

Jiang et al. found that Merit40−/− mice displayed marked hypersensitivity to DNA interstrand cross-links (ICLs) but not whole-body irradiation. This study implicates MERIT40 in the earliest stages of ICL repair and defines specific functional interactions between RAP80 complex-dependent ubiquitin recognition and the FA–BRCA ICL repair network.
Keywords: MERIT40, BRCA2, DNA2, ubiquitination, interstrand cross-link repair
Abstract
MERIT40 is an essential component of the RAP80 ubiquitin recognition complex that targets BRCA1 to DNA damage sites. Although this complex is required for BRCA1 foci formation, its physiologic role in DNA repair has remained enigmatic, as has its relationship to canonical DNA repair mechanisms. Surprisingly, we found that Merit40−/− mice displayed marked hypersensitivity to DNA interstrand cross-links (ICLs) but not whole-body irradiation. MERIT40 was rapidly recruited to ICL lesions prior to FANCD2, and Merit40-null cells exhibited delayed ICL unhooking coupled with reduced end resection and homologous recombination at ICL damage. Interestingly, Merit40 mutation exacerbated ICL-induced chromosome instability in the context of concomitant Brca2 deficiency but not in conjunction with Fancd2 mutation. These findings implicate MERIT40 in the earliest stages of ICL repair and define specific functional interactions between RAP80 complex-dependent ubiquitin recognition and the Fanconi anemia (FA)–BRCA ICL repair network.
Seminal insights into BRCA1 and BRCA2 function were revealed by their dynamic entry into discrete Rad51-containing foci in response to DNA double-strand break (DSB) formation (Scully et al. 1996, 1997; Chen et al. 1998) and essential roles in homology-directed repair (Sharan et al. 1997; Moynahan et al. 1999). This physiologic role of BRCA proteins and their interacting partners is phenotypically evident in the rare hereditary developmental disorder Fanconi anemia (FA). Germline mutations to both copies of a network of DNA repair genes—including BRCA1, BRCA2, and several associated proteins—result in DNA interstrand cross-link (ICL) hypersensitivity, characteristic developmental anomalies, and cancer susceptibility (D'Andrea and Grompe 2003; Deans and West 2011; Kim and D'Andrea 2012). In the case of BRCA1, a missense mutation within the BRCA1 C-terminal (BRCT) repeats disrupts interaction with ICL repair proteins BRIP1/FancJ and CtIP and with the RAP80 ubiquitin recognition complex that targets BRCA1 to DNA damage foci, resulting in a specific subtype of FA and extremely early onset breast and ovarian cancer (Domchek et al. 2013; Sawyer et al. 2015). This syndrome appears to be less severe in BRCA1 BRCT mutants than in FA patients with BRCA2 mutations. It is also noteworthy that BRCA1 BRCT mutant patients did not show bone marrow failure, which is present in patients with mutations in most other FA alleles (Domchek et al. 2013; Kottemann and Smogorzewska 2013; Garaycoechea and Patel 2014; Sawyer et al. 2015).
Large BRCA foci exist as a result of DSB-induced H2AX phosphorylation (γH2AX) and ensuing recruitment of E3 ubiquitin ligases RNF8 and RNF168. Extensive RNF168-dependent ubiquitination provides a recognition platform for DNA repair proteins that span megabase stretches of chromatin surrounding DSBs (Messick and Greenberg 2009; Gudjonsson et al. 2012; Mattiroli et al. 2012; Jackson and Durocher 2013). The large BRCA1 foci require interaction with the five-member RAP80 complex (RAP80, Abraxas, MERIT40, BRCC45, and BRCC36). Tandem RAP80 ubiquitin interaction motifs (UIMs) display specificity for non-proteasome-directed Lys63-linked ubiquitin, as does the associated deubiquitinating (DUB) enzyme BRCC36 (Sobhian et al. 2007; Sato et al. 2009; Sims and Cohen 2009). RAP80 also contains several SUMO-interacting motifs just N-terminal to the UIMs and displays ∼80-fold higher affinity for hybrid SUMO2,3–K63–ubiquitin chains, suggesting that both ubiquitin and SUMO chain recognition directs BRCA1–RAP80 complex foci formation (Guzzo et al. 2012). Abraxas directly binds to the BRCA1 BRCT repeats and is also required for BRCC36 DUB activity in vitro (Kim and Yu 2007; Wang et al. 2007; Feng et al. 2010; Patterson-Fortin et al. 2010). Less well understood is MERIT40, which is not required for DUB activity in vitro but maintains interaction with the other subunits and is required for their stability in cells and localization to DNA damage foci (Feng et al. 2009; Shao et al. 2009; Wang et al. 2009). In contrast to BRCA1 nullizygosity, deficiency in either RAP80 or Abraxas is well tolerated in mice (Wu et al. 2012; Yin et al. 2012; Castillo et al. 2014), indicating that the most prominent component of DSB foci formation contributes to only a subset of BRCA1 DNA repair function (Huen et al. 2009; Jiang and Greenberg 2015).
Recent evidence suggests a physiologic role for the RAP80 ubiquitin recognition complex in ICL repair. Abraxas-null cells were sensitive to mitomycin C (MMC), and this was associated with reduced homologous recombination (HR) (Castillo et al. 2014). BRCA1 has been reported to mediate several processes during ICL repair, where it functions in a HR-independent manner to promote unloading of the replication-initiating CMG complex during early stages of ICL recognition (Long et al. 2014). BRCA1, in association with BRCA2 and Rad51, is also thought to promote HR at ICL lesions that have undergone replication-dependent processing into DSBs. RAP80 is recruited during the initial stages of ICL recognition in a ubiquitin-dependent manner, possibly implicating its function in the BRCA1-dependent CMG clearance. This step in ICL repair is important for subsequent ICL recognition by the FANCD2-I complex (Long et al. 2014).
Replication fork convergence on FANCD2-I-bound ICL lesions leads to cross-link incision and DSB formation following translesion synthesis, coupled to BRCA2–Rad51 HR repair of the resultant DSB. Evidence also exists in mammalian cells for alternative mechanisms of cross-link responses. Single-molecule imaging approaches have revealed the existence of an ICL bypass mechanism that relies on FANCM to translocate replication forks beyond psoralen cross-links (Huang et al. 2013). In addition, ICL hypersensitivity of cells deficient in the canonical D2-I pathway can be circumvented by reduced end resection as a result of DNA2 loss (Karanja et al. 2014). Proposed mechanisms underlying this genetic interaction invoke fork reversal and template switch-mediated ICL bypass. These observations suggest several alternative possibilities for ICL repair, emphasizing complexity within the FA–BRCA network that is further highlighted by the overlapping yet variable phenotypes observed in patients with mutations in different FA genes.
This study uses Merit40 deficiency as a focal point to determine the importance of ubiquitin recognition in the ICL recognition and repair process. Surprisingly, we reveal unique positioning of MERIT40 within the FA–BRCA network at early stages of ICL repair, thus distinguishing its interactions with FANCD2-I ICL recognition from BRCA2-dependent HR. These studies further emphasize the nonlinear nature of ICL recognition and repair and indicate that ubiquitin recognition plays a role distinct from that of canonical aspects of the FA–BRCA network.
Results
Merit40-deficient mice are hypersensitive to ICL agents
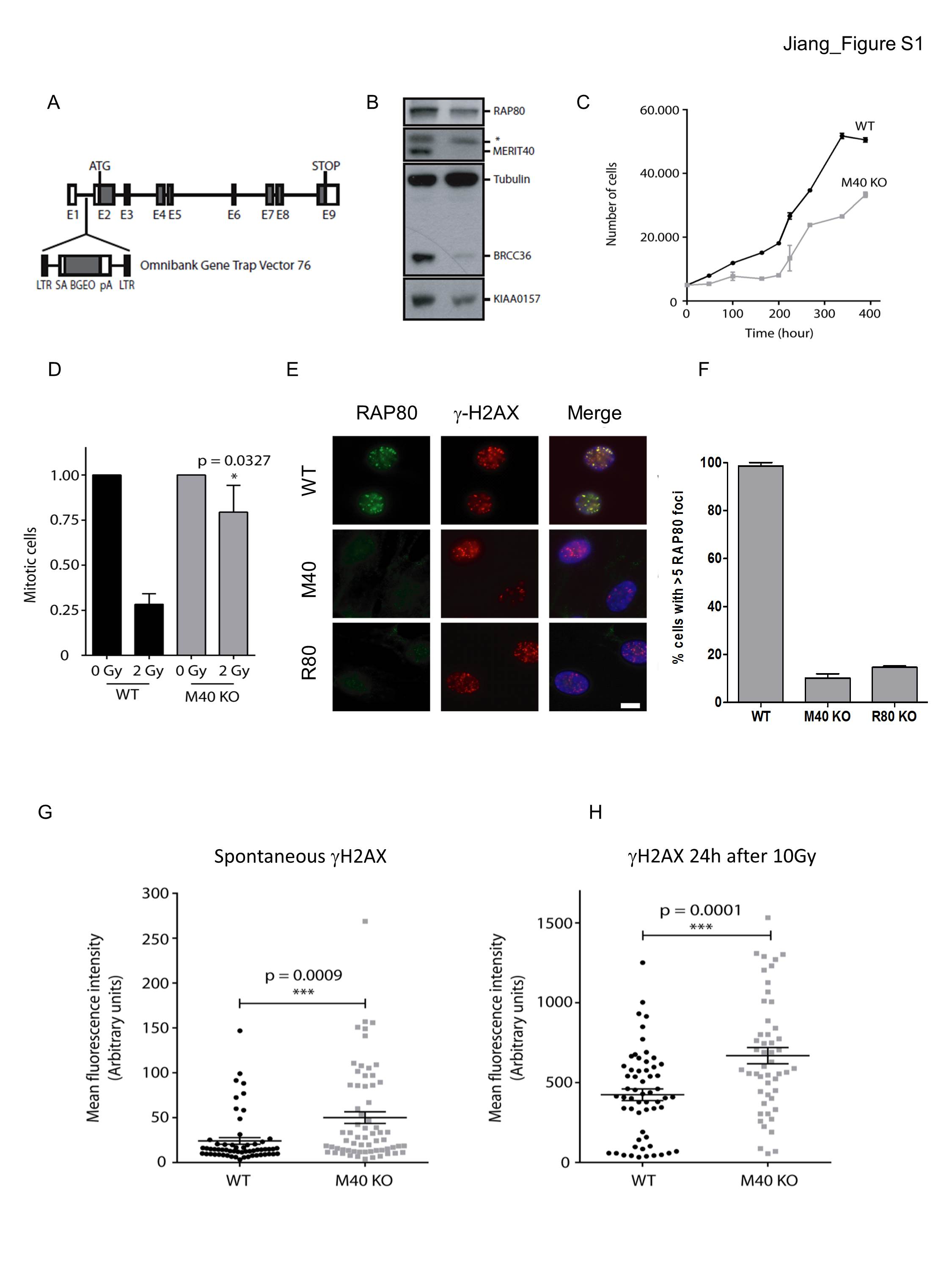
Merit40−/− mice were generated by gene trap insertion between exons 1 and 2 prior to the start codon (Supplemental Fig. S1A), as recently described (Rozenova et al. 2015). Decreased transcript level of MERIT40 mRNA was detected across seven exons by RT–PCR (data not shown), and MERIT40 protein was undetectable in Merit40−/− mouse embryonic fibroblasts (MEFs), as determined by Western blotting (Supplemental Fig. S1B). Merit40−/− mice were viable and fertile and did not display overt phenotypes in the absence of exogenous genotoxic stress (Rozenova et al. 2015; this study). However, as predicted by prior studies (Feng et al. 2009; Shao et al. 2009; Wang et al. 2009), Merit40−/− MEFs displayed reduced protein levels of binding partners and proliferative rates, impaired G2/M checkpoint function, and loss of BRCA1 and RAP80 foci after ionizing radiation (IR) (Fig. 1A,B; Supplemental Fig. S1C-F). Consistent with DNA repair deficiency, increased spontaneous and persistent DSBs were observed in Merit40−/− MEFs, as measured by γH2AX foci (Supplemental Fig. S1G,H).
Figure 1.

Merit40-deficient mice are hypersensitive to MMC but not IR. (A) Representative images of MEFs of the indicated genotypes at 8 h after IR. Bar, 10 μm. (B) Quantification of foci number from A. Error bars represent standard error of the mean (SEM). Significance was analyzed with two-tailed paired Student's t-test: (*) P < 0.05; (**) P < 0.01; (***) P < 0.001; (****) P < 0.0001. (C) Six-week-old to 12-wk-old wild-type (WT) and Merit40−/− (M40) littermates were subjected to 8.76 Gy of IR. Mice were monitored daily, and survival rate is displayed as indicated. (D) MEFs were treated with 4 μM DNA-PK inhibitor or DMSO for 1 h before being subjected to IR. DNA-PK inhibitor-treated wild-type MEFs were used as a positive control. Three independent experiments were conducted. Statistical analysis was performed between wild type and M40 and between wild type and wild type + PKi at the same IR dose. (E) Mice were subjected to a one-time intraperitoneal injection of 12 mg/kg MMC, and survival for each genotype was monitored as in C. (F) Merit40−/− MEFs displayed reduced survival in response to escalating doses of MMC, as assessed by clonogenic assay. Student's t-tests were performed between wild-type and M40 cells of the indicated types at the same drug dose. (G) Representative field of TUNEL staining in wild-type and M40 mice (200×). Bar, 500 μm. (H) TUNEL-positive cells were quantified by counting four fields per mouse and three mice per genotype at day 3 after MMC.
To investigate the physiologic contribution of MERIT40 to different forms of DNA damage, Merit40−/− mice were exposed to IR, which creates DSBs in all cell cycle phases, or MMC, which produces ICL lesions that are primarily repaired in S phase. Surprisingly, no significant difference in mortality occurred between Merit40−/− and wild-type littermate controls following 8.76 Gy of IR (Fig. 1C) and Merit40−/− MEFs were not hypersensitive to IR (Fig. 1D). Conversely, a highly significant (P = 0.0027) increase in mortality rate was observed following treatment with the ICL-inducing agent MMC in Merit40−/− mice when compared with wild-type littermate controls (Fig. 1E). Similarly, Merit40−/− MEFs displayed increased sensitivity to MMC, as did U2OS cells following knockdown of RAP80 complex members MERIT40 and Abraxas (Fig. 1F; Supplemental Fig. S2A). Significantly higher numbers of chromatid breaks and radial chromosome structures were also observed in Merit40−/− MEFs and primary splenocytes (Supplemental Fig. S2B,C), suggestive of an ICL repair deficit. Consistent with impaired S-phase DNA repair, apoptotic rates were elevated in Merit40−/− mice in highly proliferative organ systems 3 d after intraperitoneal injection of MMC but not in cardiac tissue, which largely exists in a post-mitotic state (Fig. 1G,H; Supplemental Fig. S2D,E). Together, these data highlight an important physiologic role of MERIT40 in the repair of S-phase lesions in cycling cells.
To further examine the importance of MERIT40 in S-phase DNA damage responses, wild-type and Merit40−/− MEFs were labeled with 5-bromo-2′-deoxyuridine (BrdU) and then incubated for 1 h with MMC or hydroxyurea (HU) to halt S-phase progression. Following removal of MMC or HU, cells were chased with EdU to evaluate the ability of S-phase cells (BrdU-positive) to restart replication (Ragland et al. 2013). Merit40−/− MEFs displayed significantly reduced recovery of DNA replication in comparison with wild-type MEFs after HU or MMC treatment (Fig. 2A–C). A similar phenotype was observed when using aphidicolin (APH) to inhibit replication in both wild-type and Merit40−/− MEFs as well as in MCF10A cells with knockdown of three members of the RAP80 complex (Supplemental Fig. S3A,B). We further investigated this phenomenon using DNA fiber analysis to examine single-replication track responses in wild-type and Merit40−/− MEFs. In agreement with flow cytometry-based results, Merit40−/− MEFs displayed reduced replication restart efficiency upon transient replication blocks from MMC or HU (Fig. 2D,E). Merit40−/− MEFs were more sensitive to cisplatin, MMC, and PARP inhibitors (Supplemental Fig. S3C–E), suggestive of a broad deficiency in replication-associated DNA damage responses. Despite the significant impairment in replication restart, Merit40−/− MEFs displayed very mild sensitivity to HU, but significant elevation in chromosomal abnormalities was observed in Merit40−/− MEFs after HU treatment (Supplemental Fig. S3F,G). This pattern of mild sensitivity and increased chromosome aberrations is in agreement with prior results for Fanca mutant cells (Schlacher et al. 2012).
Figure 2.

MERIT40 mediates replication fork recovery in response to S-phase damage. (A) Schematic illustration of replication fork restart experiments. Cells were pulsed with BrdU for 30 min before HU or MMC treatment to label cells that are in S phase; HU or MMC was added for 1 h to induce replication fork stalling, followed by EdU treatment for 1 h to detect fork recovery for cells in S phase. BrdU+ cells were gated, and further analysis was performed on the population to detect EdU incorporation following replication fork recovery. (B) Representative image of replication fork restart. M40 cells have a lower rate of replication fork restart. (C) Quantification of B based on three independent experiments of HU or MMC treatment. (D) Representative image of single-fiber analysis. (Red tracks) IdU; (green tracks) CldU. Example of a stalled fork and a restarted fork. (E) Schematic depicting experiment design. The percentage of restarted forks equals the number of restarted forks divided by the sum of the stalled forks and restarted forks.
In addition to its role as an essential member of the RAP80 complex, MERIT40 is critical for the stability and function of the related BRISC (BRCC36 isopeptidase complex) DUB complex, which we recently implicated in promoting inflammatory cytokine receptor signaling (Cooper et al. 2009; Zheng et al. 2013). KIAA0157 is a homolog to Abraxas and is uniquely present in the BRISC. KIAA0157 knockout cells lose the BRISC while retaining the nuclear RAP80 complex. Importantly, Kiaa0157−/− MEFs showed proper recruitment of BRCA1 and RAP80 to IR-induced foci, and KIAA0157 knockout mice were not sensitive to IR or MMC (Supplemental Fig. S4). This indicates that MMC sensitivity in the absence of MERIT40 was due to deficiency in the RAP80 complex and was not related to deficiency in the BRISC.
The RAP80 complex requires PARsylation and ubiquitylation for ICL recognition
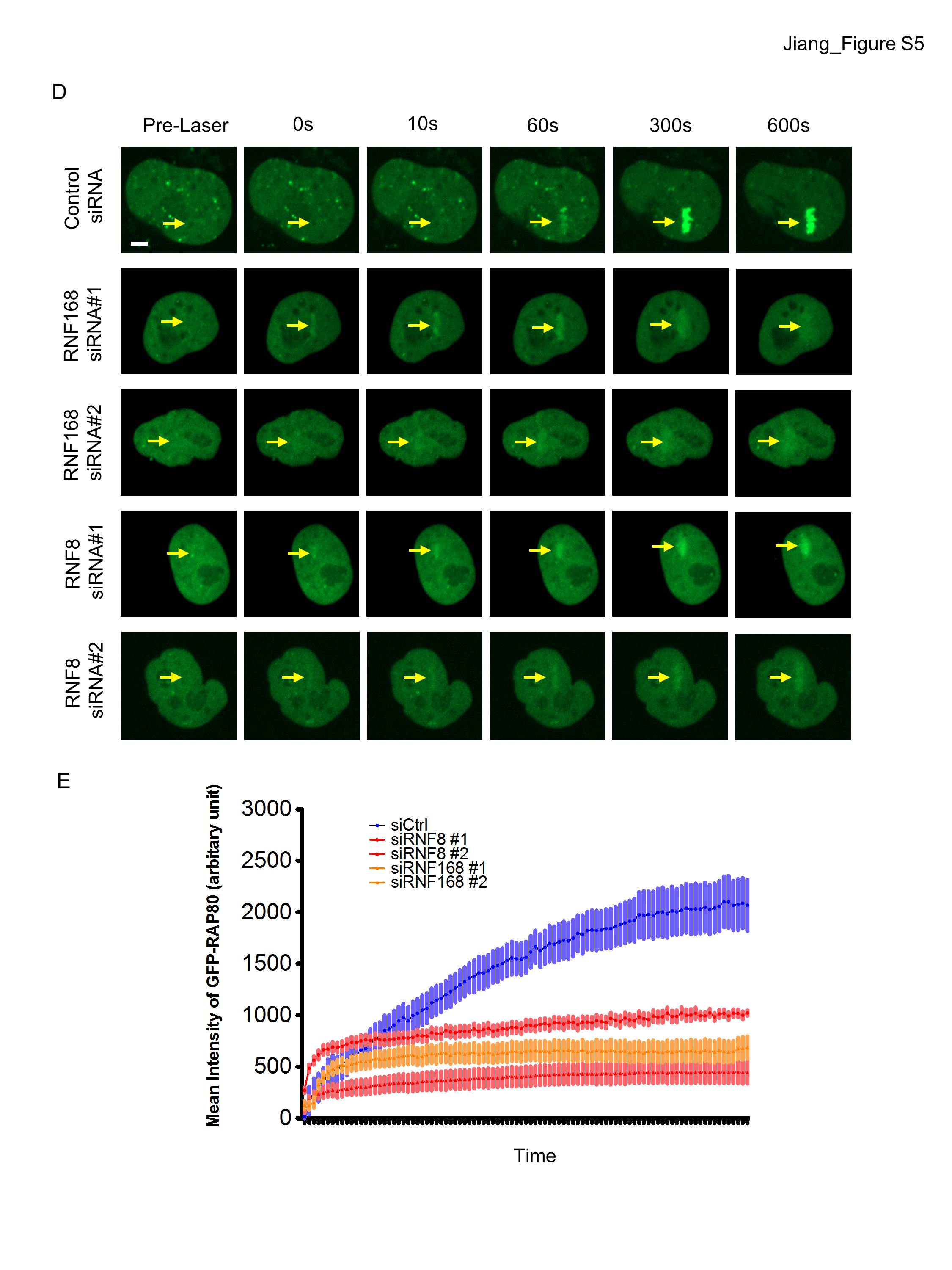
MERIT40 localizes to DSBs as a constituent of the RAP80 complex. DSB recognition requires a phosphorylation-to-ubiquitylation signaling cascade initiated by MDC1 recognition of γH2AX and subsequent recruitment of E3 ubiquitin ligases RNF8 and RNF168. To understand whether similar requirements exist for ICL repair, MERIT40 localization was monitored at UV-induced psoralen cross-links and MMC-induced cross-links (Fig. 3A; Supplemental Fig. S5A,B). The accumulation of MERIT40 at psoralen cross-links was abolished in HeLa cells upon deletion of RNF8 and RNF168 (Fig. 3A,B). Additionally, deletion of the first UIM domain eliminated the accumulation of RAP80 at psoralen cross-links (Fig. 3C,D), confirming the importance of ubiquitin binding for localization. RAP80-GFP recruitment was specific to ICL damage, as no recruitment was observed without psoralen administration prior to laser activation (Supplemental Fig. S5C). Interestingly, the initial recruitment (within 30 sec) of RAP80 was not affected by depletion of RNF8 or RNF168, indicating that rapid accumulation of RAP80 depends on other events (Supplemental Fig. S5D,E). PAR-dependent mechanisms have been implicated in the rapid localization of BRCA1 to DNA damage sites (Li and Yu 2013), and RAP80 has recently been reported to specifically bind PAR (Hu et al. 2014). Indeed, either of the PARP inhibitors NU1025 or Olaparib abrogated early phase GFP-RAP80 recruitment (<1 min) to laser-induced ICL damage (Fig. 3E,F). These data indicate that MERIT40 is recruited to ICLs in association with RAP80 in a biphasic manner. PARP activity is required for the initial recognition, while ubiquitin binding mediates stable accumulation of the complex.
Figure 3.

Biphasic recruitment of the RAP80 complex to ICL lesions is dependent on PARP1 and ubiquitination. (A,B) Representative images and quantification of MERIT40 at laser-induced psoralen cross-links in HeLa cells. Cross-linking experiments were performed at 48 h after transfection with the indicated siRNAs. The arrows depict positive recruitment signals. Three independent experiments were conducted. (C) Representative images of EYFP-RAP80 and a mutant lacking the first UIM domain at 5 min after laser-induced psoralen cross-linking. (D) Live imaging of EYFP-RAP80 and RAP80 mutant recruitment to psoralen cross-linking. (E) GFP-tagged RAP80 protein was recruited to psoralen-induced ICLs at various time points after laser activation in HeLa cells. PARP inhibitors Olaparib (5 μM) and NU1025 (500 μM) were added to the cells at 1 h prior to ICL damage induction. (F) Quantification of the mean intensity of recruitment signals at each time point after laser induction of ICL was quantified.
Merit40 deficiency reduces unhooking efficiency and homology-directed repair of ICL lesions
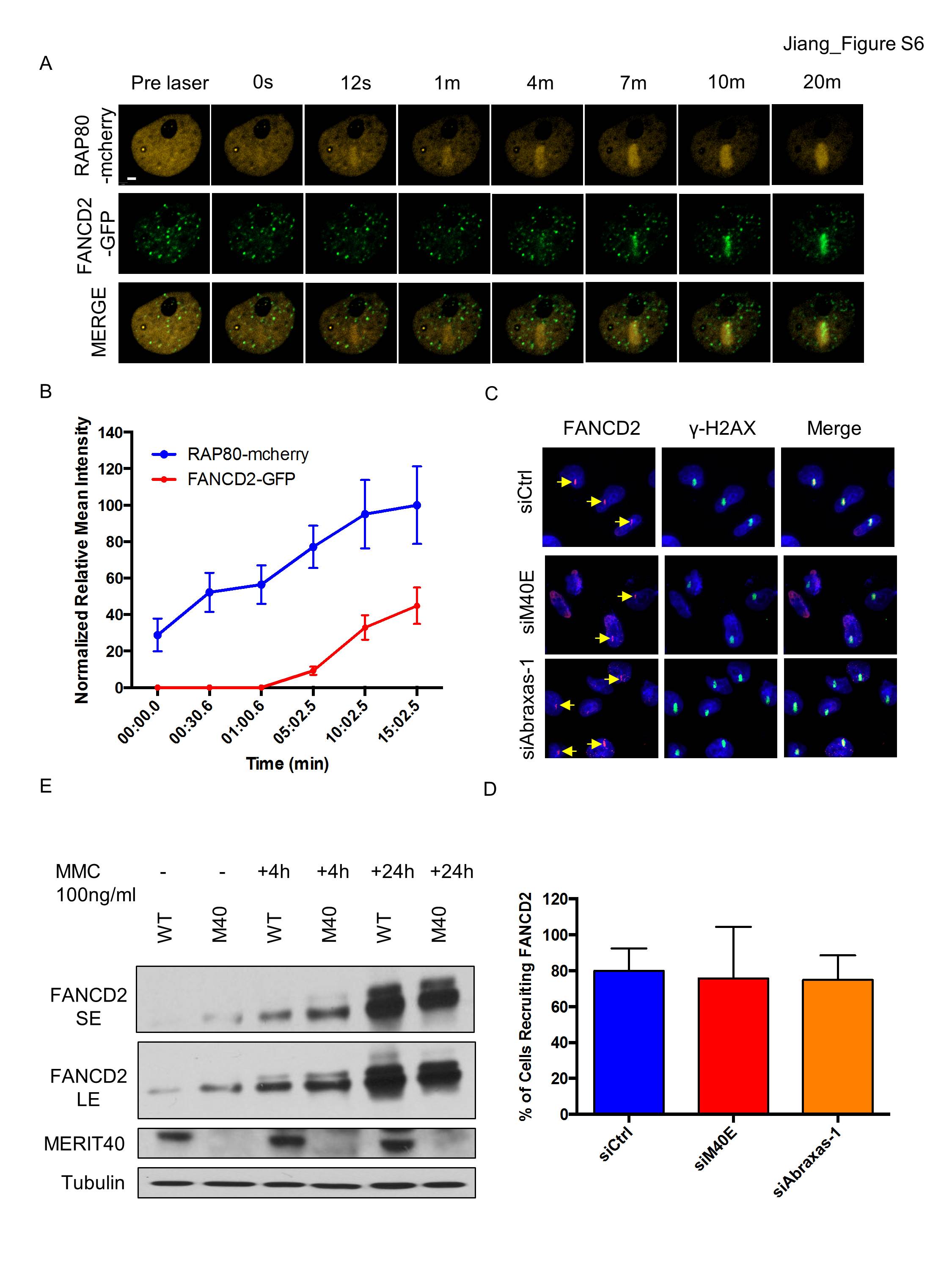
The preceding data suggest a specific role for MERIT40 in ICL repair. As the RAP80 complex is rapidly recruited to ICL sites, we asked whether RAP80 affects FANCD2 recruitment and downstream function. RAP80 accumulated at psoralen cross-links at ∼1 min, while FANCD2 required nearly 10 min for maximal ICL localization (Fig. 4A,B). Live-cell imaging experiments confirmed that mcherry-RAP80 ICL recruitment preceded that of GFP-FANCD2 (Supplemental Fig. S6A,B). FANCD2 monoubiquitination is essential for localization subsequent to repair of ICLs. Despite the faster ICL recruitment, knockdown of the RAP80 complex did not affect FANCD2 localization to ICL sites at 20 min after UV activation of psoralen (Supplemental Fig. S6C,D). Additionally, splenocytes from wild-type and Merit40−/− mice did not show an obvious difference with respect to FANCD2 monoubiquitination at either 4 or 24 h after MMC (Supplemental Fig. S6E).
Figure 4.

Merit40 mutation reduces ICL unhooking efficiency and HR repair of ICL damage. (A,B) Representative images and quantification of RAP80 and FANCD2 at laser-induced psoralen cross-links in HeLa cells. The arrows indicate positive recruitment signals. (C) Cells were treated for 1 h with 50 μM cisplatin followed by cisplatin removal. Samples were taken at different time points after cisplatin treatment, and unhooking of ICL was measured using a modified comet assay (please see the Materials and Methods for a detailed description). Immediately before analysis, cells collected from different time points were exposed to 12.5 Gy of IR. DNA interstrand cross-linking was expressed as percentage decrease in tail moment compared with irradiated controls calculated by the following formula: percent decrease in Olive tail moment = [1 − (TMdi − TMcu)/(TMci − TMcu)] × 100, where TMdi is the tail moment of the cisplatin-treated, irradiated sample; TMcu is the tail moment of the untreated, unirradiated control; and TMci is the tail moment of the untreated, irradiated control. (D) MEFs were labeled with BrdU for two cell cycles and then treated with 20 ng/mL MMC for the last 24 h prior to harvesting for metaphase analysis of sister chromatid exchanges. Quantification was derived from four independent experiments. (E–G) HeLa cells depleted of MERIT40 by three individual siRNAs show reduced recruitment of BRCA1 (E), Rad51 (F), and RPA (G) to psoralen-induced ICLs. The mean intensities of protein colocalized to ICL stripes were quantified in single cells at 20 min after laser treatment.
The aforementioned results do not exclude the possibility that early recruitment of MERIT40 to ICL damage is important in the initial phases of ICL processing. A modified comet assay can be used to assess ICL unhooking efficiency in mammalian cells at the single-cell level (Hartley et al. 1999; Spanswick et al. 1999; Clauson et al. 2013). The presence of ICLs limits DNA migration into comet tails, leading to decreased tail length. The degree of ICLs present was determined by comparing the tail moment of irradiated, cisplatin-treated samples with irradiated samples that were not treated with cisplatin (please see the legend for Fig. 4; Materials and Methods for a detailed description). The decrease in comet tail length peaked at 6 h in wild-type MEFs, followed by a sustained increase in length through 40 h. The peak in comet tail length decrease occurred at 9 h after cisplatin treatment in Merit40−/− MEFs and remained delayed in comparison with wild-type MEFs for 30 h (Fig. 4C). These data reveal that loss of MERIT40 reduces the efficiency of ICL unhooking and support a functional importance to the rapid ICL recruitment of the RAP80 complex.
To investigate subsequent steps in ICL repair, we investigated whether loss of MERIT40 affected HR efficiency and protein recruitment. Merit40−/− cells showed modestly reduced sister chromatid exchange (SCE) after MMC treatment (Fig. 4D; Supplemental Fig. S7A). MERIT40 knockdown was achieved by three different siRNAs in HeLa cells, and the intensity of the repair protein level was examined at psoralen cross-links. The accumulation of BRCA1 and Rad51 was reduced upon MERIT40 knockdown (Fig. 4E,F; Supplemental Fig. S7B,C). Additionally, the mean intensity of RPA recruitment was reduced upon depletion of MERIT40 at psoralen cross-links (Fig. 4G; Supplemental Fig. S7D), and Merit40−/− cells displayed significantly reduced p-RPA levels after MMC treatment (Supplemental Fig. S7E). Similar reductions in RPA and Rad51 at MMC-induced foci were also observed in U2OS cells following MERIT40 knockdown (Supplemental Fig. S7F,G). These results suggest that HR deficiency in Merit40-null cells results from a cumulative reduction in end resection and Rad51 nucleofilament formation.
In contrast, several prior reports demonstrate that loss of RAP80 leads to increased end resection and HR in response to nuclease- or etoposide-induced DSBs in human cancer cell lines (Coleman and Greenberg 2011; Hu et al. 2011; Adamson et al. 2012; Goldstein and Kastan 2015). This could occur as a consequence of the RAP80 complex imparting a differential impact on the processing of DSBs in comparison with ICLs or, alternatively, due to differences between human cancer cell lines and primary mouse cells. Consistent with prior data, we detected an ∼30% increase in SCEs in Merit40−/− MEFs after etoposide-induced DSBs (Supplemental Fig. S7H). Furthermore, a 2.5-fold increase in ssDNA levels was observed in Merit40−/− splenocytes compared with wild-type counterparts, as measured by a nondenaturing BrdU signal in S/G2-phase cells at 2 h after IR (Supplemental Fig. S5I–K; Bunting et al. 2012). These data reveal that loss of MERIT40 differentially affects end resection at DSBs and ICL damage and suggest unique contributions of the RAP80 complex to each repair process.
Merit40 deficiency does not exacerbate MMC sensitivity in Fancd2-null mice.
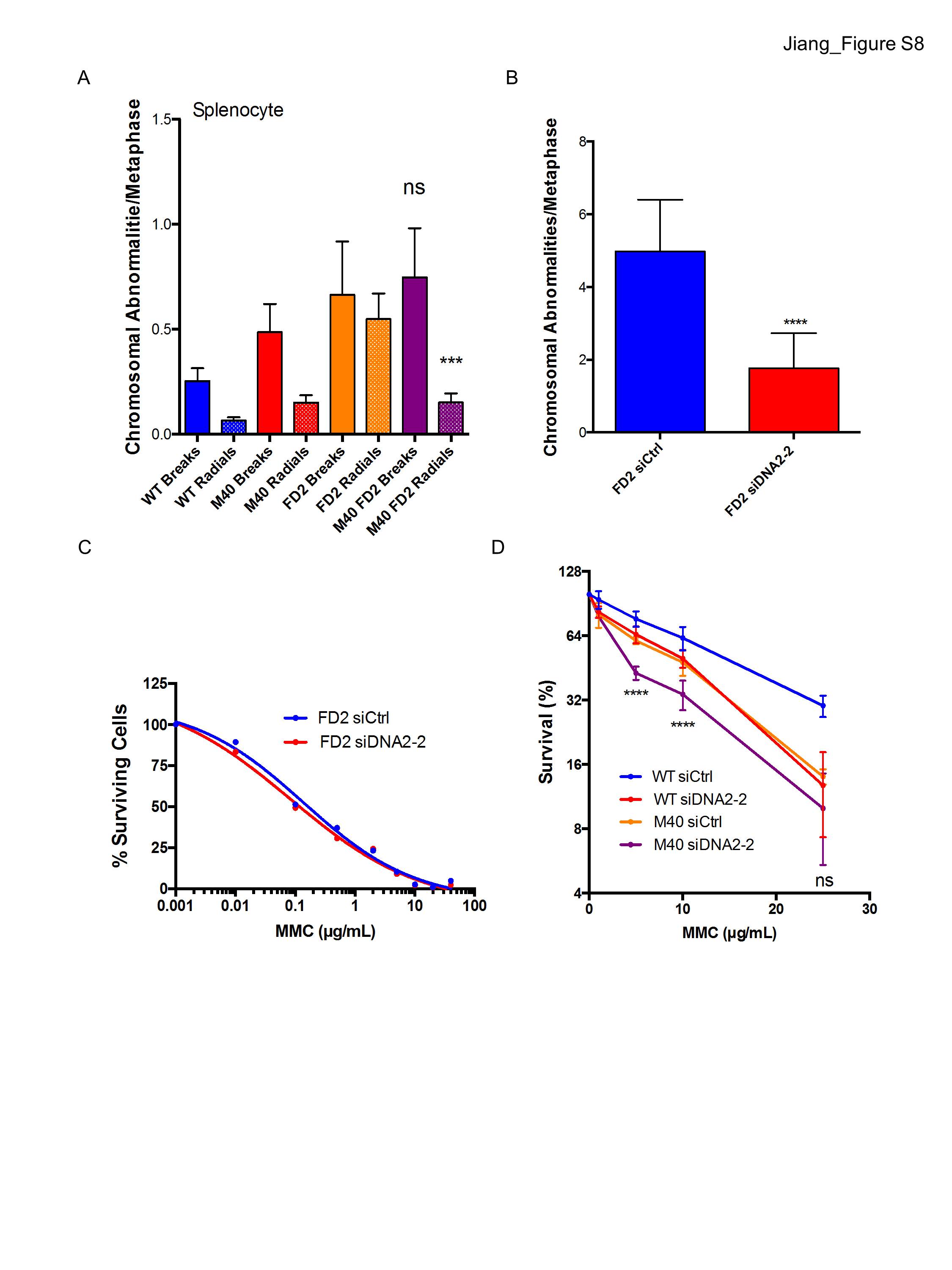
A genetic approach was undertaken to understand how ubiquitin recognition by the RAP80 complex intersected with the major components of ICL repair. Merit40−/− mice were bred to genetic backgrounds harboring mutations in the major arms of ICL repair, Fancd2, or conditional Brca2 (floxed exon 11). MEF cell lines were established from these crosses. Merit40, Fancd2 double-knockout mice are viable and do not show reduced body weight or other additional compromises in comparison with Fancd2-null mice. Moreover, loss of MERIT40 did not affect the sensitivity of Fancd2−/− MEFs to MMC (Fig. 5A). Additionally, Merit40−/− Fancd2−/− MEFs or splenocytes did not show significantly different levels of chromosomal breaks or radial chromosomes in comparison with Fancd2−/− MEF cells (Fig. 5B). RPA levels at psoralen cross-links and SCEs in response to MMC were also similar in Merit40−/− Fancd2−/− MEFs when compared with Fancd2−/− MEFs (Fig. 5C–E). However, while double-knockout Merit40−/− Fancd2−/− splenocytes did not show significant differences in total chromosomal aberrations when compared with Fancd2−/− cells, there was a trend toward increased chromosomal breaks, and a significant reduction in radial chromosomes occurred in the splenocytes of double knockouts (Supplemental Fig. S8A). These data suggest that MERIT40 and FANCD2 are not functionally redundant during ICL repair.
Figure 5.

Combined Merit40 and FancD2 deficiency in mice does not increase genomic instability in response to MMC. (A) Wild-type (WT), M40, FANCD2 knockout (FD2), and M40 FD2 double-knockout MEFs were exposed to the indicated doses of MMC, and survival was assessed by clonogenic assay in two independent experiments. In each experiment, six repeats were analyzed at each drug concentration for each genotype. (B) Chromosome abnormalities were quantified in MEFs derived from wild-type, M40, FD2, and M40 FD2 mice. Chromosomal abnormalities per metaphase, including chromatid and chromosomal breaks (breaks) and radial chromosomes (radials), were quantified in three independent experiments. n > 100 metaphases per genotype. (C,D) Representative images and quantification of RPA recruitment to laser-induced psoralen cross-links. The mean intensity of protein colocalized to ICL stripes was quantified in single cells at 10 min after laser treatment. Bar, 7 μm. (E) SCEs were quantified in wild-type, M40, FD2, and M40 FD2 MEFs treated with 20 ng/mL MMC for 24 h. Student's t-tests in this panel were performed between FD2 and M40 FD2 cells.
Cooperative interactions between MERIT40 and BRCA2 in response to ICLs
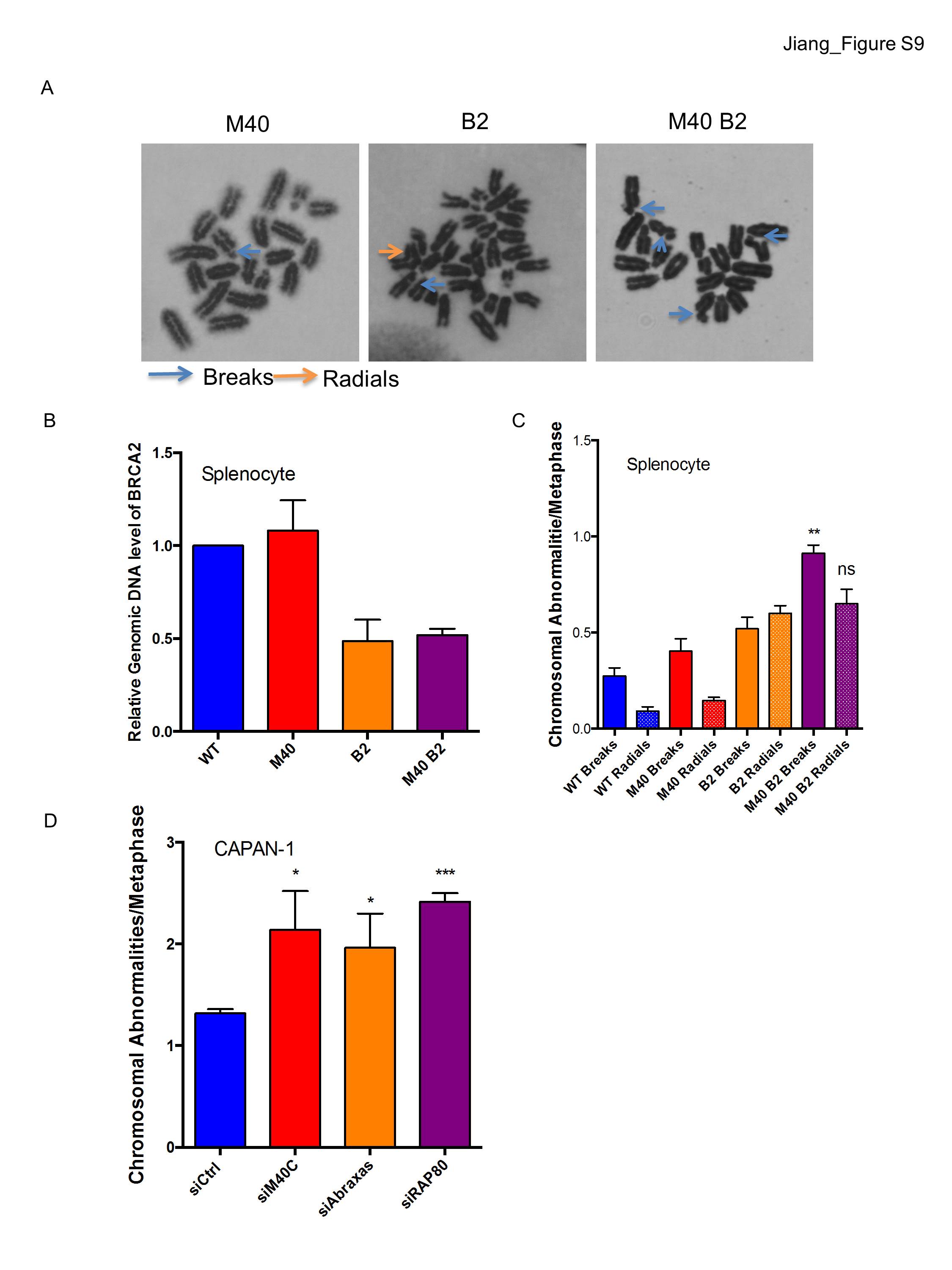
Given the minimal genetic interaction between MERIT40 and FANCD2, we asked whether MERIT40 would instead contribute in a more substantial manner to ICL repair in the context of impairment in other aspects of the FA–BRCA ICL repair network. Conditional deletion of BRCA2 was achieved in MEFs by 4-hydroxy tamoxifen (4-OHT)-induced activation of the CreERT2 fusion protein, which was constitutively expressed in cells harboring a homozygous floxed BRCA2 exon 11 allele. Wild-type and Merit40−/− MEFs expressing the CreERT2 fusion protein were used in all experiments to control for the potential off-target Cre-induced DNA damage (Silver and Livingston 2001). Interestingly, the plating efficiency of Merit40−/− Brca2−/− MEFs was significantly lower compared with MEFs harboring either Merit40−/− or Brca2−/− in isolation (Fig. 6A,B). Combined Brca2 and Merit40 deficiency demonstrated increased sensitivity to ICL damage, as measured by elevated cytogenetic aberrations in both MEFs and primary splenocytes (Fig. 6C; Supplemental Fig. S9A-C). Merit40−/− Brca2−/− cells showed a greater than additive increase in chromosomal breaks compared with Merit40−/− or Brca2−/− in MEFs, while double-knockout splenocytes showed an approximately additive increase in chromosome aberrations compared with the single knockouts. Moreover, knockdown of RAP80, MERIT40, or Abraxas in pancreatic adenocarcinoma CAPAN-1 cells, which contain a truncated form of the BRCA2 protein (Goggins et al. 1996), resulted in an approximately twofold higher level of chromosomal aberrations compared with control siRNA transfected cells (Supplemental Fig. S9D). The data are consistent with the presence of a strong genetic interaction in cells lacking MERIT40 and BRCA2 and suggest an enhanced requirement for BRCA2 in the absence of MERIT40. To understand the mechanism of how MERIT40 and BRCA2 cooperate in response to ICLs, we examined the HR efficiency by SCE analysis in response to MMC. A significantly reduced amount of SCEs was observed in Merit40−/− Brca2−/− compared with Brca2−/− cells (Fig. 6D), suggesting that MERIT40 and BRCA2 work in part to coordinately promote HR in response to ICL damage.
Figure 6.

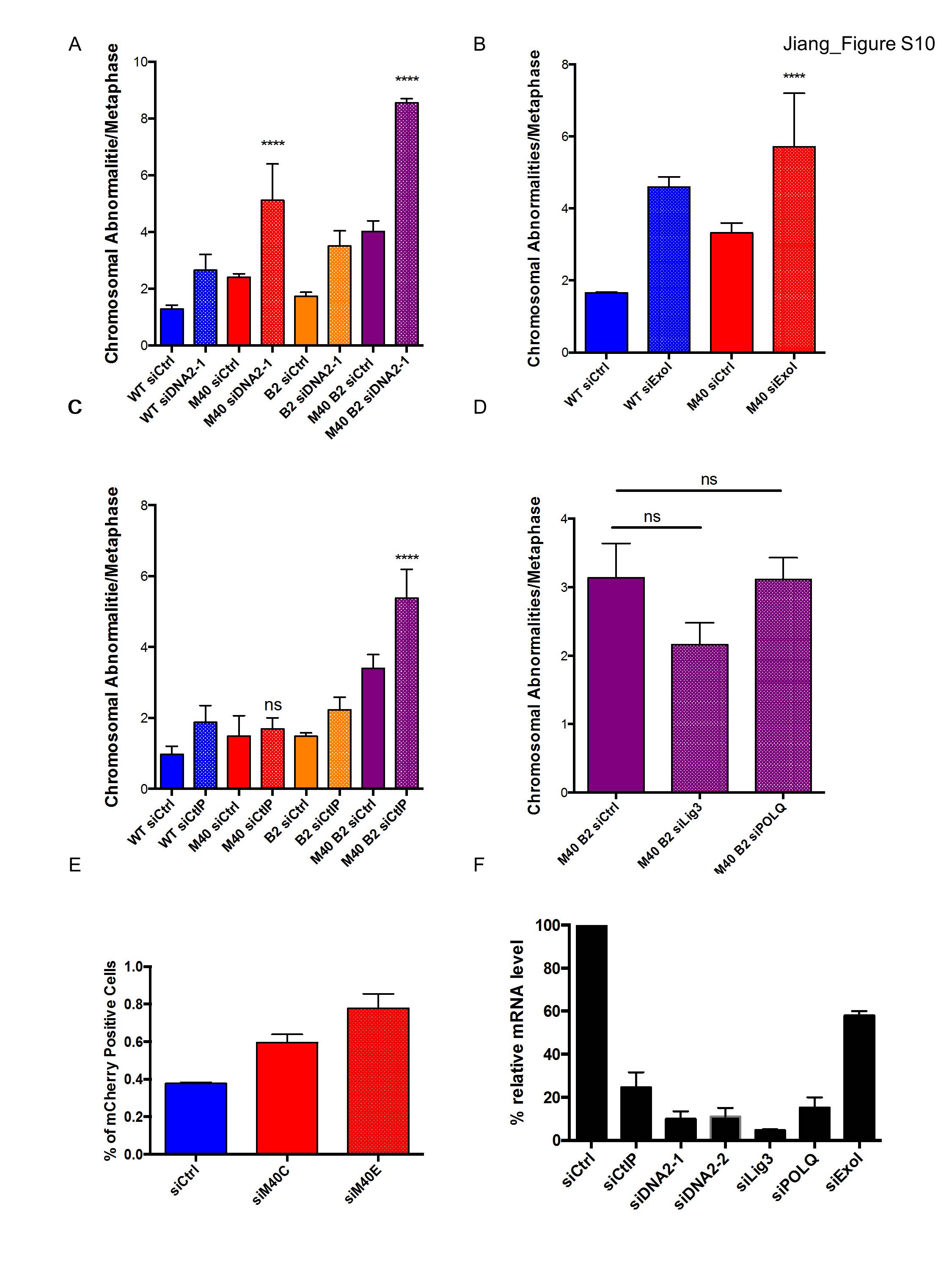
Cooperative genetic interactions occur between MERIT40 and BRCA2 in response to ICL damage. (A,B) Plating efficiency of wild type (WT), M40, two clones of inducible BRCA2f/fCre-ERT2 (B2A and B2B), and three clones of inducible MERIT40 BRCA2f/fCre-ERT2 MEFs (M40B2A, M40B2B, and M40B2C). MEFs were treated with 500 nM 4-OHT for 48 h and then plated at 500 cells per 60-mm plate. Crystal violet-stained colonies were counted 10 d after plating. Student's t-tests were performed between wild type and M40A and also between B2A and three clones of M40 B2 MEFs. (C) Wild-type, M40, B2f/fCre-ERT2, and M40 B2f/fCre-ERT2 MEFs were treated with 4-OHT for 48 h to induce BRCA2 incision. Cells were then treated with 20 ng/mL MMC for an additional 24 h prior to metaphase harvest. n > 100 metaphases per genotype. Student's t-tests were performed between B2 and M40 B2 cells. (D) MEFs were incubated with 4-OHT for 48 h to induce BRCA2 incision and then treated as in Figure 4D. Student's t-tests were performed between B2 and M40 B2 cells, and quantification was derived from four independent experiments. (E) Loss of DNA2 exacerbates levels of chromosomal abnormalities in M40 B2 MEFs. Two independent experiments were conducted with two different siRNAs. The data for siDNA2-1 are shown in Supplemental Figure 10A. (F) SCEs were analyzed for the indicated cells. Student's t-tests were performed between M40 B2 siCtrl and M40 B2 siDNA2-2. Two independent experiments were conducted.
DNA2 differentially affects ICL repair in Merit40- and FancD2-null cells
The preceding results reveal contrasting genetic interactions between MERIT40 and canonical arms of the FA–BRCA ICL repair network: FANCD2 and BRCA2. These observations necessitated additional investigations into whether MERIT40 and ubiquitin recognition is functionally redundant with the FANCD2-I arm of ICL repair or, alternatively, constitutes a distinct component of the ICL repair network. DNA2 was previously shown to be responsible for overresection in Fancd2−/− cells, and knockdown of DNA2 suppressed ICL hypersensitivity in Fancd2−/− cells (Karanja et al. 2014). Given that ICL hypersensitivity was not increased in Merit40, Fancd2-double-null cells, we asked whether DNA2 knockdown would also restore ICL resistance in Merit40−/− cells. If MERIT40 affects ICL repair in a common mechanism with FANCD2, a prediction is that concomitant DNA2 deficiency would similarly impact ICL repair in either a Fancd2-null or Merit40-null background. In agreement with previous reports (Karanja et al. 2014), DNA2 knockdown reduced genomic instability in MMC-treated Fancd2−/− cells (Supplemental Fig. S8B,C). Conversely, transfection of two independent siRNAs targeting DNA2 strongly increased chromosome aberrations in MMC-treated Merit40−/− cells and reduced cell survival in response to MMC (Fig. 6E; Supplemental Figs. S8D, S10A). These data reveal that MERIT40's and FANCD2's contributions to ICL repair can be differentiated by their genetic interactions with DNA2.
DNA2 knockdown also increased genomic instability in Merit40−/− Brca2−/− cells (Fig. 6E; Supplemental Fig. S10A). Similar results were obtained following transfection of siRNA targeting CtIP in double-knockout cells and by Exo1 knockdown in Merit40−/− MEFs, consistent with an increased requirement for end resection in MERIT40-null cells (Supplemental Fig. S10B,C). Comparable SCE numbers were observed before and after DNA2 knockdown in Merit40−/− Brca2−/− cells (Fig. 6F), suggesting that DNA2-dependent resection contributes to an alternative, HR-independent repair function in the context of dual deficiency for BRCA2 and MERIT40. Knockdown of essential microhomology-mediated end-joining (MMEJ) proteins Lig3 or POLQ did not exacerbate chromosomal abnormalities (Supplemental Fig. S10D–F), suggesting that other resection-dependent repair events are invoked in Merit40−/− Brca2−/− cells.
Discussion
The physiologic importance of RAP80 complex ubiquitin foci association has remained elusive despite considerable investigation into DNA damage recognition mechanisms and its roles in cancer susceptibility (Nikkilä et al. 2009; Antoniou et al. 2010; Bolton et al. 2010; Solyom et al. 2012; Castillo et al. 2014). This study reveals that a major function of the RAP80 complex is in DNA replication-associated repair. Surprisingly, in contrast to Rap80−/− and Abraxas−/− mice (Wu et al. 2012; Yin et al. 2012; Castillo et al. 2014), Merit40−/− mice or cells were not significantly hypersensitive to IR even though they exhibited G2 checkpoint impairment. These issues notwithstanding, Abraxas deficiency was also reported to confer MMC hypersensitivity (Castillo et al. 2014). Our results are consistent with these published reports in that deficiency of multiple RAP80 complex members showed similar deficits in ICL responses.
ICL hypersensitivity was associated with reduced ssDNA generation and homology-directed repair in Merit40−/− cells despite elevation of these DNA damage-associated markers in response to etoposide-, IR-, or nuclease-induced breaks (Coleman and Greenberg 2011; Hu et al. 2011; Goldstein and Kastan 2015). The opposing reduction in ssDNA formation in response to ICL damage may be a consequence of delayed ICL unhooking in Merit40−/− cells, consistent with involvement of MERIT40 in the earliest stages of ICL recognition and processing. Several nucleases have been suggested to process ICL lesions, including the SLX4 complex and FAN1 (Kratz et al. 2010; MacKay et al. 2010; Smogorzewska et al. 2010; Tsai et al. 2010; Crossan et al. 2011; Kim et al. 2011; Stoepker et al. 2011; Kim and D'Andrea 2012; Kottemann and Smogorzewska 2013). Future investigation is warranted as to whether MERIT40 affects their activities at ICLs.
Merit40−/− MEFs also displayed difficulty in restarting replication forks after DNA polymerase stalling (Fig. 2; Supplemental Fig. S3A,B), indicating that, in addition to ICL repair, the primary role of MERIT40 function is more broadly applicable to S-phase genotoxic stress. Multiple FA and BRCA pathway proteins have been implicated in replication fork maintenance or restart (Petermann et al. 2010; Schlacher et al. 2011, 2012; Neelsen and Lopes 2015; Zellweger et al. 2015). The function that MERIT40 exerts in S phase might be through its direct role in replication fork restart or through recruitment of BRCA1, which has been established to protect replication forks from nucleases (Schlacher et al. 2012). DNA2 has also been shown to mediate fork restart after MMC treatment by degrading reversed replication forks and preventing the formation of aberrant replication fork intermediates (Thangavel et al. 2015). The fact that knockdown of DNA2 in Merit40−/− MEFs led to lower plating efficiency without any genotoxic treatment (data not shown) indicates that MERIT40 and DNA2 might work in independent processes to maintain replication fork stability during ICL repair.
Repair of ICL lesions in S phase requires a multistep process that entails ICL recognition, dual incisions on either side of the cross-link, DNA synthesis through the cross-link by TLS polymerases and BRCA–Rad51-dependent HR repair of the ensuing DSB. The ubiquitinated D2-I complex plays a critical role in early ICL recognition and processing steps, whereas BRCA1 and BRCA2 have been implicated in both early and later aspects of ICL repair. Our extensive genetic interaction data are supportive of epistasis between MERIT40 and FANCD2. MERIT40 was required for timely ICL unhooking, and, importantly, double-knockout Merit40−/−, FancD2−/− mice and cells did not show any phenotypic or ICL repair exacerbations beyond that of FancD2 alone. In stark contrast, Merit40−/− and Brca2−/− alleles produced at least additive increases in genomic instability in response to MMC coupled with strongly reduced cell viability. However, we posit that MERIT40 contributes to multiple aspects of ICL repair (Fig. 7) in a manner that is not completely overlapping with FANCD2-I, thus accounting for differences in its genetic interactions with DNA2. These genetic interactions enhance our understanding of the multifactorial response to ICL damage and suggest targeting MERIT40 or other components of the RAP80 complex as an additional means to reduce viability in BRCA2 mutant cells.
Figure 7.
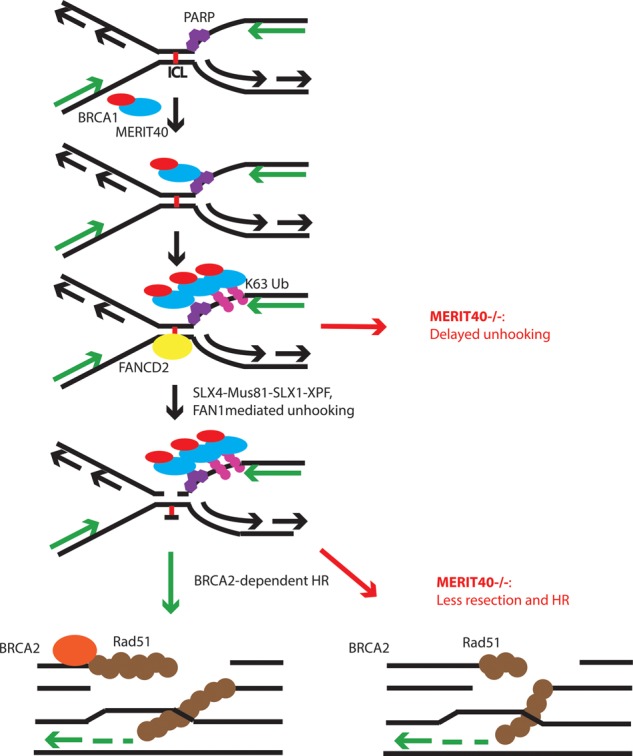
Model for contributions of MERIT40 to ICL repair. MERIT40 uses PAR and ubiquitination to rapidly recognize ICL damage prior to FANCD2. Merit40−/− cells have delayed unhooking coupled with reduced end resection and HR in response to ICLs. Merit40 deficiency exacerbates genomic instability in BRCA2 mutant cells but not in the context of FANCD2 mutation.
Materials and methods
Mice
C57BL/6J (strain 000664) and B6(Cg)-TyrC-2J/J (strain 000058) were purchased from the Jackson Laboratory. The homozygous conditional BRCA2tm1Brn mouse strain (01XB9) was purchased from the National Cancer Institute Mouse Repository. The Cre-ERT2 B6.Cg-Tg(UBC-cre/ERT2)1EjB/J strain was generously provided by Dr. Eric J. Brown (University of Pennsylvania, Philadelphia). The FancD2 knockout mouse was generously provided by Dr. Alan D'Andrea (Dana-Farber Cancer Institute, Harvard Medical School).
Generation of MERIT40 knockout mice, genotyping, and MEFs
MERIT40 (RIKEN cDNA 5430437P03 gene MGI: 1915501) knockout mice were generated by microinjecting an embryonic stem cell clone provided by Texas A&M Institute for Genomic Medicine (IST11207B9) into C57BL/6 host mouse blastocysts. The MERIT40 locus was disrupted by insertion of the Omnibank gene trap vector 76 into the MERIT40 first intron upstream of exon 2 containing the ATG translation start site as described in Rozenova et al. (2015) and on the Texas A&M Institute for Genomic Medicine Web site (http://www.tigm.org). Chimeric mice were generated and bred to B6(Cg)-TyrC-2J/J, and black pups were selected for genotyping. The resulting heterozygous mice were crossed to generate Merit40−/− mice.
Genotyping
Genotyping was performed by PCR on genomic DNA isolated from mouse tails by digestion overnight at 55°C in a lysis buffer containing 100 mM Tris (pH 8.0), 5 mM EDTA, 0.2% SDS, 200 mM NaCl, and 100 µg/mL proteinase K. DNA was precipitated with isopropanol and washed twice with 70% ethanol before being resuspended in TE buffer, quantified, and used for PCR reactions. The following primers were mixed for genotyping: M40F (5′-GGGAAACTTCAACTGTATCTTTTCGGCTCC-3′), M40R (5′-AAGACAGAAGATGGGCACGAGCCCCTTAC-3′), and V76R (5′-CTTGCAAAATGGCGTTACTTAAGC-3′). Primer pair M40F/M40R gave a band of 480 base pairs (bp) corresponding to the wild-type allele, and PCR from primer pair M40F/V76R gave a band of 320 bp corresponding to the disrupted allele. PCR conditions did not allow formation of the product corresponding to the wild-type allele when containing the gene trap vector. Amplifications were performed using GoTaq Green master mix (Promega) in a S1000 thermal cycler (Bio-Rad). BRCA1Δ11co/Δ11co mice were genotyped according to the Mouse Repository instructions.
The siRNA targets for human MERIT40 were M40 C (5′-CAGAGAACGUGCAGACGAUdTdT-3′), M40 D (5′-GCUC UGUAGCUGCCUCUAUdTdT-3′), and M40 E (5′-GGAGA UGAGUUGGAAGGAUdTdT-3′). The siRNA target mouse genomes were CtIP (UGUGAUCGCUGUGCAGUAAdTdT), DNA2-1 (GCAACAACGGUGUUUCGAUdTdT), DNA2-2 (CG GUACAAUUCUCCACGAAdTdT), siExoI (GGAUGUACUCU ACCUUCUAdTdT), siLig3 (GAGCUGGAAGAUAACGAAAd TdT), and siPOLQ (CCCAUCAAAUGUGAACGUAdTdT). siRNA transfections were performed using Lipofectamine RNAiMax (Invitrogen) according to the manufacturer's instructions, and cells were assayed 48 h after transfection.
G2/M checkpoint assay
The G2/M checkpoint assay was performed by assessing the percentage of mitotic cells at 2 h after irradiation with either 0 or 2 Gy. Cells were collected 2 h later and processed for flow cytometry as previously described. A rabbit polyclonal antibody against phosphorylated histone H3 (Upstate Biotechnology) was used to detect mitotic cells.
Irradiation
All radiation exposures were performed with a Gammacell 40 irradiator (Nordion International), which uses cesium-137 as the radiation source.
Mouse survival assay
To assess radiosensitivity and MMC sensitivity in vivo, age-matched (between 6 and 12 wk) wild-type and Merit40−/− littermates were subjected to 8.76 Gy of IR or 12 mg/kg MMC. MMC was resuspended in PBS and injected intraperitoneally. Mice were monitored daily to establish Kaplan-Meier curves. A log-rank test was used for statistical analysis.
Clonogenic survival assay
Cells were induced with 500 nM 4-OHT for 48 h and then seeded in a 60-mm dish. The following day, cells were treated with MMC at the indicated dose for 24 h. The drug was washed out, and cells were allowed to recover for 1–2 wk. Plates were stained with 0.5% crystal violet for 1 h, and colonies were counted. Each genotype and drug dose was done in triplicate in three independent experiments.
Cell culture
All human cell lines and MEFs were maintained in DMEM (Invitrogen) with 10% calf serum and penicillin and streptomycin. Rap80+/+ and Rap80−/− cell lines were provided by Dr. Junjie Chen (M.D. Anderson Cancer Center). MEFs were immortalized after transfection with an shRNA directed against p19Arf. Splenocytes were maintained in RPMI glutamax with β-mercaptethanol, 10% FBS, 1% sodium pyruvate, 1% nonessential amino acid, and 10 µg/mL LPS.
Antibodies
A MERIT40 rabbit polyclonal antibody raised against mouse MERIT40 was generated against a GST-MERIT40 fusion protein and used at 1:200 for immunoblotting and 1:100 for immunofluorescence as previously described (Shao et al. 2009). γ-H2AX (Millipore, JBW301) was used at 1:2000 for immunofluorescence and 1:1000 for immunoblotting. BRCA1 was detected by immunofluorescence with a homemade rabbit polyclonal antibody raised against the exon 11 region of mouse BRCA1 at 1:100. Homemade Rap80 rabbit polyclonal antibody was used for immunoblotting at 1:500 and immunofluorescence at 1:150. BRCC36 was detected for immunoblotting with a rabbit polyclonal antibody (Sobhian et al. 2007) at 1:1000. KIAA0157 was detected with a rabbit polyclonal antibody (LSBio, LS-C102632) diluted at 1:1000 (Zheng et al. 2013). α-Tubulin (1:10,000; Cell Signaling, DM1A) was used as a loading control on immunoblots. p-RPA (S4/S8) (Bethyl Laboratories, A300-245a) was used for immunoblotting at 1:1000. Total RPA (Novus Biologicals, NB600-565) was used for immunoblotting at 1:200. FANCD2 (Abcam, ab108928) was used of immunoblotting at 1:200. RPA32/RPA2 (Abcam, ab61184) was used for immunofluorescence in mouse cells at 1:200.
Immunofluorescence
Cells were washed with PBS and pre-extracted in a buffer containing 10 mM Pipes, 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, and 0.5% Triton X-100 for 5 min at 4°C or fixed directly in a solution containing 3% paraformaldehyde and 2% sucrose for 10 min at room temperature. Cells were subsequently permeabilized with 0.5% Triton-X100, 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 5 mM EDTA, 0.05% NP-40, and 0.25% gelatin IV for 10 min at 4°C and then incubated in a blocking solution (10% goat serum diluted in PBST) prior to incubation with the appropriate primary antibody for 1 h at 37°C. Cells were then washed with PBST and next incubated with secondary antibody diluted in PBST for 20 min at 37°C. After three washes with PBST, coverslips were mounted onto glass slides using Vectashield mounting medium containing DAPI (Vector Laboratories) and visualized using a Nikon Eclipse 80i fluorescent microscope.
Metaphase spreads
Cells were treated with 0.5 μM nocodazole for 3 h and swollen in 75 mM KCl for 25 min at 37°C. Cells were fixed on ice with a 3:1 methanol/acetic acid solution. Metaphases were dropped onto slides preheated at 42°C, allowed to dry, and stained with Giemsa. The numbers of sister chromatid breaks and chromosome breaks per metaphase were counted. For SCE analysis, cells were pretreated for 48 h with 10 μM BrdU and collected as above. After metaphases were dropped onto slides, they were stained with 10 μg/mL Hoechst 33258 in PBS for 20 min. The slides were rinsed in McIlvaine solution for 10 min and treated with 365-nm UV for 30 min. Slides were incubated in 1× SSC for 1 h at 55°C and then stained with Giemsa and analyzed for the number of SCEs per chromosome.
DNA fiber assay
Cells were labeled with 50 μM IdU or 100 μM CldU. DNA fibers were spread as described (Jackson and Pombo 1998) and stained with primary and fluorescent secondary antibody (primary antibodies: α-IdU [Becton Dickinson, 347580] and α-CldU [Abcam, ab6326]). Fibers were imaged with a 60× objective and analyzed using ImageJ software.
Native BrdU detection
Exponentially growing splenocytes were pulsed with 1 mM BrdU (Sigma) for 30 h, irradiated (30 Gy; 2 h of recovery at 37°C), and then fixed with 70% ethanol. Cells were blocked in 3% BSA in PBS for 30 min and then stained with anti-BrdU monoclononal antibody (1:50; Invitrogen, Mobu-1) for 1 h. Before analysis, propidium iodide was added to a final concentration of 50 μg/mL.
Modified alkaline comet assay
Cells were treated with 50 μM cisplatin for 1 h. After treatment, cells were either immediately harvested (0 h) or incubated in fresh medium before harvesting at 4, 6, 7.5, 9, 17, or 40 h after cisplatin was washed off. Control cells were not treated with cisplatin. Immediately before analysis, cells were irradiated (12.5 Gy) to get a fixed number of DNA strand breaks. We performed an alkaline comet assay based on the protocol from CometAssay kit (Trevigen, 42150-050-K). Olive tail moment was obtained by using the OpenComet plug-in in ImageJ. The percent decrease in Olive tail moment = [1 − (TMdi − TMcu)/(TMci − TMcu)] × 100, where TMdi is the tail moment of the cisplatin-treated, irradiated sample; TMcu is the tail moment of the untreated, unirradiated control; and TMci is the tail moment of the untreated, irradiated control. The percent decrease in Olive tail moment at the early time point demonstrates peaks when ICLs in cells get unhooked.
Supplementary Material
Acknowledgments
We thank A. D'Andrea (Dana-Farber Cancer Institute) for providing FancD2-null mice, J. Chen (M.D. Anderson Cancer Center) for Rap80+/+ and Rap80−/− cell lines, E. Brown (University of Pennsylvania) for Cre-ERT2 mice, and R. Ragland (University of Pennsylvania) for advice on replication restart experiments. This work was supported by National Institutes of Health grants GM101149, CA138835, and CA17494 to R.A.G., who is also supported by funds from the Abramson Family Cancer Research Institute and Basser Research Center for BRCA. This research was supported in part by the Intramural Research Program of the National Institutes of Health National Institute on Aging (ZO1 AG000746-08).
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.264192.115.
References
- Adamson B, Smogorzewska A, Sigoillot FD, King RW, Elledge SJ. 2012. A genome-wide homologous recombination screen identifies the RNA-binding protein RBMX as a component of the DNA-damage response. Nat Cell Biol 14: 318–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoniou AC, Wang X, Fredericksen ZS, McGuffog L, Tarrell R, Sinilnikova OM, Healey S, Morrison J, Kartsonaki C, Lesnick T, et al. 2010. A locus on 19p13 modifies risk of breast cancer in BRCA1 mutation carriers and is associated with hormone receptor-negative breast cancer in the general population. Nat Genet 42: 885–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton KL, Tyrer J, Song H, Ramus SJ, Notaridou M, Jones C, Sher T, Gentry-Maharaj A, Wozniak E, Tsai Y-Y, et al. 2010. Common variants at 19p13 are associated with susceptibility to ovarian cancer. Nat Genet 42: 880–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting SF, Callén E, Kozak ML, Kim JM, Wong N, López-Contreras AJ, Ludwig T, Baer R, Faryabi RB, Malhowski A, et al. 2012. BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Mol Cel 46: 125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo A, Paul A, Sun B, Huang TH, Wang Y, Yazinski SA, Tyler J, Li L, You MJ, Zou L, et al. 2014. The BRCA1-interacting protein Abraxas is required for genomic stability and tumor suppression. Cell Rep 8: 807–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Silver DP, Walpita D, Cantor SB, Gazdar AF, Tomlinson G, Couch FJ, Weber BL, Ashley T, Livingston DM, et al. 1998. Stable interaction between the products of the BRCA1 and BRCA2 tumor suppressor genes in mitotic and meiotic cells. Mol Cel 2: 317–328. [DOI] [PubMed] [Google Scholar]
- Clauson C, Scharer OD, Niedernhofer L. 2013. Advances in understanding the complex mechanisms of DNA interstrand crosslink repair. Cold Spring Harb Perspect Med 5: a012732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman KA, Greenberg RA. 2011. The BRCA1–RAP80 complex regulates DNA repair mechanism utilization by restricting end resection. J Biol Chem 286: 13669–13680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper EM, Cutcliffe C, Kristiansen TZ, Pandey A, Pickart CM, Cohen RE. 2009. K63-specific deubiquitination by two JAMM/MPN+ complexes: BRISC-associated Brcc36 and proteasomal Poh1. EMBO J 28: 621–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossan GP, van der Weyden L, Rosado IV, Langevin F, Gaillard P-HL, McIntyre RE, Gallagher F, Kettunen MI, Lewis DY, Brindle K, et al. 2011. Disruption of mouse Slx4, a regulator of structure-specific nucleases, phenocopies Fanconi anemia. Nat Genet 43: 147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Andrea AD, Grompe M. 2003. The Fanconi anaemia/BRCA pathway. Nat Rev Cancer 3: 23–34. [DOI] [PubMed] [Google Scholar]
- Deans AJ, West SC. 2011. DNA interstrand crosslink repair and cancer. Nat Rev Cancer 11: 467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domchek SM, Tang J, Stopfer J, Lilli DR, Hamel N, Tischkowitz M, Monteiro ANA, Messick TE, Powers J, Yonker A, et al. 2013. Biallelic deleterious BRCA1 mutations in a woman with early-onset ovarian cancer. Cancer Discov 3: 399–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Huang J, Chen J. 2009. MERIT40 facilitates BRCA1 localization and DNA damage repair. Genes Dev 23: 719–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Wang J, Chen J. 2010. The Lys63-specific deubiquitinating enzyme BRCC36 is regulated by two scaffold proteins localizing in different subcellular compartments. J Biol Chem 285: 30982–30988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garaycoechea JI, Patel KJ. 2014. Why does the bone marrow fail in Fanconi anemia? Blood 123: 26–34. [DOI] [PubMed] [Google Scholar]
- Goggins M, Schutte M, Lu J, Moskaluk CA, Weinstein CL, Petersen GM, Yeo CJ, Jackson CE, Lynch HT, Hruban RH, et al. 1996. Germline BRCA2 gene mutations in patients with apparently sporadic pancreatic carcinomas. Cancer Res 56: 5360–5364. [PubMed] [Google Scholar]
- Goldstein M, Kastan MB. 2015. Repair versus checkpoint functions of BRCA1 are differentially regulated by site of chromatin binding. Cancer Res 75: 2699–2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudjonsson T, Altmeyer M, Savic V, Toledo L, Dinant C, Grøfte M, Bartkova J, Poulsen M, Oka Y, Bekker-Jensen S, et al. 2012. TRIP12 and UBR5 suppress spreading of chromatin ubiquitylation at damaged chromosomes. Cell 150: 697–709. [DOI] [PubMed] [Google Scholar]
- Guzzo CM, Berndsen CE, Zhu J, Gupta V, Datta A, Greenberg RA, Wolberger C, Matunis MJ. 2012. RNF4-dependent hybrid SUMO–ubiquitin chains are signals for RAP80 and thereby mediate the recruitment of BRCA1 to sites of DNA damage. Sci Signal 5: ra88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley JM, Spanswick VJ, Gander M, Giacomini G, Whelan J, Souhami RL, Hartley JA. 1999. Measurement of DNA cross-linking in patients on ifosfamide therapy using the single cell gel electrophoresis (comet) assay. Clin Cancer Res 5: 507–512. [PubMed] [Google Scholar]
- Hu Y, Scully R, Sobhian B, Xie A, Shestakova E, Livingston DM. 2011. RAP80-directed tuning of BRCA1 homologous recombination function at ionizing radiation-induced nuclear foci. Genes Dev 25: 685–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Petit SA, Ficarro SB, Toomire KJ, Xie A, Lim E, Cao SA, Park E, Eck MJ, Scully R, et al. 2014. PARP1-driven poly-ADP-ribosylation regulates BRCA1 function in homologous recombination-mediated DNA repair. Cancer Discov 4: 1430–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Liu S, Bellani MA, Thazhathveetil AK, Ling C, de Winter JP, Wang Y, Wang W, Seidman MM. 2013. The DNA translocase FANCM/MHF promotes replication traverse of DNA interstrand crosslinks. Mol Cel 52: 434–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huen MSY, Sy SMH, Chen J. 2009. BRCA1 and its toolbox for the maintenance of genome integrity. Nat Rev Mol Cell Biol 11: 138–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SP, Durocher D. 2013. Regulation of DNA damage responses by ubiquitin and SUMO. Mol Cel 49: 795–807. [DOI] [PubMed] [Google Scholar]
- Jackson DA, Pombo A. 1998. Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J Cell Biol 140: 1285–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Greenberg RA. 2015. Deciphering the BRCA1 tumor suppressor network. J Biol Chem 290: 17724–17732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karanja KK, Lee EH, Hendrickson EA, Campbell JL. 2014. Preventing over-resection by DNA2 helicase/nuclease suppresses repair defects in Fanconi anemia cells. Cell Cycle 13: 1540–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, D'Andrea AD. 2012. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev 26: 1393–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Yu X. 2007. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science 316: 1202–1205. [DOI] [PubMed] [Google Scholar]
- Kim Y, Lach FP, Desetty R, Hanenberg H, Auerbach AD, Smogorzewska A. 2011. Mutations of the SLX4 gene in Fanconi anemia. Nat Genet 43: 142–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottemann MC, Smogorzewska A. 2013. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 493: 356–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratz K, Schöpf B, Kaden S, Sendoel A, Eberhard R, Lademann C, Cannavo E, Sartori AA, Hengartner MO, Jiricny J. 2010. Deficiency of FANCD2-associated nuclease KIAA1018/FAN1 sensitizes cells to interstrand crosslinking agents. Cell 142: 77–88. [DOI] [PubMed] [Google Scholar]
- Li M, Yu X. 2013. Function of BRCA1 in the DNA damage response is mediated by ADP-ribosylation. Cancer Cell 23: 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long DT, Joukov V, Budzowska M, Walter JC. 2014. BRCA1 promotes unloading of the CMG helicase from a stalled DNA replication fork. Mol Cel 56: 174–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKay C, Déclais A-C, Lundin C, Agostinho A, Deans AJ, Macartney TJ, Hofmann K, Gartner A, West SC, Helleday T, et al. 2010. Identification of KIAA1018/FAN1, a DNA repair nuclease recruited to DNA damage by monoubiquitinated FANCD2. Cell 142: 65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattiroli F, Vissers JHA, van Dijk WJ, Ikpa P, Citterio E, Vermeulen W, Marteijn JA, Sixma TK. 2012. RNF168 ubiquitinates K13–15 on H2A/H2AX to drive DNA damage signaling. Cell 150: 1182–1195. [DOI] [PubMed] [Google Scholar]
- Messick TE, Greenberg RA. 2009. The ubiquitin landscape at DNA double-strand breaks. J Cell Biol 187: 319–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moynahan ME, Chiu JW, Koller BH, Jasin M. 1999. Brca1 controls homology-directed DNA repair. Mol Cel 4: 511–518. [DOI] [PubMed] [Google Scholar]
- Neelsen KJ, Lopes M. 2015. Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat Rev Mol Cell Biol 16: 207–220. [DOI] [PubMed] [Google Scholar]
- Nikkilä J, Coleman KA, Morrissey D, Pylkäs K, Erkko H, Messick TE, Karppinen S-M, Amelina A, Winqvist R, Greenberg RA. 2009. Familial breast cancer screening reveals an alteration in the RAP80 UIM domain that impairs DNA damage response function. Oncogene 28: 1843–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson-Fortin J, Shao G, Bretscher H, Messick TE, Greenberg RA. 2010. Differential regulation of JAMM domain deubiquitinating enzyme activity within the RAP80 complex. J Biol Chem 285: 30971–30981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. 2010. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cel 37: 492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragland RL, Patel S, Rivard RS, Smith K, Peters AA, Bielinsky A-K, Brown EJ. 2013. RNF4 and PLK1 are required for replication fork collapse in ATR-deficient cells. Genes Dev 27: 2259–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozenova K, Jiang J, Donaghy R, Aressy B, Greenberg RA, Tong W. 2015. MERIT40 deficiency expands hematopoietic stem cell pools by regulating thrombopoietin receptor signaling. Blood 125: 1730–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y, Yoshikawa A, Yamashita M, Yamagata A, Fukai S. 2009. Structural basis for specific recognition of Lys 63-linked polyubiquitin chains by NZF domains of TAB2 and TAB3. EMBO J 28: 3903–3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer SL, Tian L, Kahkonen M, Schwartzentruber J, Kircher M, Majewski J, Dyment DA, Innes AM, Boycott KM, Moreau LA, et al. 2015. Biallelic mutations in BRCA1 cause a new Fanconi anemia subtype. Cancer Discov 5: 135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. 2011. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145: 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Wu H, Jasin M. 2012. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51–BRCA1/2. Cancer Cell 22: 106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully R, Ganesan S, Brown M, De Caprio JA, Cannistra SA, Feunteun J, Schnitt S, Livingston DM. 1996. Location of BRCA1 in human breast and ovarian cancer cells. Science 272: 123–126. [DOI] [PubMed] [Google Scholar]
- Scully R, Chen J, Plug A, Xiao Y, Weaver D, Feunteun J, Ashley T, Livingston DM. 1997. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell 88: 265–275. [DOI] [PubMed] [Google Scholar]
- Shao G, Patterson-Fortin J, Messick TE, Feng D, Shanbhag N, Wang Y, Greenberg RA. 2009. MERIT40 controls BRCA1–Rap80 complex integrity and recruitment to DNA double-strand breaks. Genes Dev 23: 740–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharan SK, Morimatsu M, Albrecht U, Lim DS, Regel E, Dinh C, Sands A, Eichele G, Hasty P, Bradley A. 1997. Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. Nature 386: 804–810. [DOI] [PubMed] [Google Scholar]
- Silver DP, Livingston DM. 2001. Self-excising retroviral vectors encoding the Cre recombinase overcome Cre-mediated cellular toxicity. Mol Cel 8: 233–243. [DOI] [PubMed] [Google Scholar]
- Sims JJ, Cohen RE. 2009. Linkage-specific avidity defines the lysine 63-linked polyubiquitin-binding preference of Rap80. Mol Cel 33: 775–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smogorzewska A, Desetty R, Saito TT, Schlabach M, Lach FP, Sowa ME, Clark AB, Kunkel TA, Harper JW, Colaiácovo MP, et al. 2010. A genetic screen identifies FAN1, a Fanconi anemia-associated nuclease necessary for DNA interstrand crosslink repair. Mol Cel 39: 36–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobhian B, Shao G, Lilli DR, Culhane AC, Moreau LA, Xia B, Livingston DM, Greenberg RA. 2007. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science 316: 1198–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solyom S, Aressy B, Pylkäs K, Patterson-Fortin J, Hartikainen JM, Kallioniemi A, Kauppila S, Nikkilä J, Kosma V-M, Mannermaa A, et al. 2012. Breast cancer-associated Abraxas mutation disrupts nuclear localization and DNA damage response functions. Sci Transl Med 4: 122ra23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanswick VJ, Hartley JM, Ward TH, Hartley JA. 1999. Measurement of drug-induced DNA interstrand crosslinking using the single-cell gel electrophoresis (comet) assay. Methods Mol Med 28: 143–154. [DOI] [PubMed] [Google Scholar]
- Stoepker C, Hain K, Schuster B, Hilhorst-Hofstee Y, Rooimans MA, Steltenpool J, Oostra AB, Eirich K, Korthof ET, Nieuwint AWM, et al. 2011. SLX4, a coordinator of structure-specific endonucleases, is mutated in a new Fanconi anemia subtype. Nat Genet 43: 138–141. [DOI] [PubMed] [Google Scholar]
- Thangavel S, Berti M, Levikova M, Pinto C, Gomathinayagam S, Vujanovic M, Zellweger R, Zellweger R, Moore H, Moore H, et al. 2015. DNA2 drives processing and restart of reversed replication forks in human cells. J Cell Biol 208: 545–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, Chang HY. 2010. Long noncoding RNA as modular scaffold of histone modification complexes. Science 329: 689–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Matsuoka S, Ballif BA, Zhang D, Smogorzewska A, Gygi SP, Elledge SJ. 2007. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science 316: 1194–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Hurov K, Hofmann K, Elledge SJ. 2009. NBA1, a new player in the Brca1 A complex, is required for DNA damage resistance and checkpoint control. Genes Dev 23: 729–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Liu C, Chen J, Yu X. 2012. RAP80 protein is important for genomic stability and is required for stabilizing BRCA1-A complex at DNA damage sites in vivo. J Biol Chem 287: 22919–22926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Z, Menendez D, Resnick MA, French JE, Janardhan KS, Jetten AM. 2012. RAP80 is critical in maintaining genomic stability and suppressing tumor development. Cancer Res 72: 5080–5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, Vindigni A, Lopes M. 2015. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol 208: 563–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Gupta V, Patterson-Fortin J, Bhattacharya S, Katlinski K, Wu J, Varghese B, Carbone CJ, Aressy B, Fuchs SY, et al. 2013. A BRISC–SHMT complex deubiquitinates IFNAR1 and regulates interferon responses. Cell Rep 5: 180–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}