

Abstract
The importance of chronic low-grade inflammation in the pathology of numerous age-related chronic conditions is now clear. An unresolved inflammatory response is likely to be involved from the early stages of disease development. The present position paper is the most recent in a series produced by the International Life Sciences Institute's European Branch (ILSI Europe). It is co-authored by the speakers from a 2013 workshop led by the Obesity and Diabetes Task Force entitled ‘Low-grade inflammation, a high-grade challenge: biomarkers and modulation by dietary strategies’. The latest research in the areas of acute and chronic inflammation and cardiometabolic, gut and cognitive health is presented along with the cellular and molecular mechanisms underlying inflammation–health/disease associations. The evidence relating diet composition and early-life nutrition to inflammatory status is reviewed. Human epidemiological and intervention data are thus far heavily reliant on the measurement of inflammatory markers in the circulation, and in particular cytokines in the fasting state, which are recognised as an insensitive and highly variable index of tissue inflammation. Potential novel kinetic and integrated approaches to capture inflammatory status in humans are discussed. Such approaches are likely to provide a more discriminating means of quantifying inflammation–health/disease associations, and the ability of diet to positively modulate inflammation and provide the much needed evidence to develop research portfolios that will inform new product development and associated health claims.
Keywords: Low-grade inflammation, Biomarkers, Chronic diseases, Health claims
Introduction and overview of the focus of the position paper
Inflammation is a central component of innate (non-specific) immunity. In generic terms, inflammation is a local response to cellular injury that is marked by increased blood flow, capillary dilatation, leucocyte infiltration, and the localised production of a host of chemical mediators, which serves to initiate the elimination of toxic agents and the repair of damaged tissue( 1 ). It is now clear that the termination (alternatively known as resolution) of inflammation is an active process involving cytokines and other anti-inflammatory mediators, particularly lipids, rather than simply being the switching off of pro-inflammatory pathways( 2 , 3 ).
Inflammation acts as both a ‘friend and foe’: it is an essential component of immunosurveillance and host defence, yet a chronic low-grade inflammatory state is a pathological feature of a wide range of chronic conditions, such as the metabolic syndrome (MetS), non-alcoholic fatty liver disease (NAFLD), type 2 diabetes mellitus (T2DM) and CVD( 4 , 5 ). Although the association between inflammation and chronic conditions is widely recognised, the issue of causality and the degree to which inflammation contributes and serves as a risk factor for the development of disease remain unresolved. As will be discussed, part of this uncertainty is due to a general lack of sensitive and specific biomarkers of low-grade chronic inflammation that can be used in human trials( 1 ).
The present article results from an International Life Sciences Institute (ILSI) Europe Workshop held in September 2013 in Granada, Spain entitled ‘Low-grade inflammation a high grade challenge: biomarkers and modulation by dietary strategies’, and aims to serve as an update to existing reviews in the area of inflammation and health and its assessment and modulation( 1 , 6 , 7 ). In particular, the present article will focus on the latest research findings in the areas of inflammation and cardiometabolic, cognitive and gut health, and how early-life nutrition and the macronutrient and plant bioactive composition of the adult diet influence inflammatory processes. It will discuss existing and emerging methods used to quantify inflammatory status in humans. Importantly, the article will identify knowledge gaps and methodological limitations that need to be addressed.
Exploring the role of inflammation in health and chronic diseases
Low-grade inflammation in cardiometabolic disease
The role of inflammation in the early-stage pathophysiology of atherothrombotic events has been recognised for over 20 years. Leucocyte recruitment into the sub-endothelial compartment of damaged arteries initiates a cascade of events mediated by leucocyte-derived inflammatory mediators. In particular, chemokines and cytokines propagate atherosclerosis via (1) increased chemokine production and expression of endothelial adhesion molecules, stimulating further leucocyte recruitment, (2) promoting lipid-laden foam-cell formation, (3) initiating smooth muscle cell proliferation, and (4) inducing plaque instability and eventual rupture( 8 , 9 ). The ensuing thrombosis is also in large part dependent on the inflammatory status of the ruptured plaque.
In addition to a direct role on events within the arterial wall, inflammation is an important determinant of the multi-organ cardiometabolic dysfunction, and the increased risk of T2DM, NAFLD and CVD associated with obesity( 10 ). Adipose tissue hypertrophy is associated with immune cell infiltration, in particular that of macrophages and T cells, and a local pro-inflammatory milieu wherein key cytokines including TNF-α, IL-6 and IL-1β impede the insulin signalling cascade to induce insulin resistance( 11 , 12 ). This ultimately leads to a dysregulation of glucose and lipid metabolism in adipose tissue, skeletal muscle and liver. However, up to 30 % of obese individuals are considered metabolically healthy (MHO)( 13 ), and there is evidence to suggest that a lack of the typical elevation in the inflammatory profile associated with obesity may underlie this ‘protected’ MHO phenotype. For example, in morbidly obese individuals, Barbarroja and co-workers observed mean homeostatic model assessment for insulin resistance (HOMA-IR) scores (insulin sensitivity index) of 3·31 and 11·48 in subjects with MHO (BMI 55 kg/m2) or who were metabolically unhealthy obese (BMI 56 kg/m2), respectively, which was associated with a 2- to 4-fold greater adipose expression of inflammatory cytokines (TNF-α, IL-1β and IL-6) between the two obese groups( 14 ).
Inflammation plays a direct role in the progression of NAFLD, the most common liver disorder in Western countries. NAFLD comprises a spectrum of conditions ranging from benign steatosis to non-alcoholic steatohepatitis characterised by hepatocyte injury (hepatocyte ballooning and Mallory bodies) and necroinflammation, and potentially to progressive fibrosis that can lead to cirrhosis( 15 , 16 ). The pathological progression of NAFLD is considered to have a two-hit basis (Fig. 1). The first hit, hepatocyte accumulation of fat, is thought to arise due to an increased delivery of fatty acids to the hepatocyte, an increase in hepatocyte fatty acid and TAG synthesis, and decreased fatty acid oxidation. The resultant excess of fat may result in lipotoxicity and a pro-inflammatory and pro-oxidative state (the second hit), which ultimately induces cellular senescence, which, if unchecked, leads to fibrosis and cirrhosis. Hepatic inflammation is mediated via the activation of local macrophages called Kupffer cells. Currently, no medication or surgical procedure has been approved for treating NAFLD or non-alcoholic steatohepatitis with confidence. Considering the overall lack of success in curbing global trends in the prevalence of excess body weight, inflammatory processes are emerging as a strong therapeutic target to reduce the risk of T2DM, CVD and NAFLD in obese individuals.
Fig. 1.

Two-hit model of non-alcoholic fatty liver disease. (A colour version of this figure can be found online at http://www.journals.cambridge.org/bjn).
Gut–systemic inflammatory associations
With recent significant advances in the ability to characterise the gut microbiota in increasing detail, comes the recognition of the importance of the microbiota not only in gastrointestinal health, but also in systemic metabolism and cardiometabolic health, with the immune system and inflammatory processes central to gut–systemic tissue ‘cross-talk’. The human intestine contains 1 × 1013 to 1 × 1014 bacterial cells, which outnumber human cells by a factor of 10 to 1 and contain approximately 150 times as many genes as the human genome( 17 ). Increasing evidence indicates that the microbiota is significantly altered through the ageing process( 18 , 19 ) and in obesity( 18 ), with a deleterious decline in microbiota ‘richness’ and gene expression diversity evident in both situations( 18 ).
Gastrointestinal tract–microbiota interactions influence host health, and in particular immune function, by promoting the development and maintenance of the mucosal immune system, protecting against pathogen invasion and maintaining gastrointestinal tract barrier integrity( 20 ). Gut permeability to bacterial lipopolysaccharides (LPS), a potent inflammatory stimulant, appears to be an important trigger for low-grade systemic inflammation. LPS are found on the outer membrane of Gram-negative bacteria such as Proteobacteria (e.g. Escherichia coli), and serve as an endotoxin. In the elderly, a higher count of LPS-producing bacteria in the colon, along with a lower abundance of bifidobacteria( 21 , 22 ), a combination which is thought to promote increased gut permeability( 21 ), is likely to lead to higher plasma levels of LPS (termed metabolic endotoxaemia). Through the interaction with Toll-like receptor 4 on mononuclear cells, microbiota-derived LPS may be an important trigger in the development of inflammation and metabolic diseases( 23 ). In a recent dietary intervention study in male C57Bl/6 mice, the alteration in microbiota profiles as a result of a high-fat diet was strongly associated with gut permeability, endotoxaemia and adipose tissue inflammation( 24 ).
In addition to its role in low-grade inflammatory cardiometabolic conditions, emerging evidence is suggesting that the gut microbiota can influence the risk of high-grade autoimmune inflammatory conditions such as type 1 diabetes mellitus, coeliac disease, inflammatory bowel disease and rheumatoid arthritis( 25 – 27 ), the incidence of which has risen dramatically since the 1940s. These conditions are now thought to affect 5–10 % of those in Western societies( 28 ). Certain members of the gut microbiota have been shown to induce mimics of human antigens and trigger the production of autoantibodies responsible for aberrant immune responses to normal human proteins and hormones including leptin, peptide YY and ghrelin( 29 ). It is not unreasonable to speculate that the adverse impact of the energy-dense, nutrient-poor Western-style diet on human gut microbiota and immune system, which have both been finely tuned and honed by high-fibre, high-polyphenol traditional diets over the millennia, may therefore be an important contributor to the environmental stimuli that trigger and progress autoimmune conditions( 30 ). A possible starting point when discussing the underlying mechanisms by which diets rich in whole plant foods or fermentable fibres can have an impact on immune function and tolerance may be the recent demonstration that butyrate, an important fermentation end product produced by the gut microbiota from fibre, controls human dendritic cell maturation, a key process in immune homeostasis, since dendritic cells are considered as ‘gate keepers’ of the immune system( 31 , 32 ). In addition, butyrate induces murine peripheral regulatory T-cell generation( 33 ), acetate affects neutrophil chemotaxis and oxidative burst, butyrate inhibits adipocyte–macrophage inflammatory interactions( 34 ), and propionate reduces the inflammatory output of adipose tissue( 35 ). Probiotic, fibre or polyphenol up-regulation of microbial activities that control both the quantity and profile of bile acids returning to the liver via the enterohepatic circulation with their subsequent regulation of farnesoid X receptor and TGR5 is also emerging as an important pathway linking the gut microbiota with extra-intestinal physiological/immune function( 33 , 36 , 37 ).
Low-grade systemic inflammation and neuroinflammation
Communication between the systemic immune system and the central nervous system (CNS) is a critical but often overlooked component of the inflammatory response to tissue injury, disease or infection. Activation of highly conserved neuronal and hormonal communication pathways in mammals drives diverse CNS-regulated components of the inflammatory response, including fever, neurogenic inflammation, descending anti-inflammatory mechanisms and a coordinated set of metabolic and behavioural changes, including fatigue, anhedonia, depression and mild cognitive impairment. These behavioural changes are collectively referred to as ‘sickness behaviour’( 38 – 40 ). Experimental studies have provided evidence that activation of microglia, the macrophages of the CNS, as well as the cerebral vasculature, plays a key role in the development of these behavioural changes, by inducing pro-inflammatory mediators, such as IL-1β, TNF-α and PGE2 in the CNS( 38 , 41 , 42 ).
Much of what we know is derived from studies using mimetics of bacterial and viral infection. Depending on the stimulus used, these mimetics induce a transient response in otherwise healthy subjects; for example, administration of LPS results in enhanced production of IL-6 (approximately 80-fold) and IL-1β (approximately 4-fold), peaking at 3 h after a challenge and returning to baseline at 24 h( 32 ). CNS responses to (patho)physiological stimuli, such as genuine infections or low-grade inflammation as a result of the MetS, are less well described.
Development of sickness behaviour in response to an infection is part of the normal response to fighting infection, and can occur during low-grade sub-pyrogenic inflammation( 41 ); however, these adaptive responses are not always harmless. Microglia have a very low turnover, and it has been suggested that these long-lived cells have an innate memory, resulting in a prolonged and heightened response under neuroinflammatory conditions( 43 ). A normal part of the homeostatic signalling from the periphery to the brain, therefore, has the potential to have a profound impact on brain disease initiation or progression( 44 , 45 ). In a recent prospective clinical study, Alzheimer's disease patients were followed for 6 months and assessed for the presence of circulating cytokines, episodes of microbial infection and cognitive decline. Patients with both high levels of TNF-α (>4·2 pg/ml) at baseline and microbial infection during the assessment period showed a 4-fold greater cognitive decline, relative to patients with low levels of TNF-α ( < 2·4 ng/ml) at baseline and no infections( 46 ). Raised serum levels of TNF-α and IL-6, but not CRP, are also associated with increased frequency of other common neuropsychiatric symptoms observed in Alzheimer's disease patients, including apathy, anxiety, depression and agitation( 47 ).
Recently, the effects of LPS and a real infection (Salmonella typhimurium) on cerebral endothelial and microglial activation were compared. While LPS administration resulted in a robust but transient neuroinflammatory response, a genuine infection induced a prolonged pro-inflammatory cytokine response in the CNS, leading to microglial priming( 48 ).
A detailed consideration of the impact and mechanistic basis for the association between neuroinflammation and neuronal and overall CNS function, cognition and the risk of age-related cognitive decline and dementia is outside the scope of the present review, and has been the topic of many recent expert review articles( 49 – 54 ).
Collectively, these data highlight inflammatory pathways as important targets for strategies promoting healthy brain ageing and reducing the risk of age-related cognitive decline.
Dietary modulation of low-grade inflammation
There is a substantial amount of evidence to suggest that many foods, nutrients and non-nutrient food components modulate inflammation both acutely and chronically( 1 , 6 ). However, dietary studies have been typically limited to measuring a small number of blood markers of inflammation, often in the fasting state, and these may not necessarily reflect inflammation in tissue compartments or what happens in response to inflammatory challenges. This presents a significant limitation to our understanding of diet/nutrient–inflammation interactions. Previous ILSI Europe activities have dealt extensively with the food/nutrition–inflammation interaction( 6 , 7 ), and it is beyond the scope of the present review to provide a systematic or extensive coverage of this area. Instead, some specific examples will be discussed.
Dietary fats and inflammation
Dietary fatty acids may affect inflammatory processes through effects on body weight and adipose tissue mass and via an impact on membrane and lipid raft composition and function. Within the cell, membrane-derived fatty acids and their derivatives can influence inflammation by serving as modulators of NF-κB and PPAR-α/γ transcription factor pathways( 55 ), and as precursors for a host of eicosanoid and docosanoid oxidation products produced via the action of epoxygenases, lipoxygenases and cyclo-oxygenases( 56 ). Also, recent advances in the field have uncovered NLRP3 (NACHT, LRR and PYD domains-containing protein 3) inflammasome activation and IL-1β signalling as a key sensor of SFA-mediated metabolic stress in obesity and T2DM( 57 ) and EPA- and DHA-derived resolvins and protectins that actively ameliorate a pro-inflammatory state( 58 ). Obesity significantly reduced DHA-derived 17-hydroxydocosahexaenoic acid, a resolvin D1 precursor, and protectin D1 in adipose tissue, which may in turn have pro-inflammatory consequences( 59 ). Also, dietary EPA/DHA supplementation within an obesogenic dietary challenge restored endogenous adipose resolvin and protectin biosynthesis, concomitant with attenuated adipose inflammation and insulin resistance( 59 ). An elegant human study showed that a relatively high dose of LC n-3 PUFA augmented anti-inflammatory eicosanoid secretion and attenuated inflammatory gene expression in the subcutaneous adipose tissue of severely obese non-diabetic patients( 60 ). Thus, there is much recent information on novel mechanisms of action by which dietary fatty acids of different classes influence inflammatory processes, some acting in pro-inflammatory and others in anti-inflammatory or inflammation-resolving ways.
There is some evidence, albeit not always consistent, for pro-inflammatory effects of dietary SFA( 1 ). Much of this evidence comes from either in vitro or cross-sectional studies, and there are limited randomised controlled trial (RCT) examining changes in SFA intake and inflammation in humans. The LIPGENE RCT investigated the effects of substituting dietary SFA with MUFA or as part of a low-fat diet, with or without LC n-3 PUFA supplementation, in subjects with the MetS( 61 ). While a low-fat n-3 PUFA-enriched diet significantly reduced the risk of the MetS( 62 ), modifying dietary fat had no significant effect on key biomarkers of cardiometabolic risk including insulin sensitivity and the plasma inflammatory markers assessed( 63 ). However, there was clear modulation of NF-κB-mediated inflammation and oxidative stress in the postprandial state according to lipid composition( 64 , 65 ). This lack of impact of LC n-3 PUFA on the fasting plasma inflammasome in humans( 66 ) is in line with previous human studies( 63 , 67 ), but contradicts the effects observed in a wide variety of cell and animal models. However, as will be discussed in the section ‘Translating research into public health benefit and novel products’, it is difficult to know whether the output from these RCT truly demonstrates a lack of efficacy or reflects insufficient dose and/or duration or poor selection of fasting plasma biomarkers of inflammation, which are insensitive to physiologically meaningful changes occurring in key metabolic tissues such as the liver and adipose tissue.
As with other common phenotypes, there is evidence emerging that the associations between dietary fat composition and inflammation are influenced by common gene variants( 68 ). In the LIPGENE study, SNP in the genes encoding the anti-inflammatory peptide adiponectin (ADIPOQ) and its receptor (ADIPOR1) have been shown to interact with SFA to modulate the effect of dietary fat modification on insulin resistance( 69 ), and using a case–control approach, it was observed that a common SNP of the C3 gene was related to the risk of the MetS, but more importantly, the impact of this was greatly accentuated by high plasma levels of SFA( 70 ). Also, the combination of polymorphisms in genes encoding IL-6, lymphotoxin α (LTA) and TNF-α had an additive effect, which interacted with plasma fatty acid status to modulate the risk of the MetS( 71 ). Grimble et al. ( 72 ) demonstrated that the ability of LC n-3 PUFA to decrease TNF-α production is influenced by inherent TNF-α production and by polymorphisms in the TNF-α and LTA genes.
Inflammation in the postprandial state is likely to contribute to the pathological impact of exaggerated postprandial lipaemia( 73 ). Although there has been some investigation of the impact of meal fatty acid composition on non-fasting inflammatory biomarkers, the data thus far remain inconsistent( 73 ). It has been reported that in overweight men, plasma IL-6, TNF-α and soluble vascular adhesion molecule-1 concentrations decreased after an n-6 PUFA-rich meal, while markers were increased after a SFA-rich meal( 74 ). In contrast, Manning et al. ( 75 ) showed that high-fat meals increased IL-6, independent of the type of fatty acid, and had no impact on IL-8 and TNF-α concentrations.
Dietary carbohydrates and inflammation
Besides postprandial lipaemia, postprandial glucose is an independent predictor of diabetes and CVD, an effect which may be mediated through oxidative stress and inflammation( 76 ). Importantly, there appears to be no glycaemic threshold for reduction of either microvascular or macrovascular complications. The progressive relationship between plasma glucose and the risk of CVD extends well below the diabetic threshold( 77 , 78 ).
Acute glucose variations from peaks to nadirs include postprandial glucose excursions that can be described by two components. The first component, which is the duration of the postprandial glucose increment, is a major contributor to chronic sustained hyperglycaemia, while the second component, which is the magnitude of the postprandial rise, is more often a reflection of glucose variability. It is difficult to discriminate between the contributions of these two components of dysglycaemia. It seems that both contribute to the two main mechanisms that lead to diabetic and cardiovascular complications, namely excessive protein glycation and activation of oxidative stress and inflammation.
Although mechanistic evidence indicates a positive correlation between the glycaemic index and load of the diet and low-grade inflammation, intervention studies, to date, do not convincingly support this. Hu et al. ( 79 ) observed a stepwise relationship between dietary glycaemic index and oxidative stress markers in healthy adults. Furthermore, high-glycaemic index carbohydrates increase NF-κB activation and NF-κB binding in mononuclear cells of young, lean healthy subjects( 80 ). Diets low in glycaemic load and high in whole grains may have a protective effect against systemic inflammation in diabetic patients, as reviewed elsewhere( 81 ). Consistent with this, epidemiological studies have shown an inverse relationship between dietary fibre and CRP levels. Both the DASH diet (naturally high in fibre, i.e. 30 g fibre/d) and a fibre-supplemented usual diet (30 g psyllium fibre/d) decreased CRP concentrations in lean normotensive subjects( 82 ). In contrast, a high-carbohydrate, low-fat diet with a relatively high dietary fibre and complex carbohydrate content, within the context of a lifestyle intervention programme, has been shown to reduce diabetes incidence in the long term by 50 %( 83 ). The prominent role of the type of carbohydrate has also been illustrated in studies showing that dietary carbohydrate modification, i.e. an oat/wheat/potato diet, up-regulated sixty-two genes related to stress, cytokine–chemokine-mediated immunity and IL pathways compared with a rye–pasta diet( 84 ). These differences in the inflammatory response have been ascribed to differences in the early insulin response and the resultant late hypoglycaemia in the oat/wheat/potato group.
Taken together, studies have suggested that healthy eating patterns characterised by reduced postprandial glycaemia and lipaemia are associated with reduced concentrations of markers of low-grade inflammation.
Plant bioactive compounds and inflammation
Recent prospective cohort data suggest that improved cognitive function and a reduced risk of age-related neurodegenerative diseases, associated with increased fruit and vegetable intake( 85 – 87 ), may be in large part attributable to intake of specific flavonoids( 87 ), and may involve an effect on inflammatory processes (Table 1). In particular, increased consumption of total flavonoids was positively associated with episodic memory in middle-aged adults( 88 ) and with a reduced rate of cognitive decline in adults aged 70 years and over( 89 ). The anthocyanin group of flavonoids, with certain soft fruits providing the most significant dietary source, has emerged as being particularly potent. In the Nurses' Health Cohort, greater intakes of blueberries and strawberries were associated with slower rates of cognitive decline, with a high intake of soft fruits estimated to delay cognitive ageing by up to 2·5 years( 90 ). Furthermore, a large cross-sectional study has also indicated that total flavonoid intake is inversely correlated with serum CRP concentrations( 91 ). In support of this association, a number of dietary intervention studies have provided evidence that dietary flavonoids are capable of modulating inflammatory cytokines (e.g. TNF-α) and CRP production( 91 – 94 ). However, there are relatively few human RCT investigating the anti-inflammatory and cognitive effects of flavonoids (Table 1).
Table 1.
Dietary flavonoids and inflammation: evidence from epidemiological and intervention studies
| Reference | Subject description | Product | Dosage/duration of intake | Results |
|---|---|---|---|---|
| Chun et al. ( 91 ) | 8335 men and women, age 19 years and over | Total flavonoid | Dietary intake | Reduced plasma concentrations of CRP (25–30 %) in flavonoid consumers v. non-consumers |
| Landberg et al. ( 139 ) | 1194–1598 women | Flavonoid-rich foods | Dietary intake | Reduced plasma concentrations of CRP (21 %), IL-18 (20 %) and TNF-R2 (8 %) for ≥ 1 servings/d v. < 1 serving/month |
| Edirisinghe et al. ( 140 ) | Twenty-four overweight men and women | Strawberry anthocyanins | Single dose/6 h | Reduced concentrations of CRP (13 %) and IL-6 (16 %), 6 h following a high-carbohydrate, moderate-fat meal, but no results observed for TNF-α and IL-1β |
| Steptoe et al. ( 141 ) | Thirty-seven non-smoking men, age 18–55 years | Black tea | 1050 mg tea extract/6 weeks | Decreased platelet activation (mean 5·84 v. 6·60 %) and plasma CRP concentrations (mean 0·76 v. 0·97 mg/l) in the treatment group v. placebo group |
| Karlsen et al. ( 142 ) | 120 men and women, age 40–74 years | Anthocyanin extract from bilberries and blackcurrant | 300 mg/d for 3 weeks | Decreased plasma concentrations of IL-8 (25 %), IFN-α (15 %) and RANTES (15 %) in the treatment group v. placebo group, but no results observed for CRP |
| Oyama et al. ( 92 ) | Thirty healthy smokers, mean age 37 years | Green tea catechins | Control dose 0 mg, middle dose 80 mg and high dose 580 mg; 2-week intervention | Decreased concentrations of 8-OHdG (20 %), IL-6 (42 %) and soluble Fas (25 %) in the high-dose group v. catechin-free group at day 14, but no results observed for IL-1β |
| Widlansky et al. ( 143 ) | Sixty-six men and women, average age 54 years | Black tea | 900 ml/d for 4 weeks | No effects observed for plasma CRP and urinary 8-OHdG concentrations |
| Mellen et al. ( 144 ) | Fifty men and women with coronary disease, mean age 58 years | Muscadine grape seeds | 1300 mg/d for 4 weeks | No effects observed for plasma CRP and IL-6 concentrations |
| Heinz et al. ( 145 ) | 120 women, age 30–79 years | Quercetin | 500–1000 mg/d for 12 weeks | No effects observed for plasma IL-6 or TNF-α concentrations |
CRP, C-reactive protein; TNF-R2, TNF receptor 2; IFN-α, interferon-α; RANTES, regulated on activation, normal T-cell expressed and secreted; 8-OHdG, 8-hydroxydeoxyguanosine.
Although the effects of flavonoids were originally ascribed to an antioxidant action, it is now clear that levels achieved in biological tissues may not be sufficient to act in this way. Evidence indicates that flavonoids are capable of acting in a number of other ways that may result in their targeting of inflammation, including (1) the modulation of intracellular signalling cascades that control neuronal survival, death and differentiation; (2) an impact on gene expression and (3) interacting with the mitochondria( 95 – 98 ). In particular, emerging evidence suggests that dietary flavonoids may exert neuroprotective effects by suppressing the activation of microglia, which mediate inflammatory processes in the CNS (see the earlier section). Although rather complex, the main anti-inflammatory properties of flavonoids include (1) an inhibitory role in the release of cytokines, such as IL-1β and TNF-α, from activated microglia; (2) an inhibitory action against inducible NO synthase induction and subsequent NO production in response to glial activation; (3) an ability to inhibit the activation of NADPH oxidase and subsequent generation of reactive oxygen species in activated glia; and (4) a capacity to down-regulate the activity of pro-inflammatory transcription factors, such as NF-κB, through their influences on a number of glial and neuronal signalling pathways( 99 , 100 ). However, almost all mechanistic studies have been carried out in vitro at rather supraphysiological concentrations, with limited research on animal models and scarce data from human RCT.
Early-life nutrition and inflammation
During development, the human embryo and fetus undergo an enormously complex series of changes in both cell type and cell number. Each of these changes takes place in a strictly choreographed series, and disruption of the process can lead to dramatic and long-lasting consequences. There are many recent summaries of the processes involved( 101 ). In humans, this was most clearly demonstrated in the Second World War, when Dutch women were placed under famine conditions following a railway workers' strike (the Dutch Hunger Winter)( 102 , 103 ). Studies on the offspring of women who were pregnant at this time have shown clearly that women who were pregnant in the first trimester gave birth to babies who would go on to develop a much wider spectrum of health problems than babies born to women who were in the second or third trimester, though these offspring still would continue to show health problems( 104 ).
Factors other than undernutrition can also have both short- and long-term consequences. Of particular relevance, obese women give birth to babies with a higher risk of both small for gestational age and large for gestational age, of complications at birth and of developing the MetS( 105 , 106 ). All these cannot be explained by postnatal events, and are at least partly explained by the phenomenon known as ‘fetal programming’ or ‘developmental programming’( 107 , 108 ). This hypothesis states that nutrition-related exposures in utero ‘programme’ the baby to expect a postnatal nutritional environment, and if a different one is experienced, then there is a risk of the development of metabolic complications. There have been refinements to the basic hypothesis and to our understanding of the mechanisms involved( 109 – 112 ); however, the fundamental observations remain unchanged and unchallenged. How these associations are mediated is not yet clearly demonstrated, but several hypotheses are being tested. There is substantial support for nutrition altering the epigenetic profile of the offspring, including hypermethylation of cytokine receptors. Evidence indicates that low Fe status at birth, which is associated with impaired lung function in children, can result in reduced nephron number and decreased levels of cell-cycle enzymes( 113 ), suggesting that nutritional deficiency during a critical phase of development can inhibit organ growth. This fits with data showing that thymus growth is reduced, and that this leads to changes in the cytokine profile.
Maternal obesity also has dramatic effects on pregnancy outcome. Again, there are many detailed reviews dealing with this topic( 107 ). The mechanisms seem to involve inflammatory responses, and increased cytokine levels have been reported in the placenta and cord blood of babies born to obese mothers. Whether, in humans, the placenta alone is responsible is not clear, and it is quite likely that adipose tissue itself, which becomes infiltrated with macrophages, will produce increased amounts of pro-inflammatory cytokines( 114 ). The situation becomes more complex in obesity, because in addition to the cytokines, or possibly because of the cytokines, inflammation results in changes in Fe metabolism( 115 ), and there is abundant evidence to show that decreased Fe status during pregnancy has adverse effects on the offspring( 116 – 119 ). Obesity results in increased hepcidin production( 120 , 121 ). Hepcidin is a negative regulator of Fe absorption( 122 ), and lower Fe status in the mother before birth is associated with an increased risk of wheezing in the children (W Bright, G Devereux, HJ McArdle, unpublished results). Thus, decreased Fe status may be an additional risk factor in obese mothers.
Translating research into public health benefit and novel products
Biomarkers of inflammation in human nutrition studies
As explained previously, inflammation is a normal process, and there are a large number of cells and mediators involved; measurement of these is often used as a ‘biomarker’ of inflammation, i.e. an indicator that inflammation is occurring. These cells and mediators are largely involved in, or are produced as a result of, the inflammatory process, irrespective of the trigger or its location in the body, and are common to all inflammatory situations( 1 ). To monitor inflammation in a meaningful way, the markers used must be valid: they must reflect the inflammatory process under study and must be predictive of future health status. The range of potential biomarkers of inflammation was considered by an Expert Group of ILSI Europe, with the aim of identifying robust and predictive markers, or patterns or clusters of markers, which can be used to assess inflammation in human nutrition studies in the general population; markers indicative of a specific inflammatory pathology (e.g. rheumatoid arthritis) and/or in less accessible tissue sites (e.g. in lung lavage fluid or in intestinal biopsy material) were not considered to be relevant to more healthy populations( 7 ). Currently, there is no consensus as to which markers best represent low-grade inflammation( 6 ), or differentiate between acute and chronic inflammation or between the various phases of inflammatory responses( 7 ). Therefore, a range of blood cellular markers (e.g. total leucocytes, granulocytes and activated monocytes) and soluble mediators (cytokines and chemokines (TNF, IL-1, IL-6, IL-8, CC chemokine ligand 2 (CCL2), CCL3, CCL5), adhesion molecules (vascular cell adhesion molecule-1, intercellular adhesion molecule-1, E-selectin), adipokines (adiponectin) and acute-phase proteins (CRP, serum amyloid A, fibrinogen)) are frequently measured. Some of these are associated with future risk of CVD and with cardiometabolic health( 1 , 6 , 7 ). However, there are several key issues concerning the use of these markers as determinants of low-grade inflammation. First, they are non-specific acute-phase response and pro-inflammatory response markers, and, by themselves, do not represent metabolic low-grade inflammation. Second, even in healthy individuals, there is wide variation in the measurements made. This is because there are a number of modifying factors that affect the concentration of an inflammatory marker at a given time. These modifying factors include age, diet, body fatness, physical fitness and genetics, among others( 1 ).
One can question whether static measurements of single or complex biomarkers are truly informative about health status, reasoning from the concept that health is defined by the ability to adequately adapt to everyday challenges( 123 ). Measuring the concentration of inflammatory markers in the bloodstream under basal conditions is probably less informative and relatively insensitive compared with measurements of the concentration change in response to a challenge. A number of inflammatory challenges have been described. These include an oral glucose load( 80 ), an oral fat load( 124 , 125 ), acute exercise, administration of bacterial LPS( 126 ), exposure to UV irradiation( 127 , 128 ) and vaccination. Although each of these challenges has been used in nutritional studies, many are poorly standardised, limiting the comparisons that can be made. Most often, the markers measured in response to challenges are those mentioned earlier in the context of static basal measurements. Currently, a number of large European consortia, i.e. PhenFlex (http://www.nugo.org/everyone/42 701/7/0/30), NutriTech (http://www.nugo.org/nutritech) and BioClaims (http://bioclaims.uib.es), are developing and validating the metabolic challenge test concept for application in the assessment of health status, including the study of inflammatory process markers( 129 ).
The past decade has seen huge growth in innovation in ‘omics’ technologies that provide enormous opportunities for high-throughput biological sample characterisation, with patterns and clusters of markers (signatures or fingerprints) emerging as robust biomarkers of inflammation( 89 , 130 ). The enormous challenge in this era of big data is making biological sense of different levels of data, including the transcriptome, proteome, metabolome and clinical chemistry data. Novel data analysis methodologies, such as machine learning, offer large potential for identifying relevant data for specific biological outcomes based on complex multidimensional datasets( 131 ). In addition, bioinformatic tools have been developed to interpret these complex data in the context of existing biological knowledge in the literature and databases, also termed network biology( 132 , 133 ). These technologies will be instrumental to the discovery of relevant biomarker signatures that reflect ‘low-grade inflammation’ based on inflammatory response networks connected to organ-specific metabolic derailment.
With the coming of age of the ‘omics’ technologies and bioinformatic tools, a large increase in the number, specificity and sensitivity of candidate biomarkers of inflammation can be expected in the next decade( 134 ). A screening of the ‘Thomson Reuters IntegritySM Biomarker Database’ reveals that as of May 2014, 945 candidate biomarkers of inflammation have been described, of which only seventeen, including CRP, TNF-α, serotransferrin and erythrocyte sedimentation rate, have been developed into biomarker assays approved and recommended by regulatory bodies for use in clinical studies. This represents the classical biomarker gap: many candidate biomarkers are identified based on preclinical and clinical studies; however, due to relatively limited efforts in validation and assay development, these are subsequently not further developed( 135 ). To accelerate biomarker development, a paradigm shift in this area is needed; instead of single companies developing a single biomarker assay, pre-competitive collaborations between different industrial, academic, and research and technology organisations have the advantage of a more efficient development process time- and cost-wise, by combining a wide diversity of expertise, in the development of a harmonised, standardised and accepted assay. In these consortia, ideally, companies from nutrition, pharma and diagnostics join forces in a pre-competitive way.
A major concerted effort should comprise (1) the discovery of context-based biomarker signatures for the assessment of the status of low-grade inflammation, (2) the development of challenge tests that determine the inflammatory response functionality in the context of metabolic stress-induced low-grade inflammation, and (3) the development of the identified biomarkers towards application in a clinically accepted assay, with normative data.
Low-grade inflammation and health claims
The European Food Safety Authority (EFSA) guidance document on scientific requirements for health claims related to gut and immune function( 136 ) specifically states that chronic inflammation is associated with the development of a number of diseases, and that ‘altering levels of markers of inflammation might indicate a beneficial effect in the context of “a reduction of disease risk claim”, if it can be demonstrated that altering the levels of inflammatory markers is accompanied by a reduced incidence of a disease for a specific dietary intervention’. No additional specificity is added for chronic low-grade inflammation. At present, the European Union health claim register (http://ec.europa.eu/nuhclaims) does not contain any authorised or non-authorised health claims that specifically address the health benefit area of suppression or control of low-grade inflammation.
To build strong health claims on nutrition for improving inflammation control in the future, one of the key focus areas should be the need for clinically relevant prognostic marker(s) or marker signatures that reflect the inflammatory state in a context-specific manner, which have been well validated and for which a robust standardised assay is available. The lack of health claims is probably attributable to the fact that, although numerous biologically plausible mechanisms have been established to explain inflammation–disease associations, no single biomarker or cluster of biomarkers of inflammation has yet been robustly demonstrated to be sufficiently predictive of future disease. Based on the EFSA guidance on this topic( 136 ) and the classification of candidate biomarkers as described by the expert group of ILSI Europe( 137 ), the suggested strategy for building a EFSA health claim dossier (Fig. 2) comprises (1) a definition of the composition of the product; (2) a well-founded selection of the target population; (3) the selection of a clinically relevant composite biomarker panel representing inflammation as well as the selected health benefit (or disease risk) endpoints; and (4) a number of sufficiently powered and well-controlled human studies assessing the effect of the test material (nutrient, food, product) on the relevant biomarkers in the relevant target population.
Fig. 2.
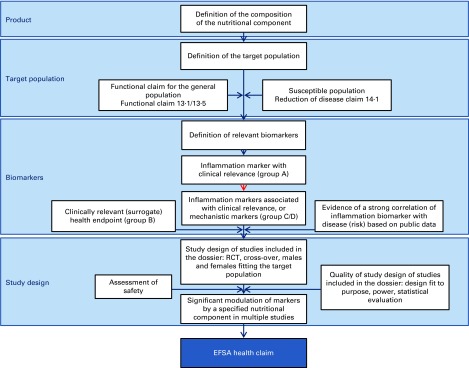
Schematic of topics to be addressed when building a dossier for a European Food Safety Authority (EFSA) health claim on control of chronic low-grade inflammation. The blue boxes indicate the main topics to be addressed; the white boxes state the actual content topics. Building a strong EFSA health claim dossier requires (1) a definition of the composition of the nutritional component including manufacturing procedures in scope and out of scope for the claim, (2) a clear definition of the target population, being the general population or a specific subpopulations at risk, including the defining parameters, (3) a definition of biomarkers measured to assess the health effects of the nutritional component, including a description of the proof of clinical relevance, or the clinical validity of the combination of inflammation biomarkers and related clinically relevant biomarkers for health benefit endpoints associated with the health claim, and (4) a full description of clinical study design for all studies included in the dossier, including statistical power analysis and safety evaluation. The red arrow indicates the primary hurdle for functional health claims in the area of chronic low-grade inflammation, which is the lack of (combinations of) inflammation biomarkers with established and therefore accepted clinical relevance. This is primarily the consequence of inflammatory responses being non-specific normal physiological responses to tissue damage, and discrimination between normal and abnormal levels or combinations has not been well established in relation to chronic low-grade inflammation. The description of the classification of clinical relevance of biomarkers (categories A–D) was adapted from Albers et al. ( 137 ). RCT, randomised controlled trial. (A colour version of this figure can be found online at http://www.journals.cambridge.org/bjn).
Summary and suggestions for the way forward
Inflammation is a normal component of host defence; however, elevated unresolved chronic inflammation is a core perturbation in a range of chronic diseases and is an important determinant of the pathological impact of excess adiposity. Cell, animal and human epidemiological studies have identified a number of potential diet derived anti- and pro-inflammatory components, some of which have been discussed here; this topic has been dealt with more extensively elsewhere( 1 , 6 , 7 ). Available human RCT evidence is more limited and sometimes conflicting or inconsistent, in part attributable to under-powered studies where inflammation was not specified as a primary study outcome. Furthermore, research tends to take a reductionist approach and examine the impact of individual dietary components in isolation, despite the identification of numerous potential diet-derived anti-inflammatory and inflammation-resolving bioactive compounds, with likely additive or synergistic effects. There is a need to take a more holistic approach and consider the impact of combinations of components of foods and dietary patterns, with a likely greater overall benefit than each single component might have on its own. Moreover, although it is evident that the inflammatory response is highly variable, a full understanding of the source of heterogeneity is distinctly lacking. More extensive profiling of participants in human studies and consideration of potential key variables such as age, sex, genotype and lifestyle factors in statistical models is needed in order to help understand the aetiology of the variation in both inflammation itself and in its response to dietary change. This approach will also allow for the identification of population subgroups that may particularly benefit from interventions that target inflammation.
Establishing and quantifying reliable, precise diet–inflammation–health associations is reliant on the availability of approved, standardised biomarkers with normative data for use in human observation studies and RCT. Biomarker research is a highly active area with significant advances to be expected in the coming years( 138 ). Rather than rely on a limited number of generic markers common to both acute and low-grade chronic inflammation, future inflammation ‘testing’ is likely to involve quantifying clusters or signatures of markers with some tissue specificity. Such biomarkers should generally be measured in the challenged state( 1 ), with the choice of the physiological stressor dependent on the tissue, and research question of interest. The biomarkers assessed are likely to include those already typically measured (cytokines, chemokines, soluble adhesion molecules, etc.), but are also likely to include tissue-specific markers and fingerprints based on gene expression profiles (e.g. in blood mononuclear cells), cell or plasma proteomics, and microRNA.
The research focus on the establishment of a robust diet–inflammation–health association is justifiable, considering the substantial role of low-grade inflammation in the pathology of numerous chronic diseases, thereby making it a key future preventative and therapeutic target.
Acknowledgements
The present review results from a workshop organised by the European Branch of ILSI Europe. This publication was coordinated by Dr Peter Putz, Scientific Project Manager at ILSI Europe. The workshop was funded by the ILSI Europe Obesity and Diabetes Task Force, the ILSI Europe Metabolic Imprinting Task Force, ILSI Brazil, ILSI North America and ILSI Southeast Asia Region. Industry members of the task forces are listed on the ILSI Europe website at http://www.ilsi.eu. For further information about ILSI Europe, please email info@ilsieurope.be or call +32 2 771 00 14. The opinions expressed herein and the conclusions of this publication are those of the authors and do not necessarily represent the views of ILSI Europe nor those of its member companies.
The authors thank the members of the Organising Committee: Professor Jean-Louis Bresson and Professor Ascension Marcos for their invaluable contribution to this work through their enthusiastic and generous participation. The authors are also grateful to Ms Belinda Antonio, Ms Toula Aslanidis, Ms Ruth Marquet, Mr Pierre Mouelhi and Mr Alex Rankin for their administrative support. The authors thank Dr Lorraine Gambling, Dr Helen Hayes and Ms Val Stevens for technical assistance.
The authors' contributions are as follows: A. M. M. and P. C. C. had responsibility for producing the final version of the manuscript. All authors contributed to the discussion and had input into the writing of the manuscript.
L. S. is an employee of Newtricious and S. V. is an employee of Mondelēz International. A. B. was an employee of ILSI Europe. The remaining authors have no conflicts of interest.
Abbreviations: CNS, central nervous system; ILSI, International Life Sciences Institute; LPS, lipopolysaccharide; MetS, metabolic syndrome; NAFLD, non-alcoholic fatty liver disease; RCT, randomised controlled trial; T2DM, type 2 diabetes mellitus
References
- 1. Calder PC, Ahluwalia N, Albers R, et al. (2013) A consideration of biomarkers to be used for evaluation of inflammation in human nutritional studies. Br J Nutr 109, S1–S34. [DOI] [PubMed] [Google Scholar]
- 2. Ortega-Gomez A, Perretti M & Soehnlein O (2013) Resolution of inflammation, an integrated view. EMBO Mol Med 5, 661–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Serhan CN, Chiang N & Dyke TEV (2008) Resolving inflammation, dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol 8, 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hotamisligil GS (2006) Inflammation and metabolic disorders. Nature 444, 860–867. [DOI] [PubMed] [Google Scholar]
- 5. Libby P (2002) Inflammation in atherosclerosis. Nature 420, 868–874. [DOI] [PubMed] [Google Scholar]
- 6. Calder PC, Ahluwalia N, Brouns F, et al. (2011) Dietary factors and low-grade inflammation in relation to overweight and obesity. Br J Nutr 106, S1–S78. [DOI] [PubMed] [Google Scholar]
- 7. Calder PC, Albers R, Antoine JM, et al. (2009) Inflammatory disease processes and interactions with nutrition. Br J Nutr 101, 1–45. [DOI] [PubMed] [Google Scholar]
- 8. Hallenbeck JM, Hansson GK & Becker KJ (2005) Immunology of ischemic vascular disease, plaque to attack. Trends Immunol 26, 550–556. [DOI] [PubMed] [Google Scholar]
- 9. Hansson GK (2005) Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med 352, 1685–1695. [DOI] [PubMed] [Google Scholar]
- 10. Harford KA, Reynolds CM, McGillicuddy FC, et al. (2011) Fats, inflammation and insulin resistance, insights to the role of macrophage and T-cell accumulation in adipose tissue. Proc Nutr Soc 70, 408–417. [DOI] [PubMed] [Google Scholar]
- 11. Lumeng CN, DelProposto JB, Westcott DJ, et al. (2008) Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes 57, 3239–3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Weisberg SP, McCann D, Desai M, et al. (2003) Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112, 1796–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wildman RP, Muntner P, Reynolds K, et al. (2008) The obese without cardiometabolic risk factor clustering and the normal weight with cardiometabolic risk factor clustering, prevalence and correlates of 2 phenotypes among the US population (NHANES 1999–2004). Arch Intern Med 168, 1617–1624. [DOI] [PubMed] [Google Scholar]
- 14. Barbarroja N, Lopez-Pedrera R, Mayas MD, et al. (2010) The obese healthy paradox, is inflammation the answer? Biochem J 430, 141–149. [DOI] [PubMed] [Google Scholar]
- 15. Fujii H & Kawada N (2012) Inflammation and fibrogenesis in steatohepatitis. J Gastroenterol 47, 215–225. [DOI] [PubMed] [Google Scholar]
- 16. Smith BW & Adams LA (2011) Nonalcoholic fatty liver disease and diabetes mellitus, pathogenesis and treatment. Nat Rev Endocrinol 7, 456–465. [DOI] [PubMed] [Google Scholar]
- 17. Gill SR, Pop M, Deboy RT, et al. (2006) Metagenomic analysis of the human distal gut microbiome. Science 312, 1355–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Power SE, O'Toole PW, Stanton C, et al. (2014) Intestinal microbiota, diet and health. Br J Nutr 111, 387–402. [DOI] [PubMed] [Google Scholar]
- 19. Toole PJO & Claesson MJ (2010) Gut microbiota, changes throughout the lifespan from infancy to elderly. Int Dairy J 20, 281–291. [Google Scholar]
- 20. Kurashima Y, Goto Y & Kiyono H (2013) Mucosal innate immune cells regulate both gut homeostasis and intestinal inflammation. Eur J Immunol 43, 3108–3115. [DOI] [PubMed] [Google Scholar]
- 21. Cani PD & Delzenne NM (2009) Interplay between obesity and associated metabolic disorders, new insights into the gut microbiota. Curr Opin Pharmacol 9, 737–743. [DOI] [PubMed] [Google Scholar]
- 22. Collado MC, Isolauri E, Laitinen K, et al. (2008) Distinct composition of gut microbiota during pregnancy in overweight and normal-weight women. Am J Clin Nutr 88, 894–899. [DOI] [PubMed] [Google Scholar]
- 23. Miller SI, Ernst RK & Bader MW (2005) LPS, TLR4 and infectious disease diversity. Nat Rev Microbiol 3, 36–46. [DOI] [PubMed] [Google Scholar]
- 24. Serino M, Luche E, Gres S, et al. (2012) Metabolic adaptation to a high-fat diet is associated with a change in the gut microbiota. Gut 61, 543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Belkaid Y & Hand TW (2014) Role of the microbiota in immunity and inflammation. Cell 157, 121–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. de Sousa MLF, Grzeskowiak LM, de Sales Teixeira TF, et al. (2014) Intestinal microbiota and probiotics in celiac disease. Clin Microbiol Rev 27, 482–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dunne JL, Triplett EW, Gevers D, et al. (2014) The intestinal microbiome in type 1 diabetes. Clin Exp Immunol 177, 30–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Miller FW, Pollard KM, Parks CG, et al. (2012) Criteria for environmentally associated autoimmune diseases. J Autoimmun 39, 253–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fetissov SO, Sinno MH, Coeffier M, et al. (2008) Autoantibodies against appetite-regulating peptide hormones and neuropeptides, putative modulation by gut microflora. Nutrition 24, 348–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tuohy KM, Fava F & Viola R (2014) ‘The way to a man's heart is through his gut microbiota’ – dietary pro- and prebiotics for the management of cardiovascular risk. Proc Nutr Soc 73, 172–185. [DOI] [PubMed] [Google Scholar]
- 31. Liu L, Li L, Min J, et al. (2012) Butyrate interferes with the differentiation and function of human monocyte-derived dendritic cells. Cell Immunol 277, 66–73. [DOI] [PubMed] [Google Scholar]
- 32. Millard AL, Mertes PM, Ittelet D, et al. (2002) Butyrate affects differentiation, maturation and function of human monocyte-derived dendritic cells and macrophages. Clin Exp Immunol 130, 245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Arpaia N, Campbell C, Fan X, et al. (2013) Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504, 451–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ohira H, Fujioka Y, Katagiri C, et al. (2013) Butyrate attenuates inflammation and lipolysis generated by the interaction of adipocytes and macrophages. J Atheroscler Thromb 20, 425–442. [DOI] [PubMed] [Google Scholar]
- 35. Al-Lahham SH, Roelofsen H, Priebe M, et al. (2010) Regulation of adipokine production in human adipose tissue by propionic acid. Eur J Clin Invest 40, 401–407. [DOI] [PubMed] [Google Scholar]
- 36. Boesjes M & Brufau G (2014) Metabolic effects of bile acids in the gut in health and disease. Curr Med Chem 21, 2822–2829. [DOI] [PubMed] [Google Scholar]
- 37. Shen W, Gaskins HR & McIntosh MK (2014) Influence of dietary fat on intestinal microbes, inflammation, barrier function and metabolic outcomes. J Nutr Biochem 25, 270–280. [DOI] [PubMed] [Google Scholar]
- 38. Dantzer R & Kelley KW (2007) Twenty years of research on cytokine-induced sickness behavior. Brain Behav Immun 21, 153–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dantzer R, O'Connor JC, Freund GG, et al. (2008) From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 9, 46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hart BL (1990) Behavioral adaptations to pathogens and parasites: five strategies. Neurosci Biobehav Rev 14, 273–294. [DOI] [PubMed] [Google Scholar]
- 41. Teeling JL, Felton LM, Deacon RM, et al. (2007) Sub-pyrogenic systemic inflammation impacts on brain and behavior, independent of cytokines. Brain Behav Immun 21, 836–850. [DOI] [PubMed] [Google Scholar]
- 42. Teeling JL, Cunningham C, Newman TA, et al. (2010) The effect of non-steroidal anti-inflammatory agents on behavioural changes and cytokine production following systemic inflammation: implications for a role of COX-1. Brain Behav Immun 24, 409–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Krstic D, Madhusudan A, Doehner J, et al. (2012) Systemic immune challenges trigger and drive Alzheimer-like neuropathology in mice. J Neuroinflamm 9, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Perry VH, Cunningham C & Holmes C (2007) Systemic infections and inflammation affect chronic neurodegeneration. Nat Rev Immunol 7, 161–167. [DOI] [PubMed] [Google Scholar]
- 45. Perry VH & Teeling J (2013) Microglia and macrophages of the central nervous system: the contribution of microglia priming and systemic inflammation to chronic neurodegeneration. Semin Immunopathol 35, 601–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hennessy AA, Barrett E, Ross RP, et al. (2012) The production of conjugated alpha-linolenic, gamma-linolenic and stearidonic acids by strains of bifidobacteria and propionibacteria. Lipids 47, 313–327. [DOI] [PubMed] [Google Scholar]
- 47. Holmes C, Cunningham C, Zotova E, et al. (2011) Proinflammatory cytokines, sickness behavior, and Alzheimer disease. Neurology 77, 212–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Puntener U, Booth SG, Perry VH, et al. (2012) Long-term impact of systemic bacterial infection on the cerebral vasculature and microglia. J Neuroinflamm 9, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Thiel A, Cechetto DF, Heiss WD, et al. (2014) Amyloid burden, neuroinflammation, and links to cognitive decline after ischemic stroke. Stroke 45, 2825–2829. [DOI] [PubMed] [Google Scholar]
- 50. Niranjan R (2013) Molecular basis of etiological implications in Alzheimer's disease: focus on neuroinflammation. Mol Neurobiol 48, 412–428. [DOI] [PubMed] [Google Scholar]
- 51. Liu L & Chan C (2014) The role of inflammasome in Alzheimer's disease. Ageing Res Rev 15, 6–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ferreira ST, Clarke JR, Bomfim TR, et al. (2014) Inflammation, defective insulin signaling, and neuronal dysfunction in Alzheimer's disease. Alzheimers Dement 10, S76–S83. [DOI] [PubMed] [Google Scholar]
- 53. Heneka MT, Kummer MP & Latz E (2014) Innate immune activation in neurodegenerative disease. Nat Rev Immunol 14, 463–477. [DOI] [PubMed] [Google Scholar]
- 54. Heneka MT, Golenbock DT & Latz E (2015) Innate immunity in Alzheimer's disease. Nat Immunol 16, 229–236. [DOI] [PubMed] [Google Scholar]
- 55. Calder PC (2013) Long chain fatty acids and gene expression in inflammation and immunity. Curr Opin Clin Nutr Metab Care 16, 425–433. [DOI] [PubMed] [Google Scholar]
- 56. Wall R, Ross RP, Fitzgerald GF, et al. (2010) Fatty acids from fish: the anti-inflammatory potential of long-chain omega-3 fatty acids. Nutr Rev 68, 280–289. [DOI] [PubMed] [Google Scholar]
- 57. Vandanmagsar B, Youm YH, Ravussin A, et al. (2011) The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med 17, 179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Norling LV & Serhan CN (2010) Profiling in resolving inflammatory exudates identifies novel anti-inflammatory and pro-resolving mediators and signals for termination. J Intern Med 268, 15–24. [DOI] [PubMed] [Google Scholar]
- 59. Neuhofer A, Zeyda M, Mascher D, et al. (2013) Impaired local production of proresolving lipid mediators in obesity and 17-HDHA as a potential treatment for obesity-associated inflammation. Diabetes 62, 1945–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Itariu BK, Zeyda M, Hochbrugger EE, et al. (2012) Long-chain n-3 PUFAs reduce adipose tissue and systemic inflammation in severely obese nondiabetic patients: a randomized controlled trial. Am J Clin Nutr 96, 1137–1149. [DOI] [PubMed] [Google Scholar]
- 61. Shaw DI, Tierney AC, McCarthy S, et al. (2009) LIPGENE food-exchange model for alteration of dietary fat quantity and quality in free-living participants from eight European countries. Br J Nutr 101, 750–759. [DOI] [PubMed] [Google Scholar]
- 62. Paniagua JA, Perez-Martinez P, Gjelstad IM, et al. (2011) A low-fat high-carbohydrate diet supplemented with long-chain n-3 PUFA reduces the risk of the metabolic syndrome. Atherosclerosis 218, 443–450. [DOI] [PubMed] [Google Scholar]
- 63. Tierney AC, McMonagle J, Shaw DI, et al. (2011) Effects of dietary fat modification on insulin sensitivity and on other risk factors of the metabolic syndrome – LIPGENE: a European randomized dietary intervention study. Int J Obes (Lond) 35, 800–809. [DOI] [PubMed] [Google Scholar]
- 64. Cruz-Teno C, Perez-Martinez P, Delgado-Lista J, et al. (2012) Dietary fat modifies the postprandial inflammatory state in subjects with metabolic syndrome: the LIPGENE study. Mol Nutr Food Res 56, 854–865. [DOI] [PubMed] [Google Scholar]
- 65. Pena-Orihuela P, Camargo A, Rangel-Zuniga OA, et al. (2013) Antioxidant system response is modified by dietary fat in adipose tissue of metabolic syndrome patients. J Nutr Biochem 24, 1717–1723. [DOI] [PubMed] [Google Scholar]
- 66. Robinson LE & Mazurak VC (2013) n-3 Polyunsaturated fatty acids: relationship to inflammation in healthy adults and adults exhibiting features of metabolic syndrome. Lipids 48, 319–332. [DOI] [PubMed] [Google Scholar]
- 67. Vessby B, Uusitupa M, Hermansen K, et al. (2001) Substituting dietary saturated for monounsaturated fat impairs insulin sensitivity in healthy men and women: The KANWU Study. Diabetologia 44, 312–319. [DOI] [PubMed] [Google Scholar]
- 68. Madden J, Williams CM, Calder PC, et al. (2011) The impact of common gene variants on the response of biomarkers of cardiovascular disease (CVD) risk to increased fish oil fatty acids intakes. Annu Rev Nutr 31, 203–234. [DOI] [PubMed] [Google Scholar]
- 69. Ferguson JF, Phillips CM, Tierney AC, et al. (2010) Gene–nutrient interactions in the metabolic syndrome: single nucleotide polymorphisms in ADIPOQ and ADIPOR1 interact with plasma saturated fatty acids to modulate insulin resistance. Am J Clin Nutr 91, 794–801. [DOI] [PubMed] [Google Scholar]
- 70. Phillips CM, Goumidi L, Bertrais S, et al. (2009) Complement component 3 polymorphisms interact with polyunsaturated fatty acids to modulate risk of metabolic syndrome. Am J Clin Nutr 90, 1665–1673. [DOI] [PubMed] [Google Scholar]
- 71. Phillips CM, Goumidi L, Bertrais S, et al. (2010) Additive effect of polymorphisms in the IL-6, LTA, and TNF-α genes and plasma fatty acid level modulate risk for the metabolic syndrome and its components. J Clin Endocrinol Metab 95, 1386–1394. [DOI] [PubMed] [Google Scholar]
- 72. Grimble RF, Howell WM, O'Reilly G, et al. (2002) The ability of fish oil to suppress tumor necrosis factor alpha production by peripheral blood mononuclear cells in healthy men is associated with polymorphisms in genes that influence tumor necrosis factor alpha production. Am J Clin Nutr 76, 454–459. [DOI] [PubMed] [Google Scholar]
- 73. Jackson KG, Poppitt SD & Minihane AM (2012) Postprandial lipemia and cardiovascular disease risk: interrelationships between dietary, physiological and genetic determinants. Atherosclerosis 220, 22–33. [DOI] [PubMed] [Google Scholar]
- 74. Masson CJ & Mensink RP (2011) Exchanging saturated fatty acids for (n-6) polyunsaturated fatty acids in a mixed meal may decrease postprandial lipemia and markers of inflammation and endothelial activity in overweight men. J Nutr 141, 816–821. [DOI] [PubMed] [Google Scholar]
- 75. Manning PJ, Sutherland WH, McGrath MM, et al. (2008) Postprandial cytokine concentrations and meal composition in obese and lean women. Obesity (Silver Spring) 16, 2046–2052. [DOI] [PubMed] [Google Scholar]
- 76. Blaak EE, Antoine J, Benton D, et al. (2012) Impact of postprandial glycaemia on health and prevention of disease. Obes Rev 13, 923–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Diabetes Control and Complications Trial Research Group (1996) The absence of a glycemic threshold for the development of long-term complications: the perspective of the Diabetes Control and Complications Trial. Diabetes 45, 1289–1298. [PubMed] [Google Scholar]
- 78. Diabetes Control and Complications Trial Research Group (1995) The relationship of glycemic exposure (HbA1c) to the risk of development and progression of retinopathy in the diabetes control and complications trial. Diabetes 44, 968–983. [PubMed] [Google Scholar]
- 79. Hu Y, Block G, Norkus EP, et al. (2006) Relations of glycemic index and glycemic load with plasma oxidative stress markers. Am J Clin Nutr 84, 70–77. [DOI] [PubMed] [Google Scholar]
- 80. Dickinson S, Hancock DP, Petocz P, et al. (2008) High-glycemic index carbohydrate increases nuclear factor-κB activation in mononuclear cells of young, lean healthy subjects. Am J Clin Nutr 87, 1188–1193. [DOI] [PubMed] [Google Scholar]
- 81. Qi L & Hu FB (2007) Dietary glycemic load, whole grains, and systemic inflammation in diabetes: the epidemiological evidence. Curr Opin Lipidol 18, 3–8. [DOI] [PubMed] [Google Scholar]
- 82. King DE, Egan BM, Woolson RF, et al. (2007) Effect of a high-fiber diet vs a fiber-supplemented diet on C-reactive protein level. Arch Intern Med 167, 502–506. [DOI] [PubMed] [Google Scholar]
- 83. den Boer AT, Herraets IJ, Stegen J, et al. (2013) Prevention of the metabolic syndrome in IGT subjects in a lifestyle intervention: results from the SLIM study. Nutr Metab Cardiovasc Dis 23, 1147–1153. [DOI] [PubMed] [Google Scholar]
- 84. Kallio P, Kolehmainen M, Laaksonen DE, et al. (2007) Dietary carbohydrate modification induces alterations in gene expression in abdominal subcutaneous adipose tissue in persons with the metabolic syndrome: the FUNGENUT Study. Am J Clin Nutr 85, 1417–1427. [DOI] [PubMed] [Google Scholar]
- 85. Sofi F, Abbate R, Gensini GF, et al. (2010) Accruing evidence on benefits of adherence to the Mediterranean diet on health: an updated systematic review and meta-analysis. Am J Clin Nutr 92, 1189–1196. [DOI] [PubMed] [Google Scholar]
- 86. Tangney CC, Kwasny MJ, Li H, et al. (2011) Adherence to a Mediterranean-type dietary pattern and cognitive decline in a community population. Am J Clin Nutr 93, 601–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Barberger-Gateau P, Raffaitin C, Letenneur L, et al. (2007) Dietary patterns and risk of dementia: the Three-City cohort study. Neurology 69, 1921–1930. [DOI] [PubMed] [Google Scholar]
- 88. Kesse-Guyot E, Fezeu L, Andreeva VA, et al. (2012) Total and specific polyphenol intakes in midlife are associated with cognitive function measured 13 years later. J Nutr 142, 76–83. [DOI] [PubMed] [Google Scholar]
- 89. Bakker GC, van Erk MJ, Pellis L, et al. (2010) An antiinflammatory dietary mix modulates inflammation and oxidative and metabolic stress in overweight men: a nutrigenomics approach. Am J Clin Nutr 91, 1044–1059. [DOI] [PubMed] [Google Scholar]
- 90. Devore EE, Kang JH, Breteler MM, et al. (2012) Dietary intakes of berries and flavonoids in relation to cognitive decline. Ann Neurol 72, 135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Chun OK, Chung SJ, Claycombe KJ, et al. (2008) Serum C-reactive protein concentrations are inversely associated with dietary flavonoid intake in U.S. adults. J Nutr 138, 753–760. [DOI] [PubMed] [Google Scholar]
- 92. Oyama J, Maeda T, Sasaki M, et al. (2010) Green tea catechins improve human forearm vascular function and have potent anti-inflammatory and anti-apoptotic effects in smokers. Intern Med 49, 2553–2559. [DOI] [PubMed] [Google Scholar]
- 93. Ueda H, Yamazaki C & Yamazaki M (2004) A hydroxyl group of flavonoids affects oral anti-inflammatory activity and inhibition of systemic tumor necrosis factor-alpha production. Biosci Biotechnol Biochem 68, 119–125. [DOI] [PubMed] [Google Scholar]
- 94. Zern TL, Wood RJ, Greene C, et al. (2005) Grape polyphenols exert a cardioprotective effect in pre- and postmenopausal women by lowering plasma lipids and reducing oxidative stress. J Nutr 135, 1911–1917. [DOI] [PubMed] [Google Scholar]
- 95. Mandel SA, Amit T, Kalfon L, et al. (2008) Cell signaling pathways and iron chelation in the neurorestorative activity of green tea polyphenols: special reference to epigallocatechin gallate (EGCG). J Alzheimers Dis 15, 211–222. [DOI] [PubMed] [Google Scholar]
- 96. Schroeter H, Spencer JP, Rice-Evans C, et al. (2001) Flavonoids protect neurons from oxidized low-density-lipoprotein-induced apoptosis involving c-Jun N-terminal kinase (JNK), c-Jun and caspase-3. Biochem J 358, 547–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Schroeter H, Bahia P, Spencer JPE, et al. (2007) ( − )Epicatechin stimulates ERK-dependent cyclic AMP response element activity and upregulates GLUR2 in cortical neurons. J Neurochem 101, 1596–1606. [DOI] [PubMed] [Google Scholar]
- 98. Vauzour D, Vafeiadou K, Rice-Evans C, et al. (2007) Activation of pro-survival Akt and ERK1/2 signalling pathways underlie the anti-apoptotic effects of flavanones in cortical neurons. J Neurochem 103, 1355–1367. [DOI] [PubMed] [Google Scholar]
- 99. González-Gallego J, García-Mediavilla MV, Sánchez-Campos S, et al. (2010) Fruit polyphenols, immunity and inflammation. Br J Nutr 104, S15–S27. [DOI] [PubMed] [Google Scholar]
- 100. Spencer JP, Vafeiadou K, Williams RJ, et al. (2012) Neuroinflammation: modulation by flavonoids and mechanisms of action. Mol Aspects Med 33, 83–97. [DOI] [PubMed] [Google Scholar]
- 101. British Nutrition Foundation (editor). (2013) Nutrition and Development: Short and Long Term Consequences for Health. Oxford: Wiley-Blackwell. [Google Scholar]
- 102. Franzek EJ, Sprangers N, Janssens AC, et al. (2008) Prenatal exposure to the 1944–45 Dutch ‘hunger winter’ and addiction later in life. Addiction 103, 433–438. [DOI] [PubMed] [Google Scholar]
- 103. Heijmans BT, Tobi EW, Stein AD, et al. (2008) Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci U S A 105, 17046–17049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Roseboom TJ, van der Meulen JH, Ravelli AC, et al. (2001) Effects of prenatal exposure to the Dutch famine on adult disease in later life: an overview. Mol Cell Endocrinol 185, 93–98. [DOI] [PubMed] [Google Scholar]
- 105. Cottrell EC & Ozanne SE (2008) Early life programming of obesity and metabolic disease. Physiol Behav 94, 17–28. [DOI] [PubMed] [Google Scholar]
- 106. McMillen IC, Rattanatray L, Duffield JA, et al. (2009) The early origins of later obesity: pathways and mechanisms. Adv Exp Med Biol 646, 71–81. [DOI] [PubMed] [Google Scholar]
- 107. Frias AE & Grove KL (2012) Obesity: a transgenerational problem linked to nutrition during pregnancy. Semin Reprod Med 30, 472–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Hales CN, Barker DJ, Clark PM, et al. (1991) Fetal and infant growth and impaired glucose tolerance at age 64. BMJ 303, 1019–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Hales CN & Barker DJ (2001) The thrifty phenotype hypothesis. Br Med Bull 60, 5–20. [DOI] [PubMed] [Google Scholar]
- 110. Gluckman PD, Hanson MA, Morton SM, et al. (2005) Life-long echoes – a critical analysis of the developmental origins of adult disease model. Biol Neonate 287, 127–139. [DOI] [PubMed] [Google Scholar]
- 111. McMullen S, Langley-Evans SC, Gambling L, et al. (2012) A common cause for a common phenotype: the gatekeeper hypothesis in fetal programming. Med Hypotheses 78, 88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Gluckman PD, Cutfield W, Hofman P, et al. (2005) The fetal, neonatal, and infant environments – the long-term consequences for disease risk. Early Hum Dev 81, 51–59. [DOI] [PubMed] [Google Scholar]
- 113. Swali A, McMullen S, Hayes H, et al. (2011) Cell cycle regulation and cytoskeletal remodelling are critical processes in the nutritional programming of embryonic development. PLoS One 6, e23189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Lazar MA (2005) How obesity causes diabetes: not a tall tale. Science 307, 373–375. [DOI] [PubMed] [Google Scholar]
- 115. McClung JP & Karl JP (2009) Iron deficiency and obesity: the contribution of inflammation and diminished iron absorption. Nutr Rev 67, 100–104. [DOI] [PubMed] [Google Scholar]
- 116. Gambling L, Danzeisen R, Gair S, et al. (2001) Effect of iron deficiency on placental transfer of iron and expression of iron transport proteins in vivo and in vitro . Biochem J 356, 883–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Gambling L, Dunford S, Wallace DI, et al. (2003) Iron deficiency during pregnancy affects postnatal blood pressure in the rat. J Physiol 552, 603–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Georgieff MK (2011) Long-term brain and behavioral consequences of early iron deficiency. Nutr Rev 69, S43–S48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Siddappa AM, Georgieff MK, Wewerka S, et al. (2004) Iron deficiency alters auditory recognition memory in newborn infants of diabetic mothers. Pediatr Res 7, 7. [DOI] [PubMed] [Google Scholar]
- 120. Bekri S, Gual P, Anty R, et al. (2006) Increased adipose tissue expression of hepcidin in severe obesity is independent from diabetes and NASH. Gastroenterol 131, 788–796. [DOI] [PubMed] [Google Scholar]
- 121. Zafon C, Lecube A & Simo R (2010) Iron in obesity. An ancient micronutrient for a modern disease. Obes Rev 11, 322–328. [DOI] [PubMed] [Google Scholar]
- 122. Laftah AH, Ramesh B, Simpson RJ, et al. (2004) Effect of hepcidin on intestinal iron absorption in mice. Blood 103, 3940–3944. [DOI] [PubMed] [Google Scholar]
- 123. Huber M, Knottnerus JA, Green L, et al. (2011) How should we define health? BMJ 343, d4163. [DOI] [PubMed] [Google Scholar]
- 124. Nappo F, Esposito K, Cioffi M, et al. (2002) Postprandial endothelial activation in healthy subjects and in type 2 diabetic patients: role of fat and carbohydrate meals. J Am Coll Cardiol 39, 1145–1150. [DOI] [PubMed] [Google Scholar]
- 125. Esposito K, Nappo F, Giugliano F, et al. (2003) Meal modulation of circulating interleukin 18 and adiponectin concentrations in healthy subjects and in patients with type 2 diabetes mellitus. Am J Clin Nutr 78, 1135–1140. [DOI] [PubMed] [Google Scholar]
- 126. Michaeli B, Berger MM, Revelly JP, et al. (2007) Effects of fish oil on the neuro-endocrine responses to an endotoxin challenge in healthy volunteers. Clin Nutr 26, 70–77. [DOI] [PubMed] [Google Scholar]
- 127. Rhodes LE, Darby G, Massey KA, et al. (2013) Oral green tea catechin metabolites are incorporated into human skin and protect against UV radiation-induced cutaneous inflammation in association with reduced production of pro-inflammatory eicosanoid 12-hydroxyeicosatetraenoic acid. Br J Nutr 110, 891–900. [DOI] [PubMed] [Google Scholar]
- 128. Pilkington SM, Massey KA, Bennett SP, et al. (2013) Randomized controlled trial of oral omega-3 PUFA in solar-simulated radiation-induced suppression of human cutaneous immune responses. Am J Clin Nutr 97, 646–652. [DOI] [PubMed] [Google Scholar]
- 129. Wopereis S, Wolvers D, van Erk M, et al. (2013) Assessment of inflammatory resilience in healthy subjects using dietary lipid and glucose challenges. BMC Med Genomics 6, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Rudkowska I, Paradis AM, Thifault E, et al. (2013) Transcriptomic and metabolomic signatures of an n-3 polyunsaturated fatty acids supplementation in a normolipidemic/normocholesterolemic Caucasian population. J Nutr Biochem 24, 54–61. [DOI] [PubMed] [Google Scholar]
- 131. Swan AL, Mobasheri A, Allaway D, et al. (2013) Application of machine learning to proteomics data: classification and biomarker identification in postgenomics biology. OMICS 17, 595–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Kelder T, Conklin BR, Evelo CT, et al. (2010) Finding the right questions: exploratory pathway analysis to enhance biological discovery in large datasets. PLoS Biol 8, e1000472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Barabasi AL & Oltvai ZN (2004) Network biology: understanding the cell's functional organization. Nat Rev Genet 5, 101–113. [DOI] [PubMed] [Google Scholar]
- 134. van Gool AJ, Henry B & Sprengers ED (2010) From biomarker strategies to biomarker activities and back. Drug Discov Today 15, 121–126. [DOI] [PubMed] [Google Scholar]
- 135. Kumar C & van Gool AJ (2013) Introduction: biomarkers in translational and personalized medicine. In Comprehensive Biomarker Discovery and Validation for Clinical Application, 1st ed., pp. 3–39 [Horvatovich P and Bischoff R, editors]. London: Royal Society of Chemistry. [Google Scholar]
- 136. EFSA Panel on Dietetic Products, Nutrition and Allergies (NDA) (2011) Guidance on the scientific requirements for health claims related to gut health and immune function. EFSA J 9, 1984–1996. [Google Scholar]
- 137. Albers R, Bourdet-Sicard R, Braun D, et al. (2013) Monitoring immune modulation by nutrition in the general population: identifying and substantiating effects on human health. Br J Nutr 110, S1–S30. [DOI] [PubMed] [Google Scholar]
- 138. de Vries J, Antoine JM, Burzykowski T, et al. (2013) Markers for nutrition studies: review of criteria for the evaluation of markers. Eur J Nutr 52, 1685–1699. [DOI] [PubMed] [Google Scholar]
- 139. Landberg R, Sun Q, Rimm EB, et al. (2011) Selected dietary flavonoids are associated with markers of inflammation and endothelial dysfunction in U.S. women. J Nutr 141, 618–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Edirisinghe I, Banaszewski K, Cappozzo J, et al. (2011) Strawberry anthocyanin and its association with postprandial inflammation and insulin. Br J Nutr 106, 913–922. [DOI] [PubMed] [Google Scholar]
- 141. Steptoe A, Gibson EL, Vuononvirta R, et al. (2007) The effects of chronic tea intake on platelet activation and inflammation: a double-blind placebo controlled trial. Atherosclerosis 193, 277–282. [DOI] [PubMed] [Google Scholar]
- 142. Karlsen A, Retterstol L, Laake P, et al. (2007) Anthocyanins inhibit nuclear factor-κB activation in monocytes and reduce plasma concentrations of pro-inflammatory mediators in healthy adults. J Nutr 137, 1951–1954. [DOI] [PubMed] [Google Scholar]
- 143. Widlansky ME, Duffy SJ, Wiseman S, et al. (2005) Effects of black tea consumption on plasma catechins and markers of oxidative stress and inflammation in patients with coronary artery disease. Free Radic Biol Med 38, 499–506. [DOI] [PubMed] [Google Scholar]
- 144. Mellen PB, Daniel KR, Brosnihan KB, et al. (2010) Effect of muscadine grape seed supplementation on vascular function in subjects with or at risk for cardiovascular disease: a randomized crossover trial. J Am Coll Nutr 29, 469–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Heinz SA, Henson DA, Nieman DC, et al. (2010) A 12-week supplementation with quercetin does not affect natural killer cell activity, granulocyte oxidative burst activity or granulocyte phagocytosis in female human subjects. Br J Nutr 104, 849–857. [DOI] [PubMed] [Google Scholar]