

Abstract
Glial cell line-derived neurotrophic factor (GDNF) promotes PNS development and kidney morphogenesis via a receptor complex consisting of the glycerophosphatidylinositol (GPI)-anchored, ligand binding receptor GDNF family receptor α1 (GFRα1) and the receptor tyrosine kinase Ret. Although Ret signal transduction in vitro is augmented by translocation into lipid rafts via GFRα1, the existence and importance of lipid rafts in GDNF–Ret signaling under physiologic conditions is unresolved. A knock-in mouse was produced that replaced GFRα1 with GFRα1–TM, which contains a transmembrane (TM) domain instead of the GPI anchor. GFRα1–TM still binds GDNF and promotes Ret activation but does not translocate into rafts. In Gfrα1TM/TM mice, GFRα1–TM is expressed, trafficked, and processed at levels identical to GFRα1. Although Gfrα1+/TM mice are viable, Gfrα1TM/TM mice display bilateral renal agenesis, lack enteric neurons in the intestines, and have motor axon guidance deficits, similar to Gfrα1−/− mice. Therefore, the recruitment of Ret into lipid rafts by GFRα1 is required for the physiologic functions of GDNF in vertebrates.
SIGNIFICANCE STATEMENT Membrane microdomains known as lipid rafts have been proposed to be unique subdomains in the plasma membrane that are critical for the signaling functions of multiple receptor complexes. Their existence and physiologic relevance has been debated. Based on in vitro studies, lipid rafts have been reported to be necessary for the function of the Glial cell line-derived neurotrophic factor (GDNF) family of neurotrophic factors. The receptor for GDNF comprises the lipid raft-resident, glycerophosphatidylinositol-anchored receptor GDNF family receptor α1 (GFRα1) and the receptor tyrosine kinase Ret. Here we demonstrate, using a knock-in mouse model in which GFRα1 is no longer located in lipid rafts, that the developmental functions of GDNF in the periphery require the translocation of the GDNF receptor complex into lipid rafts.
Keywords: GDNF, lipid raft, neurotrophic factor, Ret, spinal motor neuron, transgenic
Introduction
Glial cell line-derived neurotrophic factor (GDNF) is a growth factor that is critical for the development of the nervous system and kidneys (Airaksinen and Saarma, 2002). The high-affinity binding receptor for GDNF is GDNF family receptor α1 (GFRα1), a glycerophosphatidylinositol (GPI)-anchored cell-surface protein (Treanor et al., 1996; Cacalano et al., 1998). During GDNF binding to GFRα1, which causes the dimerization of two GFRα1 molecules, this complex then binds to and activates signal-transducing receptors (Bespalov and Saarma, 2007). The receptor tyrosine kinase Ret is necessary for the functions of GDNF in the PNS, such as motor neuron axon guidance (Enomoto et al., 2000; Kramer et al., 2006; Runeberg-Roos and Saarma, 2007; Dudanova et al., 2010; Bonanomi et al., 2012). GDNF/GFRα1-mediated Ret activation is also required for kidney morphogenesis and for the proliferation, migration, and differentiation of enteric precursors that form the enteric nervous system (Baloh et al., 2000).
Lipid rafts are membrane microdomains that are enriched in cholesterol and sphingolipids, forming more ordered lipid bilayers than the surrounding plasma membrane (Simons and van Meer, 1988; Patra, 2008; Simons and Sampaio, 2011). Lipid rafts are enriched with GPI-anchored proteins and proteins that are modified with saturated lipids, such as Src family kinases. In cultured cells, GFRα1 is enriched highly in lipid rafts because of its GPI anchor, but Ret is excluded from lipid rafts under basal, unactivated conditions (Tansey et al., 2000; Paratcha et al., 2001). During the exposure of primary neurons to GDNF in vitro, Ret is translocated rapidly into lipid rafts in a GFRα1-dependent manner, which is important for downstream signal transduction (Tansey et al., 2000; Encinas et al., 2001; Paratcha et al., 2001; Pierchala et al., 2006). Investigations of the importance of lipid rafts in Ret signal transduction is based on biochemical experiments using detergent insolubility or buoyancy on density gradients. However, it has been argued that cooling cells down and using detergents for cell lysis may coalesce membrane microdomains that do not exist normally (Munro, 2003). Direct evidence for the existence of lipid rafts in living cells using imaging techniques, such as fluorescence energy transfer, have provided varied results (Simons and Ikonen, 1997; Munro, 2003; Simons and Sampaio, 2011). Identifying the cellular functions of lipid rafts typically relies on their disruption via the depletion of cholesterol or sphingomyelin in the plasma membrane of cultured cells, which likely also affects non-raft-dependent functions (Brown and London, 1998; Galbiati et al., 2001; Kenworthy, 2002; Munro, 2003). Because these in vitro methods and treatments cannot be applied to complex, multicellular organisms, evidence for the physiological relevance of lipid rafts at an organismal level remains lacking.
We report here the production of a knock-in mouse containing a GFRα1 gene replacement such that GDNF still promotes Ret activation but not its translocation into lipid rafts. These knock-in mice displayed renal agenesis, a loss of the enteric nervous system, and motor neuron axon pathfinding defects reminiscent of GFRα1 knock-out mice, providing evidence for the physiologic importance of lipid rafts in neurotrophic factor signaling in vivo.
Materials and Methods
Production of GFRα1–TM knock-in mice.
The cDNA for human GFRα1–TM was kindly provided by Jeffrey Milbrandt (Washington University, St. Louis, MO) and subcloned into the second exon of the Gfrα1 gene. The plasmid encoding the genomic region flanking exon 2, along with other constructs for homologous recombination, were generously provided by Hideki Enomoto (RIKEN Center for Developmental Biology, Kobe, Japan). Homologous recombination of ES cell clones was confirmed by Southern blotting using a probe outside of the targeting construct region, and subsequent offspring were genotyped by PCR (Fig. 1C). The PCR primers are as follows: common forward, 5′-CTTCCAGGTTGGGTCGGAACTGAACCC; wild-type reverse, 5′-AGAGAGCTCAGCGTGCAGAGATC; and mutant reverse, 5′-CATGCTCCAGTAGATACGCAGACA.
Figure 1.

Production of knock-in mice in which GFRα1 is excluded from lipid rafts. A, Stable Neuro2A cell lines expressing either GFRα1 or GFRα1-TM were stimulated with GDNF (50 ng/ml) for 15 min before detergent extraction. The extracts were immunoblotted with antibodies to phosphorylated Ret (PY1062Ret), total Ret51, and actin. GDNF activates Ret to a similar extent in Neuro2A cells expressing GFRα1–TM as those expressing GFRα1. B, Targeting strategy for the production of GFRα1–TM knock-in mice. Insertion of the cDNA for human GFRα1–TM into exon 2 of Gfrα1 eliminated expression of the native gene. C, Confirmation of homologous recombination of the targeting vector into the Gfrα1 locus was confirmed by Southern blotting using a probe outside of the targeted region (left). Genotyping of F2 offspring by PCR identified mice that were Gfrα1+/+, Gfrα1+/TM, and Gfrα1TM/TM (right). D, GFRα1–TM is expressed at levels similar to GFRα1 in knock-in mice. Whole cellular extracts from transfected HEK293 cells (right) or cerebral cortices (left) of Gfrα1+/+, Gfrα1+/TM, and Gfrα1TM/TM mice were immunoblotted with antibodies to GFRα1 that detects both normal and mutant GFRα1. Actin immunoblotting of the extracts confirmed similar loading of protein among the different samples.
Two independent knock-in lines from two different ES cell clones were analyzed and had identical phenotypes. An AC–Cre cassette was used in the targeting construct that allowed the expression of Cre in somatic cells but self-excises in the male germ line (Bunting et al., 1999). The original two founder lines were backcrossed seven generations into C57BL/6 mice, and these mice were used for all the studies described. For the examination of spinal motor neuron projections, Gfrα1TM/+ mice were bred with Hb9–GFP mice (The Jackson Laboratories), and embryos were imaged directly under a stereoscope with fluorescent illumination (Discovery V8; Zeiss).
Primary sympathetic neurons.
Sympathetic neurons of the superior cervical ganglion (SCG) were dissected, enzymatically dissociated, and maintained in vitro as described previously (Tsui and Pierchala, 2010). The SCGs from each mouse were isolated and cultured separately, and their genotype ascertained from extracted tail DNA. Thus, individual conditions typically represented extracts produced from sympathetic neurons derived from individual mice. For the pharmacologic inhibition of matrix metalloproteinases and phospholipases, TNF-α protease inhibitor 1 (TAPI-1; 10 μm; Calbiochem) and U73122 (1-[6[[(17β)-3-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-1H-pyrrole-2,5-dione; 2 μm; Sigma), respectively, were applied to the cultures for 48 h. Vehicle alone (DMSO; Sigma) served as the negative control, and phosphatidylinositol-specific phospholipase C enzyme (PI-PLC; 2 U/ml; Sigma) served as a positive control.
Tissue extractions and immunoprecipitations.
Whole embryos [embryonic day 14.5 (E14.5) to E18.5] or organs dissected from E19.5 mouse pups were homogenized using a Tissuelyser II with steel grinding jars (Qiagen). These homogenates were then detergent extracted (10% glycerol, 1% Nonidet P-40, and sodium orthovanidate in Tris-buffered saline, pH 7.4). Insoluble debris was removed by centrifugation, and the supernatant was then used for immunoprecipitations or was used directly for immunoblotting. For immunoprecipitations, crude extracts were precleared by incubation with protein A and protein G (Roche) for 1 h at 4°C. The protein A/G was then removed by centrifugation, and Ret antibodies (Ret C19-G and Ret C20 mixed; Santa Cruz Biotechnology) or GFRα1 antibodies (Cell Sciences), along with fresh protein A/G, were added and allowed to incubate in the extracts overnight at 4°C with gentle agitation. Immunoprecipitates were then washed, and the protein complexes were denatured by boiling them in SDS-PAGE sample buffer (2% SDS, 1% β-mercaptoethanol, 10% glycerol, and bromophenol blue in Tris, pH 6.8). For the analysis of soluble and secreted proteins, sympathetic neurons were maintained in culture for 5–7 d. The medium was then replaced with serum-free medium, and the neurons were maintained for an additional 4 d with two medium changes (4 ml total). The conditioned medium was centrifuged for 30 min at 4°C (16,000 × g) to remove cellular debris, and the proteins in the conditioned medium supernatants were precipitated using saturated trichloroacetic acid (TCA). The protein pellets were washed twice with ice-cold ethanol/ether (1:1 v/v) to remove any remaining TCA and were then analyzed by immunoblotting. Equal volumes of conditioned medium from equal numbers of neurons were analyzed in each condition.
Cell-surface biotinylation.
Biotinylation of cell-surface proteins of primary neurons was performed as described previously (Tsui and Pierchala, 2010). Biotinylated proteins were isolated from intracellular proteins by detergent extraction of the neurons, followed by the selective isolation of biotinylated proteins using streptavidin agarose (Pierce Thermo Fisher Scientific). Isolated proteins were then analyzed by immunoblotting.
Immunoblotting analysis.
Denatured protein extracts were subjected to SDS-PAGE and blotted onto PVDF membranes (Immobilon P; Millipore). Membranes were incubated in either 5% milk or 3% BSA for 1 h before an overnight incubation in the primary antibody at 4°C. The blots were then washed, incubated in the appropriate HRP-linked secondary antibody, and visualized by using a chemiluminescent substrate (Pierce Thermo Fisher Scientific). The antibodies used, and their working dilutions from the stock, were as follows: anti-GFRα1 (1:1000; R&D Systems), anti-phosphotyrosine (1:2000, clone 4G10; Millipore), anti-actin and anti-Ret51 (1:1000 each; Santa Cruz Biotechnology), anti-caveolin-1 (1:500; Sigma), anti-transferrin receptor (1:1000; Invitrogen), and anti-multi-ubiquitin (1:1000, clone FK2; Enzo Life Sciences). Quantification of immunoblots was performed using NIH ImageJ.
Acetylcholinesterase histochemistry and tyrosine hydroxylase immunohistochemistry.
Sections of small and large intestine were dissected from E18.5 embryos, and the mesenteric attachments were removed. These tissues were incubated in a glycine buffer, pH 5.6, containing ethopropazine HCl, aceytlthiocholine iodide, cupric sulfate, and sodium acetate (all chemicals from Sigma) for 30 min. The color was developed by incubation in sodium sulfide for 2–3 min. The intestines were then mounted in glycerol and imaged using a stereoscope with polarized lighting (Discovery V8; Zeiss). Whole-mount immunohistochemistry for tyrosine hydroxylase was performed as described previously (Enomoto et al., 2001). After immunostaining, the embryos were dissected into an “open-book” preparation for stereoscope imaging to reveal the sympathetic chain ganglia and projections.
Results
Production of mice that selectively lack lipid raft-mediated GDNF signaling
To determine whether lipid rafts are required for the developmental functions of GDNF, knock-in mice were produced in which the GFRα1 gene was replaced with a cDNA encoding GFRα1–TM. GFRα1–TM is a fusion protein in which GFRα1 lacking the amino acids that target GPI anchorage were replaced with the transmembrane (TM) domain of HLA–B44 (Hansbrough et al., 1991; Tansey et al., 2000). This results in a GFRα1 molecule that is not GPI anchored and instead has a TM domain that targets GFRα1 to the cell surface but not to lipid rafts (Hansbrough et al., 1991; Tansey et al., 2000). GFRα1–TM still binds to GDNF and promotes efficient Ret activation but not the translocation of this signaling complex into lipid rafts (Fig. 1A; Tansey et al., 2000). For the generation of the targeting construct, GFRα1–TM was inserted into exon 2 of Gfrα1, thereby disrupting exon 2 and resulting in a complete loss of GFRα1 expression (Fig. 1B, C), as was done previously to make GFRα1 knock-out mice (Enomoto et al., 1998).
To determine whether GFRα1–TM protein in Gfrα1TM/TM mice was expressed at levels similar to native GFRα1, cellular extracts were produced from the cerebral cortex (Fig. 1D) of postnatal day 0 mice and analyzed by immunoblotting. GFRα1–TM from Gfrα1TM/TM mice was expressed at levels similar to native GFRα1 from Gfrα1+/+ mice compared with the levels of actin. In transfection studies, the vast majority of GFRα1–TM migrates at a similar molecular weight to GPI-anchored GFRα1 by SDS-PAGE (Fig. 1D). Thus, the production of Gfrα1TM/TM knock-in mice using this strategy did not result in a hypomorphic allele that could confound the phenotypic analysis.
Plasma membrane targeting is normal in Gfrα1TM/TM mice
To confirm that the replacement of the GPI anchor of GFRα1 with a TM domain did not impair its trafficking to the plasma membrane, primary sympathetic neurons from Gfrα1+/+, Gfrα1+/TM, and Gfrα1TM/TM mice were subjected to cell-surface biotinylation. Cell-surface proteins were then isolated from other proteins using immobilized streptavidin and analyzed by immunoblotting (Fig. 2A). This analysis revealed that a similar amount of GFRα1–TM was localized to the plasma membrane of Gfrα1TM/TM neurons compared with GPI-anchored GFRα1 from Gfrα1+/+ neurons, indicating that intracellular trafficking of GFRα1–TM to the plasma membrane was not altered.
Figure 2.
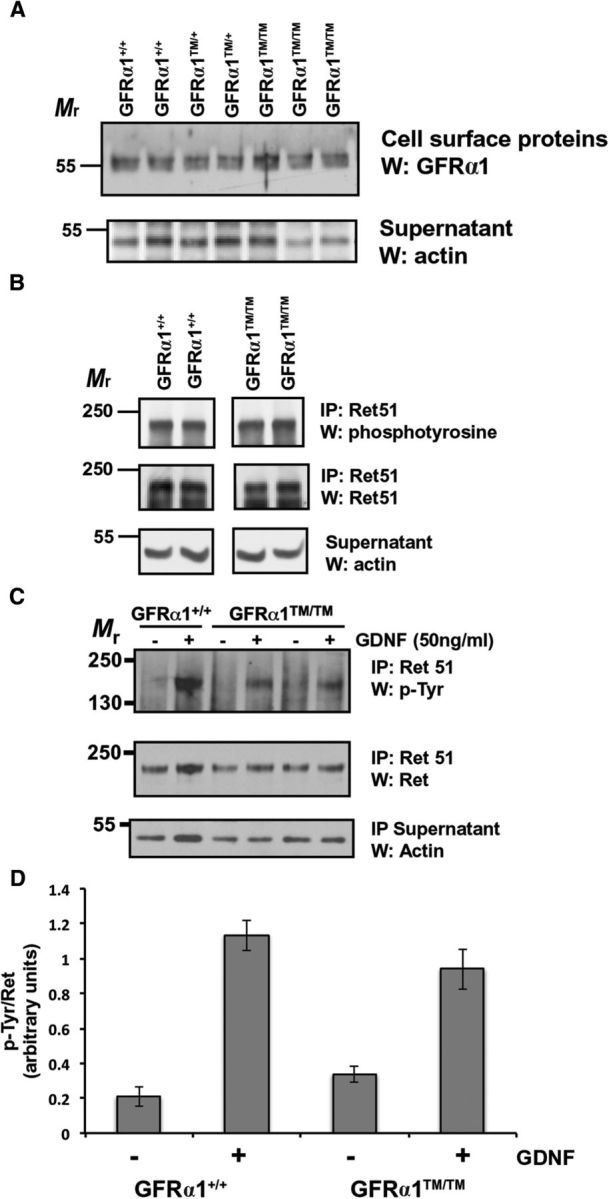
The localization of GFRα1 in lipid rafts is not necessary for the subcellular trafficking or activation of Ret. A, Primary neurons were subjected to cell-surface biotinylation, followed by detergent extraction and isolation of the biotinylated, cell-surface proteins. The cell-surface proteins were subjected to GFRα1 immunoblotting (top), and actin immunoblotting of the remaining supernatant fractions served as protein loading controls (bottom). B, E15.5 Gfrα1+/+ and Gfrα1TM/TM mice were homogenized and detergent extracted. Ret was isolated from these extracts by immunoprecipitation, and the level of activation was determined by phosphotyrosine immunoblotting. The amount of Ret in each sample was ascertained by reprobing the blots with Ret51 antibodies, and actin immunoblotting of supernatants served to confirm the analysis of equal amounts of protein. These experiments were performed three times with similar results. C, Primary neurons from Gfrα1+/+ and Gfrα1TM/TM mice were stimulated with GDNF (50 ng/ml) or medium alone for 10 min. The neurons were then detergent extracted, Ret51 was immunoprecipitated from the extracts, and its level of activation was determined by phosphotyrosine immunoblotting (top). The level of Ret in each condition was ascertained by reprobing these blots with Ret51 antibodies (middle), and actin immunoblotting of the immunoprecipitation supernatants served as a loading control (bottom). D, The experiments shown in C were quantified and graphed as the mean ± range (n = 2) or SEM (n = 3). Similar results were obtained from neurons derived from two Gfrα1+/+ and three Gfrα1TM/TM mice from two separate cultures.
Ret activation is unaffected in Gfrα1TM/TM mice
The GFRα1–TM fusion does not alter the region of GFRα1 that binds to GDNF and, therefore, does not affect formation of the GDNF/GFRα1/Ret complex or the autophosphorylation of Ret (Fig. 1A; Tansey et al., 2000). To confirm that this was also true in GFRα1–TM knock-in mice, whole E15.5 Gfrα1+/+ and Gfrα1TM/TM mice were homogenized, and Ret was isolated from these extracts (Fig. 2B). Phosphotyrosine immunoblotting of the Ret immunoprecipitations revealed that there was no difference in the amount of autophosphorylated Ret between these two genotypes, suggesting that GFRα1–TM is fully capable of forming an activated GDNF/Ret complex in vivo. To further confirm that GDNF family ligand (GFL)-initiated Ret autophosphorylation was not impaired in neurons expressing GFRα1–TM, sympathetic neurons from the SCG were isolated and cultured from Gfrα1+/+ and Gfrα1TM/TM mice. These neurons were then stimulated with GDNF, or medium alone, and the extent of Ret activation was determined by Ret immunoprecipitation, followed by phosphotyrosine immunoblotting (Fig. 2C, D). GDNF activated Ret to a similar extent in neurons derived from Gfrα1TM/TM mice as it did neurons from Gfrα1+/+ mice (Fig. 2C, D), indicating that GFRα1–TM is capable of activating Ret during GDNF stimulation.
GFRα1–TM is shed from the plasma membrane to a similar extent as wild-type GFRα1
GFRα1 can be shed from the plasma membrane of cells maintained in vitro and from tissues in vivo, and soluble GFRα1 can participate in signaling events in trans (Paratcha et al., 2001). If cleavage from the membrane occurs via a lipase that cleaves the GPI anchor of GFRα1, then Gfrα1TM/TM mice would be deficient in the membrane shedding of GFRα1–TM. Therefore, the extent of membrane shedding was examined in primary sympathetic neurons from Gfrα1+/+, Gfrα1+/TM, and Gfrα1TM/TM mice. Conditioned medium was not collected until after 7 d in vitro, well after the initial period of cellular injury and death attributable to the enzymatic dissociation of the ganglia. On day 8 in vitro, conditioned medium was collected every 2 d for 4 d and was centrifuged thoroughly to remove any cellular debris. Soluble GFRα1 or GFRα1–TM was then analyzed from the medium by immunoblotting (Fig. 3A, B). Interestingly, there was just as much secreted GFRα1 from the Gfrα1TM/TM neurons as there was GFRα1 secreted from the Gfrα1+/+ and Gfrα1TM/+ neurons, and quantification of the immunoblots confirmed that there were no significant differences between the three genotypes (Fig. 3B). Immunoblotting of brain extracts from Gfrα1+/+ and Gfrα1−/− mice with the GFRα1 antibody confirmed the specificity of our immunoblotting analysis (Fig. 3A). These data suggest that GFRα1 was shed from the plasma membrane by proteolysis, most likely by an extracellular protease, such as metalloproteinases, rather than by a phospholipase. To examine this possibility, wild-type primary sympathetic neurons were exposed to an inhibitor of matrix metalloproteinases (TAPI-1) or an inhibitor of phospholipases (U73122). After 48 h, the culture medium was collected, and the amount of GFRα1 shedding was analyzed by immunoblotting, as before. Inhibition of either metalloproteinases or phospholipases reduced significantly the amount of GFRα1 that was shed into the culture medium (Fig. 3C, D). Exposure of the cells to purified PI-PLC for 2 h resulted in GFRα1 cleavage and release into the culture medium as well, confirming this immunoblotting assay. Treatment of neurons with both TAPI-1 and U73122 did not have an additive effect on the inhibition of GFRα1 membrane shedding, suggesting that there may be other enzymes that can liberate GFRα1 from the cell surface (Fig. 3C, D). Thus, matrix metalloproteinases contribute to the shedding of GFRα1 from the surface of primary neurons. Together, Gfrα1TM/TM mice appeared to have normal levels of cell-surface GFRα1–TM shedding and would presumably be capable of normal levels of signaling in trans.
Figure 3.

Shedding of GFRα1 is dependent on matrix metalloproteinases. A, The conditioned medium from Gfrα1+/+, Gfrα1+/TM, and Gfrα1TM/TM neurons was collected and cleared of cellular debris, and the proteins were precipitated. The presence of soluble GFRα1/GFRα1–TM was analyzed by GFRα1 immunoblotting (top). Actin immunoblotting of cellular extracts from the neurons that conditioned the medium confirmed the analysis of similar amounts of neurons (bottom). GFRα1 immunoblotting of brain extracts from Gfrα1+/+, Gfrα1+/−, and Gfrα1−/− mouse embryos confirmed the specificity of the band that we identified as GFRα1 from conditioned medium (right). B, The immunoblots shown in A were quantified and graphed as the mean ± SEM. There were no significant differences among any of the genotypes. Three to five mice of each genotype from at two separate cultures were analyzed. C, Primary sympathetic neurons were exposed to TAPI-1 (metalloproteinase inhibitor), U73122 (phospholipase inhibitor), both TAPI-1 and U73122, or medium containing vehicle alone, for 48 h. As a positive control, a separate set of neurons was exposed to PI-PLC for 2 h. The amount of released GFRα1 was determined as in A. D, The experiments displayed in C were quantified and graphed as the mean ± SEM. There was a significant reduction in the amount of released GFRα1 during TAPI-1 and U73122 treatment compared with neurons exposed to vehicle alone (p < 0.05). Exposure to both TAPI-1 and U73122 did not have any additional inhibitory effect. This analysis represents three to four separate dishes of each condition from three independent cultures.
Ret does not translocate into lipid rafts during GDNF stimulation in Gfrα1TM/TM mice
As a proof of concept that the GDNF/GFRα1/Ret complex does not translocate into lipid rafts in Gfrα1TM/TM mice, lipid raft translocation experiments were performed. Detergent-resistant membranes were isolated from primary sympathetic neurons from Gfrα1+/+ and Gfrα1TM/TM mice. When Gfrα1+/+ neurons were stimulated with GDNF, Ret translocated rapidly into lipid rafts (Fig. 4A). This was in dramatic contrast to Gfrα1TM/TM neurons in which GDNF exposure resulted in a markedly reduced movement of Ret into the detergent-resistant, lipid raft fraction (Fig. 4A). The small portion of Ret that did translocate into lipid rafts during GDNF stimulation may be attributable to Ret kinase-dependent translocation of Ret into rafts that occurs with slower kinetics (Tansey et al., 2000; Paratcha et al., 2001). Overall, quantification of these experiments indicated that there was a significant, 75% reduction in the movement of the Ret receptor complex into lipid rafts during GDNF exposure in Gfrα1TM/TM neurons (Fig. 4B).
Figure 4.

Ret does not translocate into lipid rafts during GFL activation in Gfrα1TM/TM neurons. A, Primary sympathetic neurons isolated from Gfrα1+/+ and Gfrα1TM/TM mice were maintained in vitro for several days. The neurons were then treated with GDNF or medium alone for 15 min. Detergent-resistant membranes were isolated from the neurons and analyzed by immunoblotting for Ret51. Immunoblotting for caveolin and transferrin receptor confirmed relative purity of the detergent-resistant and detergent-soluble fractions, respectively. B, The experiments shown in A, were quantified and graphed as the mean ± SEM. There was a statistically significant decrease in the amount of Ret51 that translocated into lipid rafts during GDNF stimulation in Gfrα1TM/TM neurons compared with Gfrα1+/+ neurons (p < 0.05). This experiment was performed four times with similar results.
Gfrα1TM/TM mice have deficits in kidney morphogenesis and enteric nervous system development
We observed that Gfrα1TM/TM mice, but not Gfrα1TM/+ or Gfrα1+/+ littermate mice, died perinatally, with <10% of Gfrα1TM/TM mice surviving >24 h after birth. This was reminiscent of Gfrα1−/− mice that die perinatally as a result of renal agenesis (Enomoto et al., 1998). To determine whether there were deficits in kidney development, the urogenital tracts of Gfrα1+/+, Gfrα1TM/+, and Gfrα1TM/TM mice were analyzed. Gfrα1TM/TM mice displayed bilateral renal agenesis, in contrast to Gfrα1TM/+ and Gfrα1+/+ mice that had apparently normal kidneys (Fig. 5A, B). This renal morphogenesis phenotype was completely penetrant, and no Gfrα1TM/TM mice had kidneys, although other urogenital structures, such as the bladder and male and female gonads, appeared macroscopically normal. The examination of Gfrα1TM/+ mice did not reveal any abnormalities, and renal development appeared grossly normal (Fig. 5A, B), suggesting that GFRα1–TM does not act in a dominant inhibitory manner in the presence of wild-type GFRα1. These data suggest that the presence of GFRα1 in lipid rafts is required for the ureteric bud branching initiated by GDNF and Ret to promote kidney morphogenesis.
Figure 5.
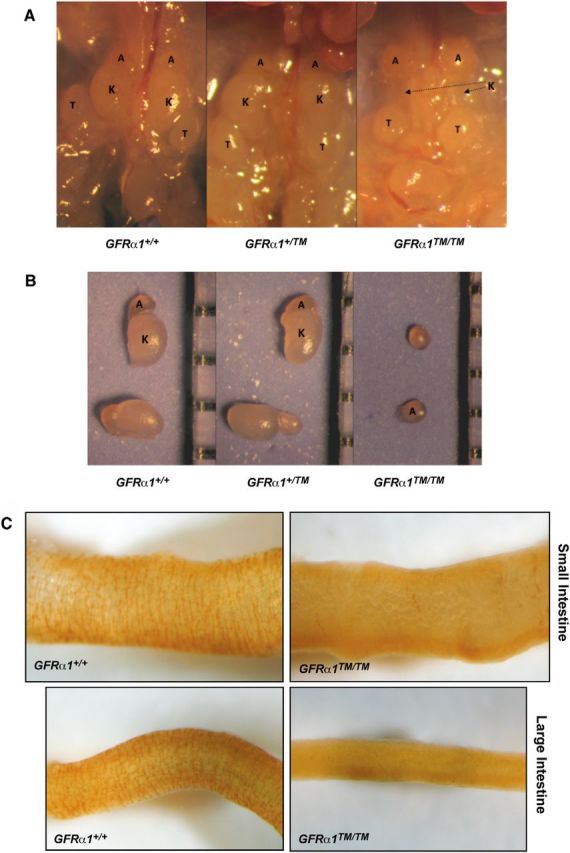
Localization of the GDNF signaling complex in lipid rafts is required for kidney morphogenesis and enteric nervous system development. A, The urogenital tracts of Gfrα1+/+, Gfrα1+/TM, and Gfrα1TM/TM mice were examined at P0. All Gfrα1TM/TM mice lacked kidneys, whereas Gfrα1+/+ and Gfrα1+/TM mice had normal, bilateral kidney development. B, The size and pallor of the kidneys from Gfrα1+/TM mice were similar to Gfrα1+/+ mice. Note that development of the adrenal glands appeared normal in all genotypes. The renal agenesis of Gfrα1TM/TM mice was highly penetrant (128 of 128 Gfrα1TM/TM mice had bilateral renal agenesis). In both A and B, A in the images denotes the adrenal glands, K denotes the kidneys, and T indicates the testes. C, Visualization of the myenteric plexes was performed using acetylcholinesterase histochemistry of the small intestines (top 2 panels) and large intestines (bottom panels) of E18.5 Gfrα1+/+ and Gfrα1TM/TM mice. There was no enteric nervous system in the large intestines of Gfrα1TM/TM mice (100%, 11 of 11 Gfrα1TM/TM mice). The small intestines of most Gfrα1TM/TM mice also lacked enteric neurons (82%, 9 of 11 mice), although a small minority of Gfrα1TM/TM mice (18%, 2 of 11 mice) appeared to have enteric nervous system development in the proximal third of the small intestine. Eight to 11 mice of each genotype from four different litters were analyzed.
A second dramatic developmental abnormality in Gfrα1−/− mice is the loss of enteric neurons distal to the stomach (Enomoto et al., 1998). During embryonic development, neural crest derivatives populate the gut, initiating their migration and proliferation in the stomach and continuing through both the small and large intestines. This process requires GDNF, which signals via GFRα1 and Ret. GDNF, GFRα1, and Ret null mice all display an identical loss of enteric neurons in both the small and large intestines (Schuchardt et al., 1994; Moore et al., 1996; Pichel et al., 1996; Sánchez et al., 1996; Cacalano et al., 1998; Enomoto et al., 1998). Acetylcholinesterase staining of both the small and large intestines of E18.5 Gfrα1+/+, Gfrα1TM/+, and Gfrα1TM/TM mice indicated that there was a dramatic loss of enteric neurons in the small and large intestines of Gfrα1TM/TM mice (Fig. 5C). All of the Gfrα1TM/TM mice analyzed lacked enteric neurons in the large intestine compared with the normal presence of neurons in Gfrα1+/+ and Gfrα1TM/+ mice. The small intestines of Gfrα1TM/TM mice were also typically devoid of enteric neurons, although some Gfrα1TM/TM mice (18%, 2 of 11 Gfrα1TM/TM mice) displayed initial enteric nervous system development in the proximal small intestine closest to the stomach. Overall, the enteric nervous system deficits of the Gfrα1TM/TM mice were somewhat less severe than in Gfrα1−/− mice, which have a complete loss of enteric neurons in both the small and large intestines (Cacalano et al., 1998; Enomoto et al., 1998). These data suggested that GDNF-mediated proliferation and migration of enteric precursors requires signaling of the Ret complex in lipid rafts.
Spinal motor neuron, but not sympathetic neuron, axonal projections are aberrant in Gfrα1TM/TM mice
The axonal projections of motor neurons into the hindlimb is a highly stereotyped process. Neurons from the lateral division of the lateral motor column (LMCL) project into the dorsal portion of the limb and contribute to the peroneal nerve, whereas the medial division of the LMC projects into the ventral limb. The axon guidance mechanism that, at least in part, directs LMCL neurons dorsally is the combinatorial activation of Ret via both a low level of GFRα1–GDNF signaling along with reverse signaling of EphA receptors expressed in the dorsal mesenchyme onto Ephrin-As expressed on the motor axons (Kramer et al., 2006; Dudanova et al., 2010, 2012; Bonanomi et al., 2012). Although it has been proposed that the “coincidence detection” via Ret from both GFRα1 and Ephrin-As requires their localization in lipid rafts (Bonanomi et al., 2012), there has not been a means to test this in vivo. To determine whether the dorsal projection of LMCL neurons require the localization of GFRα1 in lipid rafts, Gfrα1TM/TM mice were crossed with an Hb9–GFP reporter mouse that selectively labels all spinal motor neuron cell bodies and projections (Bonanomi et al., 2012). The dorsal–ventral projections of LMC motor neurons from the limbs of E13.5 mice were examined via direct imaging of GFP fluorescence of motor projections in the hindlimbs. In Hb9–GFP;Gfrα1+/+ mice, both dorsal and ventral projections into the hindlimbs were clearly observed, in contrast to Hb9–GFP;Gfrα1TM/TM mice in which the dorsal projection was either greatly reduced or absent entirely (Fig. 6A, B). The peroneal nerves of Hb9–GFP;Gfrα1TM/+ mice appeared normal (data not shown), suggesting that GFRα1–TM was not dominantly inhibitory to the remaining allele of GFRα1 in regards to spinal motor axon guidance. Therefore, GDNF requires GFRα1/Ret signaling from lipid rafts for coincidence detection via ephrin-A reverse signaling that is necessary for the axonal guidance of spinal motor neurons in the hindlimb.
Figure 6.
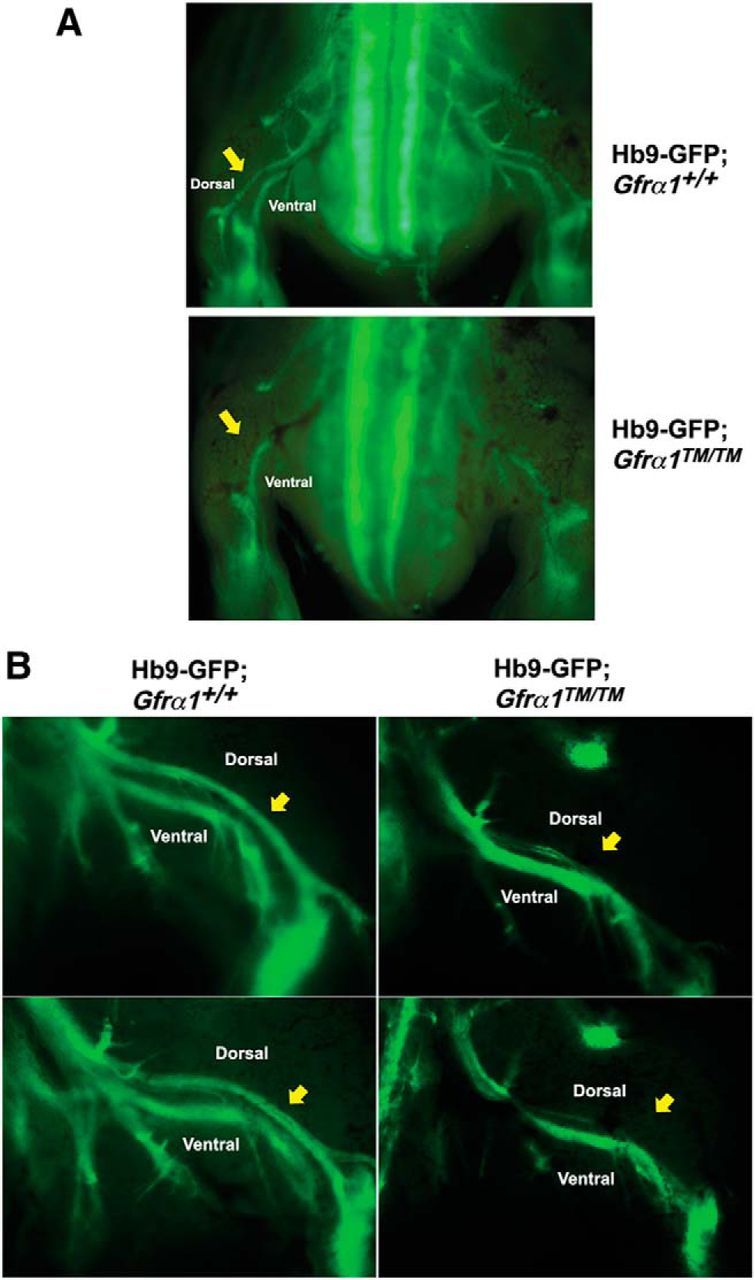
Spinal motor projections are deficient in Gfrα1TM/TM mice. A, Hb9–GFP;Gfrα1TM/TM and Hb9–GFP;Gfrα1+/+ embryos were imaged directly in an open-book preparation. Dorsal and ventral projections in the hindlimb are clearly visible, and yellow arrows indicate the dorsal projection that is absent in Hb9–GFP;Gfrα1TM/TM mice. B, Sagittal view of the hindlimbs of Hb9–GFP;Gfrα1+/+ and Hb9–GFP;Gfrα1TM/TM embryos with the dorsal hindlimb motor projection indicated by yellow arrows.
During embryonic development of sympathetic neurons, migration of the sympathetic chain ganglia, as well as axonal projections along the vasculature, requires Ret signaling in response to artemin via GFRα3 (Nishino et al., 1999; Enomoto et al., 2001; Honma et al., 2002). To determine whether GFRα1–TM could impair the signaling of other GFLs, we examined the development of the sympathetic nervous systems of Gfrα1+/+ and Gfrα1TM/TM mice. No abnormalities were observed in the migration or the positioning of the sympathetic chain, including the SCG and stellate ganglion (STG), in Gfrα1TM/TM mice (Fig. 7). Furthermore, axonal projections of the sympathetic chain ganglia were also normal in Gfrα1TM/TM mice. Last, the size and projections of the SCG and STG were not altered in Gfrα1TM/TM mice compared with Gfrα1+/+ mice (Fig. 7). Therefore, eliminating the localization of GFRα1 from lipid rafts did not alter GFRα1-independent functions, suggesting that the GFRα1–TM knock-in strategy did not globally affect the signaling of other GFLs in vivo.
Figure 7.

Development of the sympathetic nervous system is phenotypically normal in Gfrα1TM/TM mice. Whole-mount tyrosine hydroxylase immunohistochemistry of P0 mice demonstrates normal positioning of the SCG (left) and the normal positioning and caudal axonal projections to structures of the STG (middle). The sympathetic chain ganglia (right) in Gfrα1+/+ and Gfrα1TM/TM mice are also phenotypically normal and display similar axonal projections. Identical phenotypes were observed from three to five individual mice of each genotype from two to three independent litters.
Ret ubiquitination is altered in the brains of Gfrα1TM/TM mice
Signal transduction studies of primary neurons in vitro suggest that lipid rafts sequester activated Ret away from the ubiquitination/degradation machinery present in non-ordered regions, thus prolonging the half-life of activated Ret (Pierchala et al., 2006). To determine whether Ret degradation was increased in Gfrα1TM/TM mice, Ret immunoprecipitation studies were conducted from the brains of E15.5 Gfrα1+/+, Gfrα1TM/+, and Gfrα1TM/TM mice. We chose to examine the brain because other anatomic structures that lipid raft-mediated Ret signaling appears to be important for, such as the kidneys and enteric nervous system, are completely lost in Gfrα1TM/TM mice. Several CNS populations express Ret, such as cholinergic basal forebrain neurons and dopaminergic midbrain neurons, but are not lost in Ret−/− mice, making them tractable populations to study. When the levels of Ret ubiquitination were compared between Gfrα1+/+, Gfrα1TM/+, and Gfrα1TM/TM brains, Ret isolated from the brains of Gfrα1TM/TM mice was significantly more highly ubiquitinated than Ret isolated from Gfrα1+/+ brains (Fig. 8A, B). A comparison of the level of Ret ubiquitination between Gfrα1+/+ mice and Gfrα1−/− mice confirmed that most of the Ret ubiquitination we observed in the brain was attributable to activation by GDNF/GFRα1 (Fig. 8A). However, the total level of Ret was not significantly different between Gfrα1+/+ and Gfrα1TM/TM mice (Fig. 8A, C). The detection of a decrease in Ret protein may be difficult if only a small proportion of the total amount of Ret expressed in the brain, presumably activated Ret, is actively being degraded. Surprisingly, there was a higher level of Ret autophosphorylation in the brains of Gfrα1TM/TM mice compared with Gfrα1+/+ mice (Fig. 8A, D). Together, these results suggest that Ret may be degraded more rapidly, or may be trafficked aberrantly after its activation, when associated in a receptor complex with GFRα1–TM.
Figure 8.
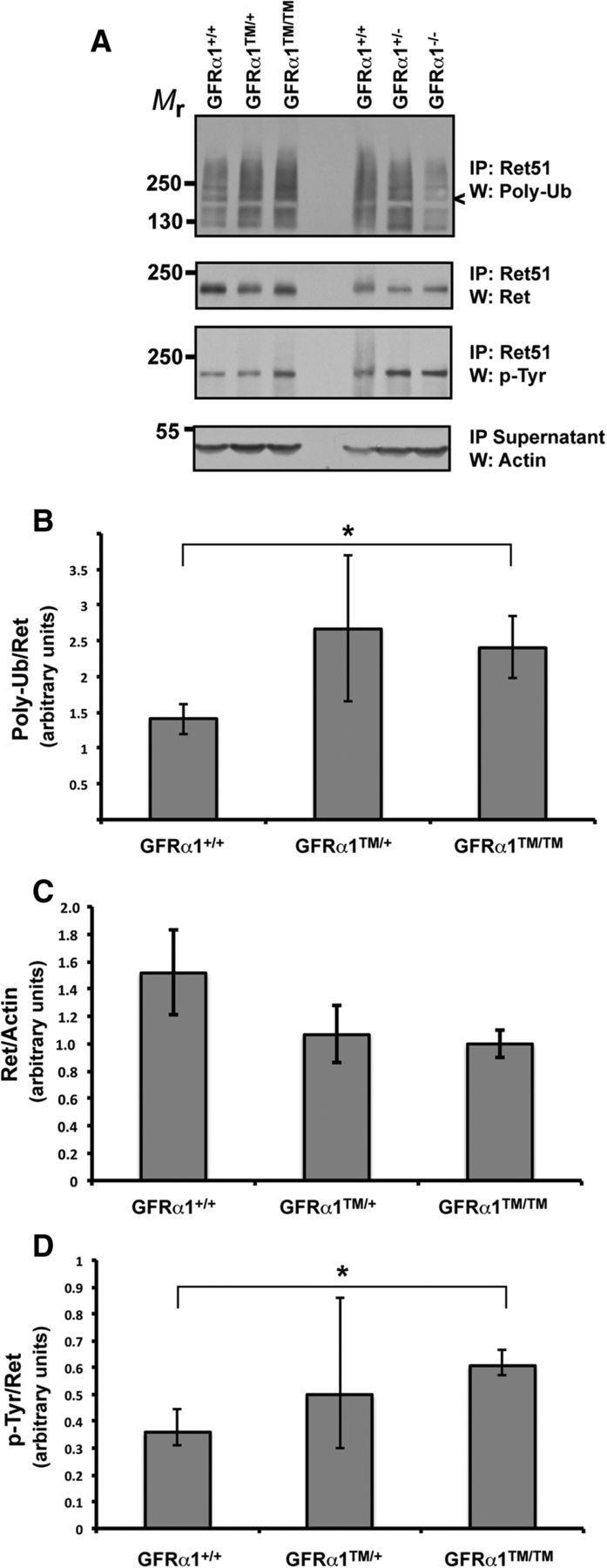
Ret ubiquitination is increased in the brains of Gfrα1TM/TM mice. A, The whole brains of E15.5 Gfrα1+/+, Gfrα1TM/+, and Gfrα1TM/TM mice were detergent extracted, and Ret was immunoprecipitated from them. The extent of ubiquitination and Ret protein levels were ascertained by poly-ubiquitin and Ret immunoblotting, respectively. The level of Ret autophosphorylation was determined by phosphotyrosine immunoblotting of the immunoprecipitates, and actin immunoblotting of the supernatants served as a loading control. These experiments were quantified and graphed as the mean ± SEM for Ret ubiquitination (B), Ret protein levels (C), and Ret autophosphorylation (D). *p < 0.05. These experiments were conducted on three to five mice of each genotype from three separate litters.
Discussion
Lipid rafts are proposed to be nanoscale membrane microdomains that are enriched in cholesterol and sphingolipids, which endows them with greater order and rigidity than non-raft regions of the lipid bilayer (Simons and Sampaio, 2011). By genetically replacing GFRα1 with GFRα1–TM, which is incapable of translocating the GDNF/Ret signaling complex into lipid rafts (Tansey et al., 2000), the hypothesis was tested that lipid rafts are physiologic membrane structures in vertebrates that are critical for the developmental functions of GDNF. Gfrα1TM/TM mice displayed a markedly reduced translocation of activated Ret into lipid rafts during GDNF stimulation, while at the same time having normal protein levels and cell-surface localization of GFRα1–TM. Phenotypic analysis of Gfrα1TM/TM mice revealed that they have bilateral renal agenesis, a nearly complete lack of enteric nervous system development, and spinal motor axon guidance abnormalities. All of these deficits are observed in Gfrα1−/− mice (Cacalano et al., 1998; Enomoto et al., 1998; Kramer et al., 2006; Bonanomi et al., 2012), suggesting that lipid rafts are critical for the GDNF/GFRα1/Ret signal transduction cascades that are necessary for the morphogenesis, proliferation, migration, and axon guidance functions of GDNF in vivo. Gfrα1TM/+ mice were similar to Gfrα1+/+ mice and had no abnormalities in any of these peripheral structures. These data suggest that GFRα1–TM does not interfere with the formation of GFRα1/Ret signaling complexes, likely because of their separation between raft and non-raft domains. However, we consistently observed a behaviorally aggressive phenotype of Gfrα1TM/+ mice compared with littermate Gfrα1+/+ mice, suggesting that there may be CNS defects in neural circuits involved in reward and/or aggressive behaviors.
The severity of the phenotypes we observed in Gfrα1TM/TM mice raises the question of whether the use of a TM domain in GFRα1 causes other signaling deficits that cannot be explained by the lack of lipid raft signaling via Ret. GFRα1–TM was examined thoroughly for alterations in its normal function and processing. The overall expression, trafficking to the cell surface, and membrane shedding of GFRα1–TM all occurred to a similar extent as GFRα1. Although the observation that GFRα1–TM was shed from primary neurons was initially surprising, the fact that a significant proportion of GFRα1 shedding is attributable to the activity of metalloproteinases explains this observation. Importantly, the three developmental processes that were examined, kidney morphogenesis, enteric nervous system development, and spinal motor axon guidance, are not impaired in mice lacking GFRα1 expressed in trans (Enomoto et al., 2004), making it unlikely that GFRα1 shedding would be important for these developmental processes. The initial Ret autophosphorylation by GDNF/GFRα1–TM also appeared to be similar to GDNF/GFRα1, which was reported previously for the GFRα1–TM fusion protein (Tansey et al., 2000). However, it is still a possibility that yet other aspects of GFRα1–TM functions are aberrant because of this TM addition, rather than lipid raft localization, making it premature to say definitively that lipid rafts are required for these developmental functions of GFRα1.
Lipid rafts enhance the signal transduction capacity of many receptor complexes by providing a platform for clustering and concentrating signaling molecules close to activated receptors (Simons and Toomre, 2000; Tsui-Pierchala et al., 2002). Indeed, in the case of GFL-mediated Ret signal transduction, lipid rafts enhance downstream signaling cascades by bringing Ret within close proximity to raft-enriched signaling molecules, such as Src and fibroblast growth factor receptor substrate 2 (Tansey et al., 2000; Encinas et al., 2001; Paratcha et al., 2001). Lipid rafts also enhance GFL/Ret signal transduction by sequestering activated Ret away from ubiquitin ligases and other molecules involved in receptor degradation, which are primarily absent from lipid rafts (Pierchala et al., 2006). The examination of Ret ubiquitination revealed that Ret isolated from the brains of Gfrα1TM/TM mice was more highly ubiquitinated than Ret from Gfrα1+/+ mice (Fig. 8). Whether altered Ret ubiquitination, which would ultimately lead to changes in the internalization, trafficking, and degradation of Ret, contributes to the developmental abnormalities observed in these mice requires additional investigation. There are other potential functions of lipid rafts that may be critical for GDNF/GFRα1/Ret signal transduction. One possibility is that intracellular trafficking is altered, given that the internalization of receptors from lipid rafts is typically not clathrin mediated, as it often is outside of rafts (Nichols, 2003; Kirkham and Parton, 2005). A simpler explanation for the importance of rafts in Ret function is as a means of segregating GFRα1 receptors away from Ret in the absence of ligand, to avoid the pre-complexing of these receptors. Indeed, overexpression of GFRαs and Ret in cell lines results in their extensive interaction in the absence of ligand, which may have deleterious effects on signaling, such as ligand-independent activation or plasma membrane clearance. It is likely that several of these mechanisms work in a combinatorial manner to promote Ret-dependent signaling that accounts for the importance of lipid rafts in GDNF function. A limitation to this signaling analysis is the general absence of anatomic structures that require GDNF/Gfrα1/Ret signaling for development, such as kidneys and the enteric nervous system, in Gfrα1TM/TM mice, precluding their biochemical examination. The observation that Gfrα1TM/TM mice were deficient in several GDNF/GFRα1-dependent developmental events in the periphery emphasizes the need to identify signaling properties of Ret in vivo that are raft dependent and those that are not and what their role is in Ret ubiquitination, signal transduction, and turnover. In addition to Ret, GDNF/GFRα1 complexes can signal via NCAM and syndecan-3 (Paratcha et al., 2003; Pozas and Ibanez, 2005; Bespalov et al., 2011), which are critical for synaptogenic and migration functions of GDNF in the brain. The extent to which lipid rafts are necessary for these functions in vivo are not known and represents an important future direction as a means of determining whether lipid rafts are required for all GDNF/GFRα1-dependent developmental functions, regardless of the signal-transducing receptor. In conclusion, we propose that the function of the GPI anchor, as opposed to TM domains, is to assemble receptor complexes in lipid raft microdomains to maximize their signaling functions.
Footnotes
This research was supported by National Institutes of Health Grants R01 NS058510 (B.A.P.), K08 DK084210 (C.C.T.), and T-32-GM007315 (N.A.G.). We thank Drs. Wenqin Luo, Jeffrey Milbrandt, and Hideki Enomoto for providing tissues, plasmids, and helpful scientific discussions.
The authors declare no competing financial interests.
References
- Airaksinen MS, Saarma M. The GDNF family: signalling, biological functions and therapeutic value. Nat Rev. 2002;3:383–394. doi: 10.1038/nrn812. [DOI] [PubMed] [Google Scholar]
- Baloh RH, Enomoto H, Johnson EM, Jr, Milbrandt J. The GDNF family ligands and receptors-implications for neural development. Curr Opin Neurobiol. 2000;10:103–110. doi: 10.1016/S0959-4388(99)00048-3. [DOI] [PubMed] [Google Scholar]
- Bespalov MM, Saarma M. GDNF family receptor complexes are emerging drug targets. Trends Pharmacol Sci. 2007;28:68–74. doi: 10.1016/j.tips.2006.12.005. [DOI] [PubMed] [Google Scholar]
- Bespalov MM, Sidorova YA, Tumova S, Ahonen-Bishopp A, Magalhães AC, Kulesskiy E, Paveliev M, Rivera C, Rauvala H, Saarma M. Heparan sulfate proteoglycan syndecan-3 is a novel receptor for GDNF, neurturin, and artemin. J Cell Biol. 2011;192:153–169. doi: 10.1083/jcb.201009136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonanomi D, Chivatakarn O, Bai G, Abdesselem H, Lettieri K, Marquardt T, Pierchala BA, Pfaff SL. Ret is a multifunctional coreceptor that integrates diffusable- and contact-axon guidance signals. Cell. 2012;148:568–582. doi: 10.1016/j.cell.2012.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DA, London E. Functions of lipid rafts in biological membranes. Annu Rev Cell Dev Biol. 1998;14:111–136. doi: 10.1146/annurev.cellbio.14.1.111. [DOI] [PubMed] [Google Scholar]
- Bunting M, Bernstein KE, Greer JM, Capecchi MR, Thomas KR. Targeting genes for self-excision in the germ line. Genes Dev. 1999;13:1524–1528. doi: 10.1101/gad.13.12.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacalano G, Fariñas I, Wang LC, Hagler K, Forgie A, Moore M, Armanini M, Phillips H, Ryan AM, Reichardt LF, Hynes M, Davies A, Rosenthal A. GFRalpha1 is an essential receptor component for GDNF in the developing nervous system and kidney. Neuron. 1998;21:53–62. doi: 10.1016/S0896-6273(00)80514-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudanova I, Gatto G, Klein R. GDNF acts as a chemoattractant to support ephrinA-induced repulsion of limb motor axons. Curr Biol. 2010;20:2150–2156. doi: 10.1016/j.cub.2010.11.021. [DOI] [PubMed] [Google Scholar]
- Dudanova I, Kao TJ, Herrmann JE, Zheng B, Kania A, Klein R. Genetic evidence for a contribution of EphA:ephrinA reverse signaling to motor axon guidance. J Neurosci. 2012;32:5209–5215. doi: 10.1523/JNEUROSCI.5707-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encinas M, Tansey MG, Tsui-Pierchala BA, Comella JX, Milbrandt J, Johnson EM., Jr c-Src is required for glial cell line-derived neurotrophic factor (GDNF) family ligand-mediated neuronal survival via a phosphatidylinositol-3 kinase (PI-3K)-dependent pathway. J Neurosci. 2001;21:1464–1472. doi: 10.1523/JNEUROSCI.21-05-01464.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto H, Araki T, Jackman A, Heuckeroth RO, Snider WD, Johnson EM, Jr, Milbrandt J. GFR alpha1-deficient mice have deficits in the enteric nervous system and kidneys. Neuron. 1998;21:317–324. doi: 10.1016/S0896-6273(00)80541-3. [DOI] [PubMed] [Google Scholar]
- Enomoto H, Heuckeroth RO, Golden JP, Johnson EM, Jr, Milbrandt J. Development of cranial parasympathetic ganglia requires sequential actions of GDNF and neurturin. Development. 2000;127:4877–4889. doi: 10.1242/dev.127.22.4877. [DOI] [PubMed] [Google Scholar]
- Enomoto H, Crawford PA, Gorodinsky A, Heuckeroth RO, Johnson EM, Jr, Milbrandt J. RET signaling is essential for migration, axonal growth and axon guidance of developing sympathetic neurons. Development. 2001;128:3963–3974. doi: 10.1242/dev.128.20.3963. [DOI] [PubMed] [Google Scholar]
- Enomoto H, Hughes I, Golden J, Baloh RH, Yonemura S, Heuckeroth RO, Johnson EM, Jr, Milbrandt J. GFRalpha1 expression in cells lacking RET is dispensable for organogenesis and nerve regeneration. Neuron. 2004;44:623–636. doi: 10.1016/j.neuron.2004.10.032. [DOI] [PubMed] [Google Scholar]
- Galbiati F, Razani B, Lisanti MP. Emerging themes in lipid rafts and caveolae. Cell. 2001;106:403–411. doi: 10.1016/S0092-8674(01)00472-X. [DOI] [PubMed] [Google Scholar]
- Hansbrough JR, Lublin DM, Roth KA, Birkenmeier EA, Gordon JI. Expression of a liver fatty acid binding protein/human decay- accelerating factor/HLA-B44 chimeric gene in transgenic mice. Am J Physiol. 1991;260:G929–G939. doi: 10.1152/ajpgi.1991.260.6.G929. [DOI] [PubMed] [Google Scholar]
- Honma Y, Araki T, Gianino S, Bruce A, Heuckeroth R, Johnson EM, Jr, Milbrandt J. Artemin is a vascular-derived neurotrophic factor for developing sympathetic neurons. Neuron. 2002;35:267–282. doi: 10.1016/S0896-6273(02)00774-2. [DOI] [PubMed] [Google Scholar]
- Kenworthy A. Peering inside lipid rafts and caveolae. Trends Biochem Sci. 2002;27:435–437. doi: 10.1016/S0968-0004(02)02178-3. [DOI] [PubMed] [Google Scholar]
- Kirkham M, Parton RG. Clathrin-independent endocytosis: new insights into caveolae and non-caveolar lipid raft carriers. Biochim Biophys Acta. 2005;1746:349–363. doi: 10.1016/j.bbamcr.2005.11.005. [DOI] [PubMed] [Google Scholar]
- Kramer ER, Knott L, Su F, Dessaud E, Krull CE, Helmbacher F, Klein R. Cooperation between GDNF/Ret and ephrinA/EphA4 signals for motor-axon pathway selection in the limb. Neuron. 2006;50:35–47. doi: 10.1016/j.neuron.2006.02.020. [DOI] [PubMed] [Google Scholar]
- Moore MW, Klein RD, Fariñas I, Sauer H, Armanini M, Phillips H, Reichardt LF, Ryan AM, Carver-Moore K, Rosenthal A. Renal and neuronal abnormalities in mice lacking GDNF. Nature. 1996;382:76–79. doi: 10.1038/382076a0. [DOI] [PubMed] [Google Scholar]
- Munro S. Lipid rafts: elusive or illusive? Cell. 2003;115:377–388. doi: 10.1016/S0092-8674(03)00882-1. [DOI] [PubMed] [Google Scholar]
- Nichols B. Caveosomes and endocytosis of lipid rafts. J Cell Sci. 2003;116:4707–4714. doi: 10.1242/jcs.00840. [DOI] [PubMed] [Google Scholar]
- Nishino J, Mochida K, Ohfuji Y, Shimazaki T, Meno C, Ohishi S, Matsuda Y, Fujii H, Saijoh Y, Hamada H. GFR alpha3, a component of the artemin receptor, is required for migration and survival of the superior cervical ganglion. Neuron. 1999;23:725–736. doi: 10.1016/S0896-6273(01)80031-3. [DOI] [PubMed] [Google Scholar]
- Paratcha G, Ledda F, Baars L, Coulpier M, Besset V, Anders J, Scott R, Ibáñez F. Released GFRa1 potentiates downstream signaling, neuronal survival, and differentiation via a novel mechanism of recruitment of c-Ret to lipid rafts. Neuron. 2001;29:171–184. doi: 10.1016/S0896-6273(01)00188-X. [DOI] [PubMed] [Google Scholar]
- Paratcha G, Ledda F, Ibáñez CF. The neural cell adhesion molecule NCAM is an alternative signaling receptor for GDNF family ligands. Cell. 2003;113:867–879. doi: 10.1016/S0092-8674(03)00435-5. [DOI] [PubMed] [Google Scholar]
- Patra SK. Dissecting lipid raft facilitated cell signaling pathways in cancer. Biochim Biophys Acta. 2008;1785:182–206. doi: 10.1016/j.bbcan.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Pichel JG, Shen L, Sheng HZ, Granholm AC, Drago J, Grinberg A, Lee EJ, Huang SP, Saarma M, Hoffer BJ, Sariola H, Westphal H. Defects in enteric innervation and kidney development in mice lacking GDNF. Nature. 1996;382:73–76. doi: 10.1038/382073a0. [DOI] [PubMed] [Google Scholar]
- Pierchala BA, Milbrandt J, Johnson EM., Jr Glial cell line-derived neurotrophic factor-dependent recruitment of Ret into lipid rafts enhances signaling by partitioning Ret from proteasome-dependent degradation. J Neurosci. 2006;26:2777–2787. doi: 10.1523/JNEUROSCI.3420-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozas E, Ibáñez CF. GDNF and GFRalpha1 promote differentiation and tangential migration of cortical GABAergic neurons. Neuron. 2005;45:701–713. doi: 10.1016/j.neuron.2005.01.043. [DOI] [PubMed] [Google Scholar]
- Runeberg-Roos P, Saarma M. Neurotrophic factor receptor RET: structure, cell biology, and inherited diseases. Ann Med. 2007;39:572–580. doi: 10.1080/07853890701646256. [DOI] [PubMed] [Google Scholar]
- Sánchez MP, Silos-Santiago I, Frisén J, He B, Lira SA, Barbacid M. Renal agenesis and the absence of enteric neurons in mice lacking GDNF. Nature. 1996;382:70–73. doi: 10.1038/382070a0. [DOI] [PubMed] [Google Scholar]
- Schuchardt A, D'Agati V, Larsson-Blomberg L, Costantini F, Pachnis V. Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature. 1994;367:380–383. doi: 10.1038/367380a0. [DOI] [PubMed] [Google Scholar]
- Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- Simons K, Sampaio JL. Membrane organization and lipid rafts. Cold Spring Harb Perspect Biol. 2011;3:a004697. doi: 10.1101/cshperspect.a004697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1:31–39. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- Simons K, van Meer G. Lipid sorting in epithelial cells. Biochemistry. 1988;27:6197–6202. doi: 10.1021/bi00417a001. [DOI] [PubMed] [Google Scholar]
- Tansey MG, Baloh RH, Milbrandt J, Johnson EM., Jr GFRa-mediated localization of RET to lipid rafts is required for effective downstream signaling, differentiation, and neuronal survival. Neuron. 2000;25:611–623. doi: 10.1016/S0896-6273(00)81064-8. [DOI] [PubMed] [Google Scholar]
- Treanor JJ, Goodman L, de Sauvage F, Stone DM, Poulsen KT, Beck CD, Gray C, Armanini MP, Pollock RA, Hefti F, Phillips HS, Goddard A, Moore MW, Buj-Bello A, Davies AM, Asai N, Takahashi M, Vandlen R, Henderson CE, Rosenthal A. Characterization of a multicomponent receptor for GDNF. Nature. 1996;382:80–83. doi: 10.1038/382080a0. [DOI] [PubMed] [Google Scholar]
- Tsui CC, Pierchala BA. The differential axonal degradation of Ret accounts for cell-type-specific function of glial cell line-derived neurotrophic factor as a retrograde survival factor. J Neurosci. 2010;30:5149–5158. doi: 10.1523/JNEUROSCI.5246-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui-Pierchala BA, Encinas M, Milbrandt J, Johnson EM., Jr Lipid rafts in neuronal signaling and function. Trends Neurosci. 2002;25:412–417. doi: 10.1016/S0166-2236(02)02215-4. [DOI] [PubMed] [Google Scholar]