

Abstract
Permeable reactive barriers (PRBs) consist of a labile carbon source that is positioned to intercept nitrate-laden groundwater to prevent eutrophication. Decomposition of carbon in the PRB drives groundwater anoxic, fostering microbial denitrification. Such PRBs are an ideal habitat to examine microbial community structure under high-nitrate, carbon-replete conditions in coastal aquifers. We examined a PRB installed at the Waquoit Bay National Estuarine Research Reserve in Falmouth, MA. Groundwater within and below the PRB was depleted in oxygen compared to groundwater at sites upgradient and at adjacent reference sites. Nitrate concentrations declined from a high of 25 μM upgradient and adjacent to the barrier to <0.1 μM within the PRB. We analyzed the total and active bacterial communities filtered from groundwater flowing through the PRB using amplicons of 16S rRNA and of the 16S rRNA genes. Analysis of the 16S rRNA genes collected from the PRB showed that the total bacterial community had high relative abundances of bacteria thought to have alternative metabolisms, such as fermentation, including candidate phyla OD1, OP3, TM7, and GN02. In contrast, the active bacteria had lower abundances of many of these bacteria, suggesting that the bacterial taxa that differentiate the PRB groundwater community were not actively growing. Among the environmental variables analyzed, dissolved oxygen concentration explained the largest proportion of total community structure. There was, however, no significant correlation between measured environmental parameters and the active microbial community, suggesting that controls on the active portion may differ from the community as a whole.
INTRODUCTION
Humans are adding excess nitrogen (N) to coastal ecosystems at a high rate (1), which has contributed to the moderate to high degree of eutrophication found in 65% of U.S. estuaries examined (2). Nitrogen is generally the limiting nutrient in coastal ecosystems, so anthropogenic nitrogen inputs stimulate both primary production and benthic nutrient recycling (3, 4). These N inputs increase macrophyte and phytoplankton growth and can change the community composition of primary producers (5). When primary producers die, microbial degradation can lead to hypoxia and the decline of secondary producers (6).
Non-point source anthropogenic N inputs associated with agriculture, urbanization, and wastewater treatment are prevalent causes of eutrophication in estuaries (7). In many coastal systems underlain by unconsolidated sediments, groundwater is a major delivery mechanism for non-point sources of N (8). Nitrate (NO3−) also leaches from wastewater treatment plant percolation basins and septic system drain fields (9) into groundwater, which then flows into estuaries, exacerbating the harmful effects of eutrophication. In fact, groundwater-borne NO3− can comprise as much as 71 to 97% of nitrogen loading to estuaries in southern New England (10, 11).
Since groundwater is an important source of non-point anthropogenic NO3− to estuaries, development of strategies for removal of excess NO3− from groundwater is important for maintaining coastal ecosystem health. One inexpensive yet efficient method of removing NO3− is through the installation of permeable reactive barriers (PRBs) designed to foster denitrification (12). PRBs are installed to intercept groundwater NO3− flowing to the shore, and they contain a labile carbon source, often wood chips, that provides a large source of oxidizable organic matter to the otherwise low-carbon aquifer (13, 14). The added carbon enhances heterotrophic decomposition, resulting in an increase in biological oxygen demand and hypoxia or anoxia in the groundwater. In the absence of oxygen, denitrifying bacteria can use the large supply of anthropogenic NO3− in groundwater as a terminal electron acceptor to metabolize organic matter. Denitrifying bacteria are often limited by carbon availability (13); by removing carbon limitation and creating low-oxygen conditions favorable to denitrifying microbes, the PRBs promote NO3− removal before it can induce eutrophication in receiving waters (14).
PRB-induced anoxia likely also promotes other changes in bacterial community composition. The PRBs are designed to increase the amount of labile carbon to support denitrification, but this carbon can also support other types of anaerobic respiration (e.g., sulfate and iron reduction), as well as fermentation. Bacterial communities in estuaries, oceans, and sediments often vary with dissolved oxygen supply, carbon availability, salinity, nutrients, temperature, light, and predation (15–20). If the PRB alters any of these environmental parameters relative to typical aquifer conditions, these changes might also result in local shifts in bacterial community composition. Although PRBs have been used in freshwater systems (14, 21, 22), this is the first PRB installed in the mixing zone between fresh groundwater and adjacent marine waters, an area referred to as the subterranean estuary (23). The subterranean estuary is a particularly dynamic region of the aquifer, with a large number of electron acceptors, including nitrate-rich groundwater and sulfate-rich estuarine water, but with relatively small amounts of organic carbon to support heterotrophic respiration. The PRBs are therefore a good model system to study how an additional source of organic matter in brackish coastal aquifers affects bacterial community structure.
To determine if the PRB alters bacterial community composition, activity, and aquifer geochemistry relative to adjacent systems with no PRB present, we analyzed bacterial sequence diversity from the groundwater of three sites: upgradient of the PRB, within and directly downgradient of the PRB, and at a reference site adjacent to the PRB (Fig. 1). We also measured a suite of geochemical parameters, including salinity, dissolved oxygen, NO3−, and ammonium (NH4+) concentrations to determine if differences in geochemistry correlate with shifts in bacterial community structure. We hypothesized that the carbon source provided by the wood chips in the PRB would promote anoxia and heterotrophic bacterial metabolisms to such an extent that even bacteria with metabolisms that have negative reduction potential, such as sulfate reduction and fermentation, could achieve high relative proportions inside the PRB.
FIG 1.

Stylized representation of the location of the PRB (brown box) relative to high NO3− groundwater flow paths and the estuary. Multidepth sampling wells are shown in cross-section and are represented by black squares for wells that are upgradient and within the wood chip barrier. The locations of the reference wells are also provided, although the discrete sampling depths (n = 2 to 4 per well) are not visible.
MATERIALS AND METHODS
Study sites, groundwater sampling, and geochemical analyses.
The PRB, located at the Waquoit Bay National Estuarine Research Reserve in Falmouth, MA, was installed in 2005 by Lombardo and Associates (24). The PRB consists of a trench ∼20 m long, 2.5 m deep, and 3.7 m wide (a total volume of ∼185 m3). The carbon content of wood chips used in the Waquoit Bay PRB was 61%. This is in stark contrast to the very low solid-phase carbon content of the Waquoit Bay aquifer, which ranges from nondetectable levels to a high of 0.075% (25). Dissolved organic carbon (DOC) concentrations in groundwater taken from the same region were also consistently low, ranging from 7 to 700 μM (26, 27). Previous sampling at the PRB indicated that DOC in the PRB was 117.2 μM (n = 11, standard error [SE] = 10.7), while upgradient sites averaged 55.4 μM (n = 14, SE = 2.7). Furthermore, dissolved inorganic carbon (DIC) in the barrier averaged 1621 μM (n = 10, SE = 103.2), compared to 733 μM (n = 5, SE = 26.6) in upgradient sites (C. Knauss, personal communication). Although these data were collected a year prior to the sampling reported here, they indicate that the presence of the wood chips enhances the availability of DOC, and the excess DIC in the barrier suggests active heterotrophic metabolism.
We collected groundwater samples from the PRB and from an adjacent reference site on 12 October 2012 (Fig. 1). Groundwater was collected from multidepth, colinear sampling wells at depths ranging from a half meter to 4 m beneath the beach surface. These wells were positioned to collect groundwater upgradient flowing toward the PRB, within the PRB itself, and in an area of unaltered beach that was adjacent to the PRB (∼50 m away) but with no PRB present (Fig. 1). To collect bacterial DNA and RNA, 2 liters of groundwater from each depth was pumped out of the well and filtered onto a Millipore Sterivex 0.22-μm-pore filter. Fifty milliliters of the filtrate was frozen for subsequent NO3− and ammonium (NH4+) measurements. Dissolved oxygen concentrations, salinity, and temperature were recorded during sample collection using a Hanna HI9828 multiparameter meter (Geoscientific, Ltd., Vancouver, British Columbia, Canada, with a resolution of 0.1 mg liter−1). NO3− and NH4+ concentrations were analyzed on a Lachat flow injection analyzer (Hach Company, Loveland, CO) based on QuikChem methods 31-107-04-1-E and 31-107-06-1-B, respectively. The lower limit of detection for both methods was 0.36 μM. We plotted these data in contour plots that were generated using the R package spatstat version 1.41-1 (28). Briefly, the area of analysis was the convex hull defined by the observation points. Spatial smoothing was performed using a Gaussian kernel with the Diggle edge correction algorithm (29) and a sigma parameter of 0.4.
Genomic DNA and RNA extraction, amplification, and sequencing.
We extracted total nucleic acids using a modification of the MoBio PowerWater Sterivex DNA isolation kit (MoBio Laboratories, Carlsbad, CA). After nucleic acid extraction, the filtrate containing all DNA and RNA was split as follows. To separate RNA from DNA, 25 μl of eluted filtrate was placed into two separate 1.5-ml centrifuge tubes. We added 75 μl of 1/10× DNase buffer and 1 μl of DNase to each tube, which was then incubated for 10 min at 37°C. We then added 1 μl of 0.5 M EDTA and incubated the tubes at 75°C for 10 min to halt the reaction. The two replicate tubes were pooled and stored at −80°C. To separate DNA from RNA, we added 1 μl of RNase A to the remaining 50 μl of eluted filtrate, incubated the samples at 37°C for 30 min, and then stored the samples at −20°C. We converted RNA to cDNA using random hexamer primers along with the Superscript III single-strand kit for reverse transcription (RT)-PCR (Life Technologies, Carlsbad, CA). The cDNA was stored at −80°C until sequencing.
To analyze bacterial community composition, we sequenced a ∼500-bp section of the V2-V3 region of the gene that encodes the small subunit of the prokaryotic ribosome (16S rRNA), which is used to infer taxonomic identity. Analysis of 16S rRNA is indicative of bacteria that are synthesizing proteins and, with caveats (30), can be used as a rough approximation for the active portions of the community (15, 31). Analysis of the 16S rRNA gene is used to characterize the structure of the total bacterial community.
We amplified fragments of 16S rRNA genes and 16S rRNA using primers 27F (AGAGTTTGATCCTGGCTCAG) and 533R (TTACCGCGGCTGCTGGCAC), along with adaptors and barcodes designed by Beckman Genomics. We performed triplicate independent PCRs with a reaction mixture (final volume of 26 μl) that contained 2 μl DNA or RNA template, 0.5 μM each primer, 1.5 mM MgCl2, 0.8 mM deoxynucleoside triphosphate (dNTP), and 0.025 U μl−1 GoTaq polymerase (Promega Corporation, Madison, WI). The thermal cycling conditions were as follows: initial denaturation at 95°C for 2 min, followed by 30 cycles of denaturation at 95°C for 30 s, annealing at 55°C for 30 s, and extension at 72°C for 30 s. This was followed by a final elongation period at 72°C for 7 min. Some samples, primarily from the reference location (2-m depth at well 0, 1- and 3-m depths at well 1, 3-m depth at well 2, 1- and 3-m depths at well 3, and 1-m depth at well 4) required a nested PCR approach, in which a 1,500-bp fragment of the V1-to-V6 region of the16S gene was first amplified with 8F-1525R primers (32). Although performing nested PCR can amplify any biases in the samples, we tested for this bias by calculating the UniFrac similarity metric for nested and nonnested samples. As the nested samples did not cluster separately from the nonnested samples, we concluded that the nested approach was appropriate for the preparation of samples that were difficult to amplify. The reaction mixture (final volume of 26 μl) for the nested PCR contained 2 μl template, 0.3 μM each primer, 1.5 mM MgCl2, 0.8 mM dNTP, and 0.025 U μl−1 GoTaq polymerase (Promega Corporation, Madison, WI). The cycling conditions were as follows: 95°C for 3 min, followed by 30 cycles of denaturing at 95°C for 1 min, annealing at 51°C for 1 min, and elongation at 72°C for 1 min 45 s, with a final elongation at 72°C for 5 min. One microliter of PCR product was then amplified with the barcoded primers, as described above. We gel purified the final barcoded PCR products using a QIAquick gel extraction kit (Qiagen,Valencia, CA) according to the manufacturer's instructions. Gel-purified PCR products were quantified on a Nanodrop 2000 (Thermo Scientific, Wilmington, DE), pooled in equimolar ratios, and sequenced at Beckman Coulter Genomics on a Roche 454 GS FLX using Titanium chemistry.
Data analysis.
We analyzed sequence output from Beckman-Coulter using QIIME (33). Sequences were demultiplexed, quality filtered, and chimera checked using default parameters. We assigned 251,686 barcoded sequences to 48 samples, with an average of 5,243 sequences per sample. We did not include four samples that had fewer than 50 sequences per sample in downstream analysis. We clustered sequences at a 97% sequence identity (33) and picked operational taxonomic units (OTU) using uclust (34). We aligned sequences to the Greengenes core reference set using PyNast (33). Before diversity and abundance analyses were performed, we rarefied all samples to 500 sequences to normalize for various depths of sequencing among samples. We calculated alpha diversity using the Chao1 richness estimator and the Shannon diversity Index. We tested for differences in average taxonomic richness and diversity in groundwater collected from wells upgradient of the PRB, within the PRB, and adjacent to the PRB using a one-way analysis of variance (ANOVA) in R (35).
We examined bacterial community structure using the weighted UniFrac distance measure in QIIME and visualized the output with principal coordinate analysis. UniFrac combines distance-based measures with phylogenetic information, making it more informative than ordinations done with a distance measure alone (18). To determine the proportion of variation in the sequence data that could be explained by the environmental variables that we measured (NO3−, salinity, and dissolved oxygen), we performed two tests: a distance-based multivariate ANOVA (adonis) and a constrained distance-based redundancy analysis (db-RDA) using the vegan package (36) in R (35). The adonis method uses a permutation approach to test for differences among replicate multivariate samples using a distance matrix. For these analyses, we used the Bray-Curtis similarity metric on the rarified data table at the genus level, the finest-scale resolution possible given the constraints of our gene fragment size. db-RDA, can be used to test whether the measured environmental variables are able to explain a significant portion of the variance observed in the distance matrix. To perform db-RDA, we calculated a Bray-Curtis similarity matrix on the rarefied data set at both the class and genus levels and selected variables via permutation. We tested for significance on the RDA data using a type III ANOVA. Geochemistry data were log transformed, and taxonomic abundances were Hellinger transformed prior to analysis.
To determine which bacteria were driving observed patterns in community composition, we generated stacked bar graphs using class-level taxonomy tables generated in QIIME, and we tested for differences in abundances at each of our sites using ANOVA in R (35). To obtain finer-scale taxonomic resolution, we also used SIMPER analysis in vegan (36) at the genus level to determine which taxa were driving differences among sites. We were particularly interested to see how the distribution of candidate phyla, many of which are inferred to have obligately fermentative metabolisms, changed as a result of the addition of carbon in the PRB, so we used the rarefied abundance table to generate heat maps of the relative abundance of each of the classes of candidate phyla in all of the samples.
In addition to documenting the relative abundances of different bacteria in groundwater as a result of the barrier, we also compared the relative activities of different taxa in groundwater collected from wells within the barrier to wells upgradient and outside the barrier. To examine the relative activities of different bacteria, we calculated the ratio of rRNA to rRNA genes for each class using the rarefied data table. We plotted the results in a heat map, with yellow and red signifying bacterial classes that had rRNA/rRNA gene ratios of >1 (a proxy for active members of the bacterial community) and blue signifying bacterial classes that had rRNA/rRNA gene ratios of <1 (a proxy for bacterial classes with a large proportion of dormant taxa). To determine if differences in abundance of rRNA and rRNA genes in each class were significant, we performed a permutation test on the abundances in each site. For these tests, we combined rRNA and rRNA gene abundances into one data set, sampled without replacement, computed differences in means between these randomized data sets, and repeated this for 1,000 permutations. The P value is the number of times the difference was greater than or equal to the observed difference. Significant P values indicate that that class was either largely active (significantly more rRNA than rRNA genes) or was largely dormant (significantly more rRNA genes than rRNA).
In order to gain more insight into which bacteria within each class were active and which were dormant, we also analyzed the 16S rRNA/rRNA gene ratio data at the genus level. We regressed 16S rRNA sequence abundance against 16S rRNA gene abundance for each genus and calculated the residuals for these points relative to a one-to-one line. Residuals that were greater than 1 standard deviation (SD) away from the one-to-one line were considered either active (if the residuals were positive) or dormant (if the residuals were negative).
Nucleotide sequence accession number.
Sequence data have been deposited in the Sequence Read Archive under accession no. SRP053303.
RESULTS
Bacterial diversity and environmental drivers of community structure.
The total bacterial community as well as the active community both showed higher α diversity in the PRB relative to the reference site (Fig. 2). A one-way ANOVA indicated that there were significantly more observed taxa and a higher Chao1 estimated richness (P = 0.04 and P = 0.03, respectively) in the PRB samples based on analysis of 16S rRNA genes. 16S rRNA also demonstrated significant differences among sites, with more observed taxa (P = 0.003) and a higher Chao1 estimated richness (P = 0.002) and Shannon diversity index (P = 0.01) in the PRB samples than in the upgradient and reference samples.
FIG 2.
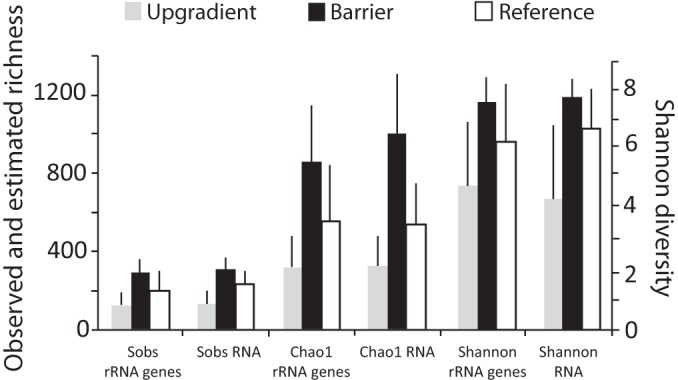
Total number of observed taxa (Sobs), Chao1 richness estimates, and Shannon diversity indices of the bacterial community composition derived from analysis of 16S rRNA and 16S rRNA genes for samples collected from upgradient (gray bars), barrier (black bars), and reference (white bars) wells. Error bars show standard errors.
The β diversity results suggest that there is a distinctly different bacterial community present in the 16S rRNA genes of the PRB sites (Fig. 3, red squares) compared to the reference (Fig. 3, blue squares), and upgradient sites (Fig. 3, purple squares). Surprisingly, that difference was not reflected as strongly in the 16S rRNA, where there was considerable overlap in community structure at PRB, upgradient, and reference sites (Fig. 3).
FIG 3.
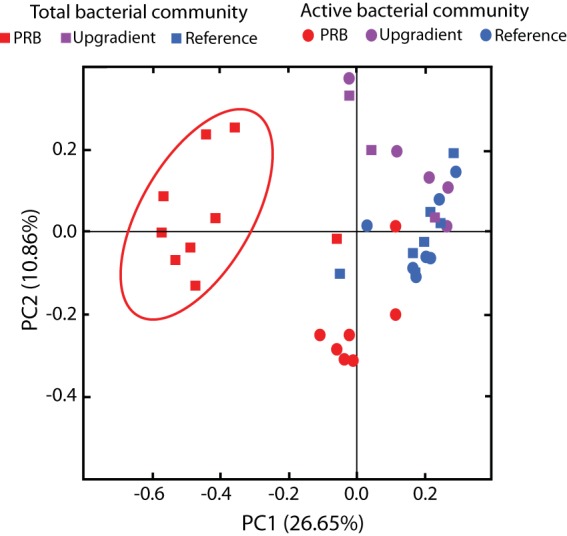
Principal coordinate analysis constructed using weighted UniFrac at the OTU level. The total bacterial community values (squares) are derived from analysis of 16S rRNA genes, and the active bacterial community values are derived from analysis of 16S rRNA (circles) for samples collected from the PRB (red), upgradient (brown), and reference (blue) locations.
We measured salinity, dissolved oxygen, NO3− and NH4+ to determine both how the PRB altered the geochemistry of groundwater percolating through the barrier and if the changes we measured correlated with changes in bacterial community structure. Our results suggest that the PRB alters the salinity regime by allowing greater salt water intrusion than in adjacent areas with no PRB (Fig. 4A). Salinity ranged from 0 to 2 ppt in wells upgradient of the barrier but ranged from 6 to 19 ppt in the PRB (Fig. 4A), reflecting its location in the mixing zone of the subterranean estuary. The reference site had lower salinities overall but also showed evidence of saltwater mixing at the lowest depths (Fig. 4B). Our data also indicate that the PRB contains a region of low-oxygen water (Fig. 4C). Groundwater in the reference and upgradient sites had dissolved oxygen concentrations of 3 to 6 mg liter−1, but as the groundwater moved through the PRB, the oxygen concentration dropped to below the limit of detection. Similarly, NO3− concentrations in upgradient and reference wells reached 25 μM (Fig. 4E), but in PRB wells, NO3− concentrations were low, typically below the detection limit (Fig. 4E). Ammonium did not show any discernible pattern among sites, suggesting the barrier did not promote dissimilatory nitrate reduction to ammonia (data not shown).
FIG 4.

Contour plots of the aquifer region upgradient and within the PRB (A, C, and E) and in a section of aquifer adjacent to the PRB that served as a reference location (B, D, and F). Sampling wells W0 to W3 correspond to areas upgradient of the PRB, W4 to W6 represent samples within the PRB, and W7 is downgradient of the PRB. The red box represents the specific location of the PRB, and the black dots represent the location of discrete-depth sampling wells. The green line indicates the surface of the beach.
Adonis results confirmed that the bulk bacterial communities (via analysis of 16S rRNA genes) were significantly different between the barrier and nonbarrier locations (F = 2.7, P = 0.001). To determine what environmental factors correlated with these differences, we performed db-RDA at the genus level, using dissolved oxygen, salinity, and nitrate as predictors in the model. Site was not included as a predictor in this analysis since it correlated with the other factors. Dissolved oxygen proved to be the only significant correlate with community composition (P = 0.047). To examine environmental correlates that might influence changes in the active community, we did the same analyses on 16S rRNA sequences. Adonis results also indicated that the active communities differed significantly among sites (F = 2.1, P = 0.013); however, none of the predictors were significant for 16S rRNA at the genus level.
Relative bacterial abundance and activity.
Comparison of the relative abundance of taxa across sites could provide insight into the organisms responsible for the proper functioning of the PRB. The most important classes present in the 16S rRNA genes of the reference and upgradient sites belonged to the phylum Proteobacteria, which accounted for, on average, 40% of all sequences in the reference site and 43% of all sequences in the upgradient sites (Fig. 5A). Classes from the Acidobacteria phylum and the candidate phylum OP3 were also important components of the non-PRB sites (Fig. 5A). Analysis of 16S rRNA gene sequences from the PRB (Fig. 5A), however, revealed a different pattern. PRB samples were dominated by classes from the candidate phylum OD1 (Parcubacteria), which accounted for, on average, 37% of the 16S rRNA gene sequences, followed by classes from the Proteobacteria, which accounted for ∼17% of all 16S rRNA gene sequences. Stacked bar plots generated using 16S rRNA sequence data from the PRB revealed a pattern distinct from the PRB 16S rRNA gene sequence data (Fig. 5B). Among the active portions of the community, the relative importance of OD1 in the PRB was considerably lower (never more than 5% of any sample) than in the bulk (Fig. 5B). Instead, classes from the Proteobacteria, which accounted for 49%, 65%, and 40% of the active community in the reference, upgradient, and PRB samples, respectively, dominated the active portion of the bacterial community. Classes from the Acidobacteria were also an important component of the active community in the reference samples, and classes from the candidate phylum OP2 were active in the PRB.
FIG 5.

Stacked bar graphs of relative abundances of bacteria with taxonomy assigned at the class level for the total microbial community via analysis of 16S rRNA genes (A) and of the active community via analysis of 16S rRNA (B).
Since many candidate phyla appear to have a considerably higher abundance in the PRB compared to upgradient and reference locations, we examined their abundance in greater detail by plotting relative abundances of different genera from each of the candidate phyla in heat maps (Fig. 6). When we examined the candidate phyla via analysis of 16S rRNA gene sequences, clear differences emerge between the reference and upgradient samples compared to the samples from the PRB. In particular, bacteria associated with the koll11 isolate from the OP3 phylum were relatively more abundant in the reference and upgradient samples than in the barrier samples (Fig. 6A). In contrast, bacteria associated with the OD1 phylum, and in particular those associated with the ABY1 and ZB2 candidate classes, were a greater proportion of the community in the PRB compared to the reference and upgradient locations (Fig. 6A), as was GN02. Unlike the clear patterns demonstrated in the 16S rRNA gene analysis, there were no clear differences in the relative abundances of different candidate phyla among the active portion of the community as determined by analysis of 16S rRNA (Fig. 6B).
FIG 6.

Abundance of different candidate phyla identified at the highest resolvable taxonomic unit in all samples based on 16S rRNA genes (A) and rRNA (B) sequences. Ref, reference; Up, upgradient.
SIMPER analysis of the 16S rRNA gene sequences indicated that all OD1 classes combined accounted for 19.4% of the difference observed between PRB and reference sites, and OP3 (Omnitrophica) and GN02 (Gracilibacteria) accounted for an additional 7.2 and 2.7%, respectively (Table 1). Thus, among the total community, candidate phyla accounted for almost 30% of the differences among sites (Table 1). In contrast, within the active portion of the community, an Acidobacteria class, a family of Helicobacteraceae, and a category of unassigned bacteria each accounted for 3.6% of the differences observed, with various families of Proteobacteria combining for an additional 12.7% (Table 1). There were no candidate phyla among the taxa driving differences among sites in the active community.
TABLE 1.
Results of SIMPER analysis that show the percentage of contribution of each taxon to total dissimilarity calculated from a Bray-Curtis dissimilarity matrix
| Gene type (community) | Classa | % of population |
|---|---|---|
| 16S rRNA gene (total) | OD1-ZB2 (c) | 9.8 |
| OD1-ABY1 (c) | 6.1 | |
| Oxalobacteraceae (f) | 5.5 | |
| OP3-koll11-GIF10-kpj58rc (o) | 4.4 | |
| OD1-unresolved (c) | 3.5 | |
| Acidobacteria 6–iii1-15 (o) | 3.5 | |
| Unidentified bacteria (p) | 3.0 | |
| OP3-koll11 (c) | 2.8 | |
| GN02–GKS2-174 (c) | 2.7 | |
| Sphingomonadaceae (f) | 1.7 | |
| 16S rRNA (active) | Acidobacteria 5 (c) | 3.6 |
| Helicobacteraceae (f) | 3.6 | |
| Unidentified bacterium (p) | 3.6 | |
| Comamonadaceae (f) | 3.0 | |
| Nitrospirales (o) | 2.3 | |
| Entotheonellaceae (f) | 2.2 | |
| Myxococcales (o) | 2.0 | |
| Gammaproteobacteria (c) | 1.9 | |
| Pseudomonas (g) | 1.9 | |
| Oxalobacteraceae (f) | 1.7 |
Each taxon was resolved to the highest taxonomic classification possible. The letters in parentheses indicate the taxonomic level: p, phylum; c, class; o, order; f, family; g, genus. The top 10 taxa each for 16S rRNA genes and rRNA are shown.
To further assess which taxa had the highest potential rates of protein synthesis, we normalized the abundance of 16S rRNA to the abundance of 16S rRNA genes and plotted the results in a heat map (Fig. 7). In the PRB, several classes of bacteria had 16S rRNA/rRNA gene ratios that were significantly higher than 1, as determined by permutation tests, indicating the highest potential for activity. These classes (demarcated with black asterisks) include Deltaproteobacteria, Epsilonproteobacteria, Lentisphaeria, and Anaerolinea (Fig. 7). Consistent with previous results, the PRB also contained classes with 16S rRNA/rRNA gene ratios significantly lower than 1 (demarcated with white asterisks). These classes include three classes of OD1 and a class of TM7. The reference sites harbored different active and dormant classes of bacteria. Significantly more active classes included Sphingobacteria and OP3 in addition to Deltaproteobacteria. The only class in the reference samples with rRNA/rRNA gene ratios that were significantly lower than 1 was a class of Acidobacteria.
FIG 7.
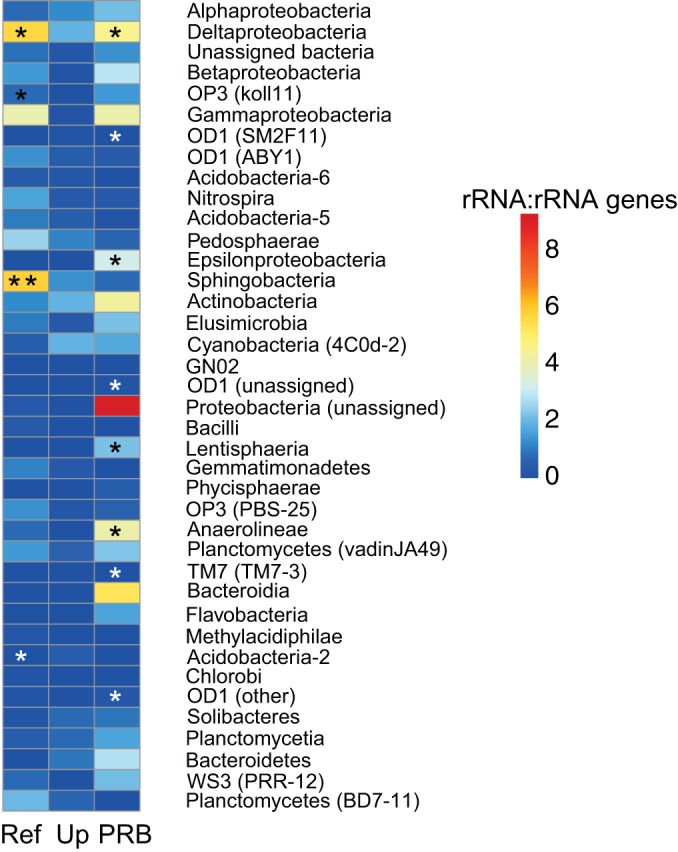
Heat map of the average ratio of 16S rRNA to rRNA genes for bacterial classes in the reference (Ref), upgradient (Up), and PRB sites. A significant result indicates that that class expressed significantly more (black asterisks) or significantly less (white asterisks) 16S rRNA relative to 16S rRNA genes. *, P < 0.05; **, P < 0.005.
We performed a similar analysis at the finest-scale taxonomic resolution (Table 2) because the class level analysis likely aggregates over both active and inactive taxa. Active bacteria (taxa whose abundance was >1 SD above a slope of unity) in the PRB included Myxococcales (Deltaproteobacteria), Victivallales, Helicobacteraceae (Epsilonproteobacteria), and Desulfobacteraceae (Deltaproteobacteria) (Table 2). Bacteria with a higher degree of dormancy (taxa whose abundance was >1 SD below a slope of unity) included multiple genera of OD1 and OP3 (Table 2). The reference site harbored active Bacteroidetes, including Sphingobacteriales and Chitinophagaceae, Deltaproteobacteria (Entotheonellaceae, Myxococcales), Gammaproteobacteria (Pseudomonas), and Nitrospira (Nitrospirales). Dormant bacteria in the reference site included OP3 (koll11, koll1-GIF10-kpj58rc), an order of Acidobacteria, and Methylacidphilales (Table 2).
TABLE 2.
Active and dormant taxa at the highest resolvable taxonomic designation
| Taxon typea | Taxab |
|---|---|
| Active | |
| Reference | Sphingobacteriales (o)* |
| Sphingobium (g)* | |
| Sphingomonas (g) | |
| Entotheonellaceae-other* | |
| Deltaproteobacteria-MIZ46 (o) | |
| Myxococcales (o)* | |
| Pseudomonas* | |
| Planctomycetes-vadinHA49 (o) | |
| Gemmatimonadales-Ellin5301 (f) | |
| Chitinophagaceae (f) | |
| Nitrospirales (o)* | |
| GN02–GKS2-174 (c)* | |
| OD1-ZB2 (c) | |
| Upgradient | Sphingobacteriales (o)* |
| Sphingomonadaceae (f)* | |
| Sphingobium (g)* | |
| Phenylobacterium (g)* | |
| Entotheonellaceae (f) | |
| Myxococcales (o)* | |
| Myxococcales-0319-6620 (f)* | |
| Cyanobacteria-4c0d (o) | |
| PRB | Myxococcales (o)* |
| Desulfobacteraceae (f) | |
| Arcobacter (g) | |
| Helicobacteraceae (f)* | |
| Victivallales (o) | |
| Dormant | |
| Reference | Acidobacteria6–iii1-15 (o)* |
| Methylacidphilales (o)* | |
| OP3-koll11 (c)* | |
| OP3-koll11-GIF10-kpj58rc (o)* | |
| Upgradient | Sphingomonas (g) |
| Bradyrhizobiaceae (f) | |
| Bosea (g) | |
| Cupriavidus (g) | |
| Herbaspirillum (g) | |
| Legionellaceae (f)* | |
| Acidobacteria 6–iii1-15 (o)* | |
| Chitinophagaceae (f) | |
| Microbacterium (g) | |
| Saprospiraceae (f) | |
| Elusimicrobiales (o) | |
| OD1-ZB2 (c) | |
| OP3-koll11 (c)* | |
| OP3-koll11-GIF10 (o)* | |
| PRB | GN02–GKS2-174 (c)* |
| OD1-ABY1 (c)* | |
| OD1-ZB2 (c)* | |
| OP3–PBS-25 (c) | |
| OP3-koll11-GIF10-kpj58rc (o) |
The differences between 16S rRNA and rRNA gene abundances were calculated for each taxon at each site. Any taxon where the residuals were ±1 SD from a slope of unity was considered active (>+1 SD) or dormant (<−1 SD).
The letters in parentheses indicate the taxonomic level: c, class; o, order; f, family; g, genus. Asterisks indicate that the taxon was >+2 SD or <−2 SD from a slope of unity.
DISCUSSION
Bacterial diversity and environmental drivers of community structure.
We hypothesized that the large supply of carbon provided by the PRB would drive changes in the bacterial community by providing additional niche space for bacteria with low-energy-yielding metabolisms. Our results indicate that both the total number of observed taxa and the estimated taxonomic richness were greater in the PRB relative to reference sites (Fig. 2) when the community was analyzed both via 16S rRNA genes (total community) and 16S rRNA (active community). The PRB was also more diverse when measured by the Shannon index, which takes into account both evenness and richness. Abundant carbon has been shown to reduce competition and allow for a greater abundance (37) and more even distribution of bacteria in soils relative to carbon-limited environments (38). The structure of the total bacterial community also supports our hypothesis that the barriers can promote metabolisms that have a negative reduction potential, such as sulfate reduction and some types of fermentation. Many of the abundant taxa found in the PRB are inferred from metagenomic studies to have fermentative metabolisms, suggesting that there is sufficient carbon available to support a wide array of metabolic niches that are not typically abundant in aerobic coastal aquifers. It remains to be seen whether the specific bacterial taxa enhanced by the PRB are also capable of carrying out denitrification, but the wide diversity of metabolisms inferred by our 16S rRNA data suggests that the PRB is likely to promote this essential ecosystem service.
Surprisingly, despite the large input of organic matter provided by the barriers, the compositions of the active portion of the bacterial community were similar among sites as assessed by analysis of 16S rRNA (Fig. 3). The only samples that were divergent in their community composition were those sequenced from the 16S rRNA genes (representative of both active and recently dead or dormant bacteria or extracellular DNA). This suggests that the taxa that made the PRB distinct from reference sites were not actively transcribing genes. We hypothesize that the difference between the 16S rRNA genes and 16S rRNA samples might result from the fact that the bacterial communities we analyzed were extracted from groundwater flowing through the barrier, rather than the bacteria that were residing in biofilms directly on the wood chips. It is possible that sampling the groundwater percolating through the barrier included both typical aquifer bacteria, similar to what would be expected from reference samples, as well as inactive (recently dead or dormant) bacteria or extracellular DNA that had been released from the PRB biofilm. Direct sampling of the wood chip biofilm could help confirm this hypothesis. If our hypothesis is true, it suggests that the area of active biogeochemical cycling is directly on the surface of the wood chips.
We measured a suite of environmental parameters in groundwater to determine if they explained differences in bacterial community structure among sites. The PRB resulted in several changes to redox chemistry in the aquifer that are both caused by and could induce changes in the bacterial community. Our data indicate that there was a sufficient amount of carbon in the PRB to completely remove oxygen and NO3− (Fig. 4C and E). The complete removal of these oxidants suggests that there was sufficient reducing power that alternative metabolisms, such as sulfate reduction and fermentation, were also likely taking place. In contrast to what we observed in the PRB, oxygen and NO3− concentrations remained high in reference samples (Fig. 4D and F).
The decrease in oxygen concentration caused by microbes inhabiting the PRB was the only geochemical parameter that correlated with the structure of the total bacterial community, and it covaried with sampling site. Surprisingly, dissolved oxygen and site were more important drivers of community composition than salinity, despite the identified importance of salinity as a driver of bacterial community structure in many other systems (15, 18, 39, 40). Bacterial community composition often changes along estuarine salinity gradients (15), but Hollister et al. (41) found that along a transect of 140 m, salinity was not important as a driver of bacterial community structure. The small size of the PRB (it covers only 20 m of length along the beach), along with the wide fluctuations in salinity that it encounters (24), may explain why salinity was not an important driver of community structure in our samples. Dissolved oxygen, however, has been identified as an important driver of community composition in wastewater treatment systems and bioreactors (39, 42). Factors like dissolved oxygen, nitrate, and salinity all vary within the PRB, so it seems likely that the collective geochemical changes the PRB creates ultimately drive differences in the bacterial community structure among sites.
Relative bacterial abundance and activity.
Our 16S rRNA gene data indicate that several classes of candidate phyla drove the broad-scale differences in total bacterial community composition among sampling sites (Fig. 3). In particular, the very large proportions of candidate phyla OD1 and OP3 in the PRB 16S rRNA genes resulted in significant differences in community structure (Fig. 5A). In fact, the proportion of OD1 in the PRB was unusually high compared with those in many other systems (43). In a compilation by Luef et al. (43), five different classes of OD1 were surveyed from 23 different habitats, and in only one sample from marine water did a class of OD1 exceed 40% of the community composition, while in the PRB, OD1 represented ∼38% of sequences (Fig. 5). Previous work has shown that OD1 is often found in high relative abundances in anoxic, reducing environments, often with high concentrations of DOC (44–46). For example, the Lost City hydrothermal vents, where OD1 was found to be abundant, had concentrations of DOC as high as 106 μM, similar to the PRB (46). Although OD1 metabolisms were not specifically identified in this study, metagenomic sequencing of these candidate phyla indicate that they are likely strict anaerobes with very small genomes, and they are thought to rely on syntrophy with other microorganisms to provide metabolites for sulfur reducing and fermentation pathways (47–49). The fact that OD1 and other candidate phyla thought to have fermentative metabolisms were present in such high relative abundances in the PRB suggests that the wood chips provide a sufficient supply of carbon that is able to support a multitude of alternative bacterial metabolisms. The low proportion of OD1 among the active portion of the community in the PRB, however, suggests that these cells were not active at the time of sampling, possibly because the cells were only active when adhered directly to the wood chips.
While OD1 was in lower relative abundance in the reference and upgradient samples compared to in the PRB, OP3 was relatively more abundant in those locations. These bacteria are also anaerobic and are commonly found in groundwater, wastewater treatment plants, bioreactors, and wetlands (50). Metagenomic analysis of OP3 indicates the genetic capacity for acetogenesis, sulfate reduction, and methanogenesis within this candidate phylum (50). The relative abundance of candidate classes, however, was both higher and more diverse in the PRB than at the reference site, which contained just the two classes of OP3 (Fig. 6). Patterns in relative abundance clearly showed a shift from moderately abundant OP3 in the reference site to highly abundant OD1, GN02, and OP3 in the PRB when examining the total bacterial community. Overall, the PRB supported a larger abundance and diversity of small, energetically inefficient candidate phyla, in keeping with our hypothesis.
The PRB supported a number of active anaerobic bacteria, including Epsilonproteobacteria and Lentisphaeria (Table 2). Epsilonproteobacteria, including Helicobacteraceae and Arcobacter, were both present and active in the PRB but not present in the reference samples (Table 2). Many Epsilonproteobacteria are found in anaerobic environments (51, 52). Some of these bacteria, including Helicobacteraceae, are related to sulfur cycling and are found in anoxic environments like deep-sea vent sulfide deposits, seep sediments, and hydrothermal fluids (53). Desulfobacteraceae, a family of Deltaproteobacteria, were present and active in the PRB and are also common in anoxic habitats, like marsh sediments and hydrothermal vents (54, 55). Victivallales, a member of the Lentisphaeria, were also active in the PRB, and strains are common in anaerobic digesters and wastewater treatment plants, suggesting they could be actively degrading the wood chips in the PRB (56).
Noticeably absent from the active bacteria in the PRB are the various classes of OD1, which were extremely abundant in the total bacterial community. Although the low proportion of OD1 among the active bacteria could be a result of their reduced genome size and fewer ribosomes compared to others (resulting in a smaller relative proportion of 16S rRNA compared to other active bacteria), we suspect that this is not the case for a number of reasons. First, the sheer magnitude of difference between the proportion of OD1 in the total community (38%) and in the active community (<5%) suggests that this is not solely an artifact of genome size. Second, SIMPER analysis indicated that OD1 played no role in determining the structure of the active community, despite accounting for nearly 20% of the dissimilarity identified in the total community (Table 1). Most importantly, however, when we normalized the abundances of the active taxa to their abundances in the total community and examined the residuals of those abundances to determine activity and dormancy, the OD1 classes ABY1 and ZB2 were more than 2 SD away from unity in the PRB samples, suggesting that there is a distinct lack of relative ribosome expression, which we interpret as a high degree of metabolic dormancy.
The primary differences among the active bacteria between the PRB and reference sites were primarily within classes of Proteobacteria and Acidobacteria. SIMPER analysis shows that the more important active taxa responsible for differences among sites include Nitrospirales, which contains one family that includes many aerobic nitrite oxidizers (57), and the family Comamonadaceae, which is known to contain many facultative denitrifiers (58). Both were relatively abundant in the reference and upgradient sites compared to the PRB sites. Active taxa in the reference site also included Sphingobium (likely aerobic carbon degraders) and Chitinophagaceae (a family of Sphingobacteriales, many of which are aerobic or facultative anaerobes) (59, 60).
In summary, even 8 years after installation, and without any maintenance, the PRB in Waquoit Bay, MA, still appears to reduce NO3− to undetectable levels, likely as a result of heterotrophic denitrification. The PRB promotes the presence of anaerobic bacteria and has a distinct overall bacterial community compared to nearby reference sites where there is no PRB. These differences are mainly due to the high relative abundance of OD1 and other bacteria with alternative metabolisms that likely persist on the large supply of degradable carbon provided by the PRB. Our data suggest, however, that many of these anaerobic bacteria are inactive (dead, dormant, or extracellular DNA), as the active component of the bacterial community in the PRB was not enriched in these anaerobic taxa. Finally, dissolved oxygen was the only environmental driver measured that explained bacterial community structure in the PRB, suggesting that the supply of carbon from the wood chips and the resultant decline in dissolved oxygen overwhelmed salinity and nitrogen supply as drivers of bacterial community composition.
ACKNOWLEDGMENTS
We thank Sarah Feinman, John Angell, Collin Knauss, and Patrick Kearns for assistance in the field. We would also like to acknowledge the use of the supercomputing facility managed by the Research Computing Department at the University of Massachusetts Boston, and Jeff Dusenberry for assistance with denoising the data set.
This work was supported by a grant from the Environmental Protection Agency to K.H. (F12E21008), a grant from MIT Sea Grant (2012-R/RC-129) to J.L.B., and a grant from NSF to K.F. and Joe Vallino (NSF CBET-0756562). The original PRB was installed by Kenneth Foreman and Joe Vallino from the Marine Biological Laboratory, and Pio Lombardo of Lombardo Associates (Newton, MA). The PRBs were supported with funding from the NOAA/UNH Cooperative Institute for Coastal and Estuarine Environmental Technology (NA04NOS190109).
External reviewers greatly improved the quality of the manuscript.
REFERENCES
- 1.Voss M, Bange HW, Dippner JW, Middelburg JJ, Montoya JP, Ward BB. 2013. The marine nitrogen cycle: recent discoveries, uncertainties, and the potential relevance of climate change. Philos Trans Royal Soc B 368:20130121. doi: 10.1098/rstb.2013.0121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bricker SB, Longstaff B, Dennison W, Jones A, Boicourt K, Wicks C, Woerner J. 2008. Effects of nutrient enrichment in the nation's estuaries: a decade of change. Harmful Algae 8:21–32. doi: 10.1016/j.hal.2008.08.028. [DOI] [Google Scholar]
- 3.Cosper EM, Dennison W, Carpenter EJ, Bricelj VM, Mitchell JG, Kuenstner SH, Colflesh DC, Dewey M. 1987. Recurrent and persistent “brown tide” blooms perturb coastal marine ecosystems. Estuaries 10:284–290. doi: 10.2307/1351885. [DOI] [Google Scholar]
- 4.Hagy JD, Boynton WR, Keefe CW, Wood KV. 2004. Hypoxia in Chesapeake Bay, 1950-2001: long term change in relation to nutrient loading and river flow. Estuaries 27:634–658. doi: 10.1007/BF02907650. [DOI] [Google Scholar]
- 5.Valiela I, McLelland J, Hauxwell J, Behr PJ, Hersch D, Foreman K. 1997. Macroalgal blooms in shallow estuaries: controls and ecophysiological and ecosystems responses. Limnol Oceanogr 42:1105–1118. doi: 10.4319/lo.1997.42.5_part_2.1105. [DOI] [Google Scholar]
- 6.Diaz RJ, Rosenberg R. 2008. Spreading dead zones and consequences for marine ecosystems. Science 321:926–929. doi: 10.1126/science.1156401. [DOI] [PubMed] [Google Scholar]
- 7.Bowen JL, Valiela I. 2001. The ecological effects of urbanization of coastal watersheds: historical increases in nitrogen loads and eutrophication of Waquoit Bay estuaries. Can J Fish Aquat Sci 58:1489–1500. doi: 10.1139/f01-094. [DOI] [Google Scholar]
- 8.Bowen JL, Kroeger KD, Tomasky G, Pabich WJ, Cole ML, Carmichael RH, Valiela I. 2007. A review of land-estuary coupling by ground water discharge: mechanisms and effects. Appl Geochem 22:175–191. doi: 10.1016/j.apgeochem.2006.09.002. [DOI] [Google Scholar]
- 9.Howes B, Kelley SW, Ramsey JS, Samimy R, Schlezinger D, Ruthven T, Eichner E. 2005. Linked watershed-embayment model to determine critical nitrogen loading thresholds for the Quashnet River, Hamblin Pond, and Jehu Pond, in the Waquoit Bay system in the towns of Mashpee and Falmouth, Massachusetts. Massachusetts Estuaries Project, Massachusetts Department of Environmental Protection, Boston, MA. [Google Scholar]
- 10.Lee V, Olsen S. 1985. Eutrophication and management initiatives for the control of nutrient inputs to Rhode Island coastal lagoons. Estuaries 8:191–202. doi: 10.2307/1352200. [DOI] [Google Scholar]
- 11.Valiela I, Costa J, Foreman K, Teal JM, Howes B, Aubrey D. 1990. Transport of groundwater-borne nutrients from watersheds and their effects on coastal waters. Biogeochemistry 10:177–197. doi: 10.1007/BF00003143. [DOI] [Google Scholar]
- 12.Schipper LA, Robertson WD, Gold AJ, Jaynes DB, Cameron SC. 2010. Denitrifying bioreactors—an approach for reducing nitrate loads to receiving waters. Ecol Eng 36:1532–1543. doi: 10.1016/j.ecoleng.2010.04.008. [DOI] [Google Scholar]
- 13.Jaynes DB, Kaspar T, Moorman T, Parkin T. 2008. In situ bioreactors and deep drain-pipe installation to reduce nitrate losses in artificially-drained fields. J Environ Qual 37:429–436. doi: 10.2134/jeq2007.0279. [DOI] [PubMed] [Google Scholar]
- 14.Robertson WD, Blowes DW, Ptacek CJ, Cherry JA. 2000. Long term performance of in situ reactive barriers for nitrate remediation. Groundwater 38:689–695. doi: 10.1111/j.1745-6584.2000.tb02704.x. [DOI] [Google Scholar]
- 15.Campbell BJ, Kirchman DL. 2013. Bacterial diversity, community structure and potential growth rates along an estuarine salinity gradient. ISME J 7:210–220. doi: 10.1038/ismej.2012.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fortunato CS, Herfort L, Zuber P, Baptitsta AM, Crump BC. 2012. Spatial variability overwhelms seasonal patterns in bacterioplankton communities across a river to ocean gradient. ISME J 6:554–563. doi: 10.1038/ismej.2011.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herlemann DPR, Labrenz M, Jürgens K, Bertilsson S, Waniek JJ, Andersson AF. 2011. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J 5:1571–1579. doi: 10.1038/ismej.2011.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lozupone CA, Knight R. 2007. Global patterns in bacterial diversity. Proc Natl Acad Sci U S A 104:11436–11440. doi: 10.1073/pnas.0611525104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gilbert JA, Field D, Swift P, Newbold L, Oliver A, Smyth T, Somerfield PJ, Huse S, Joint I. 2009. The seasonal structure of microbial communities in the Western English Channel. Environ Microbiol 11:3132–3139. doi: 10.1111/j.1462-2920.2009.02017.x. [DOI] [PubMed] [Google Scholar]
- 20.Campbell BJ, Yu L, Heidelberg JF, Kirchman DL. 2011. Activity of abundant and rare bacteria in a coastal ocean. Proc Natl Acad Sci U S A 108:12776–12781. doi: 10.1073/pnas.1101405108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schipper LA, Barkle GF, Vojvodic-Vukovic M. 2005. Maximum rates of nitrate removal in a denitrification wall. J Environ Qual 34:1270–1276. doi: 10.2134/jeq2005.0008. [DOI] [PubMed] [Google Scholar]
- 22.Robertson WD, Merkley LC. 2009. In-stream bioreactor for agricultural nitrate treatment. J Environ Qual 38:230–237. doi: 10.2134/jeq2008.0100. [DOI] [PubMed] [Google Scholar]
- 23.Moore WS. 1999. The subterranean estuary: a reaction zone of ground water and sea water. Mar Chem 65:111–125. doi: 10.1016/S0304-4203(99)00014-6. [DOI] [Google Scholar]
- 24.Vallino J, Foreman K. 2008. Effectiveness of reactive barriers for reducing N-loading to the coastal zone. NOAA/University of New Hampshire Cooperative Institute for Coastal and Estuarine Environmental Technology (CICEET), Durham, NH. [Google Scholar]
- 25.Charette MA, Sholkovitz ER, Hansel CM. 2005. Trace element cycling in a subterranean estuary. Part 1. Geochemistry of the permeable sediments. Geochim Cosmochim Acta 69:2095–2109. doi: 10.1016/j.gca.2004.10.024. [DOI] [Google Scholar]
- 26.Charette MA, Sholkovitz ER. 2006. Trace element cycling in a subterranean estuary. Part 2. Geochemistry of the pore water. Geochim Cosmochim Acta 70:811–826. doi: 10.1016/j.gca.2005.10.019. [DOI] [Google Scholar]
- 27.Pabich WJ, Valiela I, Hemond HF. 2001. Relationship between DOC concentration and vadose zone thickness and depth below water rable in groundwater of Cape Cod, USA. Biogeochemistry 55:247–268. doi: 10.1023/A:1011842918260. [DOI] [Google Scholar]
- 28.Baddeley A, Turner R. 2005. Spatstat: an R package for analyzing spatial point patterns. J Stat Softw 12:1–42. [Google Scholar]
- 29.Diggle PJ. 1985. A kernel method for smoothing point process data. J Appl Stat 34:138–147. doi: 10.2307/2347366. [DOI] [Google Scholar]
- 30.Blazewicz SJ, Barnard RL, Daly RA, Firestone MK. 2013. Evaluating rRNA as an indicator of microbial activity in environmental communities: limitations and uses. ISME J 7:2061–2068. doi: 10.1038/ismej.2013.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones SE, Lennon JT. 2010. Dormancy contributes to the maintenance of microbial diversity. Proc Natl Acad Sci U S A 107:5881–5886. doi: 10.1073/pnas.0912765107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coates JD, Michaelidou U, Bruce RA, O'Connor SM, Crespi JN, Achenbach LA. 1999. Ubiquity and diversity of dissimilatory (per) chlorate-reducing bacteria. Appl Environ Microbiol 65:5234–5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Gonzalez Pena A, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 35.R Core Team. 2014. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org/. [Google Scholar]
- 36.Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O'Hara RB, Simpson GL, Solymos P, Stevens MHH, Wagner H. 2013. vegan: community ecology package. R package version 2.0-10. R Foundation for Statistical Computing, Vienna, Austria: http://CRAN.R-project.org/package=vegan. [Google Scholar]
- 37.Dojka MA, Hugenholtz P, Haack SK, Pace NR. 1998. Microbial diversity in a hydrocarbon and chlorinated solvent contaminated aquifer undergoing intrinsic bioremediation. Appl Environ Microbiol 64:3869–3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou J, Xia B, Treves DS, Wu LY, Marsh TL, O'Neill RV, Palumbo AV, Tiedje TM. 2002. Spatial and resource factors influencing high microbial diversity in soil. Appl Environ Microbiol 68:326–334. doi: 10.1128/AEM.68.1.326-334.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang X, Hu M, Xia Y, Wen X, Ding K. 2012. Pyrosequencing analysis of bacterial diversity in 14 wastewater treatment systems in China. Appl Environ Microbiol 78:7042–7047. doi: 10.1128/AEM.01617-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matcher GF, Dorrington RA, Henninger TO, Froneman PW. 2011. Insights into the bacterial diversity in a freshwater-deprived permanently open Eastern Cape estuary, using 16S rRNA pyrosequencing analysis. Water SA 37:381–390. [Google Scholar]
- 41.Hollister EB, Engledow AS, Hammet AJM, Provin TL, Wilkinson HH, Gentry TJ. 2010. Shifts in microbial community structure along an ecological gradient of hypersaline soils and sediments. ISME J 4:829–838. doi: 10.1038/ismej.2010.3. [DOI] [PubMed] [Google Scholar]
- 42.Kim TS, Jeong JY, Wells GF, Park HD. 2012. General and rare bacterial taxa demonstrating different temporal dynamic patterns in an activated sludge bioreactor. Appl Microbiol Biotechnol 97:1755–1765. doi: 10.1007/s00253-012-4002-7. [DOI] [PubMed] [Google Scholar]
- 43.Luef B, Frischkorn KR, Wrighton KC, Holman HN, Birarda G, Thomas BC, Singh A, Williams KH, Siegerist CE, Tringe SG, Downing KH, Comolli LR, Banfield JF. 27 February 2015. Diverse uncultivated ultra-small bacterial cells in groundwater. Nat Commun doi: 10.1038/ncomms7372. [DOI] [PubMed] [Google Scholar]
- 44.Gies FA, Kanwar KM, Beatty JT, Hallam SJ. 2014. Illuminating microbial dark matter in meromictic Sakinaw Lake. Appl Environ Microbiol 80:6807–6818. doi: 10.1128/AEM.01774-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peura S, Eiler A, Bertilsson S, Nykänen H, Tiirola M, Jones RM. 2012. Distinct and diverse anaerobic bacterial communities in boreal lakes dominated by candidate division OD1. ISME J 6:1640–1652. doi: 10.1038/ismej.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lang SQ, Butterfield DA, Schulte M, Kelley DS, Lilley MD. 2010. Elevated concentrations of formate, acetate, and dissolved organic carbon found at the Lost City hydrothermal field. Geochim Cosmochim Acta 74:941–952. doi: 10.1016/j.gca.2009.10.045. [DOI] [Google Scholar]
- 47.Brazelton WJ, Nelson B, Schrenk MO. 2012. Metagenomic evidence for H2 oxidation and H2 production by serpentinite-hosted subsurface microbial communities. Front Microbiol 2:268. doi: 10.3389/fmicb.2011.00268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wrighton KC, Thomas BC, Sharon I, Miller CS, Castelle CJ, VerBerkmoes NC, Wilkins MJ, Hettich RL, Lipton MS, Williams KH, Long PE, Banfield JF. 2012. Fermentation, hydrogen, and sulfur metabolism in multiple uncultivated bacterial phyla. Science 337:1661–1665. doi: 10.1126/science.1224041. [DOI] [PubMed] [Google Scholar]
- 49.Rinke C, Patrick S, Sczyrba A, Ivanova NN, Anderson IJ, Cheng JF, Darling A, Malfatti S, Swan BK, Gies EA, Dodsworth JA, Hedlund BP, Tsiamis G, Sievert SM, Liu WT, Eisen JA, Hallam SJ, Kyrpides NC, Stepanauskas R, Rubin EM, Hugenholtz P, Woyke T. 2013. Insights into the phylogeny and coding potential of microbial dark matter. Nature 499:431–437. doi: 10.1038/nature12352. [DOI] [PubMed] [Google Scholar]
- 50.Glöckner J, Kube M, Shrestha PM, Weber M, Glöckner FO, Reinhardt R, Liesack W. 2010. Phylogenetic diversity and metagenomics of candidate division OP3. Environ Microbiol 12:1218–1229. doi: 10.1111/j.1462-2920.2010.02164.x. [DOI] [PubMed] [Google Scholar]
- 51.Nakagawa S, Takai K, Inagaki F, Hirayama H, Nunoura T, Horikoshi K, Sako Y. 2005. Distribution, phylogenetic diversity and physiological characteristics of epsilon-Proteobacteria in a deep-sea hydrothermal field. Environ Microbiol 7:1619–1632. doi: 10.1111/j.1462-2920.2005.00856.x. [DOI] [PubMed] [Google Scholar]
- 52.Qian PY, Wang Y, Lee OO, Lau SCK, Yang J, Lafi FF, Suwailem AA, Wong TYH. 2011. Vertical stratification of microbial communities in the Red Sea revealed by 16S rDNA pyrosequencing. ISME J 5:507–518. doi: 10.1038/ismej.2010.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huse SM, Welch DBM, Voorhis A, Shipunova A, Morrison HG, Eren AM, Sogin ML. 2014. VAMPS: a website for visualization and analysis of microbial population structures. BMC Bioinformatics 15:41. doi: 10.1186/1471-2105-15-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Youssef N, Elshahed MS, McInerny MJ. 2009. Microbial processes in oil fields: culprits, problems, and opportunities. Adv Appl Microbiol 66:141–251. doi: 10.1016/S0065-2164(08)00806-X. [DOI] [PubMed] [Google Scholar]
- 55.King JK, Kostka JE, Frischer ME, Saunders FM, Jahnke RA. 2001. A quantitative relationship that demonstrates mercury methylation rates in marine sediments are based on the community composition and activity of sulfate-reducing bacteria. Environ Sci Technol 35:2491–2496. doi: 10.1021/es001813q. [DOI] [PubMed] [Google Scholar]
- 56.Derrien M, Cho J, Hedlund BP. 2011. Family I. Victivallaceae fam. nov. The Bacteroidetes, Spirochaetes, Tenericutes (Mollicutes), Acidobacteria, Fibrobacteres, Fusobacteria, Dictyoglomi, Gemmatimonadetes, Lentisphaerae, Verrucomicrobia, Chlamydiae, and Planctomycetes, p 791 In Krieg NR, Staley JT, Brown DR, Hedlund BP, Paster BJ, Ward NL, Ludwig W, Whitman WB (ed), Bergey's manual of systematic bacteriology, 4th ed, vol 4 Springer, New York, NY. [Google Scholar]
- 57.Lücker S, Wagner M, Maixner F, Pelletier E, Koch H, Vacherie B, Rattei T, Damste JS, Spieck E, Le Paslier D, Daims H. 2010. A Nitrospira metagenome illuminates the physiology and evolution of globally important nitrite-oxidizing bacteria. Proc Nat Acad Sci U S A 107:13479–13484. doi: 10.1073/pnas.1003860107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Khan ST, Horiba Y, Yamamoto M, Hiraishi A. 2002. Members of the family Comamonadaceae as primary poly(3-hydroxybutyrate-co-3-hydroxyvalerate)-degrading denitrifiers in activated sludge as revealed by a polyphasic approach. Appl Environ Microbiol 68:3206–3214. doi: 10.1128/AEM.68.7.3206-3214.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Takeuchi M, Hamana K, Hiraishi A. 2001. Proposal of the genus Sphingomonas sensu stricto and three new genera, Sphingobium, Novosphingobium, and Sphingopyxis, on the basis of phylogenetic and chemotaxonomic analyses. Int J Syst Evol Microbiol 51:1405–1417. [DOI] [PubMed] [Google Scholar]
- 60.Kämpfer P. 2011. Order I. Sphingobacteriales ord. nov., p 330 In Krieg NR, Staley JT, Brown DR, Hedlund BP, Paster BJ, Ward NL, Ludwig W, Whitman WB (ed), The Bacteroidetes Spirochaetes, Tenericutes (Mollicutes), Acidobacteria, Fibrobacteres, Fusobacteria, Dictyoglomi, Gemmatimonadetes, Lentisphaerae, Verrucomicrobia, Chlamydiae, and Planctomycetes. Bergey's manual of systematic bacteriology, 4th ed, vol 4 Springer, New York, NY. [Google Scholar]