

Abstract
Introduction
Deuterated versions of existing drugs can exhibit improved pharmacokinetic or toxicological properties due the stronger deuterium-carbon bond modifying their metabolism. There is great interest in the current state of development of this approach.
Areas Covered
This review covers recent US patent applications and prosecutions in this area, that are based upon beneficial modifications in metabolism of deuterated versions of existing drugs. The current state of 35 U.S.C. §103 ‘obviousness’ rejections, are emphasized as is the development of strategies to overcome such rejections. Current trials and market considerations are also discussed.
Expert Opinion
Deuterated drugs collectively are worth at least a billion dollars. It would seem that the likelihood of obviousness rejections is increasing in this area. However, careful elucidation of metabolic outcomes from deuteration that would not be anticipated from the prior art, and are instead unexpected and unobvious, has enabled allowance. Showing drug deuteration alters pharmacokinetics by mechanisms not currently part of the prior art surrounding, deuterated drugs has also been successful. Development of these and other strategies, combined with developing the extensive base of issued patents will enable the field to remain commercially attractive for some time.
Keywords: Deuteration, Metabolism, Obviousness, Patent
1. Introduction
The covalent carbon-hydrogen (1H) bond is long known to be substantially weaker than an otherwise identical carbon-deuterium (2H) bond. While this differential bond-strength has many consequences and practical uses, this work will focus upon its utilization in developing new drugs. The breaking of these carbon-hydrogen bonds is a common feature of drug metabolism, for example during oxidation in Phase 1 metabolism. Breaking of an analogous carbon-deuterium bond can be more difficult, and so decrease the rate of metabolism. Therefore, replacement of hydrogen with deuterium in drug molecules can lead to significant alterations in this metabolism and thereby cause beneficial changes in the biological effects of drugs, such as their pharmacokinetics by decreasing their rate of metabolism allowing less frequent dosing. Such replacement may also have the effect of lowering toxicity by reducing the formation of a toxic metabolite. The potential for deuterium replacement to modulate drug metabolism, pharmacokinetics and toxicity has been known for some time, as witnessed by the patents of Reinhold [1] or McCarty [2] being issued in 1976 and 1978 respectively. Furthermore the magnitude of many known deuterium kinetic isotope effects is large, so that clinically significant improvements might be achievable.
Much of the current interest in this space has been driven by the business models of a number of new companies that have invested resources in developing and patenting deuterated versions of a range of existing, non-deuterated therapeutic compounds- the ‘Deuterium Switch’. This model has many advantages and can potentially: 1) avoid many of the costs of preclinical development as much has already been done, 2) benefit from significant bodies of clinical knowledge upon the non-deuterated compound to guide clinical development and lower clinical costs, 3) benefit from new patent protections upon these deuterated compounds, and, 4) lead to improved therapies and patient outcomes. Not surprisingly, there has been a lot of interest in this business model, and most large pharmaceutical companies now also claim deuterated versions of new molecules in their patent applications.
2. 35 U.S.C. §103 Obviousness and the Analogy to Chiral Switching
Although imperfect, a good analogy of the ‘deuterium switch’ is the ‘chiral switch’ in which enantiomerically-enriched versions of existing racemic drugs allowed many of these same benefits as detailed for the ‘deuterium switch’. The ‘chiral switch’ led to issued patents upon the pure enantiomer, companies such as Sepracor founded upon the ‘choral switch’ and such lucrative drugs as Nexium. As the techniques and concepts of enantiomeric enrichment and purification were developed for pharmaceutical use, the approach became increasingly adopted. However, it also became increasingly difficult to obtain patent protection upon new applications through ‘obviousness’ such as is defined in 35 U.S.C. §103 ‘A patent may not be obtained…if the differences between the subject matter sought to be patented and the prior art are such that the subject matter as a whole would have been obvious at the time the invention was made to a person having ordinary skill in the art to which said subject matter pertains.’
Although a field will initially tend to be the preserve of a few innovators working with limited tools and little available literature for guidance, as the field develops then so does the base of prior art and the abilities of a person having ordinary skill in the art. This then leads to rejections due to obviousness arguments as was the case for the ‘chiral switch’. A key question in the field of deuterated drugs is ‘Where is the deuterium switch with respect to §103 obviousness?’ and it is this question that will be analyzed here. The perspective will be that of a PhD practitioner in developing pharmaceutical uses of stable isotopes, and not that of patent examiners or counsel, although legal works are available for analysis. [3]
3. Historical uses of Deuteration
The detailed historical development of deuterated drugs has been reviewed from varying perspectives, to which the reader is directed. [4] However, it is worth noting that long-standing patents, from the major pharmaceutical industry itself, claim the therapeutic use of deuterated drugs. Thus Reinhold [1] described a deuterated fluoroalanine derivate for antimicrobial therapy with improved pharmacokinetic properties [5], while McCarty [2] described the use of deuterated halothane with lowered liver toxicity. Thus prior art upon using deuteration to achieve two key motives - to modify drug pharmacokinetics or toxicity- was available to patent examiners before even the 1980’s for obviousness rejections under 35 U.S.C. §103. A range of related patents subsequently issued in related and unrelated indications: for example in the specific field of deuterated volatile anesthetics, patents [6, [7, [8, [9] were issued from filings as late as in 1995, some eighteen years after Dow’s initial filing upon deuterated anesthetics. Inventors from Isotechnika later had several patents allowed upon drug deuteration, from 1998 to 2009 [10, [11] with the company later focusing upon voclosporin.
Subsequently, inventors such as Tung, Gant and Czarnik have filed an extensive series of patent applications claiming deuterated versions of existing non-deuterated drugs from 2005 onwards, with assignments to Concert, Auspex and Protia, Deuteria and Deuterx respectively. Recent patents with deuteration claims from these companies have issued as recently as 2013 and 2014. There has clearly been a significant measure of success for this strategy, and it has been widely reported [12, [13]. However, the field has increasingly questioned just when this approach will become seen as ‘obvious’ under 35 U.S.C. §103 by patenting authorities, as this will greatly change the nature of research and commercialization in this area [3].
4. Current State of 35 U.S.C. §103 Rejections
In this work US Patent applications were searched in April 2014 from each of the companies mentioned above as assignees with claims containing deuterium. These were then searched for patent prosecution histories at Public Pair, and the most recent applications from Concert, Auspex and Protia, Deuteria and Deuterx listed in Table 1, together with their status. It can be seen that there is an increasing trend from allowance towards rejections under 35 U.S.C. §103, using prior art upon the effects of deuteration upon aspects of drug stability or metabolism. Patent applications assigned to previously mentioned companies have now all received rejections (final or non-final) under 35 U.S.C. §103. It is also interesting to note that the Public Relations (PR) materials were used in the rejection of Rao and Zhang [14], and although this appears isolated, it is clear that companies need to balance the desirability of disclosures to drive investment, with the potential for these disclosures to later be used as prior art.
Table 1.
Status of Some Recent Drug Deuteration US Patent Applications
| Application | Assignee and Status |
|---|---|
| CONCERT | |
|
| |
| Tetrahydronaphthalene derivatives [15] | Not Yet Examined |
|
| |
| Substituted triazolophthalazine derivatives [16] |
Not Yet Examined |
|
| |
| Substituted xanthine derivatives [17]] | Not Yet Examined |
|
| |
| Pyrimidine derivatives [18] | Not Yet Examined |
|
| |
| Derivatives of pyrazole- [19] | Not Yet Examined |
|
| |
| Substituted isoindoline-1,3-dione [20] | Non-Final Rejection, No §103 |
|
| |
| Substituted triazolo-pyridazine [21] | Non-Final Rejection, No §103 |
|
| |
| Morphinan compounds [22] | Allowed |
|
| |
| Substituted triazolo-pyridazine [23] | Non-Final Rejection, No §103 |
|
| |
| Substituted dioxopiperidinyl phthalimide [24] |
Non-Final Rejection §103 and §103 based upon Tung [25], not ‘Dyck’ |
|
| |
| Analogues of cilostazol [26] | Allowed |
|
| |
| N-phenyl-2-pyrimidineamine derivatives [27] |
Non-Final Rejection Abbreviated ‘Dyck’ §103 citing Foster [28], Ito [29], Kushner [30], |
|
| |
| Fluorouracil derivatives [31] | Non-Final Rejection ‘Dyck’ §103 |
|
| |
| Novel pyrimidinecarboxamide derivatives [32] |
Not Yet Examined |
|
| |
| Heterocyclic kinase inhibitors [33] | Non-Final Rejection §103 citing Czarnik [34] |
|
| |
| Pyrazinoisoquinoline compounds [35] | Non-Final Rejection, ‘Dyck’ §103 |
|
| |
| 4-hydroxybutyric acid analogs [36] | Allowed after Non-Final Rejection §103 citing Kushner [30] |
|
| |
| Substituted derivatives of [37] | Not Yet Examined |
|
| |
| Prostacyclin derivatives [38] | Restriction Required |
|
| |
| Analogues of cilostazol [39] | Allowed |
|
| |
| Substituted isoindoline-1,3-dione [40] | Allowed |
|
| |
| Fluorinated diaryl urea [41] | Allowed |
|
| |
| Substituted xanthine derivatives [42] | Non-Final Rejection, Abbreviated ‘Dyck’ §103 citing Ando [43], Buteau [3], Foster [28] and Armstrong [44] |
|
| |
| Pyrazinoisoquinoline compounds [45] | Allowed |
|
| |
| Deuterated fingolimod [46] | Restriction Required |
|
| |
| AUSPEX | |
|
| |
| Cyclopropyl modulators of [14] | Allowed after Final Rejection §103 Citing non-’Dyck’ deuteration references, and Auspex and Concert PR Announcements. |
|
| |
| Benzoquinolone inhibitors of [47] | Non-Final Rejection Atypical ‘Dyck’ §103 |
|
| |
| Benzazepine inhibitors of [48] | Restriction Required |
|
| |
| Pyrimidinone inhibitors of [49] | Allowed |
|
| |
| 3,4-methylenedioxyphenyl inhibitors of [50] | Restriction Required |
|
| |
| Piperidine modulators of [51] | Restriction Required |
|
| |
| Morphinan modulators of [52] | Non-Final Rejection §103 citing Kushner [30], Sanderson [12] and Tung [53] |
|
| |
| Trimethoxyphenyl inhibitors of [54] | Non-Final Rejection ‘Dyck’ §103 |
|
| |
| 4,6-diaminopyrimidine stimulators of [55] | Request for continued examination |
|
| |
| Quinoline inhibitors of [56] | Final rejection, Atypical §103 citing Kushner [30] |
|
| |
| PROTIA, DEUTERIA, DEUTERX | |
|
| |
| Deuterium-enriched bupropion [57] | Allowed |
|
| |
| Deuterium-enriched lenalidomide. [58] | Allowed |
|
| |
| Deuterium-enriched donepezil [59] | Allowed |
|
| |
| 3-deutero-pomalidomide [60] | Non-Final Rejection §103 Citing Tung [25] |
|
| |
| 2,6-dioxo-3-deutero-piperdin-3-yl- [61] | Restriction Required |
|
| |
| Deuterium-enriched ruboxistaurin [62] | Restriction Required |
|
| |
| Deuterium-enriched meropenem [63] | Non-Final Rejection, ‘Dyck’ §103 |
|
| |
| Deuterium-enriched ixabepilone [64] | Non-Final Rejection §103 Citing Foster [28] |
|
| |
| Deuterium-enriched dasatinib [65] | Restriction Required |
|
| |
| Deuterium-enriched alogliptin [66] | Non-Final Rejection ‘Dyck’ §103 |
|
| |
| Deuterium-enriched tolterodine [67] | Restriction Required |
|
| |
| Deuterium-enriched doripenem [68] | Non-Final Rejection ‘Dyck’ §103 |
|
| |
| Deuterium-enriched risperidone [69] | Allowed |
|
| |
| Deuterium-enriched dapoxetine [70] | Restriction Required |
Examining these rejections shows the development of similar arguments for rejection that typically are based around firstly discussing the motivation to prepare deuterated versions of drugs to obtain versions with better pharmaceutical properties, typically supported by reference to Dyck et al. [71] ‘Thus deuterium substitution seems to be a useful strategy to enhance the pharmacological effects of a compound without significantly altering its basic chemical structure’ although other references [72, [73] from this group might also suffice, and Dyck is not cited in all such rejections. Support for the generality of this approach is then derived from referenced such as Ando et al, [43] Foster et al [28]. Gant et al [74], Ito et al [29], Kushner et al [30], or Poyten et al [75]. Secondly, the motivation to prepare deuterated drugs to study how the undeuterated drug or related compounds act in the body is discussed. This is supported by references such as Baillie [76], Browne [77], Cherrah et al [78], Gouyette [79], Haskins [80], Pieniaszek et al [81], Tonn et al [82] or Wolen [83].
After elucidation of these two branches, the rejections hold that a person of ordinary skill in the art would have motivation to prepare the deuterated version with reasonable expectation of success. Since there is always a desire to modify a compounds pharmacological effects (exemplified by deuteration improvements in half-life) without significant structure modification, or the need to improve pharmacokinetic studies, yet there being only a limited number of strategies to achieve this, obviousness of these solutions is found. Finally, these branches are integrated so that deuteration per se is shown to be a well-known way to improve a pharmaceutical and again obviousness of the approach ensues. The combination of this range of references would seem to cover many of the typical alterations in metabolism that would prove useful in deuterated drugs. We shall term such a rejection a ‘Dyck’ rejection.
The use of this type of U.S.C. §103 Rejection has (often in conjunction with other arguments) resulted in final rejections, abandonments and precluded patent issue. Thus, it would appear that in the absence of new or unexpected findings upon the biological effects of deuterating a drug, then establishing the non-obviousness of claims based around modifying metabolism may be difficult to achieve.
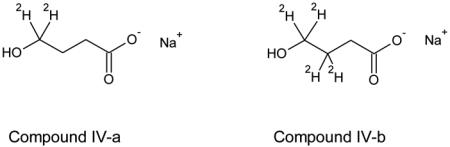
However, there are examples of successfully overcoming this kind of ‘obviousness’ rejection. A recent case of an application by Tung, Morgan and Silverman [36] received U.S.C. §103 rejection, with the obviousness of deuteration supported only by Kushner [30]. This was overcome by argument and a declaration attesting to the unexpectedly better pharmacokinetics in dogs of a less fully- deuterated version (IV-a) of gamma-hydroxybutyrate compared to the more extensively deuterated version (IV-b) as well as to the undeuterated version. In the declaration it is noted “Prior to testing IV-a I had no reason to believe that such improvement in pharmacokinetics could be realized by having two less deuterium atoms in IV-a relative to IV-b”. Although no statistical examination of the significance of the differences was given (as would be customary in current peer reviewed literature) the magnitude of the improvement is clear. Whether this data would have been sufficient had all the references used in a typical ‘Dyck’ §103 rejection is difficult to establish, however the experimental findings are unexpected in this authors opinion.
Another recent successful case against such a Dyck Rejection was made in Czarnik , regarding deuterium enriched bupropion [84]. The examiner made a non-final ‘Dyck’-type rejection (not, in this case citing Dyck, although counsel referred to this reference). The rejection was successfully overcome by a declaration and argument that the effect of deuterium in this case was not similar to those cited in the rejection (such as decreased cytochrome P-450 activity) but rather derived from a decreased half-life for racemization of the deuterated version compared to the protiated version by a factor of up to 35 fold. The requirement for breaking the carbon–hydrogen (or deuterium) bond during this tautomerization drives the large isotope effect (Figure 1). The unexpected nature of this much lower rate of racemization of the deuterated compound, was compounded by the importance of this extended half-life of racemization in the context of the half-life of elimination, and both these points were emphasized. Since references in the ‘Dyck’ rejection did not broadly teach that deuteration of drugs can modify their racemization to beneficial effect, it would appear they could not support continued U.S.C. §103 rejection.
Figure 1.

Specific deuteration greatly lowers rate of bupropion racemization
These two cases of overcoming §103 rejection that claims upon deuterated drugs give guidance on how claims might be defended against ‘Dyck’ rejections, especially when findings are unexpected in nature, and the importance of this has been emphasized by examiners in non-final rejections such as in Liu. [24] Just exactly when the continued reporting of similar ‘unexpected’ findings will tend to drive them to be seen rather as expected will be of interest to those in the field.
5. Was the Deuterium Switch Obvious to Big Pharma?
Another argument against U.S.C. §103 rejection could be envisaged based upon the idea that if drug deuteration is obvious to ones skilled in the art, then who could be more skilled and motivated than the major pharmaceutical companies. Certainly, as they make their livings directly from practicing these arts, their actions are highly indicative of those skilled in these arts. If one could establish that a patent application were filed before a certain percentage of these companies had disclosed the use of deuteration to modify drug pharmacokinetics or metabolism, then one could argue that since not all large pharmaceutical companies were yet disclosing drug deuteration in their patent applications, then the approach was non-obvious. If, as an examiner might argue, drug deuteration at the time of application was obvious, why would those most skilled in the art, major pharmaceutical companies, not attempt to claim or otherwise disclose it.
The US Patent Application data base was therefore searched using major Pharma companies as assignees, and references to metabolic stability from deuterium in their claims or specification, and the first relevant applications with language noted in Table 2. The very early patents of Reinhold [1] or McCarty [2] were not counted, as since so many patents on drug deuteration have been issued after these, their relevance as prior art must be limited. Also, because deuterated solvents are often used in NMR and other techniques, deuterium can appear in the specifications for reasons other than altering drug activity, and these were not reported as they do not pertain to obviousness of drug deuteration. There is a wide spread of dates, with such major companies as Bayer, Sanofi-Aventis and Johnson and Johnson not disclosing drug deuteration until as late as 2012 or 2013. The data is graphically shown in Fig. 2 and it can be seen that it was not until 2009-2010 that more major companies had disclosed such technologies than had not. Therefore, it would appear that such an analysis could support priority dates up to about 2009 to 2010 based upon a majority of major pharmaceutical companies (containing many such scientists skilled in the art) making their first specific drug deuteration claims, or as late as 2013 based upon the last major company to do so.
Table 2.
Major Pharma Companies Recent First Disclosure Drug Deuterium Pharmacokinetic Improvements in US Patent Applications
| Company | Year | Title |
|---|---|---|
| Pfizer | 2002 | Benzophenones and sulfones [85] |
| Merck | 2005 | Pyridazine Derivatives [86] (excludes Reinhold [1]) |
| Glaxo SmithKline | 2006 | Pyrimidine derivatives and [87] |
| Wyeth | 2007 | 41-Methoxy isotope labeled [88] |
| Novartis | 2009 | Organic Compounds [89] |
| Astra Zeneca | 2009 | Allosteric modulators of [90] |
| Roche | 2009 | Thiazolopyrimidine p13k inhibitor [91] |
| Abbott | 2010 | Compounds useful as [92] |
| Bristol-Myers Squib | 2010 | Compounds for the [93] |
| Eli Lilly | 2010 | Modified bovine g-CSF [94] |
| Bayer | 2012 | Substituted 5-fluoro-1H-pyrazolopyridines [95] |
| Sanofi-Aventis | 2012 | 9h-pyrrolo[2,3-b: 5,4-c'] dipyridine azacarboline [96] |
| Johnson and Johnson |
2013 | Process for the [97] |
Figure 2.

Time course of major pharma companies first disclosing drug deuterium pharmacokinetic improvements in US Patent applications.
6. Implications for Studies of Metabolism and Toxicity of Deuterated Drugs
Clearly, an effective way to support claims regarding deuterated versions of existing drugs is to provide evidence of unexpected modes by which deuteration exerts its effects. In the case of deuterated bupropion above, a decrease in keto-enol tautomerization induced racemization by deuteration at the specific site of this reaction was deemed sufficiently non-obvious. In the future, this patent may become to be used as prior art regarding control of such racemization, so that when taken with other reports upon deuteration isotope effects on keto-enol tautomerization [98], then similar patent applications may in future succumb to U.S.C. §103 rejection. However, in light of the many chemical reactions with potential for significant deuterium isotope effects, there remains significant scope for improving drugs through deuteration that would not be anticipated by the prior art in ‘Dyck’ rejections. There are also isotope effects other than kinetic isotope effects that deuteration could conceivably exploit. However, to avoid this Expert Opinion itself becoming prior art, these are left for others to elucidate. Fully documenting the unanticipated nature of these reactions and effects will be important, as will be the selection of references that explicitly teach away from findings, again developing the argument that one skilled in the art would not have expected the results.
7. Conclusions and Future Outlook
Significant clinical progress in the field is now being made. Trials of the Auspex compound SD-809 (deuterated tetrabenzine) are registered at Phase 3 NCT01795859 and NCT01897896 for indications in Huntington’s disease. The Concert compound CTP-499, (deuterated pentoxyfylline) [99] has a trial registered at Phase 2 NCT01487109 for indications in diabetic nephropathy. Similarly, a trial of the Avanir/Concert compound AVP-786 (deuterated dextromethorphan), has been completed at Phase 1. So there is clear progress towards regulatory approvals of deuterated drugs.
However, it is worth noting that the drug reimbursement landscape has shifted significantly since the last analogous approach -the chiral switch- that proved lucrative for example in the case of Prilosec to Nexium. These days, insurance payers and nationalized health systems increasingly demand pharmaco-economic justifications before adopting new and more expensive therapies over existing and less expensive ones. So, the developers of deuterium switch compounds will have to show significant clinical benefits over existing non-deuterated versions, independent of whether patent protection can be achieved and defended. If significant clinical benefit is achieved, it seems only reasonable that the risks taken by companies investing in deuterium switch drugs are appropriately rewarded.
8. Expert Opinion
Improving the pharmacokinetic and/or toxicological properties of existing drugs by deuteration continues to show significant potential. This is clear from the patent literature, as the major pharmaceutical companies now also claim deuterated versions of their new molecules in their ongoing patent applications. There has also been a much greater understanding of the complexity and unpredictability of the effects of deuterium replacement upon pharmacokinetics and metabolism, with excellent work now entering the peer-reviewed press from major pharmaceutical companies such as Pfizer [100] and Novartis [101]. The inability to predict deuteration effects in vivo stems from a number of variables: metabolic switching to pathways not involving C-D bond breakage, the importance of metabolism other than through cytochrome P-450, and the relative importance of metabolic as opposed to systemic clearance. Though this unpredictability is valuable in that it enables arguments against ‘obviousness’ rejection it also means that a wide range of deuterated versions of a compound must be synthesized and then have their metabolism and pharmacokinetics evaluated, entailing much work and cost. Metabolic switching is very complex, and it may occur differentially both between different animal models, and also between animal models and humans as studies translate from the preclinical to clinical stages [100]. While it will be very difficult to develop technologies to predict the pathways and consequences of metabolic switching of differing patterns of deuteration, it may be possible to develop more effective screening approaches. Currently, each deuterated form is synthesized, and then its metabolism characterized. If it was possible to make a library of different deuterated forms of a drug, the relative rates of loss of the differing forms could be determined by MS. The most stable compounds could then have their structures solved by multidimensional and accurate MS techniques, to provide leads for further work, or for advancement in development themselves.
The advancement of these compounds in clinical trials is highly promising. Compounds are being tested that address patient needs in important disease states, such as Huntington disease, and there is a good deal of excitement in the field. Since much of the groundwork for these trials has been performed upon the non-deuterated versions, there is great hope that this will enable faster, smarter and cheaper trials of the deuterated version. Much failure in clinical trials involves three main reasons: lack of efficacy, toxicity or poor pharmacokinetics: deuterated drugs benefit from avoiding the first, while they can specifically address the last two. It is also becoming clear that deuterated versions of drugs might benefit from being FDA approved through a less onerous 505(b)(2) NDA filing, which has significant advantages. However, in the case of an 505(b)(2) NDA, either patent or other exclusivity protections (e.g. 7-year orphan drug exclusivity) may delay their filing or approval, so it is imperative that a clear understanding of this landscape is obtained.
As far as the business model of the ‘deuterium switch’ - developing deuterated versions of existing drugs- there is clearly an increasing use of U.S.C. §103 ‘obviousness’ rejections during patent prosecutions in the US. Powerful arguments based upon a significant body of prior art (the ‘Dyck rejection’) have now been developed for use against claims that cover many modifications of drug metabolism by deuteration. However, there have been successful arguments made against such §103 ‘obviousness’ rejections, where carefully demonstrating that the results of deuteration were not easily anticipated from the prior art was central to allowance. It remains to be seen how rapidly these issued patents become incorporated into ‘Dyck rejection’ prior art, although the author suspects this will be done rapidly.
The first products of ‘deuterium switching’ continue to advance into and through clinical trials, and there is an extensive body of issued patents to continue replenishing this pipeline for some time to come. Current market capitalizations of companies based upon this technology suggest that the value of ‘deuterium switching’ is upwards of a billion US dollars, with potentially the most major value inflection points yet to come. Should the value of a ‘deuterium switch’ compound become sufficiently large, then litigation over patent validity might become significant, especially for more recent patents. As Vaz noted [13] “Someday…it's probably going to go to court.”
Article Highlights.
Drug deuteration, the ‘deuterium switch’, can greatly improve a given drugs pharmacokinetics or toxicity properties by altering how it is metabolized.
A number of companies have developed patent and clinical trial portfolios based around deuterium switching of known effective drugs.
Examiners are increasingly responding to deuterium switch patent applications with 35 U.S.C. §103 ‘obviousness’ rejections based upon a wide range of prior art.
It is, however, highly complex and currently unpredictable as to whether deuterium switching will have any significant effects in vivo, and if so, by how much.
Detailing the unexpected nature of many deuterium switch experimental findings appears a useful strategy to enable patent allowance.
Footnotes
Financial and competing interests disclosure
The author has no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
References
- 1.Reinhold DF. Processes for asymmetric conversion of 3-fluoro-L-alanine and 2-deutero-3-fluoro-L-alanine to their D-isomers. 1976. USPTO 3,950,411. ** Pioneering example of drug deuteration many years ahead of later developments.
- 2.McCarty LP. Compositions and methods for anesthetizing an animal using deuterated analogues of halothane and chloroform. 1978 USPTO 4,069,346. ** Pioneering example of drug deuteration many years ahead of later developments. [Google Scholar]
- 3.Buteau KC. Deuterated drugs: unexpectedly nonobvious. J High Tech L. 2009;10:22. * An interesting and useful legal analysis of the drug deuteration patent field. [Google Scholar]
- 4.Gant TG. Using Deuterium in Drug Discovery: Leaving the Label in the Drug. Journal of medicinal chemistry. 2013;57(9):3595–611. doi: 10.1021/jm4007998. * An interesting monograph review of the hisroty of the field from scientist very active in the field. [DOI] [PubMed] [Google Scholar]
- 5.Darland GK, Hajdu R, Kropp H, et al. Oxidative and defluorinative metabolism of fludalanine, 2-2H-3-fluoro-D-alanine. Drug metabolism and disposition. 1986;14(6):668–73. [PubMed] [Google Scholar]
- 6.Larsen ER, McCarty LP. Deuterated 1,1-difluoro-2,2-dihaloethyl difluoromethyl ethers. 1979. USPTO 4,154,971.
- 7.Larsen ER, McCarty LP. Deuterated 1,1-difluoro-2,2-dihaloethyl difluoromethyl esters. 1980. USPTO 4,188,405.
- 8.Baker MT, Tinker JH. Deuterated sevoflurane as an inhalational anesthetic. 1995. USPTO 5,391,579.
- 9.Baker MT, Tinker JH. Deuterated sevoflurane as an inhalational anesthetic. 1998. USPTO 5,789,450. * Interesting in that deuterated volatile anesthesia patents were still being allowed 22 years after first in the series.
- 10.Caille G, Foster RT, Lewanczuk R. Enhancement of the efficacy of nifedipine by deuteration. 1998. USPTO 5,846,514.
- 11.Naicker S, Yatscoff RW, Foster RT. Methods of making deuterated cyclosporin analogs. 2009. USPTO 7,538,189.
- 12.Sanderson K. Big interest in heavy drugs. Nature. 2009 Mar 19;458(7236):269. doi: 10.1038/458269a. [DOI] [PubMed] [Google Scholar]
- 13.Katsnelson A. Heavy drugs draw heavy interest from pharma backers. Nature medicine. 2013 Jun;19(6):656. doi: 10.1038/nm0613-656. [DOI] [PubMed] [Google Scholar]
- 14.Rao T, Zhang C. Cyclopropyl modulators of p2y12 receptor. 2012. USPTO Application 20120301458.
- 15.Jones AD, Paul BJ. Method of utilizing recycled deuterium oxide in the synthesis of deuterated compounds. 2014. USPTO Application 20140046060.
- 16.Harbeson SL. Substituted triazolophthalazine derivatives. 2014. USPTO Application 20140024652.
- 17.Tung RD. Deuterated benzo[d][1,3]-dioxol derivatives. 2014. USPTO Application 20140018390.
- 18.Tung R, Masse CE. Pyrimidine derivatives. 2014. USPTO Application 20140018379.
- 19.Pandya B, Masse CE, Silverman IR, et al. Derivatives of pyrazole-substituted amino-heteroaryl compounds. 2014. USPTO Application 20140005211.
- 20.Liu JF. Substituted isoindoline-1,3-dione derivatives. 2013. USPTO Application 20130345282.
- 21.Harbeson SL, Tung R, Liu JF. Substituted triazolo-pyridazine derivatives. 2013. USPTO Application 20130317033.
- 22.Tung R. Morphinan compounds. 2013. USPTO Application 20130310415.
- 23.Harbeson SL. Substituted triazolo-pyridazine derivatives. 2013. USPTO Application 20130225590.
- 24.Liu JF, Tung RD. Substituted dioxopiperidinyl phthalimide derivatives. 2013. USPTO Application 20130150408.
- 25.Tung R. Substituted dioxopiperidinyl phthalimide derivaties. 2010. WIPO Application WO 2010056344.
- 26.Persichetti RA, Liu JF, Masse CE, et al. Analogues of cilostazol. 2013. USPTO Application 20130131021.
- 27.Harbeson SL, Liu JF, Tung R. N-phenyl-2-pyrimidineamine derivatives. 2013. USPTO Application 20130121963.
- 28.Foster RT. Method of using deuterated calcium channel blockers. 2001. USPTO 6,221,335.
- 29.Ito N, Maesawa T, Muto K, et al. Method for deuteration of a heterocyclic ring. 2009. USPTO US7517990 B2.
- 30.Kushner D, Baker A, Dunstall T. Pharmacological uses and perspectives of heavy water and deuterated compounds. Can J Physiol Pharmacol. 1999;77(2):79–88. [PubMed] [Google Scholar]
- 31.Persichetti RA, Liu JF. Fluorouracil derivatives. 2013. USPTO Application 20130109707.
- 32.Tung R. Novel pyrimidinecarboxamide derivatives. 2013. USPTO Application 20130109660.
- 33.Tung RD. Heterocyclic kinase inhibitors. 2013. USPTO Application 20130095100.
- 34.Czarnik AW. Deuterium-enriched dasatinib. 2009. USPTO Application 20090076025.
- 35.Liu JF, Tung R, Harbeson SL. Pyrazinoisoquinoline compounds. 2013. USPTO Application 20130029997.
- 36.Tung RD, Morgan AJ, Silverman IR. 4-hydroxybutyric acid analogs. 2013. USPTO Application 20130012565.
- 37.Tung RD, Masse CE. Substituted derivatives of bicyclic [4.3.0] heteroaryl compounds. 2012. USPTO Application 20120295925.
- 38.Masse CE, Harbeson SL, Tung RD. Prostacyclin derivatives. 2012. USPTO Application 20120270934.
- 39.Liu JF, Persichetti RA. Analogues of cilostazol. 2012. USPTO Application 20120264721.
- 40.Liu JF. Substituted isoindoline-1,3-dione derivatives. 2012. USPTO Application 20120252864.
- 41.Liu JF, Tung RD, Harbeson SL. Fluorinated diaryl urea derivatives. 2012. USPTO Application 20120237474.
- 42.Tung RD, Liu JF, Harbeson SL. Substituted xanthine derivatives. 2012. USPTO Application 20120202830.
- 43.Ando K, Kawamura K. Sulfamoylheleroaryl pyrazole compounds as anti-inflammatory/analgesic agents. 2003. USPTO.
- 44.Armstrong R, Watson J, Yeadon M. Combination of a PDE4 inhibitor and tiotropium or derivative thereof for treating obstructive airways and other inflammatory diseases. 2005. USPTO Application 20050107420.
- 45.Liu JF, Tung R, Harbeson SL. Pyrazinoisoquinoline compounds. 2012. USPTO Application 20120149709.
- 46.Liu JF, Persichetti RA. Deuterated fingolimod. 2012. USPTO Application 20120141513.
- 47.Gant TG, Zhang C, Shahbaz M. Benzoquinolone inhibitors of vmat2. 2012. USPTO Application 20120003330.
- 48.Rao T, Zhang C. Benzazepine inhibitors of gamma-secretase. 2011. USPTO Application 20110306596.
- 49.Rao T, Zhang C. Pyrimidinone inhibitors of lipoprotein-associated phospholipase a2. 2011. USPTO Application 20110306552.
- 50.Rao T, Zhang C. 3,4-methylenedioxyphenyl inhibitors of gaba aminotransferase and/or gaba reuptake transporter inhibitor. 2011. USPTO Application 20110257260.
- 51.Zhang C. Piperidine modulators of dopamine receptor. 2011. USPTO Application 20110206782.
- 52.Gant TG, Sarshar S, Zhang C. Morphinan modulators of nmda receptors, sigma1 receptors, sigma2 receptors, and/or a3b4 nicotinic receptors. 2011. USPTO Application 20110206780.
- 53.Tung R. Morphinan Compounds. 2008. USPTO Application 20080280936.
- 54.Zhang C, Sommer A. Trimethoxyphenyl inhibitors of tyrosine kinase. 2011. USPTO Application 20110206661.
- 55.Rao T, Zhang C. 4,6-diaminopyrimidine stimulators of soluble guanylate cyclase. 2011. USPTO Application 20110201626.
- 56.Zhang C. Quinoline inhibitors of tyrosine kinase. 2011. USPTO Application 20110195066.
- 57.Czarnik AW. Deuterium-enriched bupropion. 2014. USPTO Application 20140018436.
- 58.Czarnik AW. Deuterium-enriched lenalidomide. 2013. USPTO Application 20130245067.
- 59.Czarnik AW. Deuterium-enriched donepezil. 2013. USPTO Application 20130090357.
- 60.DeWitt S. 3-deutero-pomalidomide. 2012. USPTO Application 20120302605.
- 61.DeWitt S. 2,6-dioxo-3-deutero-piperdin-3-yl-isoindoline compounds. 2012. USPTO Application 20120252844.
- 62.Czarnik AW. Deuterium-enriched ruboxistaurin. 2012. USPTO Application 20120136037.
- 63.Czarnik AW. Deuterium-enriched meropenem. 2012. USPTO Application 20120122833.
- 64.Czarnik AW. Deuterium-enriched ixabepilone. 2012. USPTO Application 20120035227.
- 65.Czarnik AW. Deuterium-enriched dasatinib. 2012. USPTO Application 20120015956.
- 66.Czarnik AW. Deuterium-enriched alogliptin. 2011. USPTO Application 20110312983.
- 67.Czarnik AW. Deuterium-enriched tolterodine. 2011. USPTO Application 20110281952.
- 68.Czarnik AW. Deuterium-enriched doripenem. 2011. USPTO Application 20110281927.
- 69.Czarnik AW. Deuterium-enriched risperidone. 2011. USPTO Application 20110212976.
- 70.Czarnik AW. Deuterium-enriched dapoxetine. 2011. USPTO Application 20110201690.
- 71.Dyck LE, Durden DA, Boulton AA. Effects of deuterium substitution on the catabolism of beta-phenylethylamine: an in vivo study. Journal of neurochemistry. 1986 Feb;46(2):399–404. doi: 10.1111/j.1471-4159.1986.tb12982.x. ** Highly cited by examiners developing §103 rejections. When combined with several other general references and some compound specific ones, forms that basis of a powerful argument against allowance. [DOI] [PubMed] [Google Scholar]
- 72.Dewar KM, Dyck LE, Durden DA, et al. Effect of deuterium substitution on the penetration of beta-phenylethylhydrazine into the rat brain. Biochemical pharmacology 1988 Jul. 1;37(13):2703–4. doi: 10.1016/0006-2952(88)90266-3. [DOI] [PubMed] [Google Scholar]
- 73.Dyck LE, Boulton AA. Effect of deuterium substitution on the disposition of intraperitoneal tryptamine. Biochemical pharmacology. 1986 Sep 1;35(17):2893–6. doi: 10.1016/0006-2952(86)90482-x. [DOI] [PubMed] [Google Scholar]
- 74.Gant TG, Sarshar S. Inhibitors of the gastric H+, K+-atpase with enhanced therapeutic properties. 2007. USPTO Application 20070082929.
- 75.Poyten MC, Josyula KVB, Gao P, et al. Stabilized deuteroborane-tetrahydrofuran complex. 2007. USPTO Application 20070197695.
- 76.Baillie TA. The use of stable isotopes in pharmacological research. Pharmacological Reviews. 1981;33(2):81–132. [PubMed] [Google Scholar]
- 77.Browne TR. Stable isotope techniques in early drug development: an economic evaluation. The Journal of Clinical Pharmacology. 1998;38(3):213–20. doi: 10.1002/j.1552-4604.1998.tb04418.x. [DOI] [PubMed] [Google Scholar]
- 78.Cherrah Y, Falconnet J, Desage M, et al. Study of deuterium isotope effects on protein binding by gas chromatography/mass spectrometry. Caffeine and deuterated isotopomers. Biological mass spectrometry. 1987;14(11):653–57. doi: 10.1002/bms.1200141115. [DOI] [PubMed] [Google Scholar]
- 79.Gouyette A. Synthesis of deuterium-labelled elliptinium and its use in metabolic studies. Biological mass spectrometry. 1988;15(5):243–47. doi: 10.1002/bms.1200150502. [DOI] [PubMed] [Google Scholar]
- 80.Haskins N. The application of stable isotopes in biomedical research. Biological mass spectrometry. 1982;9(7):269–77. doi: 10.1002/bms.1200090702. [DOI] [PubMed] [Google Scholar]
- 81.Pieniaszek HJ, Mayersohn M, Adams MP, et al. Moricizine bioavailability via simultaneous, dual, stable isotope administration: bioequivalence implications. The Journal of Clinical Pharmacology. 1999;39(8):817–25. doi: 10.1177/00912709922008489. [DOI] [PubMed] [Google Scholar]
- 82.Tonn G, Mutlib A, Abbott F, et al. Simultaneous analysis of diphenhydramine and a stable isotope analog (2H10) diphenhydramine using capillary gas chromatography with mass selective detection in biological fluids from chronically instrumented pregnant ewest. Biological mass spectrometry. 1993;22(11):633–42. doi: 10.1002/bms.1200221103. [DOI] [PubMed] [Google Scholar]
- 83.Wolen RL. The application of stable isotopes to studies of drug bioavailability and bioequivalence. The Journal of Clinical Pharmacology. 1986;26(6):419–24. doi: 10.1002/j.1552-4604.1986.tb03551.x. [DOI] [PubMed] [Google Scholar]
- 84.Czarnik AW. Deuterium-enriched bupropion. 2013. USPTO 8,524,780.
- 85.Lowe J. Benzophenones and sulfones as inhibitors of glycine uptake. 2002. USPTO Application 20020052401. * An early example of major pharmaceutical companies re-asserting drug deuteration strategies to modify drug DMPK.
- 86.Eggenweiler HM, Wolf M. Pyridazine derivatives. 2005. USPTO Application 20050176714.
- 87.Eatherton A. Pyrimidine derivatives and their use as CB2 modulators. 2006. USPTO Application 20060293354.
- 88.Gu J, Ruppen M. 41-Methoxy isotope labeled rapamycin 42-ester. 2007. USPTO Application 20070105888.
- 89.McCarthy C, Press NJ. Organic Compounds. 2009. USPTO Application 20090023769.
- 90.Grahames C, Mallinder P, McIntosh F, et al. Assays for Allosteric Modulators of G-Protein Coupled Receptors (GPCRs) 2009. USPTO Application 20090305321.
- 91.Castanedo GM, Gunzner JL, Malesky K, et al. Thiazolopyrimidine p13k inhibitor compounds and methods of use. 2009. USPTO Application 20090118275.
- 92.Vasudevan A, Brown BS, Keddy RG, et al. Compounds useful as inhibitors of protein kinases. 2010. USPTO Application 20100035919.
- 93.Pracitto R, Kadow JF, Bender JA, et al. Compounds for the Treatment of Hepatitis C. 2010. USPTO Application 20100184800.
- 94.Putnam AMAH, Knudsen N, Norman T, et al. Modified Bovine G-CSF Polypeptides And Their Uses. 2010. USPTO Application 20100035812.
- 95.Follmann M, Stasch JP, Redlich G, et al. Substituted 5-fluoro-1H-pyrazolopyridines and their use. 2012. USPTO Application 20120022084.
- 96.Babin D, Bedel O, Gouyon T, et al. 9h-pyrrolo[2,3-b: 5,4-c'] dipyridine azacarboline derivatives, preparation thereof, and therapeutic use thereof. 2012. USPTO Application 20120208809.
- 97.Gong Y, Zinser HB. Process for the preparation of sulfamide derivatives. 2013. USPTO Application 20130085176.
- 98.Cederstav AK, Novak BM. Investigations into the chemistry of thermodynamically unstable species. The direct polymerization of vinyl alcohol, the enolic tautomer of acetaldehyde. Journal of the American Chemical Society. 1994;116(9):4073–74. [Google Scholar]
- 99.Braman V, Graham P, Cheng C, et al. A Randomized Phase I Evaluation of CTP-499, a Novel Deuterium-Containing Drug Candidate for Diabetic Nephropathy. Clinical Pharmacology in Drug Development. 2013;2(1):53–66. doi: 10.1002/cpdd.3. * Report on a recent Phase 1 trial of one of the 'new wave' of deuterated drugs. [DOI] [PubMed] [Google Scholar]
- 100.Sharma R, Strelevitz TJ, Gao H, et al. Deuterium isotope effects on drug pharmacokinetics. I. System-dependent effects of specific deuteration with aldehyde oxidase cleared drugs. Drug metabolism and disposition. 2012;40(3):625–34. doi: 10.1124/dmd.111.042770. ** An excellent elaboration of the complexities of metabolic switching, including species differences. A must read. [DOI] [PubMed] [Google Scholar]
- 101.Manley PW, Blasco F, Mestan J, et al. The kinetic deuterium isotope effect as applied to metabolic deactivation of imatinib to the des-methyl metabolite, CGP74588. Bioorganic & medicinal chemistry. 2013;21(11):3231–39. doi: 10.1016/j.bmc.2013.03.038. [DOI] [PubMed] [Google Scholar]