

Abstract
The p16INK4a protein (p16) has been reported to be a tumor suppressor gene that suppresses the proliferation of cells through the direct inhibition of cell cycle progression. Accordingly, p16 is a potential target for cancer gene therapy. In the present study, the arginine 22, 131 and 138 residues of p16 were found to be methylation sites, as the mutation of these arginine residues to lysine resulted in the hypomethylation of p16. Furthermore, the protein arginine methyltransferases (PRMTs), such as PRMT1, PRMT4 and PRMT6, were determined to be involved in the methylation of the p16 arginine residues. PRMT6 effectively reduced the intensity of the association between p16 and CDK4, and also weakened the function of p16 in preventing cell proliferation. In addition, the p16 protein was found to be phosphorylated in various cell lines, and mutations in the serine residues weakened the cell cycle arrest and induction of apoptosis mediated by p16. Preliminarily, the crosstalk between the phosphorylation and arginine methylation modification of p16 was examined. These findings predict a role for serine phosphorylation against arginine methylation of p16.
Keywords: p16, phosphorylation, methylation, protein arginine methyltransferase, serine, arginine
Introduction
The p16INK4a protein (p16) is a tumor suppressor protein that functions as an inhibitor of cyclin-dependent kinase (CDK) 4 and CDK6, the CDKs that initiate the phosphorylation of the retinoblastoma protein (pRb) (1). Thus, p16 has the capacity to arrest the cell cycle at the G1 phase through the cyclin D-CDK4-Rb pathway (2).
It has been reported that the expression of p16 is regulated primarily at the transcriptional level, and various types of transcription factors, including Sp1 and Ets1, are involved in its transcriptional regulation (2,3). In previous studies, the histone acetyltransferase p300 has been found to be involved in the activation of p16 expression through recruitment by Sp1, while histone deacetylases 3 and 4 inhibited the activity of the p16 promoter through Yin Yang 1 and zinc-binding protein-89 (4–6).
In addition, it has been reported that phosphorylation of p16 protein at Ser152 promotes the association between p16 and CDK4, while phosphorylation at Ser8 abolished the CDK4-inhibitory activity of p16 (7). A previous study identified that hypomethylation of p16 protein exhibited a potentiated function in preventing cell proliferation (8). These results indicated that the post-translational modification of p16 plays an important role in the regulation of the function of p16.
Arginine residues are methylated by protein arginine methyltransferases (PRMTs). Type I PRMTs include PRMT1, PRMT3, coactivator-associated arginine methyltransferase 1, also termed PRMT4, PRMT6 and PRMT8, which catalyze the formation of ω-monomethylarginine and asymmetric dimethylarginine (9,10). In a previous study, methylation at specific arginine residues of the p16 protein by PRMT6 was found to be critical for the activity of p16 (8). However, the existence of crosstalk between arginine methylation and phosphorylation of p16 has not been elucidated.
The present study aimed to determine the role of phosphorylation and arginine methylation in the functional regulation of p16, and identify potential crosstalk between these mechanisms.
Materials and methods
Cell culture and transient transfection
The HeLa, BT549, 293T and A549 cell lines (Cell Bank of Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai, China) were cultured in appropriate media supplemented with 10% fetal bovine serum, 100 U/ml penicillin and 100 µg/ml streptomycin (Sigma-Aldrich, St Louis, MO, USA), and maintained in a humidified atmosphere containing 5% CO2 at 37°C. For the transient transfection of HeLa and A549 cells, the cells were seeded into six-well plates and cultured for 1 day. Transient transfection was then performed using Lipofectamine 2000 (Invitrogen Life Technologies, Carlsbad, CA, USA). Subsequent to 48 h, the cells were harvested.
Plasmids
The human plasmid expressing p16-EGFP-N1 was provided by Dr Jun Chen (New York Medical College, Valhalla, NY, USA). The specific site mutations for the four serine residues were introduced into the p16 cDNA region using a two-step polymerase chain reaction (PCR) procedure, as previously described (11). Two simultaneous PCR reactions were performed using p16-EGFP-N1 as a template. Amplified fragments from each PCR reaction were purified, mixed and subjected to a second round of PCR using two external primers. The mutagenic sequence for the Ser7 and 8 residues was AGC (Serine) mutated to GCC (Alanine). For the Ser140 residues, the sequence was AGT (Serine) mutated to GCT (Alanine). For the Ser152 residues, the sequence was TCA (Serine) mutated to GCA (Alanine). The amplified PCR products were inserted into the HindIII and BamHI sites of the EGFP-N1 vector, and the correct insertion was verified by DNA sequencing. The arginine mutations in the p16 vectors were previously described (8). The PRMT1-myc plasmid was provided by Professor Mark A. Wainberg (McGill AIDS Centre, Lady Davis Institute, Jewish General Hospital, Montréal, QC, Canada) (12). Plasmids expressing human PRMT4 were provided by Professor Mark T. Bedford (The University of Texas MD Anderson Cancer Center, Science Park, Department of Molecular Carcinogenesis, Smithville, TX, USA) (13). PRMT6 plasmids were provided by Dr Stephane Richard (Lady Davis Institute) (14).
Western blotting and co-immunoprecipitation (CoIP)
Exogenous expression of p16 or PRMTs was detected by western blotting. The A549 cells were harvested 48 h subsequent to transfection with the relevant plasmids. In total, 1×106 cells were digested and lysed with lysis buffer consisting of 50 mm Tris/HCl, 1% Nonidet P (NP)-40, 150 mm NaCl, 1 mm EDTA and 1 mm phenylmethanesulfonyl fluoride for 30 min at 4°C. Total cell extracts were separated using 12% SDS-PAGE, and then transferred to polyvinylidene fluoride (PVDF) membranes (EMD Millipore, Billerica, MA, USA). The membranes were incubated with the following antibodies: Polyclonal rabbit anti-human PRMT6 (catalog no. P6495; Sigma-Aldrich, St Louis, MO, USA; dilution, 1:1,000); polyclonal rabbit anti-human PRMT1 (catalog no. 07-404; EMD Millipore; dilution, 1:1,000), polyclonal rabbit anti-human PRMT4 (catalog no. 09-818; EMD Millipore; dilution, 1:1,000); monoclonal mouse anti-human p16 (catalog no. SAB3300036; Sigma-Aldrich; dilution, 1:2,000); monoclonal mouse anti-green fluorescent protein (GFP; catalog no. ab1218; Abcam, Cambridge, MA, USA; dilution, 1:3,000); or monoclonal mouse anti-human β-actin (catalog no. A1978; Sigma-Aldrich; dilution, 1:4,000). The membranes were then visualized using the chemiluminescent substrate method and the SuperSignal West Pico kit (Pierce Co, Rockford, IL, USA). β-actin was used as an internal control for normalizing the loading materials.
CoIP was performed using the A549 cells. The cells were lysed in lysis buffer consisting of 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 0.5% NP-40, 1 mM EDTA and protease inhibitor cocktail. Total cell extracts were incubated with gentle agitation with the antibodies against GFP or p16 overnight at 4°C. This was followed by the addition of 40 µl of Protein A agarose (Upstate Biotechnology, Lake Placid, NY, USA) and incubation for another 3 h. The pellets were collected by centrifugation at 500 × g for 3 min at 4°C and washed twice with Buffer A consisting of 20 mM Tris-HCl (pH 8.0), 10 mM NaCl, 0.5% NP-40 and 1 mM EDTA (Sigma-Aldrich). The beads were suspended in 50 µl of 5X loading buffer and boiled for 10 min. The proteins were separated on a 12% SDS-PAGE gel and then transferred to a PVDF membrane for immunoblotting with antibodies against p16, GFP, phosphoserine (polyclonal rabbit anti-human; catalog no. AB1603; EMD Millipore; dilution, 1:1,000) or dimethyl-arginine, asymmetric (ASYM24; polyclonal rabbit anti-human; catalog no. 07-414; EMD Millipore; dilution, 1:1,000).
Immunofluorescence and terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) assay
Apoptosis of A549 cells was measured using TUNEL stain (Nanjing KeyGen Biotech Co., Ltd., Nanjing, Jiangsu, China). The treated A549 cells grown on coverslips were washed twice with phosphate-buffered saline (PBS), fixed in 4% paraformaldehyde for 30 min and endogenous peroxidase activity was blocked with 3% hydrogen peroxide diluted in methanol for 10 min at room temperature. The cells were then incubated in 0.1% Triton X-100 for 2 min on ice. Subsequent to washing with PBS, the cells were covered with 50 µl of TUNEL reaction mixture and incubated in the solution for 60 min at 37°C in a humidified dark chamber. Finally, the cells were stained with DAPI prior to visualization under an Olympus FV1000 confocal microscope (Olympus Corporation, Tokyo, Japan). Images were then captured using the FV10-ASW 2.1 microscopy software (Olympus Corporation).
Flow cytometric analysis
The A549 cells were trypsinized and washed once with cold PBS and then fixed in 70% ethanol and stored at 4°C for 30 min. The fixed cells were washed with PBS and suspended in 100 µl of PBS, supplemented with 1 µl 10 mg/ml ribonuclease A and 100 µl propidium iodide (Sigma-Aldrich). The stained cells were incubated at room temperature for 30 min in the dark. The cellular DNA content was quantified by flow cytometric analysis using a Epics XL FACSCalibur flow cytometer (Beckman Coulter, Brea, CA, USA) and the and the Multicycle AV for Windows software (Beckman Coulter).
Statistical analysis
Data are expressed as the mean ± standard deviation based on ≥3 individual experiments. Treatment-related differences were evaluated using one-way analysis of variance. P<0.05 was considered to indicate a statistically significant difference.
Results
Mutant p16 protein exhibited hypomethylation and enhanced function
The EGFP-p16 expression plasmids carrying three arginine-lysine point mutations, consisting of R22/131/138K (p16KKK), were generated. Firstly, the wild-type p16 and p16KKK vectors were transfected into A549 cells, then analyzed using CoIP assays performed using the cell extracts. The extracts from the A549 cells were immunoprecipitated using antibodies against p16, and detected in immunoblotting with antibodies against p16 and asymmetric dimethylarginine (Fig. 1A). The findings of CoIP revealed that the wild-type p16 protein was methylated, while the methylation of p16KKK was prominently reduced. Next, the cell cycle distribution was detected using flow cytometric analysis (FACS). The results revealed that overexpression of the wild-type p16 protein caused an accumulation of cells in the G1 phase. However, cells overexpressing the p16KKK protein exhibited an increased accumulation of cells in the G1 phase (Fig. 1B). Finally, the A549 cells were transfected with the wild-type p16 and p16KKK vectors, and the expression was detected using a confocal laser scanning microscope. The results revealed that the wild-type p16 and p16KKK proteins were located in the nucleus (Fig. 1C). These data indicate that the arginine point mutations in the p16KKK protein led to hypomethylation of the p16 protein and reinforced the increased the ability to prevent cell proliferation.
Figure 1.

The mutant p16KKK protein exhibited hypomethylation and enhanced function. (A) The arginine 22, 131 and 138 residues in the p16 protein were methylation sites. A549 cells transfected with the p16w and p16KKK expression vectors or control EGFP-N1 empty vector. Co-immunoprecipitation was performed using the antibody against p16, and the protein expression was detected using antibodies against p16 or ASYM. (B) Flow cytometric analysis of cell cycle changes following the transfection of A549 cells with GFP, p16w or p16KKK plasmids. The A549 cells were harvested 48 h subsequent to transfection. (C) The p16KKK and p16w proteins were located in the nucleus of tbe A549 cells. The cells were transfected with the p16-wild-EGFP or p16KKK-EGFP vectors. After 48 h, the nuclei were counterstained with DAPI (blue), and then detected using confocal laser scanning microscopy. p16KKK, p16 protein carrying the R22/131/138K arginine-lysine point mutations; p16w, wild-type p16; GFP, green fluorescence protein; ASYM, asymmetric dimethylarginine; Input, proteins prior to immunoprecipitation; IP: Anti-p16, samples co-immunoprecipitated with antibodies.
PRMT6 weakened the ability of p16 to prevent cell proliferation
Identification of the protein that PRMT involves in the methylation of the arginine residues of p16 was then attempted. The present results revealed that p16 was modified with asymmetric dimethylarginine (Fig. 1A), suggesting that the type I enzymes are likely to play a role in p16 methylation. Firstly, the exogenous expression of PRMT1, PRMT4 and PRMT6 was measured by western blotting (Fig. 2A). Next, CoIP assay results revealed that the exogenous expression of PRMT1, PRMT4 and PRMT6 increased the methylation level of p16, but only PRMT6 overexpression inhibited the association of p16 and CDK4 (Fig. 2B). In addition, the results of the FACS analysis revealed that transfection with PRMT6 counteracted the arrest of A549 cells in the G1 phase induced by wild-type p16, while PRMT6 overexpression did not affect the arrest of cell cycle induced by p16KKK (Fig. 2C). However, PRMT1 and PRMT4 demonstrated no such effect. Therefore, these data revealed that PRMT6 inhibited the function of p16 through the methylation of arginine residues.
Figure 2.

PRMT6 influences the functional properties of p16. (A) Western blot analysis of the expression of the PRMT1, PRMT4 and PRMT6 proteins in A549 cells transfected with the PRMT1, PRMT4 and PRMT6 expression vectors or the control pcDNA3.1 empty vector. (B) Co-immunoprecipitation assays for arginine methylation of p16. The cell extracts were prepared and precipitated with antibody against GFP, and detected using immunoblotting with antibodies against p16, CDK4 and ASYM antibody. The cells were transfected with p16 and pcDNA3.1, PRMT1, PRMT4 or PRMT6 vectors. (C) Flow cytometric analysis of cell cycle changes following the transfection of A549 cells. The cells were harvested 48 h subsequent to transfection with wild-type p16 or p16KKK, with PRMT1, PRMT4 or PRMT6 overexpressed. The bar chart means the G1 phase percentage. Values are means ± standard deviation (n=3). Input, proteins prior to immunoprecipitation; IP: Anti-GFP, samples co-immunoprecipitated with antibodies; PRMT, protein argenine methyltransferase; ASYM, asymmetric dimethylarginine; p16KKK, p16 protein carrying the R22/131/138K arginine-lysine point mutations; GFP, green fluorescence protein.
p16 was phosphorylated in a variety of cells
The present study also investigated the effect of other post-translational modifications on the function of the p16 protein. Therefore, the status of serine phosphorylation was examined in four human cell lines derived from various cell types. CoIP assays were performed using the antibody against p16 and the protein expression was detected using the antibody against serine phosphorylation. The findings revealed that the p16 protein was phosphorylated in the human breast cancer BT549, cervical epithelioid carcinoma HeLa and embryo kidney 293T cell lines (Fig. 3). These data indicate that the serine residues of p16 are universally phosphorylated in various cell lineages.
Figure 3.
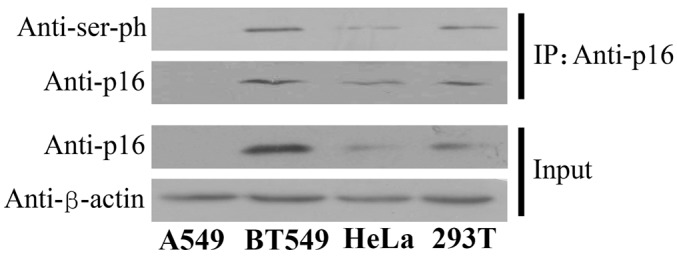
Phosphorylation of the p16 protein in a variety of cell lines. Extracts from the A549, BT549, HeLa and 293T cell lines were prepared and precipitated with anti-p16 antibody, then detected on immunoblotting with antibodies against p16 or serine phosphorylation. Input, proteins prior to immunoprecipitation; IP: Anti-p16, samples co-immunoprecipitated with antibodies; ser-ph, serine phosphorylation.
p16 protein with mutated serine residues demonstrates a decreased ability to prevent A549 cell proliferation
As a tumor suppressor, p16 prevents cell cycle progression at the G1/S checkpoint. The present study aimed to identify whether mutation of the serine residues S7, S8, S140 and S152 in the p16 protein affect the ability of p16 to arrest the cell cycle. CoIP assays demonstrated that the wild-type p16 protein was phosphorylated, while the levels of phosphorylated p16 S7A, p16 S8A, p16 S140A and p16 S152A decreased by varying amounts (Fig. 4A). The FACS analysis performed in the present study revealed that overexpression of the wild-type p16 protein resulted in an accumulation of cells in the G1 phase compared with the control (65 vs. 45%). However, cells overexpressing the mutant p16 protein exhibited a decreased ratio of cells in the G1 phase (Fig. 4B). Consequently, TUNEL staining revealed that the p16 mutant induced decreased apoptosis compared with wild-type p16 in A549 cells (Fig. 4C). These results indicated that the phosphorylation of p16 is involved in the regulation of the ability of p16 to reduce cell apoptosis.
Figure 4.

The p16 protein mutated at various serine residues exhibited a decreased ability to prevent the proliferation of A549 cells. (A) Immunoblotting of serine phosphorylation of p16 revealing that the mutation of residues 7, 8, 140 and 152 of p16 resulted in hypophosphorylation of the protein. The A549 cells were transfected with wild-type or mutant p16 expression plasmids. For the co-immunoprecipitation assay, the cells were precipitated with anti-p16 antibody and immunoblotted with antibodies against p16 or serine phosphorylation. (B) Flow cytometric analysis revealed that serine mutation reduced the proportion of A549 cells arrested in the G1 phase contrast to wild-type p16. The cells were harvested 48 h subsequent to transfection. The histogram denotes the proportion of cells in the G1 phase. (C) TUNEL assays revealing that serine phosphorylation was involved in the restraint of cell apoptosis through p16 arginine methylation. The cells were transfected with p16w or serine mutation p16. DNA fractures were stained with TRITC-dUTP (red) and then detected with confocal laser scanning microscopy. The nuclei were counterstained with DAPI (blue). IP: anti-p16, samples co-immunoprecipitated with antibodies; Input, proteins prior to immunoprecipitation; GFP, green fluorescence protein; p16w, wild-type p16; TRITC-dUTP, tetramethylrhodamine isothiocyanate-deoxyuridine triphosphate.
Crosstalk between arginine methylation and serine phosphorylation in p16
To determine whether serine phosphorylation in p16 affects arginine methylation, CoIP assays were used. The mutated serine residue and wild-type p16 vectors were transfected into A549 cells, then precipitated with anti-p16 antibody. The deposited samples were detected with antibodies against p16 and asymmetric dimethylarginine. The results revealed, as exhibited in Fig. 5, that the wild-type p16 was methylated and the level of p16 S140A methylation was increased, while p16 S152A methylation was decreased. These results indicated that an association existed between the methylation of arginine and phosphorylation of serine in p16.
Figure 5.
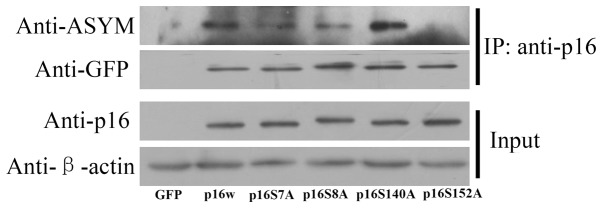
Crosstalk between the methylation of arginine and phosphorylation of serine in p16 in A549 cells. The cell extracts were prepared and precipitated with anti-p16 antibody, and then detected using immunoblotting with antibodies against GFP or serine phosphorylation. The cells were transfected with the EGFP-N1 empty vector, or the p16w or p16 mutation expression vectors. IP: anti-p16, samples co-immunoprecipitated with antibodies; Input, proteins prior to immunoprecipitation; GFP, green fluorescence protein; ASYM, asymmetric dimethylarginine; p16w, wild-type p16.
Discussion
It is well known that various post-translational modifications modulate the function and activity of target proteins by inducing changes in the structure and cellular localization of the protein (15,16). In the present study, the p16 protein was revealed to undergo arginine methylation and phosphorylation (Figs. 1 and 3). It has previously been reported that senescence in human prostatic epithelial cells not only induces an elevated level of p16 protein, but also promotes the phosphorylation of p16, which contributes to the binding affinity between CDK4/6 and p16 and cell cycle arrest in the G1 phase (17). Additional studies have demonstrated that the four specific serine sites of p16, consisting of Ser7, Ser8, Ser140 and Ser152, are phosphorylated, but only Ser152 is phosphorylated in the CDK4/6-p16 compound (7). The present results suggested that the phosphorylation of p16 played a role in regulating the ability of p16 to reduce cell apoptosis, based on the evidence that mutation of the residues from serine to alanine resulted in a decrease of the phosphorylation level of p16 and a decreased the apoptosis ratio compared with wild-type p16 in A549 cells (Fig. 4B and C). These findings support the hypothesis that phosphorylation of p16 is an important mechanism for p16 regulation. A previous study described that phosphorylation of p16 in senescent prostatic epithelial cells may facilitate the association between p16 and CDK4 and 6 (7). The phosphorylation of p16 evidently plays a role in the function of the protein, and the findings from the present study are consistent with previous data (7).
It is also recognized that one post-translational modification may enhance or prevent another post-translational modification, resulting in interplay that regulates diverse molecular processes (18,19). In the present study, hypomethylation of the p16 protein was found to enhance the ability of the protein to prevent cell proliferation, while hypophosphorylation of p16 decreased the ability of the protein to reduce cell apoptosis (Figs. 1 and 4). In addition, the present results indicated that wild-type p16 was methylated, and the amount of methylated p16 S140A was increased, while the amount of methylated p16 S152A was decreased (Fig. 5). The present results indicated that an association existed between the methylation of arginine and phosphorylation of serine in p16. A previous study reported that PRMT1 methylated forkhead box protein O1 (FOXO1) at Arg248 and Arg250, which blocked Akt-mediated phosphorylation of FOXO1 at Ser253 (18). In addition, another study revealed that epidermal growth factor receptor (EGFR) Arg1175 is methylated by PRMT5, and this modification positively modulates EGF-induced EGFR trans-autophosphorylation at Tyr1173 (19). These results are similar to the current research, promoting the further study of the function of p16.
Overall, the present study hypothesizes that crosstalk exists between phosphorylation and arginine methylation of p16. The effects of the corresponding methylation of arginine and phosphorylation of serine on the function of p16 require additional investigation.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (grant no., 31271442).
References
- 1.Sherr CJ, Roberts JM. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 2.Ohtani N, Zebedee Z, Huot TJ, Stinson JA, Sugimoto M, Ohashi Y, Sharrocks AD, Peters G, Hara E. Opposing effects of Ets and Id proteins on p16INK4a expression during cellular senescence. Nature. 2001;409:1067–1070. doi: 10.1038/35059131. [DOI] [PubMed] [Google Scholar]
- 3.Gizard F, Amant C, Barbier O, Bellosta S, Robillard R, Percevault F, Sevestre H, Krimpenfort P, Corsini A, Rochette J, et al. PPAR alpha inhibits vascular smooth muscle cell proliferation underlying intimal hyperplasia by inducing the tumor suppressor p16INK4a. J Clin Invest. 2005;115:3228–3238. doi: 10.1172/JCI22756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang X, Feng Y, Xu L, Chen Y, Zhang Y, Su D, Ren G, Lu J, Huang B. YY1 restrained cell senescence through repressing the transcription of p16. Biochim Biophys Acta. 2008;1783:1876–1883. doi: 10.1016/j.bbamcr.2008.05.015. [DOI] [PubMed] [Google Scholar]
- 5.Wang X, Pan L, Feng Y, Wang Y, Han Q, Han L, Han S, Guo J, Huang B, Lu J. P300 plays a role in p16 (INK4a) expression and cell cycle arrest. Oncogene. 2008;27:1894–1904. doi: 10.1038/sj.onc.1210821. [DOI] [PubMed] [Google Scholar]
- 6.Feng Y, Wang X, Xu L, Pan H, Zhu S, Liang Q, Huang B, Lu J. The transcription factor ZBP-89 suppresses p16 expression through a histone modification mechanism to affect cell senescence. FEBS J. 2009;276:4197–4206. doi: 10.1111/j.1742-4658.2009.07128.x. [DOI] [PubMed] [Google Scholar]
- 7.Gump J, Stokoe D, McCormick F. Phosphorylation of p16INK4A correlates with Cdk4 association. J Biol Chem. 2003;278:6619–6622. doi: 10.1074/jbc.C200622200. [DOI] [PubMed] [Google Scholar]
- 8.Wang X, Huang Y, Zhao J, Zhang Y, Lu J, Huang B. Suppression of PRMT6-mediated arginine methylation of p16 protein potentiates its ability to arrest A549 cell proliferation. Int J Biochem Cell Biol. 2012;44:2333–2341. doi: 10.1016/j.biocel.2012.09.015. [DOI] [PubMed] [Google Scholar]
- 9.Pahlich S, Zakaryan RP, Gehring H. Protein arginine methylation: Cellular functions and methods of analysis. Biochim Biophys Acta. 2006;1764:1890–1903. doi: 10.1016/j.bbapap.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 10.Di Lorenzo A, Bedford MT. Histone arginine methylation. FEBS Lett. 2011;585:2024–2031. doi: 10.1016/j.febslet.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Han L, Lu J, Pan L, Wang X, Shao Y, Han S, Huang B. Histone acetyltransferase p300 regulates the transcription of human erythroid-specific 5-aminolevulinate synthase gene. Biochem Biophys Res Commun. 2006;348:799–806. doi: 10.1016/j.bbrc.2006.07.147. [DOI] [PubMed] [Google Scholar]
- 12.Xie B, Invernizzi CF, Richard S, Wainberg MA. Arginine methylation of the human immunodeficiency virus type 1 Tat protein by PRMT6 negatively affects Tat Interactions with both cyclin T1 and the Tat transactivation region. J Virol. 2007;81:4226–4234. doi: 10.1128/JVI.01888-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frankel A, Yadav N, Lee J, Branscombe TL, Clarke S, Bedford MT. The novel human protein arginine N-methyltransferase PRMT6 is a nuclear enzyme displaying unique substrate specificity. J Biol Chem. 2002;277:3537–3543. doi: 10.1074/jbc.M108786200. [DOI] [PubMed] [Google Scholar]
- 14.Boulanger MC, Liang C, Russell RS, Lin R, Bedford MT, Wainberg MA, Richard S. Methylation of Tat by PRMT6 regulates human immunodeficiency virus type 1 gene expression. J Virol. 2005;79:124–131. doi: 10.1128/JVI.79.1.124-131.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 16.Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 17.Sandhu C, Peehl DM, Slingerland J. p16INK4A mediates cyclin dependent kinase 4 and 6 inhibition in senescent prostatic epithelial cells. Cancer Res. 2000;60:2616–2622. [PubMed] [Google Scholar]
- 18.Yamagata K, Daitoku H, Takahashi Y, Namiki K, Hisatake K, Kako K, Mukai H, Kasuya Y, Fukamizu A. Arginine methylation of FOXO transcription factors inhibits their phosphorylation by Akt. Mol Cell. 2008;32:221–231. doi: 10.1016/j.molcel.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 19.Hsu JM, Chen CT, Chou CK, Kuo HP, Li LY, Lin CY, Lee HJ, Wang YN, Liu M, Liao HW, et al. Crosstalk between Arg 1175 methylation and Tyr 1173 phosphorylation negatively modulates EGFR-mediated ERK activation. Nat Cell Biol. 2011;13:174–181. doi: 10.1038/ncb2158. [DOI] [PMC free article] [PubMed] [Google Scholar]