

Abstract
During the last decades, a wide range of fluorescent proteins (FPs) have been developed and improved. This has had a great impact on the possibilities in biological imaging and the investigation of cellular processes at the single-cell level. Recently, we have benchmarked a set of green fluorescent proteins (GFPs) and generated a codon-optimized superfolder GFP for efficient use in the important human pathogen Streptococcus pneumoniae and other low-GC Gram-positive bacteria. In the present work, we constructed and compared four red fluorescent proteins (RFPs) in S. pneumoniae. Two orange-red variants, mOrange2 and TagRFP, and two far-red FPs, mKate2 and mCherry, were codon optimized and examined by fluorescence microscopy and plate reader assays. Notably, protein fusions of the RFPs to FtsZ were constructed by direct transformation of linear Gibson assembly (isothermal assembly) products, a method that speeds up the strain construction process significantly. Our data show that mCherry is the fastest-maturing RFP in S. pneumoniae and is best suited for studying gene expression, while mKate2 and TagRFP are more stable and are the preferred choices for protein localization studies. The RFPs described here will be useful for cell biology studies that require multicolor labeling in S. pneumoniae and related organisms.
INTRODUCTION
Streptococcus pneumoniae is a human pathogen that annually kills about 1.5 million people worldwide (1). It causes harmful infections such as pneumonia, meningitis, and sepsis, especially in elderly people, young children, and immunosuppressed patients. The increase of antibiotic resistance and the lack of long-lasting vaccines against this organism make it crucial to better understand the cell biology of S. pneumoniae and gain insights into essential processes such as cell division, chromosome segregation, and cell wall synthesis. Therefore, the development of tools for cell biological studies in this organism is important.
Studies of protein localization and gene expression at the single cell level have brought new insights into the cell biology of bacteria. Nevertheless, most of our knowledge on bacterial cell biology is based on experiments performed in rod-shaped model organisms such as Escherichia coli and Bacillus subtilis, and only recently have researchers been able to examine these processes in coccoid bacteria such as Staphylococcus aureus and S. pneumoniae (2).
S. pneumoniae is a microaerophilic organism that cannot tolerate high levels of oxygen. Folding and maturation of fluorescent proteins often require sufficient concentrations of oxygen, and microaerophilic conditions might have a negative effect on maturation (3). More recently, molecular tools to study cell biology of living cells of S. pneumoniae have been published (4–6), and a set of green fluorescent proteins (GFPs) that can be used in S. pneumoniae with adequate efficiency has been described (6).
The GFP from the jellyfish Aequorea victoria was the first FP to be described (7) and cloned (8, 9). Other FP variants, such as blue, cyan, and yellow FPs, have been engineered from the jellyfish-derived GFP, whereas orange, red, and far-red variants were found in other sea animals (10–12). Most red fluorescent proteins (RFPs) naturally occur in tetrameric form, and monomerization mutants often resulted in a great loss of photostability and fluorescence intensities. However, in recent years, improved variants have been developed (11, 13). The four different RFPs that we benchmarked in this study were codon optimized for low-GC Gram-positive bacteria. We chose four monomeric RFPs that can possibly be used in protein fusions. The orange-red fluorescent protein we tested, mOrange2, is a monomeric derivative of DsRed from the coral Dictiosoma sp. with improved photostability (11). TagRFP, a monomeric derivate of eqFP578 from the sea anemone Entacmaea quadricolor (13), is an orange-red FP for which high brightness but low photostability has been reported (13). For far-red variants, we tested mCherry (10), another derivative of DsRed and one of the most widely used RFPs in bacterial research, and mKate2, derived from E. quadricolor and with excellent pH resistance and photostability (12). While the four selected RFPs share a high degree of sequence and structural similarity, mKate2 has the highest reported quantum yield when examined in vitro (12). To identify which of the four RFPs is the brightest and most rapidly maturing variant in vivo in S. pneumoniae, we placed the respective genes under control of inducible promoters PZn and PssbB and integrated them stably as a single copy within the S. pneumoniae genome. The speed of FP maturation is not always important, for instance, when the question is simply where the protein localizes. In such cases, brightness or functionality of the FP fusion might be more crucial. Therefore, we tested the properties of TagRFP, mCherry, and mKate2 when used as C-terminal protein fusions to the tubulin-like protein FtsZ and the histone-like protein HlpA. We successfully used Gibson's one-step isothermal (ISO) DNA assembly method (14) to generate several of our constructs. Importantly, using S. pneumoniae's natural competence, we show that pneumococcus can take up and integrate the linear assembly product even directly from the reaction mixture. The RFPs and the use of a fast DNA assembly method (Gibson assembly) for direct transformation presented in this study further expand the cell biological toolbox available for S. pneumoniae and related organisms.
MATERIALS AND METHODS
Bacterial strains, plasmids, media, and growth conditions.
All strains and plasmids used in the study are listed in Table 1. Streptococcus pneumoniae was grown in liquid C+Y medium at 37°C (15). Blood agar plates were prepared from Columbia agar with addition of 3% defibrinated sheep blood (Johnny Rottier, Kloosterzande, The Netherlands). The PZn promoter was induced with addition of ZnCl2 to a final concentration of 0.1 mM or 0.15 mM, and PssbB was induced by adding 100 ng/ml of competence-stimulating peptide (CSP). Escherichia coli DH5α was used for cloning and grown in LB medium at 37°C with shaking or on LB medium solidified with 1.5% (wt/vol) agar. When required, 100 μg ml−1 ampicillin (Amp) for E. coli or 1 μg ml−1 tetracycline (Tet) and 4.5 μg ml−1 chloramphenicol (Cm) for S. pneumoniae was used for selection.
TABLE 1.
Bacterial strains and plasmids
| Strain or plasmid | Relevant properties | Source or reference |
|---|---|---|
| E. coli DH5α | F− araD139 Δ(ara-leu)7696 Δ(lac)X74 galU galK hsdR2 mcrA mcrB1 rpsL | Invitrogen, CA |
| S. pneumoniae | ||
| D39 | Encapsulated strain | 25 |
| KB1-69 | D39 Tet bgaA::PZn-mOrange2 | This study |
| KB1-70 | D39 Tet bgaA::PZn-tagRFP | This study |
| KB1-71 | D39 Tet bgaA::PZn-mCherry | This study |
| KB1-72 | D39 Tet bgaA::PZn-mKate2 | This study |
| MK119 | D39 Cam hlpA hlpA-mKate2 | 18 |
| KB1-64 | D39 Cam hlpA-mCherry | This study |
| MK218 | D39 Cam hlpA hlpA-mCherry | This study |
| KB1-65 | D39 Cam hlpA hlpA-tagRFP | This study |
| KB04-03 | D39 Tet bgaA::PssbB-mOrange2 | This study |
| KB04-04 | D39 Tet bgaA::PssbB-tagRFP | This study |
| KB04-05 | D39 Tet bgaA::PssbB-mCherry | This study |
| KB04-06 | D39 Tet bgaA::PssbB-mKate2 | This study |
| DRR27 | D39 Ery ftsZ-mCherry | This study |
| DRR28 | D39 Ery ftsZ-mKate2 | This study |
| DRR54 | D39 Ery ftsZ-tagRFP | This study |
| MK125 | D39 Cam hlpA hlpA-mKate2 Tet bgaA::PZn-gfp-stkP | This study |
| MK289 | D39 Cam hlpA hlpA-mCherry Tet bgaA::PZn-gfp-stkP | This study |
| MK290 | D39 Cam hlpA, hlpA-tagRFP Tet bgaA::PZn-gfp-stkP | This study |
| MK292 | D39 Ery ftsZ-mCherry Tet bgaA::PZn-gfp-stkP | This study |
| MK293 | D39 Ery ftsZ-mKate2 Tet bgaA::PZn-gfp-stkP | This study |
| MK294 | D39 Ery ftsZ-tagRFP Tet bgaA::PZn-gfp-stkP | This study |
| Plasmids | ||
| pKB01_sfgfp(Sp) | bla tet bgaA::PZn-sfgfp(Sp) | 6 |
| pKB01_mOrange2 | bla tet bgaA::PZn-mOrange2(Sp) | This study |
| pKB01_tagRFP | bla tet bgaA::PZn-tagRFP(Sp) | This study |
| pKB01_mCherry | bla tet bgaA::PZn-mCherry(Bs) | This study |
| pKB01_mKate2 | bla tet bgaA::PZn-mKate2(Sp) | This study |
| pKB01-PssbB-mOrange2 | bla tet bgaA PssbB-mOrange2(Sp) | This study |
| pKB01-PssbB-tagRFP | bla tet bgaA PssbB-tagRFP(Sp) | This study |
| pKB01-PssbB-mCherry | bla tet bgaA PssbB-mCherry(Sp) | This study |
| pKB01-PssbB-mKate2 | bla tet bgaA PssbB-mKate2(Sp) | This study |
| pJWV25-stkP | bla tet bgaA PZn-gfp-stkP | 19 |
| pLA18 | bla tet bgaA::PssbB-luc-gfp | 26 |
Recombinant DNA techniques and oligonucleotides.
Procedures for DNA isolation, restriction, ligation, agarose gel electrophoresis, and transformation of E. coli were performed as described by Sambrook and Russell (16). Plasmid DNA and PCR products were isolated and purified using the High Pure plasmid isolation kit (Roche Applied Science, Mannheim, Germany) according to the manufacturer's instructions. Enzymes were purchased from Fermentas (Vilnius, Lithuania) and used as described by the manufacturer. For PCR amplification, Phusion and Taq DNA polymerases (Fermentas) were used. S. pneumoniae was transformed as described by Martin et al. (15). The oligonucleotides used in this study are listed in Table 2 and were purchased from Biolegio (Nijmegen, The Netherlands). Constructs were verified by sequencing.
TABLE 2.
Oligonucleotides used in this study
| Oligonucleotide | Sequence (5′→3′)a |
|---|---|
| mOrange-F+NotI | GCAGGCGGCCGCGGAGGAAAATTAATGTCTAAGGGAG |
| mOrange-R+SpeI | GCAGACTAGTCTCATGAATTCTTTCTCGAGTTAGG |
| mKate2-F+XbaI | GCAGTCTAGAGGAGGAAAATTAATGTCAGAACTTATCAAGG |
| mKate2-R+SpeI | GCAGACTAGTGCGAGATCTCATGAATTCTTTCTCG |
| tagRFP-F+XbaI | GCAGTCTAGAGGAGGAAAATTAATGTCAGAAC |
| tagRFP-R+SpeI | GCAGACTAGTCATGAATTCTTTCTCGAGTTAGGATCC |
| hlpA-up-F | AACAAGTCAGCCACCTGTAG |
| hlpA-up-R+BamHI | CTGCGGATCCTTTAACAGCGTCTTTAAGAGCTTTACCAGC |
| camR-up-F+EcoRI | ACTCGGAATTCTAATGAGCACTAGTAGGAGGCATATC |
| hlpA-down-R | CGTGGCTGACGATAATGAGG |
| Linker-mCherry-F+BamHI | CGATGGATCCGGATCTGGTGGAGAAGCTGCAGCTAAAGGAAGCAAAGGAGAAGAAGATAACATGGCAATCATC |
| mCherry-R+EcoRI | GCGCGAATTCTTATTATTTGTAAAGCTCATCCATTCCGCC |
| hlpA-F-rbs-SphI | CGTGCATGCTGGAGGAATCATTAACATGGCA |
| hlpA-R-SphI | CGCGCATGCAGACTGATTATTTAACAGCGTC |
| hlpA-linker-up-R+BglII | GCGCAGATCTTCCTTTAGCTGCAGCTTCTCC |
| mKate2-tagRFP-F+BamHI | CGCTGGATCCTCAGAACTTATCAAGG |
| tagRFP-R+EcoRI | GCATGAATTCTTATTAACGGTGTCCCAATTTACTAG |
| integration 1 | CTTGATGAAACCTACATTTG |
| integration 2 | GCTTCCATTAAGGATAGTTC |
| integration 5 | GCTATCGCTGAGCGCCGG |
| integration 6 | AGCTAGAGTTCCGCAATTGG |
| FtsK-down-RV | ATCAAAACCGAACTCACCCTGTTGAT |
| ftsZ-down_FW_eryOE | GGGAGGAAATAAGCGGCCGATGAATGTAAAAGAAAATACAGAACTTG |
| ftsZ-up-FW | CCTGTTATTGCTCGTATCGCCAAA |
| ftsZ-up-RV-mCherryOE | GCTTCTCCACCAGATCCGGAACGATTTTTGAAAAATGGAGGTG |
| ery-FW-mcherryOE | GAATGGATGAGCTTTACAAATAATAAGCCCGGAGGAATTTTCATATGAAC |
| ery-RV-ftszdOE | CAAGTTCTGTATTTTCTTTTACATTCATCGGCCGCTTATTTCCTCCC |
| ery_F_mKateOE | GTAAATTGGGACACCGTTAATAACCCGGAGGAATTTTCATATGAAC |
| mKate_RV_eryOE | GTTCATATGAAAATTCCTCCGGGTTATTAACGGTGTCCCAATTTAC |
| tagRFP_RV_eryOE | GTTCATATGAAAATTCCTCCGGGCTAGTTTATTTATGTCCCAATTTAC |
| ery_F_tagRFPOE | GTAAATTGGGACATAAATAAACTAGCCCGGAGGAATTTTCATATGAAC |
| FtsZ_checkRV | AGTGGTGCCGATATGG |
| FtsZ_checkFW | TGACAGCGAAGGCTAC |
| mCherry-RV-eryOE | GTTCATATGAAAATTCCTCCGGGCTTATTATTTGTAAAGCTCATCCATTC |
| mCherry-FW-ftszuOE | CACCTCCATTTTTCAAAAATCGTTCCGGATCTGGTGGAGAAGC |
Underlined sequences represent the endonuclease restriction sites; italic sequences represent the overlapping sequences for Gibson assembly.
Codon optimization.
mKate2, tagRFP, and mOrange2 were codon optimized for S. pneumoniae using OptimumGene (GenScript Inc., Piscataway, NJ, USA), resulting in genes mKate2(Sp), tagRFP(Sp), and mOrange2(Sp), while the sequence for a codon-harmonized variant of mCherry (based on the Bacillus subtilis codon usage table) was obtained from DSM Biotechnology Center (Delft, The Netherlands), resulting in gene mCherry(Bs). Genes were synthesized by GenScript [mKate2(Sp), tagRFP(Sp), and mOrange2(Sp)] or by GeneArt (Regensburg, Germany) [mCherry(Bs)]. Note that throughout this paper we refer to these codon-optimized genes as mKate2, tagRFP, mOrange2, and mCherry.
Construction of promoter fusion plasmids.
Different RFP genes were first fused to the Zn2+-inducible promoter PZn, resulting in construct pKB01-PZn-RFP. mKate2 and tagRFP were amplified from the synthetic genes using primer pairs mKate-F+XbaI/mKate-R-SpeI and tagRFP-F+XbaI/tagRFP-R-SpeI, respectively, and cut using XbaI and SpeI. mCherry was cut from the GeneArt plasmid pMA-mCherry using XbaI and SpeI and purified. Digested mKate2, tagRFP, and mCherry were ligated in XbaI- and SpeI-cut plasmid pJWV102 (6), resulting in plasmids pKB01-mKate2, pKB01-tagRFP, and pKB01-mCherry, respectively. mOrange2 was amplified using mOrange-F+NotI/mOrange-R+SpeI, cut using NotI and SpeI, and ligated in similarly digested pJWV102. Fragments were ligated accordingly, resulting in plasmid pKB01-mOrange2.
Subsequently, the zinc-inducible promoter was replaced by the competence-induced promoter PssbB (17), resulting in plasmid pKB01-PssbB-RFP, which was excised from pLA18 using SphI and EcoRI and introduced into pKB01-RFP plasmids cut with the same restriction enzymes.
Construction of strains with PZn- and PssbB-controlled RFP gene expression.
S. pneumoniae strains KB1-69, KB1-70, KB1-71, and KB1-72, expressing different RFP variants under control of the Zn2+-inducible promoter (PZn), were obtained by transformation of wild-type strain D39 with pKB01 derivatives. Correct integration by double crossover in the bgaA locus was tested by colony PCR using primer pairs integration 1/integration 2 and integration 5/integration 6.
Similarly, strains KB04-03, KB04-04, KB04-05, KB04-06, expressing RFPs from the competence-activated promoter PssbB, were obtained by transforming the corresponding plasmids into D39. Correct integration in the bgaA locus was checked as described above.
Construction of strains expressing translational HlpA-RFP fusions.
The construction of strain MK119 (hlpA hlpA-mKate2; Camr), containing an hlpA-mKate2 fusion (in which hlpA and mKate2 is separated by a flexible linker) and a chloramphenicol resistance gene on the same transcriptional unit as hlpA, is described elsewhere (18).
To construct strain KB1-64 (D39 hlpA-mCherry; Camr), the hlpA-up fragment (containing the hlpA gene and its promoter) was amplified from D39 chromosomal DNA using hlpA-up-F/hlpA-up-R+BamHI, and the hlpA-down fragment (containing ∼1,000 bp immediately downstream of the hlpA stop codon, including a chloramphenicol resistance marker) was amplified from strain MK119 (D39 hlpA hlpA-mKate2; Camr) using primer pair camR-up-F+EcoRI/hlpA-down-R. mCherry including a flexible linker was amplified from pMA-mCherry using linker-mCherry-F+BamHI/mCherry-R+EcoRI. Fragments were purified and cut using BamHI (fragment hlpA-up), BamHI and EcoRI (linker-mCherry), or EcoRI (hlpA-down). Cut and purified fragments were ligated. S. pneumoniae D39 was transformed with the ligation mix, and transformants were selected on plates containing chloramphenicol. A single transformant was sequence verified, and this strain was named KB1-64.
To construct strain MK218 (D39 hlpA hlpA-mCherry; Camr), an hlpA-up fragment (containing hlpA and the upstream sequence) was amplified from genomic DNA of D39 using primers hlpA-up-F and hlpA-R-SphI. The hlpA-mCherry; Camr fragment (containing the hlpA-mCherry fusion with a downstream; Camr resistance gene and the hlpA downstream sequence) was amplified from strain KB1-64 using primers hlpA-F-rbs-SphI and hlpA-down-R. The two fragments were digested with SphI and ligated. The ligation product was transformed into D39, and transformants were selected on plates containing chloramphenicol. Transformants were verified by PCR and sequencing.
To construct strain KB1-65 (D39 hlpA hlpA-tagRFP; Camr), the hlpA-linker-up fragment, containing an extra copy of hlpA including a flexible linker downstream of hlpA, was amplified from MK119 using primer pair hlpA-up-F/hlpA-linker-R+BglII, and the hlpA-down fragment was amplified as described for KB1-64. tagRFP was amplified from pUC57-tagRFP using primers mKate2-tagRFP-F+BamHI/tagRFP-R+EcoRI. Fragments were purified and cut with BglII (hlpA-linker-up), EcoRI (hlpA-down), and BamHI and EcoRI (tagRFP). The fragments were ligated and directly transformed into S. pneumoniae D39. Positive transformants were selected on plates containing chloramphenicol. A single transformant was sequence verified, and the strain was named KB1-65.
Gibson assembly for construction of FtsZ-RFP strains.
Strains expressing the different FtsZ-RFP fusions were constructed by direct transformation of linear Gibson assembly products. The parts for strain DRR27 were assembled using Gibson's one-step ISO assembly of overlapping double-stranded DNA (dsDNA) (14). The ftsZ-up and ftsZ-down fragments were amplified from chromosomal DNA using ftsZ-up-FW and ftsZ-up-RV-mCherryOE for ftsZ-up and ftsZ-down-RV and ftsZ-down_FW_eryOE for ftsZ-down. The fragment encoding erythromycin resistance was amplified from plasmid JWV502 using ery-FW-mcherryOE and ery-RV-ftszdOE. mCherry including a flexible linker was amplified from strain KB1-64 using primers mcherry-RV-eryOE and mcherry-FW-ftszuOE. All four parts included a sequence of around 20 bp (Table 2) that overlapped with the next part. Gibson one-step ISO assembly was done as described by Gibson et al. (14), and 10 μl of the reaction mixture was directly transformed into Streptococcus pneumoniae D39.
For DRR28 and DRR54, ery-ftsZdown was amplified from strain DRR27 using ery-F-mKateOE and ftsZ-down-RV for DRR28 and ery-F-tagRFPOE and ftsZ-down-RV for DRR54. mKate2 was amplified from strain MK119 using mcherry-FW-ftszuOE and mKate_RV_eryOE, and TagRFP was amplified from strain KB1-65 using mcherry-FW-ftszuOE and tagRFP_RV_eryOE. FtsZ-up was amplified the same way as for DRR27. For DRR28, ery-ftsZdown (with overlap with mKate2), ftsZ-down, and mKate2 and for DRR54, ery-ftsZdown (with overlap with TagRFP), ftsZ-down, and tagRFP were combined in equimolar amounts for Gibson assembly and directly transformed into S. pneumoniae D39. Integration via double crossover was checked in all three strains using primers FtsZ-checkRV and FtsZ-checkFW.
Construction of RFP/GFP-double-labeled strains.
Double-labeled strains GFP-StkP and FtsZ-RFP were made by transformation of plasmid pJWV25-stkP (19) into the FtsZ-RFP fusion strains DRR27, DRR28, and DRR54, to generate strains MK292 (FtsZ-mCherry, GFP-StkP), MK293 (FtsZ-mKate2, GFP-StkP), and MK293 (FtsZ-TagRFP, GFP-StkP). Double-labeled strains GFP-StkP and HlpA-RFP were made in a similar manner by transformation of plasmid pJWV25-stkP into HlpA-RFP fusion strains to generate MK125 (HlpA-mKate2, GFP-StkP), MK289 (HlpA-mCherry, GFP-StkP), and MK290 (HlpA-TagRFP, GFP-StkP).
Fluorescence measurements using a plate reader.
S. pneumoniae RFP gene-expressing strains and the D39 wild-type strain were grown at 37°C in liquid C+Y medium to an optical density at 600 nm (OD600) of 0.1 and diluted 1:100 with fresh C+Y containing 0.15 mM ZnCl2 for induction of PZn. Cells were then further grown and harvested at an OD600 of 0.15. For plate reader assays, cells were first collected by centrifugation and washed with phosphate-buffered saline (PBS). Fluorescence was measured with the following equipment and settings: Infinite 200 Pro plate reader (Tecan Group Ltd.) with I-control 1.7.1.12 software (Tecan Group Ltd.), RFP filter set (excitation at 590 nm and emission at 635 nm with a 50% dichroic mirror), and RFP signals collected as top readings with a gain setting of 100. RFP fluorescence values were corrected for background fluorescence, OD600, and negative controls (values for the wild-type strains). The OD600 levels used were corrected for the background value of the corresponding buffer.
Fluorescence microscopy.
Cells of S. pneumoniae were grown at 37°C in liquid C+Y medium to an OD600 of 0.1, diluted 1:100 with fresh C+Y, and further grown to an OD600 of 0.05 before 0.15 mM ZnCl2 was added to the cultures for induction of PZn. Cells were then further grown and collected at different time points. For microscopy, 0.4 μl of the cell suspension was directly spotted onto a microscope slide carrying a thin layer of 1.2% agarose in PBS covered with a coverslip. Images were taken with an Olympus IX71 microscope (Personal DV, Applied Precision; assembled by Imsol [Preston, United Kingdom] and Chromaphor [Münster, Germany]) using a CoolSNAP HQ2 camera (Princeton Instruments, Trenton, NJ, USA) with a 100× phase-contrast objective and Trulight (Deltavision, GE Healthcare, USA) illumination. The following standard fluorescence filter sets from Chroma Technology Corporation (Bellows Falls, VT, USA) were used to visualize RFPs: for mCherry, excitation at 575/25 nm, emission at 632/60 nm, and polychroic mirror QUAD2 (580 to 630 nm); for tetramethyl rhodamine isocyanate (TRITC), excitation at 542/27 nm, emission at 594/45 nm, and polychroic QUAD1 (550 to 630 nm). (Note that the second number indicates the bandwidth around the wavelength; e.g., 575/25 indicates excitation with light from 562.5 to 587.5 nm). For the Zn2+-inducible constructs, the exposure time was 1.4 s with 100% excitation for all RFPs. To visualize the RFP fusions to HlpA, an exposure time of 0.9 s was used. Softworx 3.6.0 (Applied Precision, Issaquah, WA, USA) software was used for image capturing. Phase-contrast images were segmented automatically, and fluorescence signal intensities were extracted using Microbetracker (20). Fluorescence levels were corrected for background fluorescence of the medium.
Western blot analysis and immunodetection.
Cells were grown in C+Y liquid medium at 37°C to an OD600 of 0.1, diluted 1:100 in C+Y containing 0.15 mM ZnCl2 for induction, and harvested at an OD600 of 0.15. Cells were harvested by centrifugation at 8,000 rpm for 10 min. For lysis, the pellet was resuspended in 100 μl of SEDS lysis buffer (150 mM NaCl, 15 mM EDTA, 0.02% SDS, 0.01% deoxycholate) and was incubated for 5 min at 37°C. Lysates were diluted in 100 μl 2× SDS loading buffer and boiled for 5 min. After separation by SDS-polyacrylamide gel electrophoresis (SDS-PAGE), proteins were transferred to a polyvinylidene difluoride (PVDF) membrane by Western blotting. Red fluorescent proteins were detected with polyclonal anti-RFP antibody (Molecular Probes) and rabbit anti-IgG–horseradish peroxidase (HRP) antibody (GE Healthcare) according to the manufacturer's instructions. Note that the polyclonal anti-RFP antibodies recognize all four RFPs due to the high structural homology.
Nucleotide sequence accession numbers.
The sequences of the mKate2, tagRFP, mOrange2, and mCherry genes have been deposited in GenBank under accession numbers KJ908191, KJ908188, KJ908189, and KJ908190, respectively.
RESULTS AND DISCUSSION
Expression of codon-optimized RFPs in S. pneumoniae.
New variants of monomeric red fluorescent protein have been discovered and developed in recent years. They are important tools for use in protein fusions to study protein localization and dynamics and for multicolor labeling, for instance, in combination with GFP, cyan fluorescent protein (CFP), or yellow fluorescent protein (YFP). In the past, the brightness of monomeric RFPs has been problematic, but more recently, improved variants have been described. We selected four of these “next-generation” variants of red fluorescent proteins and codon optimized the genes for low-GC Gram-positive bacteria (Table 3), including those for mOrange2 and mCherry from the “mFruits” palette (11), as well as TagRFP (13) and mKate2 (12). mCherry is one of the most commonly used RFPs, with a reported short maturation time (varying between 15 min [11] and 40 min [13]) but a relatively poor photostability (11). mOrange2 is an orange-red-emitting FP variant and was reported to have a high photostability and high intrinsic brightness but a relatively long maturation time (∼4.5 h) compared to the doubling time of S. pneumoniae (∼30 min under our experimental conditions) (11). TagRFP is a bright orange-red fluorescent protein with a reported maturation time of 100 min and high brightness (Table 3) (11, 13). mKate2 was also reported to have a short maturation time (<20 min) and good photostability (Table 3) (12). mKate2 and mCherry are far-red variants and therefore highly suitable for the use in combination with green and yellow fluorescent proteins. All variants are reported to be monomeric which makes them suitable for use in protein fusions.
TABLE 3.
Red fluorescent proteins
| Fluorescent protein | Maximum λex (nm) | Maximum λem (nm) | Color | Brightnessa | Reference |
|---|---|---|---|---|---|
| mOrange2 | 549 | 565 | Orange | 35 | 11 |
| TagRFP | 555 | 584 | Red | 40 | 13 |
| mCherry | 587 | 610 | Far red | 16 | 11 |
| mKate2 | 588 | 633 | Far red | 25 | 12 |
Brightness was determined as follows: (extinction coefficient × quantum yield)/1,000.
In order to directly compare the four RFPs (mOrange2, TagRFP, mCherry, and mKate2), we used plasmid pJWV102 (6), a pPP2 derivative (21) containing the Zn2+-inducible promoter PZn (4) or competence-inducible promoter PssbB (17), that integrates in the chromosome by double crossover at the nonessential bgaA locus. To exclude translational differences, the same ribosome binding site (RBS) was used for all RFPs. This allows for evaluation and unbiased comparison of the fluorescence intensities and folding times of the individual RFPs. A schematic presentation of chromosomally recombined RFP gene-carrying vectors is shown in Fig. 1.
FIG 1.
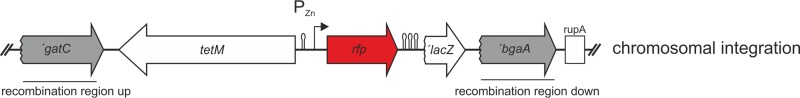
Inducible RFP expression in Streptococcus pneumoniae. The scheme for RFP gene expression strains after integration of the PZn-rfp promoter fusion at the nonessential bgaA locus by double crossover is shown. Strains were constructed by transformation with plasmids pKB01-PZn-rfp that carry PZn-rfp (mCherry, mOrange2, tagRFP, or mKate2) and a tetracycline resistance marker between the homologous recombination sites (shown in gray; ′gatC and ′bgaA). Lollipops depict transcriptional terminators. Similar constructs where the PZn promoter was replaced with the PssbB promoter were also made (see Fig. S1 in the supplemental material).
Comparison of RFP intensities at the population and single-cell levels.
The intensities of the different RFPs were analyzed at the population level using a plate reader and at the single-cell level using epifluorescence microscopy. First, to check whether all RFPs were expressed under our experimental conditions, we performed immunodetection on whole-cell lysates using an anti-RFP polyclonal antibody (Molecular Probes, Life Technologies). Cells were grown to exponential phase in the presence of Zn2+ for induction, and immunodetection was performed on whole-cell extracts, which allows for only relative quantification of protein levels. All fluorescent proteins were efficiently produced in S. pneumoniae, and only mCherry showed signs of protein degradation (Fig. 2A).
FIG 2.

Fluorescence intensities of the different red fluorescent proteins. (A) Western blot analysis of expression levels of RFPs in whole-cell extracts detected using anti-RFP antibody. (B) Mean fluorescence on the population level determined using a plate reader equipped with a standard RFP filter set. See Materials and Methods for details of the filters used. (C and D)Average fluorescence intensities on the single-cell level measured by fluorescence microscopy using an mCherry filter set and polychroic mirror QUAD2 (C) or a TRITC filter set and polychroic mirror QUAD1 (D). Values are in arbitrary units (A.U.). Single-cell fluorescence levels were determined using MicrobeTracker (20). Error bars depict the standard error of the mean. mOrange2 values are not shown in the plots since signals above background level could not be detected.
Fluorescence measurements with the plate reader were also performed on Zn2+-induced cells in exponential phase which were resuspended in PBS. The plate reader we used contains a standard RFP filter set (excitation [λex], 590 nm; emission [λem], 635 nm) which is best suitable for the far-red FPs. Not surprisingly, the signals for cells expressing TagRFP and mOrange2 hardly appeared above background. Interestingly, mCherry had the highest signal-to-noise ratio, with approximately 6-fold higher fluorescence than mKate2 (Fig. 2B), while mKate2 was reported to be brighter in vitro (12).
For fluorescence microscopy, cells were grown to an OD600 of 0.05 before expression of RFPs was induced with 0.15 mM ZnCl2. Cells were then harvested for microscopy at different time points. We used filter sets that are standard in most fluorescence microscopes: mCherry (λex, 575/25 nm; λem, 632/60 nm; QUAD 2 polychroic [580 to 630 nm]) and TRITC (λex, 542/27 nm; λem, 594/45 nm; QUAD 1 polychroic [550 to 630 nm]). The same light output and exposure times were used for all samples. Using the mCherry filter set, signals from mCherry and mKate2 were already visible after 30 min of induction (Fig. 2C). After 120 min, TagRFP also showed good fluorescence, similar in strength to that of mKate2 but still approximately 6 times less bright than mCherry (Fig. 2C). With these imaging conditions, signals for mOrange2 could not be detected. When the TRITC filter set was applied, which should be better suited for mOrange2 and TagRFP than for the far-red RFPs, mCherry still outperformed the other RFPs (Fig. 2D). With this filter set, TagRFP gave a good signal above background, with approximately half of that of mCherry for the different time points, while the signal from mKate2 was consistently very low, and mOrange2 still gave no signals above background levels after 3.5 h.
To rule out that fluorescence intensity differences depended specifically on the Zn2+ regulation, we swapped the zinc-inducible promoter PZn with the competence-induced promoter PssbB (17) and measured fluorescence intensities after 1.5 h of induction with the competence-stimulating peptide (CSP) using the same filter sets (see Fig. S1 in the supplemental material). The results are consistent with the ones we obtained for the zinc induction after 1.5 h. It should be noted that the TRITC filter set and the QUAD1 polychroic mirror might block a large amount of the emitted photons from mOrange2. Also, the slow fluorophore maturation time of mOrange2 (11) might be a probable explanation for the absence of signal and thus makes this FP unsuitable for imaging in S. pneumoniae. TagRFP gives appreciable signals with a standard TRITC filter set and is a good orange-red RFP for use in S. pneumoniae (Fig. 2D), but it might be less suitable in multicolor experiments that include yellow fluorescent proteins due to possible spectral overlap. In total, these results show that mCherry is the fastest-maturing RFP in S. pneumoniae with the highest fluorescence signal when used in promoter fusions. This is probably not simply because of more efficient protein production, since all four RFPs seem to be produced to approximate similar levels using qualitative Western blotting (Fig. 2A).
Fast assembly of protein fusion constructs by Gibson assembly.
In previous studies, mCherry has been used to tag S. pneumoniae FtsZ (22) and the capsule protein Wze (CpsD) (5), while mKate2 has been used to tag the eukaryotic-type serine/threonine kinase StkP (19) and also FtsZ (23). However, a systematic comparison of how different RFPs perform when fused to proteins for localization studies has not been made.
In order to compare the performances of different RFPs in protein fusions, the far-red and fast-folding mCherry and mKate2, as well as the red-orange TagRFP, were all fused to the C termini of FtsZ and HlpA (Hup, SPD_0997). TagRFP has been suggested as a suitable FP for protein fusions but, unlike mCherry and mKate2, has not yet been used for this purpose in S. pneumoniae. mOrange was not included in this part of the study, since we did not observe any signal when this FP was expressed alone (Fig. 2).
While fusions to hlpA were constructed by conventional methods (see Materials and Methods), the FtsZ-RFP constructs were all made using Gibson's one-step ISO assembly of overlapping dsDNA (Gibson assembly) (14) (Fig. 3A). With this method, the four different parts of the construct were amplified with 40-bp overlaps, and the different parts could then be fused without any restriction or ligation steps. Since no restriction sites are needed, it is possible to choose the optimal sequence for protein linkage. Importantly, as S. pneumoniae readily takes up linear DNA fragments via the competence pathway, we wondered whether we could directly use the Gibson assembly product, which is assembled in 1 h, to transform S. pneumoniae without any intermediate cleanup or amplification steps. Indeed, transformation with unpurified Gibson mixes readily yielded correct transformants. Thus, this direct transformation of assembly products is a fast and efficient method for strain construction and is now the preferred construction method for linear fragments in our laboratory.
FIG 3.

Fast generation of RFP fusions to FtsZ in S. pneumoniae. (A) Gibson assembly procedure (14) for construction of FtsZ-RFP-expressing strains. (B) Images show localization of FtsZ fused to TagRFP, mKate2, and mCherry. Phase-contrast (PC), RFP, and overlay (merge) images are shown. Scale bar, 2 μm. (C) Immunodetection of FtsZ-RFP fusion proteins in whole-cell extract by anti-RFP antibody. Asterisks indicate degradation products.
Characterization of RFPs in a fusion to FtsZ and HlpA.
The highly conserved tubulin-like protein FtsZ polymerizes at the septum during early cell division. The localization pattern of the FtsZ ring is well studied in bacteria, making it an excellent candidate for comparing the performances of the three red fluorescent proteins. HlpA is the only known histone-like protein encoded in the S. pneumoniae D39 genome (24) and binds unspecifically to DNA and thus localizes to the nucleoid in a fashion similar to that for the intercalating DNA dye DAPI (4′,6′-diamidino-2-phenylindole) (18). C-terminal fusions of FtsZ to tagRFP, mCherry, and mKate2 were obtained by Gibson assembly replacing wild-type FtsZ. We also attempted to construct C-terminal fusions of these fluorescent proteins to HlpA by integrating the fusion gene into the native locus. Interestingly, we obtained the hlpA-mCherry fusion strain but were unable make strains expressing the HlpA-mKate2 and HlpA-TagRFP fusions. In our attempts to make these strains, we always observed mutations in the linker sequence generating premature stop codons or deletion of (parts of) the RFP genes. Therefore, we instead constructed merodiploid strains where the hlpA fusions are present as an extra copy immediately downstream of the native hlpA gene and thus also under the control of the endogenous hlpA promoter. These strains could be readily made (Table 1) (see Materials and Methods).
Fluorescence microscopy of cells growing in mid-exponential phase showed that the total amount of fluorescence of both FtsZ and HlpA fused to TagRFP was comparable to that of the fusions with mKate2, whereas signals of the mCherry fusion proteins were lower (data not shown). Signals for the FtsZ-mKate2 and FtsZ-TagRFP strains appeared as sharp defined Z rings at midcell even before deconvolution (Fig. 3B and 4A), whereas FtsZ-mCherry demonstrated a slightly more diffuse and heterogeneous signal (Fig. 3B and 4A). The same was the case for the HlpA-fusions; the meroploid TagRFP and mKate2 fusions appeared as sharp defined nucleoid-localized spots in the cytoplasm (Fig. 4B; see Fig. S2 in the supplemental material), whereas both HlpA-mCherry strains frequently demonstrated a more diffuse and heterogeneous signal (Fig. 4B; see Fig. S2 in the supplemental material).
FIG 4.

Double labeling with RFP and GFP in S. pneumoniae. (A) Localization of FtsZ-RFP (different RFP variants in the different panels) and GFP-StkP. (B) Localization of HlpA-RFP (different RFP variants in the different panels) and GFP-StkP. Phase-contrast (PC), RFP, and GFP images are shown, in addition to an overlay of GFP and RFP (merge). Scale bars, 2 μm.
Western blot analysis of both proteins showed the presence of degradation products in the FtsZ-mCherry strain and in both HlpA-mCherry strains, whereas for fusions with mKate2 and TagRFP, no or only marginal degradation was observed (Fig. 3C; see Fig. S2B in the supplemental material). Degradation of mCherry might lead to a pool of untagged HlpA, which might be the reason why the single copy HlpA-mCherry could be made whereas this was not possible with mKate2 and TagRFP. In total, these results show that mCherry originating from our codon-optimized gene is more readily degraded than mKate2 and TagRFP in S. pneumoniae (Fig. 2A and 3C), making the latter RFPs more suitable for protein fusions. Note that protein functionality always needs to be tested, since FP fusions may not always be fully functional, as probably is the case for the HlpA-mKate2 and HlpA-TagRFP fusions.
Far-red fluorescent proteins are particularly suitable for double-labeling experiments in combination with GFP or YFP due to nonoverlapping excitation and emission spectra and thus limited chance for cross talk. We show that all three RFPs can be successfully used in combination with GFP using our standard filter sets, as exemplified by double-labeled strains showing the localization of two cell division proteins, FtsZ and StkP (Fig. 4A), or localization of the division site relative to the nucleoid (Fig. 4B).
Final remarks.
Here we benchmarked four commonly used RFPs (mOrange2, TagRFP, mCherry, and mKate2) in the important human pathogen S. pneumoniae. The genes encoding these RFPs were codon optimized, and Western blotting showed that all proteins are efficiently produced in S. pneumoniae. Under our experimental conditions, no fluorescent signals were detected with mOrange2, likely because of its long maturation time. The three other RFPs were all reported to be fast folding. However, mCherry clearly was the fastest-folding variant in our assays and generated the highest fluorescence signals, and it is thus the best RFP to use in promoter fusions. On the other hand, mCherry is also partially degraded, making it less suitable in protein fusions. It would be interesting to identify the degradation site in mCherry and mutate it to generate a superior mCherry. However, for the time being, we recommend the use of mKate2 or TagRFP as a red fluorescent localization marker protein in S. pneumoniae. Further, we showed that mKate2, TagRFP, and mCherry are suitable for dual-color imaging using standard filter sets. Finally, we demonstrated that direct transformation of Gibson assembly products could be used as a fast and straightforward cloning method in S. pneumoniae.
Supplementary Material
ACKNOWLEDGMENTS
We thank Amaya Aramendía de Goñi for initial characterization of FPs in S. pneumoniae. We thank Hans Roubos (DSM) for kindly providing the mCherry sequence.
Work in the lab of J.-W.V. is supported by the EMBO Young Investigator Programme, a VIDI fellowship (864.12.001) from the Netherlands Organization for Scientific Research, Earth and Life Sciences (NWO-ALW), and ERC starting grant 337399-PneumoCell.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02033-15.
REFERENCES
- 1.O'Brien KL, Wolfson LJ, Watt JP, Henkle E, Deloria-Knoll M, McCall N, Lee E, Mulholland K, Levine OS, Cherian T. 2009. Burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: global estimates. Lancet 374:893–902. doi: 10.1016/S0140-6736(09)61204-6. [DOI] [PubMed] [Google Scholar]
- 2.Pinho MG, Kjos M, Veening J-W. 2013. How to get (a)round: mechanisms controlling growth and division of coccoid bacteria. Nat Rev Microbiol 11:601–614. doi: 10.1038/nrmicro3088. [DOI] [PubMed] [Google Scholar]
- 3.Tsien RY. 1998. The green fluorescent protein. Annu Rev Biochem 67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- 4.Eberhardt A, Wu LJ, Errington J, Vollmer W, Veening J-W. 2009. Cellular localization of choline-utilization proteins in Streptococcus pneumoniae using novel fluorescent reporter systems. Mol Microbiol 74:395–408. doi: 10.1111/j.1365-2958.2009.06872.x. [DOI] [PubMed] [Google Scholar]
- 5.Henriques MX, Catalão MJ, Figueiredo J, Gomes JP, Filipe SR. 2013. Construction of improved tools for protein localization studies in Streptococcus pneumoniae. PLoS One 8:e55049. doi: 10.1371/journal.pone.0055049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Overkamp W, Beilharz K, Weme RDO, Solopova A, Karsens H, Kovács ÁT, Kok J, Kuipers OP, Veening J-W. 2013. Benchmarking various GFP variants in Bacillus subtilis, Streptococcus pneumoniae and Lactococcus lactis for live cell imaging. Appl Environ Microbiol 79:6481–6490. doi: 10.1128/AEM.02033-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shimomura O, Johnson FH, Saiga Y. 1962. Extraction, purification and properties of aequorin, a bioluminescent protein from the luminous hydromedusan, Aequorea. J Cell Comp Physiol 59:223–239. doi: 10.1002/jcp.1030590302. [DOI] [PubMed] [Google Scholar]
- 8.Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. 1994. Green fluorescent protein as a marker for gene expression. Science 263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- 9.Prasher DC, Eckenrode VK, Ward WW, Prendergast FG, Cormier MJ. 1992. Primary structure of the Aequorea victoria green-fluorescent protein. Gene 111:229–233. doi: 10.1016/0378-1119(92)90691-H. [DOI] [PubMed] [Google Scholar]
- 10.Shaner NC, Campbell RE, Steinbach PA, Giepmans BNG, Palmer AE, Tsien RY. 2004. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol 22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- 11.Shaner NC, Lin MZ, McKeown MR, Steinbach PA, Hazelwood KL, Davidson MW, Tsien RY. 2008. Improving the photostability of bright monomeric orange and red fluorescent proteins. Nat Methods 5:545–551. doi: 10.1038/nmeth.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shcherbo D, Murphy CS, Ermakova GV, Solovieva EA, Chepurnykh TV, Shcheglov AS, Verkhusha VV, Pletnev VZ, Hazelwood KL, Roche PM, Lukyanov S, Zaraisky AG, Davidson MW, Chudakov DM. 2009. Far-red fluorescent tags for protein imaging in living tissues. Biochem J 418:567–574. doi: 10.1042/BJ20081949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Merzlyak EM, Goedhart J, Shcherbo D, Bulina ME, Shcheglov AS, Fradkov AF, Gaintzeva A, Lukyanov KA, Lukyanov S, Gadella TWJ, Chudakov DM. 2007. Bright monomeric red fluorescent protein with an extended fluorescence lifetime. Nat Methods 4:555–557. doi: 10.1038/nmeth1062. [DOI] [PubMed] [Google Scholar]
- 14.Gibson DG. 2011. Enzymatic assembly of overlapping DNA fragments. Methods Enzymol 498:349–361. doi: 10.1016/B978-0-12-385120-8.00015-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin B, Garcia P, Castanié M-P, Claverys J-P. 1995. The recA gene of Streptococcus pneumoniae is part of a competence-induced operon and controls lysogenic induction. Mol Microbiol 15:367–379. doi: 10.1111/j.1365-2958.1995.tb02250.x. [DOI] [PubMed] [Google Scholar]
- 16.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 17.Prudhomme M, Attaiech L, Sanchez G, Martin B, Claverys J-P. 2006. Antibiotic stress induces genetic transformability in the human pathogen Streptococcus pneumoniae. Science 313:89–92. doi: 10.1126/science.1127912. [DOI] [PubMed] [Google Scholar]
- 18.Kjos M, Veening J-W. 2014. Tracking of chromosome dynamics in live Streptococcus pneumoniae reveals that transcription promotes chromosome segregation. Mol Microbiol 91:1088–1105. doi: 10.1111/mmi.12517. [DOI] [PubMed] [Google Scholar]
- 19.Beilharz K, Nováková L, Fadda D, Branny P, Massidda O, Veening J-W. 2012. Control of cell division in Streptococcus pneumoniae by the conserved Ser/Thr protein kinase StkP. Proc Natl Acad Sci U S A 109:E905–E913. doi: 10.1073/pnas.1119172109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sliusarenko O, Heinritz J, Emonet T, Jacobs-Wagner C. 2011. High-throughput, subpixel precision analysis of bacterial morphogenesis and intracellular spatio-temporal dynamics. Mol Microbiol 80:612–627. doi: 10.1111/j.1365-2958.2011.07579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Halfmann A, Hakenbeck R, Brückner R. 2007. A new integrative reporter plasmid for Streptococcus pneumoniae. FEMS Microbiol Lett 268:217–224. doi: 10.1111/j.1574-6968.2006.00584.x. [DOI] [PubMed] [Google Scholar]
- 22.Sham L-T, Barendt SM, Kopecky KE, Winkler ME. 2011. Essential PcsB putative peptidoglycan hydrolase interacts with the essential FtsXSpn cell division protein in Streptococcus pneumoniae D39. Proc Natl Acad Sci U S A 108:E1061–E1069. doi: 10.1073/pnas.1108323108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fleurie A, Manuse S, Zhao C, Campo N, Cluzel C, Lavergne J-P, Freton C, Combet C, Guiral S, Soufi B, Macek B, Kuru E, VanNieuwenhze MS, Brun YV, Di Guilmi A-M, Claverys J-P, Galinier A, Grangeasse C. 2014. Interplay of the serine/threonine-kinase StkP and the paralogs DivIVA and GpsB in pneumococcal cell elongation and division. PLoS Genet 10:e1004275. doi: 10.1371/journal.pgen.1004275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lanie JA, Ng W-L, Kazmierczak KM, Andrzejewski TM, Davidsen TM, Wayne KJ, Tettelin H, Glass JI, Winkler ME. 2007. Genome sequence of Avery's virulent serotype 2 strain D39 of Streptococcus pneumoniae and comparison with that of unencapsulated laboratory strain R6. J Bacteriol 189:38–51. doi: 10.1128/JB.01148-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Avery OT, Macleod CM, McCarty M. 1944. Studies on the chemical nature of the sustance inducing transformation of pneumococcal types: induction of transformation by a desoxyribonucleic acid fraction isolated from pneumococcus type III. J Exp Med 79:137–158. doi: 10.1084/jem.79.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Slager J, Kjos M, Attaiech L, Veening J-W. 2014. Antibiotic-induced replication stress triggers bacterial competence by increasing gene dosage near the origin. Cell 157:395–406. doi: 10.1016/j.cell.2014.01.068. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.