed] [Google Scholar]
- Kambayashi Y, Takekoshi S, Tanino Y, Watanabe K, Nakano M, Hitomi Y, Takigawa T, Ogino K, Yamamoto Y. 2007. Various molecular species of diacylglycerol hydroperoxide activate human neutrophils via PKC activation. J Clin Biochem Nutr 41: 68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SK, Kaiser D. 1991. C-factor has distinct aggregation and sporulation thresholds during Myxococcus development. J Bacteriol 173: 1722–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristofikova Z, Bockova M, Hegnerova K, Bartos A, Klaschka J, Ricny J, Ripova D, Homola J. 2009. Enhanced levels of mitochondrial enzyme 17β-hydroxysteroid dehydrogenase type 10 in patients with Alzheimer disease and multiple sclerosis. Mol BioSyst 5: 1174–1179. [DOI] [PubMed] [Google Scholar]
- Lee K, Shimkets LJ. 1994. Cloning and characterization of the socA locus which restores development to Myxococcus xanthus C-signaling mutants. J Bacteriol 176: 2200–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Shimkets LJ. 1996. Suppression of a signaling defect during Myxococcus xanthus development. J Bacteriol 178: 977–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BU, Lee K, Mendez J, Shimkets LJ. 1995. A tactile sensory system of Myxococcus xanthus involves an extracellular NAD(P)+-containing protein. Genes Dev 9: 2964–2973. [DOI] [PubMed] [Google Scholar]
- Li S, Lee BU, Shimkets LJ. 1992. csgA expression entrains Myxococcus xanthus development. Genes Dev 6: 401–410. [DOI] [PubMed] [Google Scholar]
- Liu J, Chen WL. 2009. Characterization of HetN, a protein involved in heterocyst differentiation in the cyanobacterium Anabaena sp. strain PCC 7120. FEMS Microbiol Letters 297: 17–23. [DOI] [PubMed] [Google Scholar]
- Lobedanz S, Sogaard-Andersen L. 2003. Identification of the C-signal, a contact-dependent morphogen coordinating multiple developmental responses in Myxococcus xanthus. Genes Dev 17: 2151–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Lara IM, Geiger O. 2001. The nodulation protein NodG shows the enzymatic activity of an 3-oxoacyl-acyl carrier protein reductase. Mol Plant Microbe Interact 14: 349–357. [DOI] [PubMed] [Google Scholar]
- Lorenzen W, Bozhuyuk KA, Cortina NS, Bode HB. 2014. A comprehensive insight into the lipid composition of Myxococcus xanthus by UPLC-ESI-MS. J Lipid Res 55: 2620–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin HJ, Ziemba M, Kisiela M, Botella JA, Schneuwly S, Maser E. 2011. The Drosophila carbonyl reductase sniffer is an efficient 4-oxonon-2-enal (4ONE) reductase. Chem Biol Interact 191: 48–54. [DOI] [PubMed] [Google Scholar]
- Mejia EM, Hatch GM. 2015. Mitochondrial phospholipids: role in mitochondrial function. J Bioenerg Biomembr 10.1007/s10863-015-9601-4. [DOI] [PubMed] [Google Scholar]
- Melcangi RC, Panzica GC. 2014. Allopregnanolone: state of the art. Prog Neurobiol 113: 1–5. [DOI] [PubMed] [Google Scholar]
- Muenzner J, Pletneva EV. 2014. Structural transformations of cytochrome c upon interaction with cardiolipin. Chem Phys Lipids 179: 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muirhead KE, Froemming M, Li X, Musilek K, Conway SJ, Sames D, Gunn-Moore FJ. 2010. (−)-CHANA, a fluorogenic probe for detecting amyloid binding alcohol dehydrogenase HSD10 activity in living cells. ACS Chem Biol 5: 1105–1114. [DOI] [PubMed] [Google Scholar]
- Nunomura A, Castellani RJ, Zhu X, Moreira PI, Perry G, Smith MA. 2006. Involvement of oxidative stress in Alzheimer disease. J Neuropathol Exp neurology 65: 631–641. [DOI] [PubMed] [Google Scholar]
- O'Connor KA, Zusman DR. 1991. Development in Myxococcus xanthus involves differentiation into two cell types, peripheral rods and spores. J Bacteriol 173: 3318–3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orndorff PE, Dworkin M. 1980. Separation and properties of the cytoplasmic and outer membranes of vegetative cells of Myxococcus xanthus. J Bacteriol 141: 914–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrosillo G, Moro N, Ruggiero FM, Paradies G. 2009. Melatonin inhibits cardiolipin peroxidation in mitochondria and prevents the mitochondrial permeability transition and cytochrome c release. Free Radic Biol Med 47: 969–974. [DOI] [PubMed] [Google Scholar]
- Rauschenberger K, Scholer K, Sass JO, Sauer S, Djuric Z, Rumig C, Wolf NI, Okun JG, Kolker S, Schwarz H, et al. 2010. A non-enzymatic function of 17β-hydroxysteroid dehydrogenase type 10 is required for mitochondrial integrity and cell survival. EMBO Mol Med 2: 51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolbetzki A, Ammon M, Jakovljevic V, Konovalova A, Sogaard-Andersen L. 2008. Regulated secretion of a protease activates intercellular signaling during fruiting body formation in M. xanthus. Dev cell 15: 627–634. [DOI] [PubMed] [Google Scholar]
- Schapira AH. 2006. The importance of LRRK2 mutations in Parkinson disease. Arch Neurol 63: 1225–1228. [DOI] [PubMed] [Google Scholar]
- Shimkets LJ, Kaiser D. 1982. Induction of coordinated movement of Myxococcus xanthus cells. J Bacteriol 152: 451–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simunovic V, Gherardini FC, Shimkets LJ. 2003. Membrane localization of motility, signaling, and polyketide synthetase proteins in Myxococcus xanthus. J Bacteriol 185: 5066–5075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD. 2002. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem 277: 44784–44790. [DOI] [PubMed] [Google Scholar]
- Szule JA, Fuller NL, Rand RP. 2002. The effects of acyl chain length and saturation of diacylglycerols and phosphatidylcholines on membrane monolayer curvature. Biophys J 83: 977–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takekoshi S, Kitatani K, Yamamoto Y. 2014. Roles of oxidized diacylglycerol for carbon tetrachloride-induced liver injury and fibrosis in mouse. Acta Histochem Cytochem 47: 185–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torroja L, Ortuno-Sahagun D, Ferrus A, Hammerle B, Barbas JA. 1998. scully, an essential gene of Drosophila, is homologous to mammalian mitochondrial type II L-3–hydroxyacyl-CoA dehydrogenase/amyloid-β peptide-binding protein. J Cell Biol 141: 1009–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner WJ, Kress BH, Harrison NB. 1942. α-Naphthol color test for dihydroxyacetone and hydroxymaleic acid. J Bacteriol 44: 249–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilardo E, Rossmanith W. 2013. The amyloid-β–SDR5C1(ABAD) interaction does not mediate a specific inhibition of mitochondrial RNase P. PloS one 8: e65609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilardo E, Rossmanith W. 2015. Molecular insights into HSD10 disease: impact of SDR5C1 mutations on the human mitochondrial RNase P complex. Nucleic Acids Res 43: 5112–5119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie C, Zhang H, Shimkets LJ, Igoshin OA. 2011. Statistical image analysis reveals features affecting fates of Myxococcus xanthus developmental aggregates. Proc Natl Acad Sci 108: 5915–5920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan SD, Fu J, Soto C, Chen X, Zhu H, Al-Mohanna F, Collison K, Zhu A, Stern E, Saido T, et al. 1997. An intracellular protein that binds amyloid-β peptide and mediates neurotoxicity in Alzheimer's disease. Nature 389: 689–695. [DOI] [PubMed] [Google Scholar]
- Yan Y, Liu Y, Sorci M, Belfort G, Lustbader JW, Yan SS, Wang C. 2007. Surface plasmon resonance and nuclear magnetic resonance studies of ABAD–Aβ interaction. Biochemistry 46: 1724–1731. [DOI] [PubMed] [Google Scholar]
- Yang SY, He XY, Isaacs C, Dobkin C, Miller D, Philipp M. 2014. Roles of 17β-hydroxysteroid dehydrogenase type 10 in neurodegenerative disorders. J Steroid Biochem Mol Biol 143: 460–472. [DOI] [PubMed] [Google Scholar]
- Zhang M, Mileykovskaya E, Dowhan W. 2002. Gluing the respiratory chain together. Cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. J Biol Chem 277: 43553–43556. [DOI] [PubMed] [Google Scholar]
- Zschocke J. 2012. HSD10 disease: clinical consequences of mutations in the HSD17B10 gene. J Inherit Metab Dis 35: 81–89. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.