

Abstract
There is conflicting evidence whether intermittent hypoxia in OSA influences oxidative stress. We hypothesized that withdrawal of CPAP from patients with OSA would raise markers of oxidative stress.
59 patients with CPAP-treated moderate-to-severe OSA (ODI, >20/hour) were randomized 1:1 to either stay on CPAP (n=30), or change to sham CPAP (n=29) for 2 weeks. Using samples from two similar studies at two sites, we measured early morning blood malondialdehyde (MDA, a primary outcome in one study, secondary in the other), lipid hydroperoxides, total anti-oxidant capacity, superoxide generation from mononuclear cells, and urinary F2-isoprostane. We also measured superoxide dismutase as a marker of hypoxic pre-conditioning. ‘Treatment’ effects (sham CPAP versus CPAP) were calculated via linear regression.
Sham CPAP provoked moderate-to-severe OSA (mean ODI 46/hour), but blood markers of oxidative stress did not change significantly (MDA ‘treatment’ effect [95%CIs], −0.02 [−0.23 to +0.19] μmol/l). Urinary F2-isoprostane fell significantly approximately 30% (−0.26 [−0.42 to −0.10] ng/ml), and superoxide dismutase rose similarly (+0.17 [+0.02 to +0.30] ng/ml).
We found no direct evidence of increased oxidative stress in patients experiencing a return of their moderate-to-severe OSA. The fall in urinary F2-isoprostane, and rise in superoxide dismutase, implies that hypoxic pre-conditioning may have reduced oxidative stress.
INTRODUCTION
Obstructive sleep apnoea (OSA) is a common problem and usually attended by oscillations in oxygen saturation. There is a clear association between OSA, and endothelial dysfunction,[1,2] hypertension,[3] and cardiovascular disease,[4] but the potential mechanisms by which OSA may cause cardiovascular disease are still debated.[5] There is robust evidence from randomized and controlled trials that increased catecholamine production occurs with OSA.[1,6] There is additional evidence that intermittent hypoxemia, such as that observed in OSA, may provoke endothelial dysfunction,[7] and therefore potentially the onset of atherosclerosis.[8,9]
The evidence for a link between intermittent hypoxemia and vascular damage is contradictory.[10,11] One proposed mechanism is that the repetitive oscillations in oxygen levels may provoke increased oxidative stress, via so-called ischaemia/reperfusion injury.[7,12,13] During periods of hypoxemia, antioxidant mechanisms are down regulated, such that when oxygen levels rise again, more reactive oxygen species (ROS), or free radicals, with one or more unpaired electrons, are produced in mitochondria, in excess of that which can usually be quenched (by anti-oxidants such as superoxide dismutase). These excess ROS, made up of unpaired oxygen atoms (•O2−, superoxide, primary ROS), or oxygen-containing molecules (secondary ROS, e.g. •OH), are then thought, amongst other things, to provoke systemic inflammation, endothelial damage and dysfunction – the path to atheroma.[12] There is also evidence that during hypoxic episodes, xanthine dehydrogenase is converted to xanthine oxidase; hypoxic neutrophils then consume excessive ATP with accumulation of purine catabolites (xanthine) which, following re-oxygenation, are metabolised by the increased xanthine oxidase, thus producing abnormal amounts of superoxide radical and hydrogen peroxide.[14] Part of the toxicity of primary ROS results from the generation of many further toxic molecules such as hydrogen peroxide and hydroxyl radicals (secondary ROS), and peroxynitrite (a reactive nitrogen species made from nitric oxide (NO) and superoxide). Endothelial function and vasodilation may thus be impaired, as it is dependent on NO. Increased levels of anti-oxidants have been shown to partially prevent the damage from ischaemia/reperfusion injury.[14] The majority of experimental evidence in support of this hypothesis comes from animal experiments, most of which used more severe and prolonged periods of intermittent hypoxemia than are seen in patients with OSA,[13-16] Whether the short cycle intermittent hypoxemia observed in patients with OSA (approximately 1 minute cycle lengths) activates any of these mechanisms is not clear, and the length and depth of the transient hypoxia may be critical[17,18].
We have recently developed an experimental protocol to investigate the pathophysiology of human OSA.[1] In patients with OSA, already established on continuous positive airway pressure (CPAP), comparing two weeks of CPAP-withdrawal (versus continuing CPAP) allows robust randomized controlled trials to be performed. Using this experimental protocol, we have investigated the hypothesis that two weeks of OSA will increase circulating markers of oxidative stress.
METHODS
Trial Design
These data come from two, hospital-based, randomized, controlled trials with similar protocols, that allowed us to evaluate the effects of 14 days CPAP withdrawal, versus continuing CPAP, on measures of oxidative stress in patients with prior CPAP-treated OSA. The design is similar to that reported in an earlier trial from the same units,[1] except that in the current study patients had more severe OSA.
Patients
Inclusion criteria: patients previously diagnosed with OSA, and treated with CPAP, who were registered in a database of either the Oxford Centre for Respiratory Medicine, Churchill Hospital Oxford, UK, or the Sleep Disorders Centre, University Hospital Zurich, Switzerland, were eligible for the trial if they were aged between 20-75 years, had an oxygen desaturation index (ODI, ≥4% dips) of >20/h during their original diagnostic sleep study, had been treated with CPAP for >12 months (with an average compliance of ≥4h per night), and had a residual AHI<10 on CPAP. Such patients were contacted and asked to take part in the study, the first part of which was to establish that they had an ODI>20/h during an initial pre-trial period without CPAP. Exclusion criteria are described in the data supplement.
Intervention, Randomization and Blinding
Following the initial week of overnight oximetry (3 nights on CPAP to confirm efficacy, and 4 nights off CPAP to verify the return of sufficiently severe OSA prior to trial entry), the patients returned to automatic CPAP (ResMed, Abingdon, UK) for a minimum of 2 weeks. Patients were computer-randomized 1:1 (with minimisation for OSA severity and BMI; MINIM, http://www-users.york.ac.uk/~mb55/guide/minim.htm) to either continue automatic CPAP, or switch to subtherapeutic sham CPAP for two weeks. A description of sham CPAP construction, and the limited blinding achieved, is included in the data supplement.
Sample preparation
The preparation of the samples for analysis is described in the data supplement.
Outcome measures
Continuing abolition of OSA by CPAP at baseline was further confirmed on a home (Oxford) or hospital (Zurich) sleep study the night prior to randomization (Black Shadow, Stowood Scientific Instruments, Oxford, UK or Alice5, Philips Respironics AG, Zofingen, Switzerland), and the severity of any OSA after two weeks was similarly assessed with a repeat sleep study. The additional measurements made, including blood pressure, are described in the data supplement. The main outcome of this analysis was the change in plasma MDA levels over the two weeks, CPAP withdrawal subjects versus control subjects. In Oxford the primary outcome of the study was the change in plasma MDA levels, but in Zurich the identical protocol was run with a different primary outcome (hyperaemic myocardial blood flow); we took advantage of this and collected exactly similar samples to combine with the Oxford data. Secondary outcomes were plasma oxidized low-density-lipoprotein (OxLDL), serum total anti-oxidant capacity (TAC), urinary F2-isoprostanes (fresh morning urine sample), and NADPH oxidase-derived superoxide generation from peripheral blood mononuclear cells (PBMCs).
Measurement of circulating markers
The laboratory techniques for measuring the markers of oxidative stress are described in the data supplement.
Sample size and statistical analysis
The approach to a sample size calculation is described in the data supplement. ‘Treatment’ effects (CPAP withdrawal versus control) were modelled using linear regression (IBM SPSS, version 17, Portsmouth, UK), controlling for the baseline values of the relevant dependent variable, age, BMI, statins, antihypertensives, and smoking status. The 95% confidence intervals of the observed treatment effects reveal the actual power of the study for each of the primary and secondary endpoints.
RESULTS
Fifty-nine patients were included in this study (Zurich n=45, Oxford n=14) and recruited between January 2013 and March 2014. The combination of samples from the two sites provided data from more subjects than required by the power calculation, thus allowing fewer numbers than intended to be recruited in Oxford. A full record of patients contacted, but who declined participation, was not kept. No subjects dropped out following randomisation. Figure 1 shows the subject flow chart. Baseline characteristics of the control and CPAP-withdrawal groups are shown in Table 1. The two cohorts were well matched, with no suggestion of large enough differences to bias the results.
Figure 1.
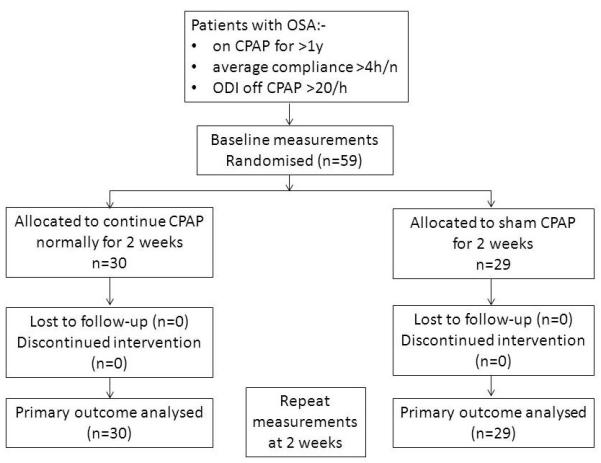
Flow chart of study subjects. OSA, obstructive sleep apnoea. CPAP, continuous positive airway pressure. ODI, oxygen desaturation index.
Table 1. Patient baseline characteristics.
| Control (30) | CPAP-withdrawal (29) | |
|---|---|---|
| Age (SD) | 59.2 (9.2) | 62.1 (10.3) |
| BMI (SD) | 34.3 (6.3) | 35.1 (7.3) |
| Males : Females (n : n) | 25 : 5 | 23 : 6 |
| Epworth Sleepiness Score (SD) | 6.5 (3.7) | 6.6 (3.5) |
| ODI on CPAP (SD) | 2.6 (2.4) | 3.9 (3.8) |
| ODI during 4 nights preliminary CPAP withdrawal (SD) | 33.4 (14.4) | 38.7 (16.2) |
| Morning laboratory systolic blood pressure (mmHg) | 133.5 (14.2) | 125.9 (14.6) |
| Morning laboratory diastolic blood pressure (mmHg) | 83.6 (13.0) | 80.8 (9.8) |
| Morning laboratory heart rate (beats/min) | 68.7 (8.9) | 68.3 (9.33) |
| On statins (n,%) | 14 (47) | 11 (38) |
| On anti-hypertensive drugs (n,%) | 17 (57) | 14 (48) |
| Smoking status (never : ex-smoker : current smoker) | 14 : 9 : 7 | 10 : 15 : 4 |
SD = standard deviation, BMI = body mass index (kg/m2), ODI = overnight oxygen desaturation index (events/hour), CPAP = continuous positive airway pressure.
Consistent with our previous study, conducted under a similar protocol,[1] in the CPAP-withdrawal group there was a significant rise in morning office systolic blood pressure (+6.1mmHg, 95%CIs +0.6 to +11.6, p=0.032), diastolic blood pressure (+4.2mmHg, 95%CIs +0.04 to +8.4, p=0.048), and heart rate (+8.3 beats/min, 95%CIs +3.9 to +12.5, p<0.001), compared to the continuing CPAP control group. Similarly, ESS rose (+3.6 points, 95%CIs +2.0 to +5.0, p<0.001), compared to the continuing CPAP control group.
Table 2 shows the results of the oxidative stress analyses. Despite the cardiovascular changes and a clear difference in the degree of intermittent hypoxia (ODI), there was no evidence of an increase in markers of oxidative stress. Interestingly, and contrary to our hypothesis, we observed a significant reduction in urinary F2-Isoprostane in the CPAP-withdrawal patients as compared to controls (p=0.002). In addition, there was a significant correlation between the ODI at 2 weeks and the fall in F2-Isoprostanes (r= −0.41, n=59, p=0.001).
Table 2. Markers of Oxidative Stress.
| ODI at 2 weeks (≥4% dips/hour) | MDA (μmol/l) | OxLDL (mU/l) | TAC (nmol/μl) | F2-ISO (ng/ml) | PBMC superoxide generation (RLU/s/cell count) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Control (n=30) | Baseline | 2 weeks | Baseline | 2 weeks | Baseline | 2 weeks | Baseline | 2 weeks | Baseline | 2 weeks | |
| Mean | 3.01 | 1.43 | 1.39 | 11.1 | 11.6 | 37.7 | 38.0 | 0.77 | 0.88 | 0.084 | 0.089 |
| SD | 3.10 | 0.34 | 0.31 | 3.72 | 4.85 | 1.71 | 2.31 | 0.30 | 0.42 | 0.044 | 0.034 |
| CPAP withdrawal (n=29) | Baseline | 2 weeks | Baseline | 2 weeks | Baseline | 2 weeks | Baseline | 2 weeks | Baseline | 2 weeks | |
| Mean | 45.7 | 1.36 | 1.38 | 13.0 | 13.2 | 37.5 | 37.1 | 0.81 | 0.63 | 0.085 | 0.082 |
| SD | 18.3 | 0.44 | 0.47 | 4.20 | 5.02 | 2.37 | 2.14 | 0.33 | 0.26 | 0.037 | 0.021 |
| ‘Treatment’ effect | +42.8 | −0.02 | −0.21 | −0.77 | −0.26 | +0.02 | |||||
| 95%CIs | +36.0 to +49.5 | −0.23 to +0.19 | −2.06 to +1.64 | −1.96 to +0.43 | −0.42 to −0.10 | −0.06 to +0.09 | |||||
| P | <0.0001 | NS (0.86) | NS (0.82) | NS (0.20) | 0.002 | NS (0.64) | |||||
SD = standard deviation, ODI = overnight oxygen desaturation index (events/hour), CPAP = continuous positive airway pressure, 95%CIs = 95% confidence intervals, MDA = plasma malondialdehyde, OxLDL = plasma oxidised low density lipoproteins, TAC = plasma total antioxidant capacity, F2-ISO = urine F2-isoprostanes, PBMC=peripheral blood mononuclear cells (13 subjects were included in this analysis, control subjects n=7, CPAP withdrawal subjects n=6). RLU=relative light units. ‘Treatment’effect was controlled for the baseline value of the relevant dependent variable, age, BMI, statins, antihypertensives, and smoking status.
DISCUSSION
This is the first randomized, controlled, study to look at the effect of two weeks CPAP-withdrawal and consequent intermittent hypoxia on markers of oxidative stress in patients with OSA. By selecting patients with moderate-to-severe OSA for entry into the CPAP-withdrawal model, we were able to induce a clear difference in the degree of intermittent hypoxia between the two groups (mean ≥4% ODI, 46/h versus 3/h at two weeks). In addition there was a clear and large effect on both systolic and diastolic blood pressure, as well as heart rate. Despite these substantial cardiovascular changes after two weeks of nocturnal intermittent hypoxia, we did not observe increased markers of oxidative stress. On the contrary, the unexpected reduction in one marker, the urinary F2-isoprostanes, implied a lessening of oxidative stress which seems counter-intuitive.
Our primary outcome, MDA levels, have been shown to be a sensitive marker of changes in oxidative stress in many clinical and experimental situations, for example following exercise,[19] statins,[20,21], and smoking.[22] The 95% CIs of our ‘treatment’ effect show that we have effectively excluded a difference of about 15% due to the intermittent nocturnal hypoxia (our study was powered to exclude a change of ≥20% with 90% power). However, we may have missed a rise in MDA of less than 15%.
Because of the unexpected finding of a fall in F2-isoprostanes, we further investigated whether intermittent hypoxia may have caused hypoxic (or ischaemic) pre-conditioning, which can protect tissues from subsequent ischaemia if the period of prior ischaemia is long enough.[14,23,24] This has been hypothesized as potentially occurring on OSA.[25-27] As an exploratory analysis, not in the original protocol, we measured levels of superoxide dismutase (SOD) by a commercial ELISA (Abcam, Cambridge, UK, Cat# ab119520). SOD is an enzyme known to be increased in association with hypoxic pre-conditioning.[28] The results of this additional analysis are shown in Table 3 and indicate a significant rise in SOD of about 30% in the CPAP withdrawal group, compared to the group continuing on CPAP. This supports the hypothesis that two weeks of intermittent nocturnal hypoxia from OSA has in some way increased defences against oxidative stress and thus potentially lessened its effects.
Table 3. Marker of hypoxic preconditioning.
| Superoxide dismutase (ng/ml) |
||
|---|---|---|
| Control (n=30) | Baseline | 2 weeks |
| Mean | 0.54 | 0.42 |
| SD | 0.30 | 0.26 |
| CPAP withdrawal (n=29) | Baseline | 2 weeks |
| Mean | 0.51 | 0.58 |
| SD | 0.20 | 0.28 |
| ‘Treatment’effect | +0.17 | |
| 95%CIs | +0.02 to +0.30 | |
| P | 0.02 | |
SD = standard deviation, 95%CIs = 95% confidence intervals, CPAP = continuous positive airway pressure.
‘Treatment’ effect was controlled for the baseline value of the relevant dependent variable, age, BMI, statins, antihypertensives, and smoking status.
Other studies of OSA or intermittent hypoxia and their possible effects on oxidative stress, have recently been well reviewed by Badr[13] and Lavie.[29] Although there are associations between OSA and markers of oxidative stress, such cross sectional studies are prone to confounding variables and cannot establish a causal relationship. Cross-sectional and uncontrolled studies of CPAP have also shown conflicting results, with some finding a relationship with OSA severity, or improvement with CPAP,[30-32] and others not.[18,33,34] The only other similarly robust study to ours from Alonso-Fernández et al, looking at the effect of 12 weeks CPAP, versus sham CPAP, in a cross-over design, which recruited 25 newly diagnosed patients with OSA, showed that plasma 8-isoprostane levels were reduced following CPAP by about 40%, and total nitrates and nitrites rose nearly threefold (suggesting increased NO production) compared to control.[35]
Animal studies of OSA have been conducted in a more rigorous fashion with appropriate experimental controls. However, such studies generally use levels of hypoxia far more severe than seen in most patients with OSA, and will also be associated with hyperventilation and hypocapnia, rather than the hypoventilation and hypercapnia seen in OSA (which will tend to promote vasodilation and preserve tissue oxygenation). Such animal studies have found evidence of increased oxidative stress from intermittent hypoxia, for example Savransky et al [36] showed that 6 months of intermittent hypoxia in mice quadrupled serum MDA levels compared to control mice. There are animal models more closely simulating OSA, using asphyxia rather than simple hypoxic gas mixtures, but these models have not been used to study oxidative stress.
Hypoxic preconditioning from intermittent hypoxia or OSA has not been as widely studied; again this is well reviewed by Lavie.[29] It is clear that intermittent hypoxia, perhaps via ROS themselves, may also up-regulate genes that control antioxidant pathways, such as Nrf2.[37] The resulting balance of ROS production versus anti-oxidant production is likely to be complex and depend on many factors such as hypoxia depth, cycle length and duration; as well as the presence of co-morbidities and pre-existing levels of anti-oxidants. However, in our model, that exactly reproduces the situation in human OSA, albeit for only two weeks, it appears that anti-oxidant capacity dominates. It may be that there was inter-individual variation in our patients, with some subjects increasing, and some decreasing, their levels of oxidative stress markers, leading to no overall average effect on MDA. This is perhaps partially supported by a non-significant increase in the SD of the MDA after two weeks of CPAP withdrawal, but this effect was not seem in the other markers.
Although we adopted an in vivo human model for the investigation of OSA, with appropriate controls, there are still some limitations. As mentioned above, the intermittent hypoxia only occurred for two weeks. It is possible that both shorter and/or longer periods might have produced different results, and might explain the difference between our results and those of the 12 week study by Alonso-Fernández et al., referred to above. [35] In measuring one-off blood markers of oxidative stress early in the morning, we have not assessed if changes might have been missed in markers with very short half-lives. This might explain why the F2-isoprostanes showed changes not reflected in the blood markers, as urine collections will represent a more integrated measure covering the last hours of sleep. In addition, of course, all these measures are indirect and may not fully represent what is happening within the mitochondria and cytosol. Although statins and antihypertensive medications can influence oxidative status, their use was corrected for in the effect size calculation, and a post-hoc analysis found no significant difference in any of the outcome measures dependant on whether these medications were being taken or not. Finally, the patient population studied will not be typical of all patients on CPAP for OSA, as they were selected for greater severity than average, had to be willing to stop CPAP for two weeks (which many patients are not prepared to do), and had to demonstrate a rapid return of moderate-to-severe OSA within 4 nights off CPAP.
In conclusion, this randomized controlled study of OSA has demonstrated a reduction in a marker of oxidative stress after two weeks of intermittent hypoxia, as evidenced by a significant decrease of urinary F2-isoprostanes. A further exploratory analysis showed a rise in plasma superoxide dismutase, which is an anti-oxidant known to be increased by hypoxic preconditioning. Our findings support the hypothesis that OSA may actually reduce oxidative stress in the short term, and highlight the targets for future investigations.
Supplementary Material
Acknowledgements
The authors would like to thank Barbara Winter SRN and Deborah Nicoll SRN, PhD, for their dedicated commitment to the running of this trial.
Sources of Funding
Primary
British Heart Foundation PG/12/80/29891
Swiss National Science Foundation 32003B_143365/1
Secondary
Oxford Health Services Research Committee
Oxford Radcliffe Charitable Funds
National Institute for Health Research (NIHR) Oxford Biomedical Research Centre, based at Oxford University Hospitals NHS Trust and University of Oxford
Footnotes
Trial registration
ISRCTN 73047833 and NCT 01797653
References
- 1.Kohler M, Stoewhas AC, Ayers L, et al. Effects of continuous positive airway pressure therapy withdrawal in patients with obstructive sleep apnea: a randomized controlled trial. Am J Respir Crit Care Med. 2011 Nov 15;184:1192–1199. doi: 10.1164/rccm.201106-0964OC. [DOI] [PubMed] [Google Scholar]
- 2.Kohler M, Craig S, Pepperell JC, et al. CPAP Improves Endothelial Function in Patients With Minimally Symptomatic OSA: Results From a Subset Study of the MOSAIC Trial. Chest. 2013 Sep;144:896–902. doi: 10.1378/chest.13-0179. [DOI] [PubMed] [Google Scholar]
- 3.Pepperell JC, Ramdassingh-Dow S, Crosthwaite N, et al. Ambulatory blood pressure after therapeutic and subtherapeutic nasal continuous positive airway pressure for obstructive sleep apnoea: a randomised parallel trial. Lancet. 2002 Jan 19;359:204–210. doi: 10.1016/S0140-6736(02)07445-7. [DOI] [PubMed] [Google Scholar]
- 4.Marin JM, Carrizo SJ, Vicente E, Agusti AG. Long-term cardiovascular outcomes in men with obstructive sleep apnoea-hypopnoea with or without treatment with continuous positive airway pressure: an observational study. Lancet. 2005 Mar 19;365:1046–1053. doi: 10.1016/S0140-6736(05)71141-7. [DOI] [PubMed] [Google Scholar]
- 5.Kohler M, Stradling JR. Mechanisms of vascular damage in obstructive sleep apnea. Nat Rev Cardiol. 2010 Dec;7:677–685. doi: 10.1038/nrcardio.2010.145. [DOI] [PubMed] [Google Scholar]
- 6.Kohler M, Pepperell JC, Casadei B, et al. CPAP and measures of cardiovascular risk in males with OSAS. Eur Respir J. 2008 Dec;32:1488–1496. doi: 10.1183/09031936.00026608. [DOI] [PubMed] [Google Scholar]
- 7.Lavie L, Polotsky V. Cardiovascular Aspects in Obstructive Sleep Apnea Syndrome - Molecular Issues, Hypoxia and Cytokine Profiles. Respiration. 2009 Sep 29;78:361–370. doi: 10.1159/000243552. [DOI] [PubMed] [Google Scholar]
- 8.Sert Kuniyoshi FH, Singh P, Gami AS, et al. Patients with obstructive sleep apnea exhibit impaired endothelial function after myocardial infarction. Chest. 2011 Jul;140:62–67. doi: 10.1378/chest.10-1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sitia S, Tomasoni L, Atzeni F, et al. From endothelial dysfunction to atherosclerosis. Autoimmun Rev. 2010 Oct;9:830–834. doi: 10.1016/j.autrev.2010.07.016. [DOI] [PubMed] [Google Scholar]
- 10.Kohler M, Stradling JR. CrossTalk proposal: Most of the cardiovascular consequences of OSA are due to increased sympathetic activity. J Physiol. 2012 Jun 15;590:2813–2815. doi: 10.1113/jphysiol.2012.229633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lavie L, Lavie P. CrossTalk opposing view: Most cardiovascular diseases in sleep apnoea are not caused by sympathetic activation. J Physiol. 2012 Jun 15;590:2817–2819. doi: 10.1113/jphysiol.2012.233833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garvey JF, Taylor CT, McNicholas WT. Cardiovascular disease in obstructive sleep apnoea syndrome: the role of intermittent hypoxia and inflammation. Eur Respir J. 2009 May;33:1195–1205. doi: 10.1183/09031936.00111208. [DOI] [PubMed] [Google Scholar]
- 13.Badran M, Ayas N, Laher I. Cardiovascular complications of sleep apnea: role of oxidative stress. Oxid Med Cell Longev. 2014;2014:985258. doi: 10.1155/2014/985258. doi: 10.1155/2014/985258. Epub@2014 Mar 6.: 985258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 15.Hoffmann MS, Singh P, Wolk R, Narkiewicz K, Somers VK. Obstructive sleep apnea and intermittent hypoxia increase expression of dual specificity phosphatase 1. Atherosclerosis. 2013 Dec;231:378–383. doi: 10.1016/j.atherosclerosis.2013.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Polak J, Shimoda LA, Drager LF, et al. Intermittent hypoxia impairs glucose homeostasis in C57BL6/J mice: partial improvement with cessation of the exposure. Sleep. 2013 Oct 1;36:1483–1490. doi: 10.5665/sleep.3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brugniaux JV, Pialoux V, Foster GE, et al. Effects of intermittent hypoxia on erythropoietin, soluble erythropoietin receptor and ventilation in humans. Eur Respir J. 2011 Apr;37:880–887. doi: 10.1183/09031936.00156009. [DOI] [PubMed] [Google Scholar]
- 18.Katsoulis K, Kontakiotis T, Spanogiannis D, et al. Total antioxidant status in patients with obstructive sleep apnea without comorbidities: the role of the severity of the disease. Sleep Breath. 2011 Dec;15:861–866. doi: 10.1007/s11325-010-0456-y. [DOI] [PubMed] [Google Scholar]
- 19.Stepanyan V, Crowe M, Haleagrahara N, Bowden B. Effects of vitamin E supplementation on exercise-induced oxidative stress: a meta-analysis. Appl Physiol Nutr Metab. 2014 Sep;39:1029–1037. doi: 10.1139/apnm-2013-0566. [DOI] [PubMed] [Google Scholar]
- 20.Moon GJ, Kim SJ, Cho YH, Ryoo S, Bang OY. Antioxidant effects of statins in patients with atherosclerotic cerebrovascular disease. J Clin Neurol. 2014 Apr;10:140–147. doi: 10.3988/jcn.2014.10.2.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Antoniades C, Bakogiannis C, Tousoulis D, et al. Preoperative atorvastatin treatment in CABG patients rapidly improves vein graft redox state by inhibition of Rac1 and NADPH-oxidase activity. Circulation. 2010 Sep 14;122:S66–S73. doi: 10.1161/CIRCULATIONAHA.109.927376. [DOI] [PubMed] [Google Scholar]
- 22.Lykkesfeldt J. Malondialdehyde as biomarker of oxidative damage to lipids caused by smoking. Clin Chim Acta. 2007 May 1;380:50–58. doi: 10.1016/j.cca.2007.01.028. [DOI] [PubMed] [Google Scholar]
- 23.Park AM, Nagase H, Kumar SV, Suzuki YJ. Effects of intermittent hypoxia on the heart. Antioxid Redox Signal. 2007 Jun;9:723–729. doi: 10.1089/ars.2007.1460. [DOI] [PubMed] [Google Scholar]
- 24.Heusch G, Botker HE, Przyklenk K, Redington A, Yellon D. Remote Ischemic Conditioning. J Am Coll Cardiol. 2015 Jan;65:177–195. doi: 10.1016/j.jacc.2014.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Almendros I, Wang Y, Gozal D. The polymorphic and contradictory aspects of intermittent hypoxia. Am J Physiol Lung Cell Mol Physiol. 2014 Jul 15;307:L129–L140. doi: 10.1152/ajplung.00089.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ozeke O, Ozer C, Gungor M, Celenk MK, Dincer H, Ilicin G. Chronic intermittent hypoxia caused by obstructive sleep apnea may play an important role in explaining the morbidity-mortality paradox of obesity. Med Hypotheses. 2011 Jan;76:61–63. doi: 10.1016/j.mehy.2010.08.030. [DOI] [PubMed] [Google Scholar]
- 27.Shah N, Redline S, Yaggi HK, et al. Obstructive sleep apnea and acute myocardial infarction severity: ischemic preconditioning? Sleep Breath. 2013 May;17:819–826. doi: 10.1007/s11325-012-0770-7. [DOI] [PubMed] [Google Scholar]
- 28.Chen CF, Tsai SY, Ma MC, Wu MS. Hypoxic preconditioning enhances renal superoxide dismutase levels in rats. J Physiol. 2003 Oct 15;552:561–569. doi: 10.1113/jphysiol.2003.045559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lavie L. Oxidative stress in obstructive sleep apnea and intermittent hypoxia - Revisited - The bad ugly and good: Implications to the heart and brain. Sleep Med Rev. 2014 Jul 24;10 doi: 10.1016/j.smrv.2014.07.003. [DOI] [PubMed] [Google Scholar]
- 30.Yamauchi M, Nakano H, Maekawa J, et al. Oxidative stress in obstructive sleep apnea. Chest. 2005 May;127:1674–1679. doi: 10.1378/chest.127.5.1674. [DOI] [PubMed] [Google Scholar]
- 31.Lavie L, Vishnevsky A, Lavie P. Evidence for lipid peroxidation in obstructive sleep apnea. Sleep. 2004 Feb 1;27:123–128. [PubMed] [Google Scholar]
- 32.Carpagnano GE, Kharitonov SA, Resta O, Foschino-Barbaro MP, Gramiccioni E, Barnes PJ. 8-Isoprostane, a marker of oxidative stress, is increased in exhaled breath condensate of patients with obstructive sleep apnea after night and is reduced by continuous positive airway pressure therapy. Chest. 2003 Oct;124:1386–1392. doi: 10.1378/chest.124.4.1386. [DOI] [PubMed] [Google Scholar]
- 33.Svatikova A, Wolk R, Lerman LO, et al. Oxidative stress in obstructive sleep apnoea. Eur Heart J. 2005 Nov;26:2435–2439. doi: 10.1093/eurheartj/ehi440. [DOI] [PubMed] [Google Scholar]
- 34.Wali SO, Bahammam AS, Massaeli H, et al. Susceptibility of LDL to oxidative stress in obstructive sleep apnea. Sleep. 1998 May 1;21:290–296. [PubMed] [Google Scholar]
- 35.Alonso-Fernandez A, Garcia-Rio F, Arias MA, et al. Effects of CPAP on oxidative stress and nitrate efficiency in sleep apnoea: a randomised trial. Thorax. 2009 Jul;64:581–586. doi: 10.1136/thx.2008.100537. [DOI] [PubMed] [Google Scholar]
- 36.Savransky V, Bevans S, Nanayakkara A, et al. Chronic intermittent hypoxia causes hepatitis in a mouse model of diet-induced fatty liver. Am J Physiol Gastrointest Liver Physiol. 2007 Oct;293:G871–G877. doi: 10.1152/ajpgi.00145.2007. [DOI] [PubMed] [Google Scholar]
- 37.Polotsky VY, Savransky V, Bevans-Fonti S, et al. Intermittent and sustained hypoxia induce a similar gene expression profile in human aortic endothelial cells. Physiol Genomics. 2010 May;41:306–314. doi: 10.1152/physiolgenomics.00091.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.